Introduction
Perfluorooctanoic acid (PFOA) is a man-made,
synthetic chemical and persistent organic pollutant, which has been
detected in several settings, including non-stick cookware,
cosmetics, bioaccumulation in food chains (1) and drinking water (2). PFOA has also been detected in the blood
in several industrialized countries. Exposure to PFOA may be
associated with several adverse effects, and recent epidemiological
studies have linked its toxicity to tumorigenic processes in the
liver, pancreas, breast and testicles (3–6).
The epithelial-mesenchymal transition (EMT) is a
complex cellular mechanism involved in tumor cell motility,
invasion and the formation of metastases (7). The downregulation or upregulation of
cell surface markers, including E-cadherin, vimentin and
serum/glucocorticoid-regulated kinase 1 (SGK1), are some of the
important alterations occurring during metastatic progression
(8–10). EMT comprises two critical steps: Loss
of E-cadherin and acquisition of vimentin. E-cadherin often
functions as a tumor suppressor, inhibiting cancer cell invasion
and metastases (11), whereas
vimentin is a major constituent of the intermediate filament family
of proteins, and its overexpression is associated with tumor growth
and invasion and poor prognosis in certain types of cancer
(12). Immunohistochemical testing
for vimentin is positive in rhabdomyosarcoma (RMS) (13). In a recent study, SGK1 was identified
and characterized as a tumor-promoting gene, with its inhibition
significantly attenuating in vitro and in vivo EMT
and metastases formation in prostate cancer, and its overexpression
promoting the invasion and migration of the tumor cells (10).
Tumor development is driven by a series of genetic
alterations caused by abnormal cell division cycles that gradually
transform normal cells into cancer cells. In the G1 phase, in which
cells respond to external signals, developmental changes in the
cell division cycle may affect cell proliferation and
differentiation. Studies of early embryonic cancer cells have shown
that differentiation can be rapidly induced in the G1 phase but not
in the S phase (14). In several
studies, the length of the G1 phase is significantly associated
with maintenance of cell differentiation. The short embryonic cycle
is characterized by a lack of active cyclin-dependent kinase (CDK)
inhibitors, and the increased levels of CDKs and cyclins,
particularly CDK2-cyclin E (15–17).
The phosphatidylinositol-3 kinase (PI3K)/protein
kinase B (AKT) pathway is often activated in various types of
cancer (18). Activation of the
PI3K/AKT pathway controls several hallmarks of cancer, including
cell cycle, survival, metabolism and motility, which serve
important roles in the regulation of EMT-associated cell surface
markers in cancer cells (19).
Apoptosis is a conserved and regulated mechanism of physiological
cell death, and the inhibition of apoptosis may induce cancer cell
growth. Evidence has confirmed that the PI3K/AKT pathway is
implicated in the regulation of cellular processes, including cell
proliferation, invasion, cell cycle progression and apoptosis
(20). Therefore, the PI3K/AKT
signaling pathway serves an important role in cancer progression
(19).
RMS is a soft tissue sarcoma of skeletal muscle
phenotype originating from a primitive mesenchymal cell. It
develops during childhood, primarily in those <5 years of age.
The incidence of RMS is inferior to that of malignant fibrous
histiocytoma and liposarcoma, ranking third of soft tissue
sarcomas. The mechanisms by which PFOA affects muscle tissues with
abundant blood supply can be examined in animal and cell models. RD
cells are a subline of RMS. Our studies have shown that PFOA is
capable of transforming RD cells into a malignant phenotype by
altering vimentin and SGK1 proteins and inducing cell migration and
invasion.
In the present study, the potential tumorigenic
activity of PFOA in RD cells was evaluated. Cell viability,
migration, invasion and apoptosis were evaluated to investigate the
effects of PFOA exposure on cell proliferation. To better evaluate
the response of RD cells to PFOA exposure, EMT and
apoptosis-related proteins E-cadherin, vimentin, SGK1, Bax, Bcl-2,
PI3K and AKT were examined. To investigate the role of the PI3K/AKT
signaling pathway in PFOA-affected RD cells, a PI3K inhibitor was
used. The findings may help to disclose the mechanisms underlying
the effects of PFOA on the development of RMS.
Materials and methods
Reagents and antibodies
PFOA (purity ≥96%) was obtained from Sigma-Aldrich;
Merck KGaA (St. Louis, MO, USA). Fetal bovine serum (FBS),
penicillin-streptomycin, Dulbecco's phosphate-buffered saline
(PBS), and Dulbecco's modified Eagle's medium (DMEM) were used from
Gibco (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Trypsin
solution (0.05%) was purchased from Gibco (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Propidium iodide (PI) was
purchased from Abcam (Cambridge, UK) and the Annexin V-FITC
Apoptosis Detection kit was purchased from BD Biosciences (San
Diego, CA, USA). All antibodies used for western blot analysis were
purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA).
BEZ235 (Selleck Chemicals, Houston, TX, USA) was used for
inhibiting the activation of PI3K.
Cell culture
The RD human embryonal RMS cell line was provided by
the Institute of Laboratory Animal Science, Chinese Academy of
Medical Sciences and Comparative Medicine Center, Peking Union
Medical College (Beijing, China). The cells were maintained in DMEM
containing 100 U/ml of streptomycin and 100 U/ml of penicillin
supplemented with 10% FBS and were cultured in a humidified
atmosphere of 5% carbon dioxide (CO2) at 37°C. The RD
cells were maintained as a monolayer in 25-cm2
polypropylene flasks with the same growth, split 1:5 every 2–3
days, and were subcultured until they reached 90% confluence prior
to being harvested with trypsin.
PFOA exposure
PFOA was dissolved in sterile deionized water to
obtain a 10-mM stock solution and added to the culture medium with
1% FBS to produce different concentrations. The RD cells were
plated in 96-well tissue culture plates (1×105
cells/well) for 12 h. The culture medium was then discarded, and
the cells were incubated with medium with 1% FBS containing PFOA
for 36 and 72 h at 37°C and subsequently washed twice with PBS.
Cell viability assay with Cell
Counting Kit-8 (CCK-8)
CCK-8 (Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan) was used to measure RD cell viability. The RD
cells were treated with different PFOA concentrations (0, 1, 10,
50, 100, 500 and 1,000 µM) for 36 and 72 h and subsequently treated
with 50 µM/l PFOA and different concentrations of BEZ235 (0, 10, 50
and 100 nM) for 72 h. Following incubation for the indicated time,
10 µl CCK-8 solution was added to each well and incubated for 2 h
at 37°C to separately examine the effects of PFOA, and PFOA and
BEZ235 on RD cell proliferation. The absorbance in each well was
measured with a Multiskan GO spectrophotometer (Thermo Fisher
Scientific, Inc.) at 450 nm. Three independent experiments were
performed in triplicate. The rate of cell growth inhibition was
calculated as an absorbance fraction compared with the control.
Cell cycle analysis
The RD cells were seeded into 6-well plates
(2.5×105 cells/well) for 12 h. The cells were treated
with PBS, PFOA, PFOA and BEZ235, or BEZ235, as described above, for
72 h, harvested in a single cell suspension, fixed in 66% ethanol,
stored at 4°C for 2 h, rehydrated with PBS, stained with PI and
incubated with RNase for 30 min. The PI fluorescence intensity was
determined via FL2 flow cytometry and 488 nM laser excitation, with
an excitation maximum of 493 nm and emission maximum of 636 nm.
Wound healing migration assays
Cell migration was assessed using wound healing
assays. The RD cells were seeded in 12-well plates for 24 h and
cultured to ~80% confluence. The cells in the monolayer were
subsequently scratched with a 10-µl pipette tip, washed twice with
DMEM with 2% FBS to remove the detached cells, and treated with
PFOA at 0 µM/l, PFOA at 50 µM/l, or PFOA at 50 µM/l and BEZ235 at
50 nM/l for 0 and 48 h. The wounded areas were observed under an
inverted microscope (Olympus CKX41), and the cell migration count
was determined in four fields using software (ImageJ 1.44p) (NIH,
Bethesda, MA, USA).
Transwell cell invasion assays
The RD cells were seeded in 12-well culture plates
at a density of 4×105 cells/well and incubated with PFOA
at 0 µM/l, PFOA at 50 µM/l, or BEZ235 at 50 µM/l and PFOA at 50
µM/l at 37°C in a 5% CO2-humidified atmosphere for 48 h,
with completely untreated cells used as a negative control. The
cells were then suspended in serum-free DMEM and plated at a
density of 2×105 cells/well in the upper chamber of
Transwell plates with polycarbonate membranes (8.0-mm pore size)
and diluted Matrigel coating (Corning, New York, NY, USA). Complete
medium (10% FBS DMEM; 600 ml) was added to the lower chamber.
Following incubation for 24 h at 37°C in a 5%
CO2-humidified atmosphere, cells that had passed through
the filters into the bottom wells were fixed in 100% methanol for
30 min at 4°C and stained with 0.5% crystal violet for 15 min at
37°C. The number of cells was counted, as previously described,
using an ImageXpress Micro XLS Widefield High-Content analysis
system (Molecular Devices, LLC, Sunnyvale, CA, USA) and SoftMax Pro
6 software (Molecular Devices, LLC).
Analysis of apoptosis
The RD cells were seeded in 6-well plates
(1×105 cells per well) for 24 h and treated with 50 µm/l
PFOA, 50 µM/l PFOA and 50 nM/l BEZ235, or 50 nM/l BEZ235 for 72 h
(with completely untreated control cells separately), washed twice
in cold PBS and resuspended in binding buffer. The Annexin-V-FITC
detection kit (10 µl) and PI (10 µl) were added and incubated for
20 min at room temperature in the dark. Following incubation, 400
µl binding buffer was added, and the cells were immediately
analyzed with a FACScan flow cytometer (FACSCalibur; BD
Biosciences) using CellQuest software©2002 (BD
Biosciences).
RNA extraction, and reverse
transcription-quantitative PCR (RT-qPCR) analysis
RT-qPCR analysis was performed to detect the mRNA
expression levels of E-cadherin, vimentin, SGK1, cyclin E2, CDK2,
p21, p27, p53, Bcl-2, Bax, PI3K and AKT, with glyceraldehyde
3-phosphate dehydrogenase (GAPDH) as a housekeeping gene. The RD
cells were treated with 0 µM/l PFOA, 50 µM/l PFOA, or 50 µM/l PFOA
and 50 nM/l BEZ235 for 72 h. Total RNA was extracted with TRIzol
reagent according to the manufacturer's protocol (Thermo Fisher
Scientific, Inc.). Following this, 1 ml of RNA was used for
concentration determination with a NanoDrop OneC Microvolume UV–Vis
Spectrophotometer with Wi-Fi. Total RNA reverse transcription was
performed with random hexamers using a reverse transcription kit
(Takara Bio, Inc., Otsu, Japan), 1 µl RNA, 4 µl 5X reaction buffer,
1 µl Ribolock RNA inhibitor, 2 µl 10 mM dNTP mix, 1 µl RevertAid
M-MulV reverse transcriptase and 11 µl water. The RNA reverse
transcription conditions were as follows: 25°C for 5 min, 45°C for
60 min and 70°C for 5 min. Real-time qPCR analysis was subsequently
performed to determine target gene expression using specific
primers (Table I) and the SYBR-Green
kit (Takara Bio, Inc.). The qPCR conditions included an initial
denaturation at 95°C for 10 min, followed by a 40-cycle
amplification consisting of denaturation at 95°C for 10 sec,
annealing at 60°C for 20 sec and extension at 72°C for 30 sec. This
was followed by melting curve analysis to verify the qPCR product
identity. The relative mRNA expression levels were evaluated using
the 2−∆∆Cq method (21).
GADPH was used as a reference gene.
 | Table I.Primer sequences used for PCR. |
Table I.
Primer sequences used for PCR.
| Gene | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| E-cadherin |
TGGAGGAATTCTTGCTTTGC |
CGCTCTCCTCCGAAGAAAC |
| Vimentin |
GGCTCGTCACCTTCGTGAAT |
GAGAAATCCTGCTCTCCTCGC |
| SGK1 |
AGGATGGGTCTGAACGACTTT |
GCCCTTTCCGATCACTTTCAAG |
| Cyclin E2 |
GGCCTATATATTGGGTTGGCG |
ACGGCTACTTCGTCTTGACA |
| P21 |
TCAGAGGAGGTGAGAGAGCG |
ACATGGCGCCTGCCG |
| P27 |
GTTCACGATGTCCGACGAGG |
ATCCACTTGCAGACCGCTT |
| CDK2 |
ATCTTTGCTGAGATGGTGACTCG |
TAAAATCTTGCCGGGCCCAC |
| P53 |
CCTGGATTGGCCAGACTGC |
TCATCCATTGCTTGGGACGG |
| Bcl-2 |
ATGTGTGTGGAGAGCGTCAA |
GAGACAGCCAGGAGAAATCAA |
| Bax |
ACGAACTGGACAGTAACATGGAG |
CAGTTTGCTGGCAAAGTAGAAAAG |
| PI3K |
AGATCGCTCTGGCCTCATTG |
TCCAGGTCATCCCCAGAGTT |
| AKT |
GAAGTCAAAGGGGCTGCCT |
TACTCCCCTCGTTTGTGCAG |
| GADPH |
ACCCATCACCATCTTCCAGGAG |
GAAGGGGCGGAGATGATGAC |
Western blot analysis
The RD cells were seeded in a 100-mm2
dish (3×105 cells/dish), adhered for 24 h, and treated
with 0 µM/l PFOA, 50 µM/l PFOA, 50 µM/l PFOA and 50 nM/l BEZ235, or
50 nM/l BEZ235 for 72 h. The cell proteins were lysed using RIPA
Lysis and Extraction Buffer (Thermo Fisher Scientific, Inc.),
centrifuged at 12,000 × g at 4°C for 30 min to retrieve proteins in
the supernatant, and collected and quantified using the
Bicinchoninic Acid Protein Assay kit. Equal quantities of protein
(30 mg) from each sample were loaded for 12% of SDS-PAGE and
transferred onto a polyvinylidene difluoride membrane. Following
blocking with 5% skim milk at room temperature for 1 h, the
membranes were subsequently probed with a primary antibody at 4°C
overnight. The antibodies used were as follows: Rabbit anti-AKT
(cat no. 4685S), PI3K p85 (cat. no. 4257T), E-cadherin (cat. no.
3195T), vimentin (cat. no. 5741T), SGK1 (cat. no. 12103S), Bcl-2
(cat. no. 4223T), Bax (cat. no. 5023T), cyclin E2 (cat. no. 4132T),
CDK2 (cat. no. 2546T), P53 (cat. no. 2527T), P21 (cat. no. 2947T)
and P27 (cat. no. 3686T), and mouse anti-GADPH (cat. no. 51332S)
and diluted at 1:1,000. The membranes were washed for 10 min with
TBS three times and incubated with peroxidase-labeled goat
anti-rabbit and anti-mouse secondary antibody at room temperature
for 1 h. The membranes were subsequently washed for 10 min with
TBST three times, and the protein bands were visualized using an
ECL western blotting kit according to the manufacturer's
instructions. Densitometry was performed using Quantity One
software 4.4.0 (Tanon 5500; Tanon, Shanghai, China).
Animal experiments
Male Balb/c mice (aged 5–6 weeks, 15–20 g) were
purchased from Beijing QuantoBio Biotechnology Co., Ltd. (Beijing,
China). The feeding environment requires a temperature of 18–29°C,
a daily temperature difference of less than 3°C, a relative
humidity of 40–70%, fresh air replacement 10 times/h, the airflow
speed of less than 18 m/s, a pressure difference of 25 Pa, a
cleanliness of 10,000, a nitrogen concentration of 15
mg/m3, a noise of less than 60dB, and the illumination
of 150–300 Lux, the clean water from the filter, add hydrochloric
acid, and the pH 2.8–3.0, changed twice a week, and feed aseptic,
high nutrition, added daily and viewed, and all experimental
manipulations were performed as previously described (22). Briefly, the mice were divided into
three groups, with six animals in each group. Their right/left
forelimb was subcutaneously injected with 1×107/cells,
and purified water + PBS, purified water + PFOA (100 mg/day), and
purified water + PFOA (100 mg/day) + BEZ235 (50 mg/day) were
separately administered for 30 days. The doses of PFOA and BEZ235
were selected based on earlier studies (22,23). All
animal treatments were approved by the Ethics Committee of the
Experimental Animal Center and by the Chinese Academy of Military
Medical Sciences (permit no. SCXK-2015-0002), in accordance with
the guiding principles for the use of animals in toxicology,
adopted by the Society of Toxicology in 1989.
Statistical analysis
All experiments were performed a minimum of three
times unless otherwise stated. Data are expressed as the mean ±
standard deviation. The statistical significance of experiments was
assessed using one-way ANOVA (parametric) or Kruskal-Wallis
(non-parametric) and a multiple comparisons test, and the LSD post
hoc test was used following ANOVA. P<0.05 was considered to
indicate a statistically significant difference. All statistical
analyses were performed with SPSS 12.0 (SPSS, Inc.), and the graphs
were produced using GraphPad Prism 7 (GraphPad Software, Inc., San
Diego, CA, USA).
Results
PFOA exposure elicits RD cell
proliferation, migration and invasion
To investigate the effect of PFOA in the development
of human RMS, RD cells were treated with different doses of PFOA
for 36 or 72 h. A CCK-8 assay was initially used to evaluate the
effect of different doses of PFOA on the RD cell survival ratio.
Exposure to PFOA at 50 and 100 µM for 72 h was found to increase
the production of CCK-8 (P<0.05) compared with that in other
groups after 72 h, but no cells were found to proliferate after 36
h (Fig. 1A and B). By contrast,
exposure to 500 and 1,000 µM concentrations inhibited cell
viability at 36 and 72 h (Fig. 1A and
B). The ability of PFOA to elicit RD cell migration and
invasion was further examined by selecting a PFOA concentration of
50 mM for all subsequent experiments. Cell migration was
investigated by treating RD cells with PFOA for 48 h in wound
healing assays. In the area of wound healing, cell migration was
significantly accelerated in RD cells treated with PFOA for 48 h
compared with that in PBS-treated controls (P<0.05; Fig. 1C and D). A Transwell assay was used to
examine cell invasion of the RD cells treated with PFOA for 48 h,
as shown in Fig. 1E and F. PFOA
exposure for 48 h was found to significantly induce RD cell
invasion. The results confirmed that PFOA exposure stimulated
proliferation, migration and invasion in RD cells.
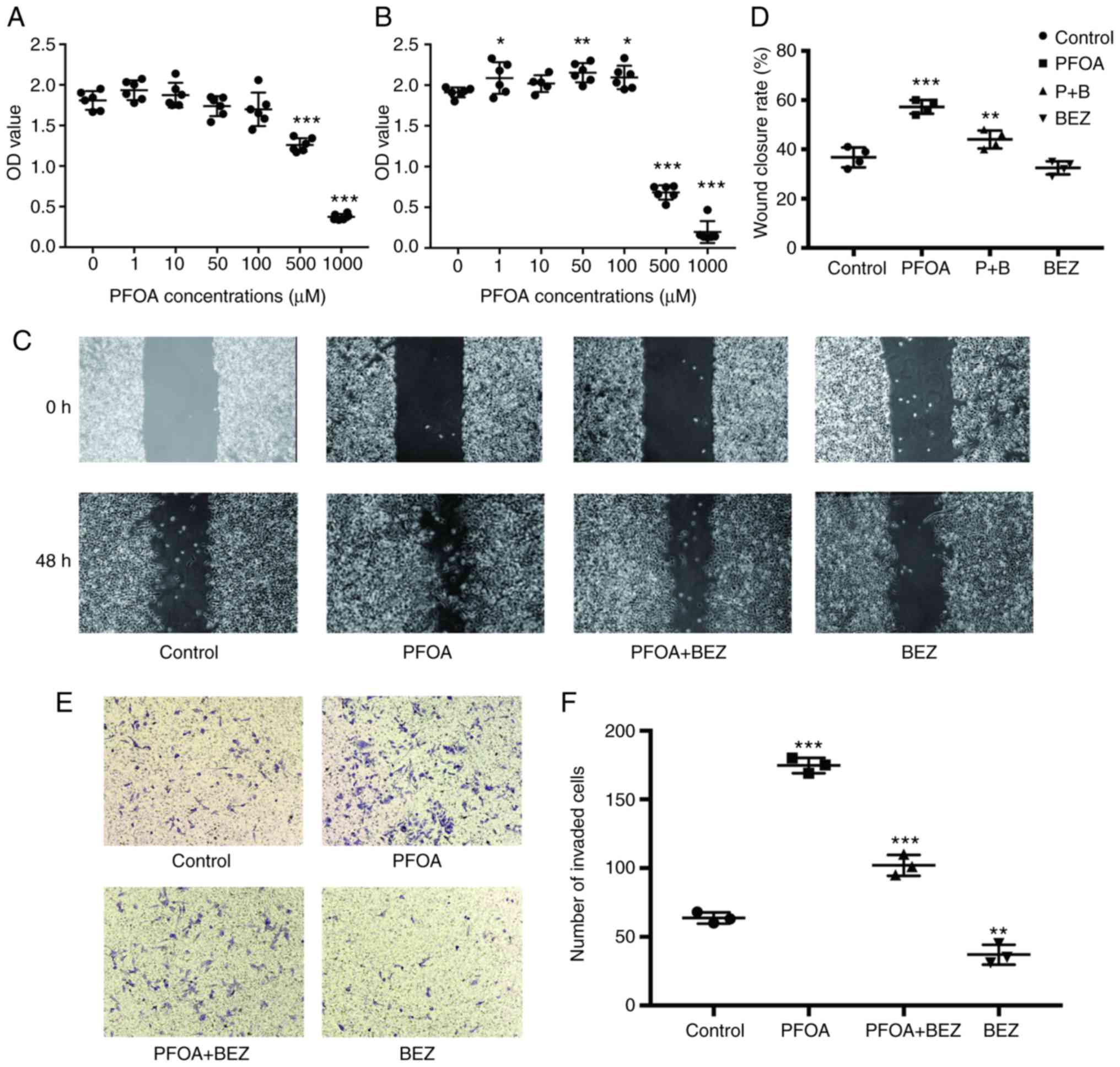 | Figure 1.PFOA elicits RD cell proliferation,
migration and invasion. (A) RD cells treated with different PFOA
concentrations for 36 h. (B) RD cells treated with different PFOA
concentrations for 72 h. Cell viability was assessed using a Cell
Counting Kit-8 assay. (C) Wound healing assays were performed in RD
cells treated with PBS, PFOA, PFOA + BEZ235, and BEZ235 for 48 h.
(D) Images of the wound healing assay. (E) In the Transwell assay,
RD cells were treated with PBS, PFOA, PFOA + BEZ235, and BEZ235 for
48 h. (Magnification, ×40). (F) Quantification of results of the
Transwell assay. Control, PBS treatment. *P<0.05, **P<0.01
and ***P<0.001 vs. control (PBS-treated). PFOA,
perfluorooctanoic acid; PBS, phosphate-buffered saline; BEZ,
BEZ235; P+B, PFOA + BEZ235. |
PFOA-induced upregulation of
EMT-related markers in RD cells
EMT is one of the key events during tumor invasion
and migration in several types of human cancer. It elicits changes
in several molecular pathways and networks, with loss of the
expression of E-cadherin and the overexpression of vimentin and
SGK1 as critical steps driving the developmental process in human
cancer (9,10,24). To
determine whether PFOA exposure elicited EMT, the expression levels
of E-cadherin, vimentin and SGK1 were investigated. Based on the
RT-qPCR and western blot analyses, PFOA exposure resulted in
significant increases in the expression of vimentin and SGK1 at the
mRNA and protein levels, respectively (Fig. 2A-E). However, despite five sets of
primers designed, E-cadherin was not detected at either the mRNA or
protein level. This suggests that PFOA exposure may promote EMT in
RD cells.
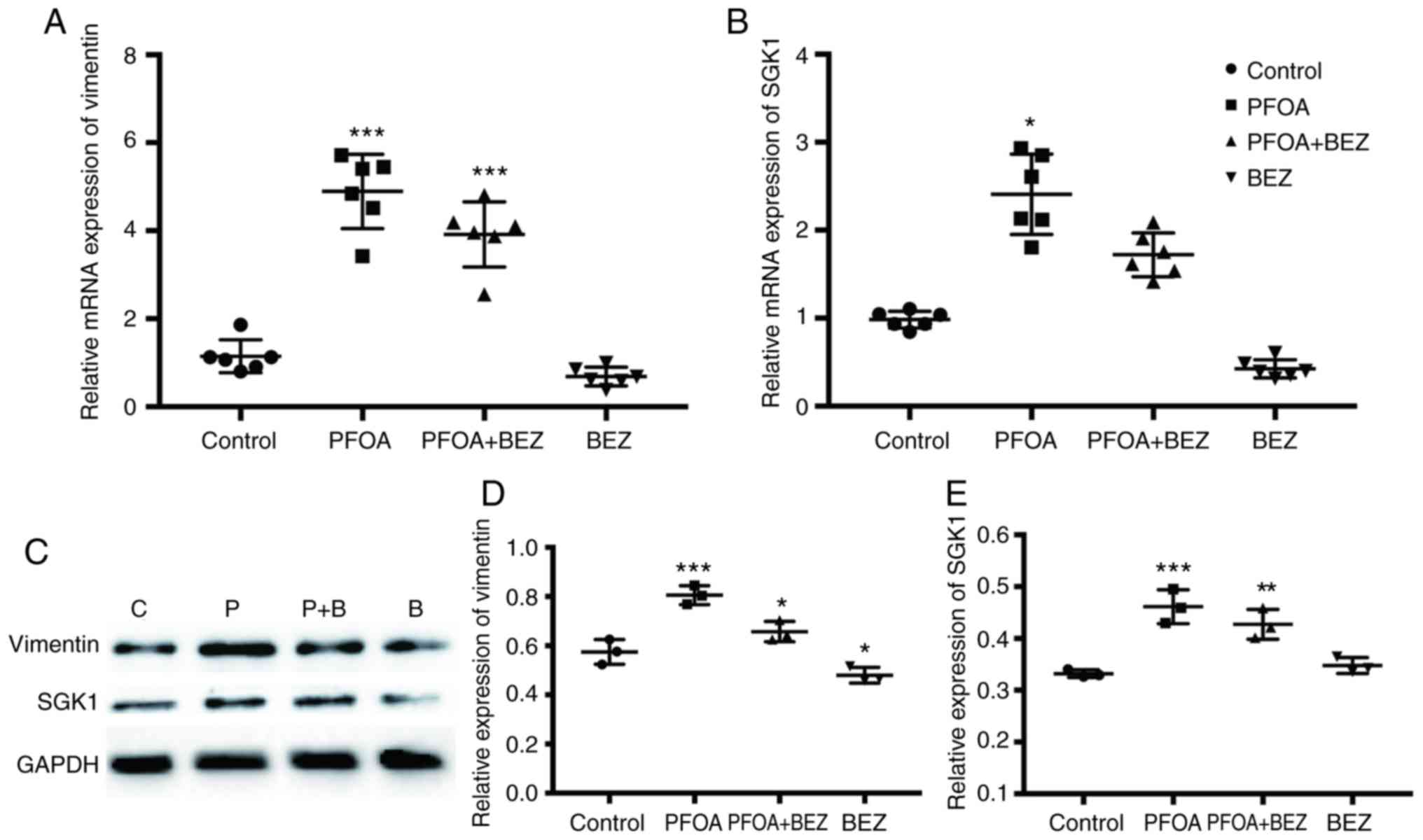 | Figure 2.Effects of PFOA treatment on the
expression of vimentin and SGK1 in RD cells. Reverse
transcription-quantitative polymerase chain reaction analysis of
(A) vimentin and (B) SGK1 in PBS-, PFOA-, PFOA + BEZ235-, and
BEZ235-treated RD cells. Expression data is normalized to GADPH.
(C) Expression levels of vimentin and SGK1 were assessed, with
GADPH used as an internal control, by western blotting. (D and E)
Quantitative measurement of vimentin and SGK1 band densities. Data
are presented as the mean ± SD. *P<0.05, **P<0.01 and
***P<0.001 vs. control (PBS-treated). PFOA, perfluorooctanoic
acid; SGK1, serum/glucocorticoid-regulated kinase 1; PBS,
phosphate-buffered saline; BEZ, BEZ235; P+B, PFOA + BEZ235; C,
control. |
PFOA inhibits apoptosis in RD
cells
To investigate the effect of PFOA exposure on cell
apoptosis, which was quantitatively determined by Annexin V-FITC
and PI fluorescence staining, flow cytometry was conducted.
Following treatment with 50 µM/l PFOA for 72 h (Fig. 3A), the percentage of total cell death
(early and late apoptosis + necrosis) was significantly decreased
from 13.74 to 9.86% (P<0.05; Fig.
3B). To further investigate the apoptotic mechanism, the
intracellular apoptotic signaling pathway was examined in RD cells
following treatment with 50 µM/l of PFOA for 72 h. The results
showed that the mRNA and protein expression levels of Vcl-2 were
significantly upregulated, whereas that of Bax was significantly
downregulated (Fig. 3C-G). These data
suggest that PFOA may inhibit RD cell apoptosis by inhibiting the
intracellular apoptotic signaling pathway.
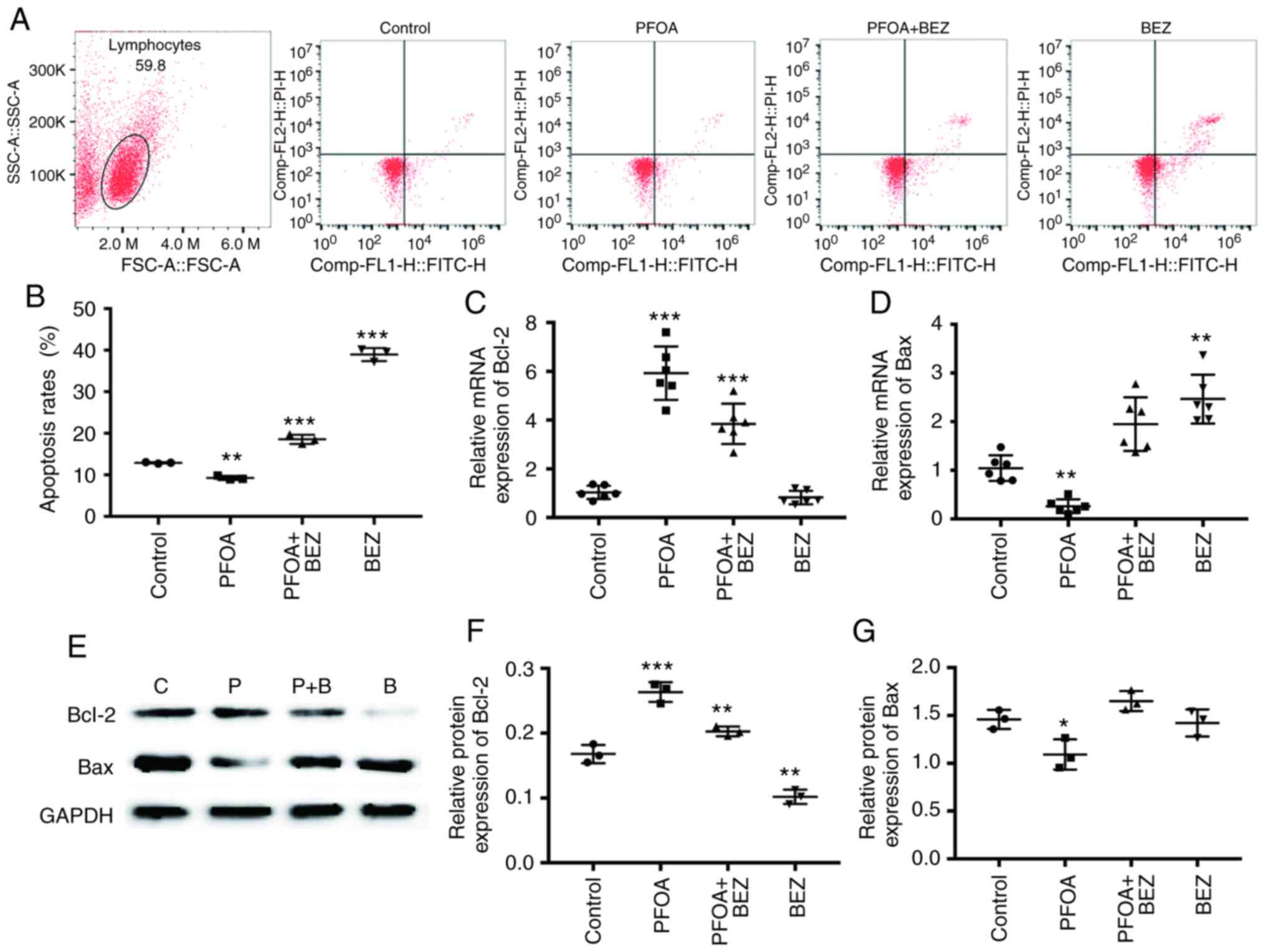 | Figure 3.PFOA inhibits RD cell apoptosis. (A)
Flow cytometry for evaluation of Annexin V FITC/PI-stained RD cells
was performed 72 h after PBS, PFOA, PFOA + BEZ235, and BEZ235
treatment. (B) Cell apoptotic rates were statistically analyzed.
The lower left quadrant represents intact viable cells
(Annexin-FITC and PI-negative). The lower right quadrant represents
early apoptotic cells (Annexin V-FITC-positive and PI-negative).
The upper right region represents late apoptotic cells or secondary
necrotic cells (Annexin-FITC- and PI-positive). Expression levels
of (C) Bcl-2 and (D) Bax were analyzed by quantitative PCR in the
PBS, PFOA, PFOA + BEZ235, and BEZ235-treated RD cells. (E) Western
blotting and quantification of (F) Bcl-2 and (G) Bax band
densities. Data are presented as the mean ± SD. *P<0.05,
**P<0.01 and ***P<0.001 vs. control (PBS-treated). Data
represent three independent experiments. PFOA, perfluorooctanoic
acid; PBS, phosphate-buffered saline; BEZ, BEZ235; P+B, PFOA +
BEZ235; C, control; PI, propidium iodide. |
PFOA can alter the proportion of the
three RD cell cycle phases and expression of associated mRNA and
protein levels
The present study found that treatment with 50 µM of
PFOA may elicit RD cell proliferation. PI staining and flow
cytometry were used to investigate the effect of PFOA exposure on
RD cells and cell cycle phase proportions. The proportion of cells
in each cell cycle phase after 72 h of PFOA exposure are shown in
Fig. 4A and B. The results showed
that, following the addition of PFOA, the proportion of cells in
the S phase was reduced and that in the G1/G2 phase was increased.
The expression of cell cycle-associated mRNAs and proteins were
subsequently examined, and cyclin E2 and CDK2 were found to
increase at the mRNA and protein levels following administration of
FPOA (Fig. 4C-G).
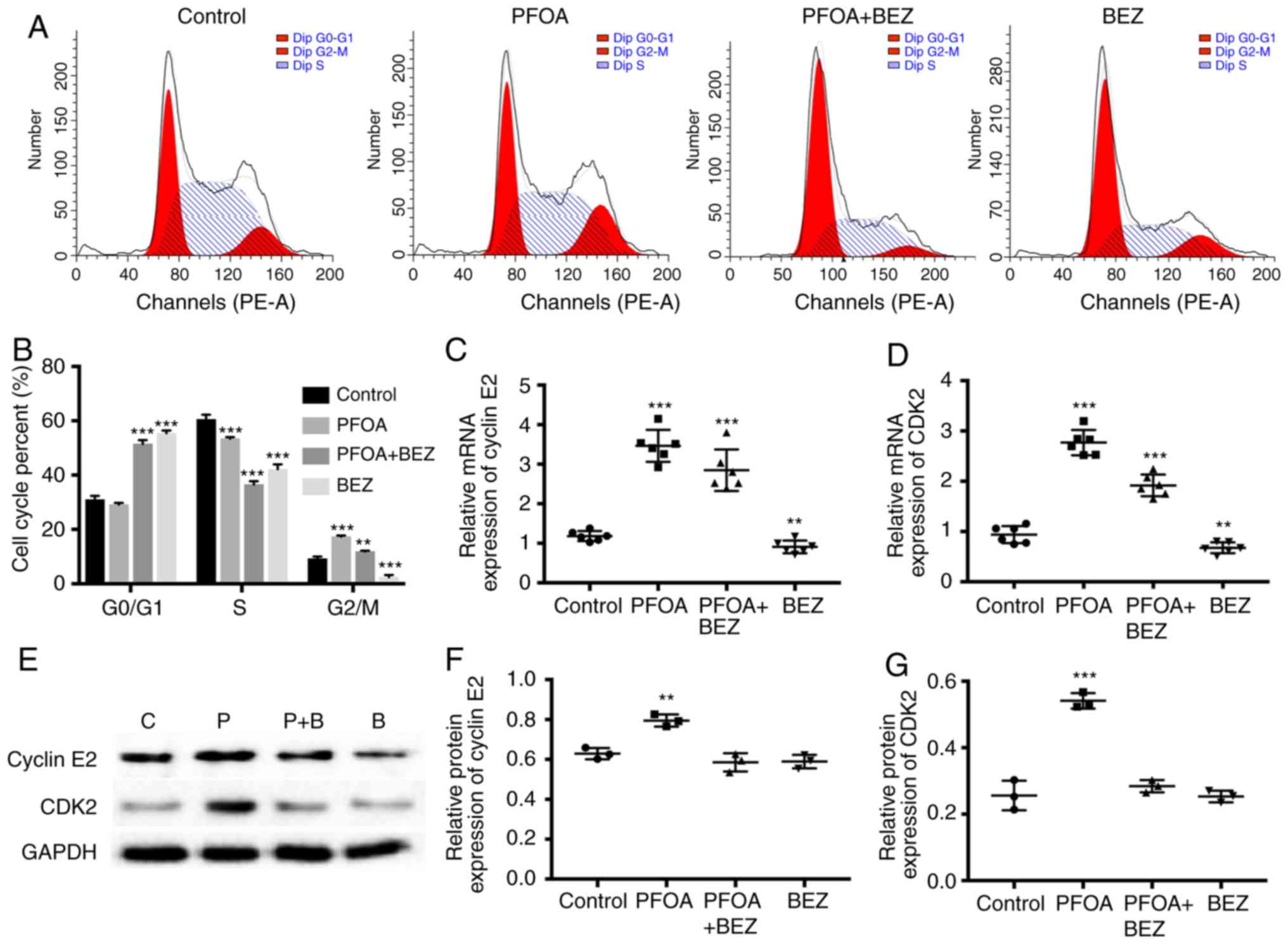 | Figure 4.Effect of PFOA on RD cell cycle. (A)
RD cells were treated with PBS, PFOA, PFOA + BEZ235, and BEZ235 for
72 h, and the proportion of cells in each cell cycle phase was
detected by flow cytometry. (B) Flow cytometry results showed that
PFOA-treated RD cells had an increased proportion of cells in the
G1/G2 phase. PFOA elicited the mRNA expression of (C) cyclin E2 and
(D) CDK2. (E) Western blotting showed that PFOA also increased the
protein expression of (F) cyclin E2 and (G) CDK2 in RD cells. No
effects on the expression of p27, p21 or p53 were observed. Data
are presented as the mean ± SD. **P<0.01 and ***P<0.001 vs.
control (PBS-treated). PFOA, perfluorooctanoic acid; PBS,
phosphate-buffered saline; BEZ, BEZ235; P+B, PFOA + BEZ235; CDK2,
cyclin-dependent kinase 2. |
PFOA exposure induces RD cell
proliferation, migration and apoptosis by activation of PI3K/AKT
signaling pathway
To investigate the mechanism induced by PFOA
exposure, the activity of the oncogenic PI3K/AKT pathway was
examined. The results showed that, compared with that in the
control group, PFOA exposure increased RD expression of PI3K and
AKT at mRNA and protein levels after 72 h (Fig. 5A-E). This suggested that PFOA enhances
proliferation, migration and invasion and inhibits apoptosis by
promoting PI3K/AKT activity in RD cells. To confirm this
hypothesis, the preferential BEZ235 concentration was initially
selected using a CCK8 assay (Fig. 5F)
and subsequently compared with single PFOA exposure and combined
PFOA and BEZ235 exposure in RD cells. The results showed that the
double PFOA and BEZ235 exposure impaired cell proliferation
(Fig. 5G), migration (Fig. 1C) and invasion (Fig. 1E), in addition to EMT (Fig. 2A-E), apoptosis (Fig. 3A-G) and cell cycle (Fig. 4A-G) following exposure to PFOA
alone.
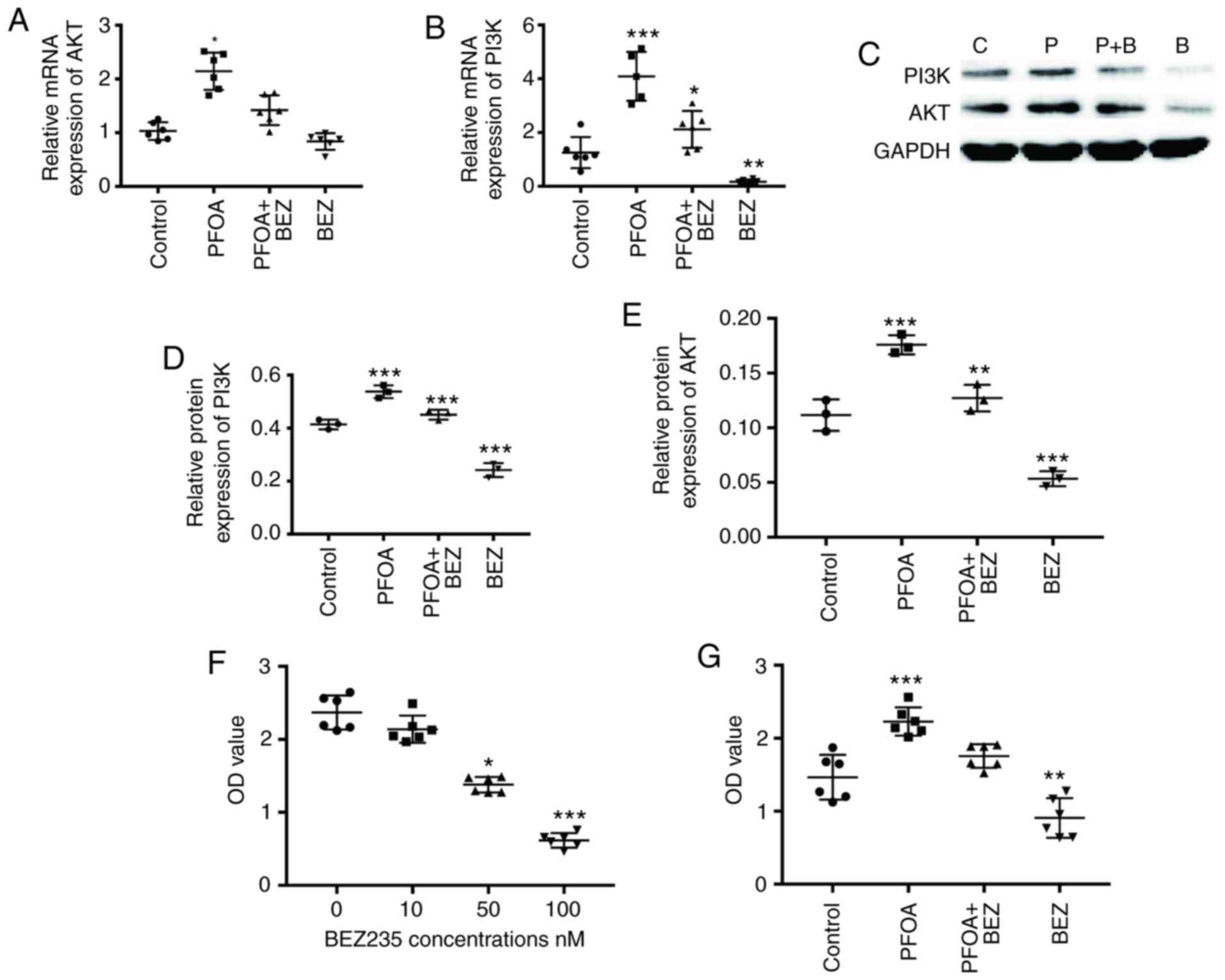 | Figure 5.PFOA impacts the RD PI3K/AKT
signaling pathway. RD cells were treated with PBS, PFOA, PFOA +
BEZ235, and BEZ235 for 72 h. The mRNA expression levels of (A) AKT
and (B) PI3K were determined by quantitative PCR analysis. (C)
Western blotting was performed and the protein levels of (D) PI3K
and (E) AKT were determined. (F) CCK-8 assay showing RD cells
treated with different concentrations of BEZ235 for 72 h. (G) CCK-8
assay showing RD cells treated with PBS, PFOA, PFOA + BEZ235, and
BEZ235 for 72 h, Data are presented as the mean ± SD. *P<0.05,
**P<0.01 and ***P<0.001 vs. control (PBS-treated). PFOA,
perfluorooctanoic acid; PI3K, phosphatidylinositol-3 kinase; AKT,
protein kinase B; PBS, phosphate-buffered saline; BEZ, BEZ235; P+B,
PFOA + BEZ235; CCK-8, Cell Counting Kit-8. |
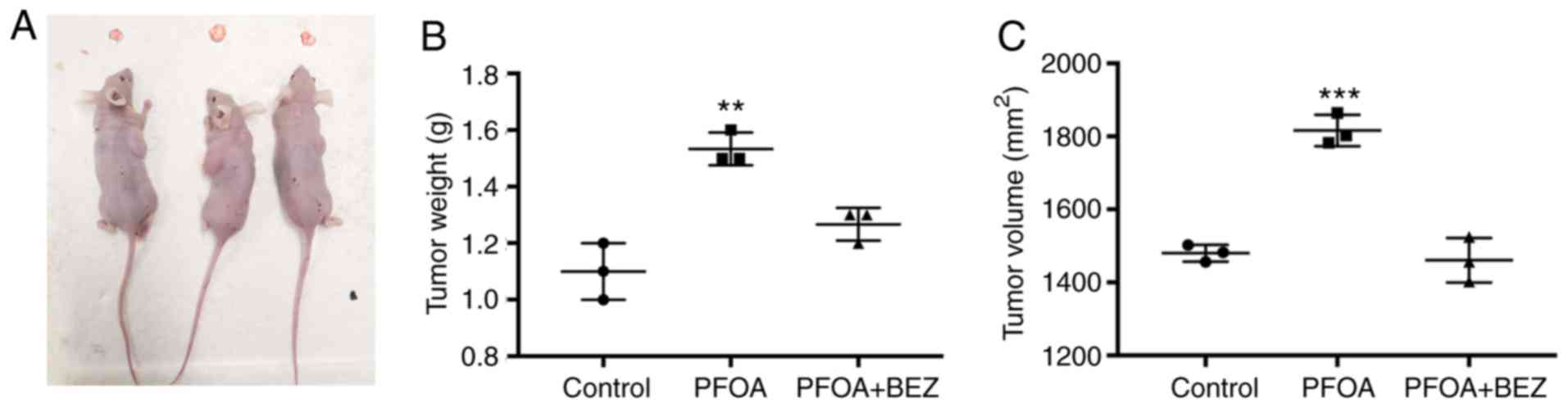
In vivo experiment confirmation that
PFOA can elicit RD cell proliferation
Our studies had comformed that PFOA promotes RD cell
proliferation and inhibits apoptosis by activating the PI3K/AKT
signaling pathway. Based on this, RD cells were used to construct a
tumor model to measure whether PFOA can promote tumor progression
in vivo and investigate whether PFOA has an impact on the
tumor PI3K/AKT signaling pathway. Compared with measurements in the
PBS control group, tumor size and weight were found to
significantly increase following PFOA administration. Subsequently,
tumor development was inhibited following BEZ235 administration
(Fig. 6A-C).
Discussion
RMS is a type of tumor mainly derived from myogenic
lineages and is thus a pediatric tumor (25), being the most frequent soft tissue
tumor of childhood under the age of 5 years. Our previous study
initially showed that PFOA can promote the migration and invasion
of RD cells. Other studies have shown that PFOA exposure is
involved in the tumor initiation process and potentially associated
with DNA damage, increased oxidative stress and the development of
several diseases, including cardiovascular, cerebrovascular and
neoplastic disease (2,3). Additionally, perfluorooctane sulfonate
(PFOS) can promote MCF-10A breast epithelial cell proliferation by
regulating cell cycle proteins and accelerating the cell cycle
process (26). Although PFOS and PFOA
are per- and polyfluoroalkyl substances which share structural and
functional properties, they have different toxicological effects
(27). The experiments performed in
the present study demonstrated that PFOA promoted RD cell
proliferation by regulating different cell cycle-related proteins.
PFOA exposure induced RD cell proliferation and upregulated the
cyclin E2 and CDK2 levels. PFOA induced RD cell migration and
invasion, suggesting that it may induce normal skeletal muscle
cells to transform into neoplastic cells.
Malignant tumors are characterized by their endless
replication and cell circulation ability. This can occur at
multiple levels, including in one or several proteins involved in
cell cycle control and progression. As acknowledged, the most
important step in tumor development is an excessive G-S phase.
Evidence shows that cyclin E2 is a cyclin E family member
potentially implicated in the G0/G1 to S transition by CDK2 binding
and Rb protein phosphorylation (16,28).
Cyclin E-CDK2 is considered an essential and master regulator of
cell cycle progression from the G1 to S phase (28). The results of the present study show
that PFOA exposure increased cyclin E2 and CDK2 at the mRNA and
protein levels. This may explain how PFOA promotes RD cell
proliferation. Experiments on RD cells showed that CDK and cyclin
regulation significantly inhibited cell proliferation (29). These results support the hypothesis
that RD cell proliferation is elicited by cyclin E-CDK2
induction.
Two major CKI families are implicated in cell cycle
regulation. One is the Cip/kip family (including p27 and p21),
which may inhibit cyclin E-CDK (30).
The downregulation or inactivation of p27 and p21, which normally
cause G1 arrest, leads to abnormal cell cycle regulation, increases
cell proliferation and causes malignant tumor formation (31). The P53 negative cell cycle regulator
is mutated in several human tumors. Activation of the expression of
p21 may inhibit cell cycle progression and control cell cycle at
the apoptotic stage (32). A study on
A2780 ovarian cells showed that p21 not only directly inhibited the
activity of CDK but also increased the level of p27 by stabilizing
p27 protein (33). However, the
desired results were not obtained using the PI staining method to
detect the proportion of cells in the three cell cycle phases. RD
cell exposure to PFOA showed that cyclin E2 and CDK2 increased at
the mRNA level, whereas no changes in p27, p21 or p53 were
observed. Further investigation of the effects of PFOA on cell
cycle gene and protein alterations in RD cells are required in
order to gain further insights into its respective mechanism of
action.
The metastatic process of tumor cells is complex,
with invasion and migration being key steps. However, EMT is
essential for malignant tumor cells to acquire invasion and
migration ability. EMT is acknowledged as a complex cellular
mechanism, associated with invasion and metastatic progression in
various cancer cells. E-cadherin is an important epithelial marker
and EMT-initiating factor, with EMT being activated when the
expression of E-cadherin is downregulated, inhibited or lost
(34). Although decitabine can induce
the expression of E-cadherin, E-cadherin was not detected at the
mRNA or protein levels in the present study. It was found that PFOA
exposure increased the expression of vimentin and SGK1 in RD cells,
suggesting that PFOA may elicit EMT. Vimentin is a typical EMT
marker and a conserved 57-kD protein, expressed in several
mesenchymal cells (35) and
overexpressed in epithelial tumors, positively correlating with
tumor growth and invasion acceleration and with prognosis. SGK1 is
a tumor-promoting gene with elevated expression in several
malignancies, including prostate (36), gastric (37) and breast (38) cancer. Studies have suggested that SGK1
can promote metastases formation and it has been shown to serve a
key role in promoting EMT in colorectal (39) and prostate (10) cancer. Therefore, it was crucial to
investigate whether PFOA promoted RD cell migration and invasion in
the present study. PFOA was shown to promote RD cell migration and
invasion in wound healing and Transwell assays. PFOA may increase
the expression of vimentin and SGK1 at the mRNA and protein
levels.
Apoptosis refers to programmed cell death, and
defects in this process can cause cancer or autoimmunity, whereas
enhanced apoptosis may lead to degenerative diseases (40). The antiapoptotic protein Bcl-2 may
help tumor cells resist apoptosis, whereas the apoptotic protein
Bax may elicit tumor cell apoptosis. Bcl-2 and Bax are present in
the mitochondria and form a heterodimer. The overexpression of
anti-apoptotic Bcl-2 or Bcl-xL occurs in >50% of cancer cases
(41). Bax directly affects
apoptosis, being one of the most well-known downstream p53
transcriptional targets. P53 is able to inhibit the anti-apoptotic
function of Bcl-2 and Bcl-xL on mitochondria during apoptosis
(42). In the present study, PFOA
exposure inhibited the expression of Bax and elicited the
expression of Bcl-2 at the mRNA and protein levels.
In the present study, PFOA exposure promoted EMT and
inhibited apoptosis. As recognized, the PI3K/AKT pathway serves an
important role in cell growth by inhibiting apoptosis in several
types of cancer (43,44). PI3K modulates signaling pathways
implicated in cell growth, apoptosis, or both, which may modulate
the activity of AKT. Activating AKT may induce cell cycle
progression, survival and migration through the phosphorylation of
certain physiological factors. It has been reported that the
activation of PI3K and AKT may lead to ovarian, thyroid, pancreatic
and breast cancer (45). Based on
these findings, using RT-qPCR and western blot analyses, the
present study showed that PFOA increased the expression of PI3K and
AKT. The results suggest that PFOA may enhance cell migration,
invasion and proliferation and inhibit apoptosis through activation
of the PI3K/AKT signaling pathway.
Further experiments were conducted to confirm this
hypothesis. BEZ235, the PI3K inhibitor, reduced PFOA-induced
proliferation, with the levels of formazan production significantly
lower than those in RD cells exposed to PFOA alone. PFOA exposure
affected cell proliferation through PI3K regulation of the cell
cycle; treatment with BEZ235 and PFOA downregulated the mRNA
expression levels of cyclin E2 and CKD2 in RD cells compared with
those following treatment with PFOA alone. In the wound healing and
Transwell invasion assays, the cells treated with BEZ235 and PFOA
significantly reduced RD cells' migration and invasion compared
with that in cells treated with PFOA alone. PFOA also promoted the
expression of vimentin and SGK1, as determined by RT-qPCR and
western blot assays. Although the expression of E-cadherin was not
detected at the mRNA or proteins levels, further investigations are
warranted to elucidate why. Treatment with BEZ235 and PFOA reversed
the single PFOA treatment-induced upregulation of vimentin and
SGK1. Similar results were found for the apoptotic Bcl-2 and Bax
proteins. The mRNA and protein expression levels of Bax were higher
following BEZ235 and PFOA treatment those that following treatment
with PFOA alone, and the mRNA and protein expression levels of
Bcl-2 were lower following treatment with BEZ235 and PFOA compared
with those treated with to PFOA only. These results indicated that
PFOA treatment induces EMT and inhibits apoptosis in RD cells, also
requiring activation of the PI3K/AKT signaling pathway.
Taken together, the results of the present study
suggest that PFOA regulates RD cell proliferation, migration,
invasion and apoptosis through activation of the PI3K/AKT signaling
pathway, potentially eliciting tumorigenesis. The in vivo
experiments allowed analysis of the effects of PFOA on the tumors,
showing that it led to increased tumor volume and weight. In future
investigations, further in vivo experiments are to be
developed to investigate the effects of PFOA on the tumor and on
the tumorigenic mechanism, through analysis of cell cycle, EMT and
apoptosis. A clearer understanding of the role of PFOA in
tumorigenesis can assist in preventing and treating tumors elicited
by this agent.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation Of China (grant no. 61827806).
Availability of data and materials
The datasets used during the present study are
available from the corresponding anthor upon reasonable
request.
Authors' contributions
Conceptualization of the study was achieved by QZ
and CW, the research methodology was designed by QZ, JW, CC, YK,
HY, JD and CW. Formal analysis of the data was conducted by QZ and
YK. Funding acquisition was accomplished by CW. Project
administration was carried out by CW, and study resources were
obtained by QZ and CW. Software analysis of data and figures were
conducted by QZ, YK, JW and CC, supervision of the research was
conducted by XN and CW. Writing of the original draft was
undertaken by QZ, JW and CC, and writing, review and editing of the
manuscript were carried out by QZ, JD, YS, XW and CW. All authors
read and approved the manuscript and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
All animal treatments were approved by the Ethics
Committee of the Experimental Animal Center and by the Chinese
Academy of Military Medical Sciences (permit no. SCXK-2015-0002),
in accordance with the guiding principles for the use of animals in
toxicology, adopted by the Society of Toxicology in 1989.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bartell SM, Calafat AM, Lyu C, Kato K,
Ryan PB and Steenland K: Rate of decline in serum PFOA
concentrations after granular activated carbon filtration at two
public water systems in Ohio and West Virginia. Environ Health
Perspect. 118:222–228. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shin HM, Vieira VM, Ryan PB, Steenland K
and Bartell SM: Retrospective exposure estimation and predicted
versus observed serum perfluorooctanoic acid concentrations for
participants in the C8 health project. Environ Health Perspect.
119:1760–1765. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Barry V, Winquist A and Steenland K:
Perfluorooctanoic acid (PFOA) exposures and incident cancers among
adults living near a chemical plant. Environ Health Perspect.
121:1313–1318. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lau C, Anitole K, Hodes C, Lai D,
Pfahles-Hutchens A and Seed J: Perfluoroalkyl acids: A review of
monitoring and toxicological findings. Toxicol Sci. 99:366–394.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pierozan P, Jerneren F and Karlsson O:
Perfluorooctanoic acid (PFOA) exposure promotes proliferation,
migration and invasion potential in human breast epithelial cells.
Arch Toxicol. 92:1729–1739. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wielsøe M, Kern P and Bonefeld-Jørgensen
EC: Serum levels of environmental pollutants is a risk factor for
breast cancer in Inuit: a case control study. Environ Health.
16:562017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cha YH, Yook JI, Kim HS and Kim NH:
Catabolic metabolism during cancer EMT. Arch Pharm Res. 38:313–320.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Natarajan J, Chandrashekar C and
Radhakrishnan R: Critical biomarkers of epithelial-mesenchymal
transition in the head and neck cancers. J Cancer Res Ther.
10:512–518. 2014.PubMed/NCBI
|
|
9
|
Myong NH: Loss of E-cadherin and
acquisition of vimentin in epithelial-mesenchymal transition are
noble indicators of uterine cervix cancer progression. Korean J
Pathol. 46:341–348. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu W, Wang X, Wang Y, Dai Y, Xie Y, Ping
Y, Yin B, Yu P, Liu Z, Duan X, et al: SGK1 inhibition-induced
autophagy impairs prostate cancer metastasis by reversing EMT. J
Exp Clin Cancer Res. 37:732018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jeanes A, Gottardi CJ and Yap AS:
Cadherins and cancer: How does cadherin dysfunction promote tumor
progression? Oncogene. 27:6920–6929. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Satelli A and Li S: Vimentin as a
potential molecular target in cancer therapy or Vimentin, an
overview and its potential as a molecular target for cancer
therapy. Cell Mol Life Sci. 68:3033–3046. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ibrahim U, Saqib A, Mohammad F, Ding J,
Salman B, Collado FK and Dhar M: Embryonal rhabdomyosarcoma of the
cervix: A rare disease at an uncommon age. Cureus.
9:e18642017.PubMed/NCBI
|
|
14
|
Mummery CL, van den Brink CE and de Laat
SW: Commitment to differentiation induced by retinoic acid in P19
embryonal carcinoma cells is cell cycle dependent. Dev Biol.
121:10–19. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Filipczyk AA, Laslett AL, Mummery C and
Pera MF: Differentiation is coupled to changes in the cell cycle
regulatory apparatus of human embryonic stem cells. Stem Cell Res.
1:45–60. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Santamaria D and Ortega S: Cyclins and
CDKS in development and cancer: Lessons from genetically modified
mice. Front Biosci. 11:1164–1188. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stead E, White J, Faast R, Conn S,
Goldstone S, Rathjen J, Dhingra U, Rathjen P, Walker D and Dalton
S: Pluripotent cell division cycles are driven by ectopic Cdk2,
cyclin A/E and E2F activities. Oncogene. 21:8320–8333. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Courtney KD, Corcoran RB and Engelman JA:
The PI3K pathway as drug target in human cancer. J Clin Oncol.
28:1075–1083. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fruman DA and Rommel C: PI3K and cancer:
Lessons, challenges and opportunities. Nat Rev Drug Discov.
13:140–156. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Meng X, Dong X, Wang W, Yang L, Zhang X,
Li Y, Chen T, Ma H, Qi D and Su J: Natural borneol enhances
paclitaxel-induced apoptosis of ESCC cells by inactivation of the
PI3K/AKT. J Food Sci. 83:1436–1443. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yan S, Wang J, Zhang W and Dai J:
Circulating microRNA profiles altered in mice after 28 d exposure
to perfluorooctanoic acid. Toxicol Lett. 224:24–31. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang F, Qian XJ, Qin W, Deng R, Wu XQ, Qin
J, Feng GK and Zhu XF: Dual phosphoinositide 3-kinase/mammalian
target of rapamycin inhibitor NVP-BEZ235 has a therapeutic
potential and sensitizes cisplatin in nasopharyngeal carcinoma.
PLoS One. 8:e598792013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ma Z, Liu X, Li F, Wang Y, Xu Y, Zhang M
and Zhang X, Ying X and Zhang X: Perfluorooctanoic acid induces
human Ishikawa endometrial cancer cell migration and invasion
through activation of ERK/mTOR signaling. Oncotarget.
7:66558–66568. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Blum JM, Añó L, Li Z, Van Mater D, Bennett
BD, Sachdeva M, Lagutina I, Zhang M, Mito JK, Dodd LG, et al:
Distinct and overlapping sarcoma subtypes initiated from muscle
stem and progenitor cells. Cell Rep. 5:933–940. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pierozan P and Karlsson O: PFOS induces
proliferation, cell-cycle progression, and malignant phenotype in
human breast epithelial cells. Arch Toxicol. 92:705–716. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tsuda S: Differential toxicity between
perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA).
J Toxicol Sci 41 (Special). SP27–SP36. 2016. View Article : Google Scholar
|
|
28
|
Hwang HC and Clurman BE: Cyclin E in
normal and neoplastic cell cycles. Oncogene. 24:2776–2786. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Marampon F, Ciccarelli C and Zani BM:
Down-regulation of c-Myc following MEK/ERK inhibition halts the
expression of malignant phenotype in rhabdomyosarcoma and in non
muscle-derived human tumors. Mol Cancer. 5:312006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Le Page C, Huntsman DG, Provencher DM and
Mes-Masson AM: Predictive and prognostic protein biomarkers in
epithelial ovarian cancer: Recommendation for future studies.
Cancers (Basel). 2:913–954. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bali A, O'Brien PM, Edwards LS, Sutherland
RL, Hacker NF and Henshall SM: Cyclin D1, p53, and p21Waf1/Cip1
expression is predictive of poor clinical outcome in serous
epithelial ovarian cancer. Clin Cancer Res. 10:5168–5177. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Milde-Langosch K and Riethdorf S: Role of
cell-cycle regulatory proteins in gynecological cancer. J Cell
Physiol. 196:224–244. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
He G, Kuang J, Huang Z, Koomen J,
Kobayashi R, Khokhar AR and Siddik ZH: Upregulation of p27 and its
inhibition of CDK2/cyclin E activity following DNA damage by a
novel platinum agent are dependent on the expression of p21. Br J
Cancer. 95:1514–1524. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qin Y, Guo H, Tang B and Yang SM: The
non-reverse transcriptase activity of the human telomerase reverse
transcriptase promotes tumor progression (review). Int J Oncol.
45:525–531. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Satelli A and Li S: Vimentin in cancer and
its potential as a molecular target for cancer therapy. Cell Mol
Life Sci. 68:3033–3046. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yao Y, Jiang Q, Jiang L, Wu J, Zhang Q,
Wang J, Feng H and Zang P: Lnc-SGK1 induced by Helicobacter pylori
infection and highsalt diet promote Th2 and Th17 differentiation in
human gastric cancer by SGK1/Jun B signaling. Oncotarget.
7:20549–20560. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Szmulewitz RZ, Chung E, Al-Ahmadie H,
Daniel S, Kocherginsky M, Razmaria A, Zagaja GP, Brendler CB,
Stadler WM and Conzen SD: Serum/glucocorticoid-regulated kinase 1
expression in primary human prostate cancers. Prostate. 72:157–164.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jo A, Yun HJ, Kim JY, Lim SC, Choi HJ,
Kang BS, Choi BY and Choi HS: Prolyl isomerase PIN1 negatively
regulates SGK1 stability to mediate tamoxifen resistance in breast
cancer cells. Anticancer Res. 35:785–794. 2015.PubMed/NCBI
|
|
39
|
Gulhati P, Bowen KA, Liu J, Stevens PD,
Rychahou PG, Chen M, Lee EY, Weiss HL, O'Connor KL, Gao T and Evers
BM: mTORC1 and mTORC2 regulate EMT, motility, and metastasis of
colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Res.
71:3246–3256. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hassan M, Watari H, AbuAlmaaty A, Ohba Y
and Sakuragi N: Apoptosis and molecular targeting therapy in
cancer. Biomed Res Int. 2014:1508452014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Amundson SA, Myers TG, Scudiero D, Kitada
S, Reed JC and Fornace AJ Jr: An informatics approach identifying
markers of chemosensitivity in human cancer cell lines. Cancer Res.
60:6101–6110. 2000.PubMed/NCBI
|
|
42
|
Mihara M, Erster S, Zaika A, Petrenko O,
Chittenden T, Pancoska P and Moll UM: p53 has a direct apoptogenic
role at the mitochondria. Mol Cell. 11:577–590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Franke TF, Hornik CP, Segev L, Shostak GA
and Sugimoto C: PI3K/Akt and apoptosis: Size matters. Oncogene.
22:8983–8998. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cantley LC and Neel BG: New insights into
tumor suppression: PTEN suppresses tumor formation by restraining
the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA.
96:4240–4245. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Blanco-Aparicio C, Renner O, Leal JF and
Carnero A: PTEN, more than the AKT pathway. Carcinogenesis.
28:1379–1386. 2007. View Article : Google Scholar : PubMed/NCBI
|