Introduction
Neuroblastoma (NB) is the most common extracranial
solid tumor in children; it is derived from the primary neural
crest. NB accounts for 6–10% of all cancers diagnosed in children,
and causes 9–15% of all cancer-related deaths in childhood
(1–4).
The biological behavior of NB is dramatically heterogeneous in
clinical settings, spanning from spontaneous regression of low-risk
NB to aggressive progression to high-risk clinical stage (5,6). There are
various biological prognostic markers that are used to predict poor
outcomes following the diagnosis of NB, including the amplification
of the proto-oncogene N-MYC, dysregulation of anaplastic lymphoma
kinase (ALK) and genetic aberrations. The amplification of N-MYC
usually predicts an unfavorable prognosis in patients with NB
(7–12). Although intensive therapies have been
utilized to treat this malignancy, the recurrence and metastatic
rates of high-risk NB are still extremely high and are responsible
for 50% of the mortality of NB patients (2). Given the poor prognosis of high-risk NB,
there is an urgent need to further investigate the mechanisms
underlying the initiation and aggressive progression of NB, hoping
to identify more effective therapeutic targets.
β-adrenergic receptor (β-AR) signaling plays an
important role in the regulation of physiological and pathological
processes (13,14). For instance, it is significantly
involved in the modulation of the cardiovascular system, lipid
metabolism and immune homeostasis (15–18). β-AR
signaling is activated by the binding of β-AR with catecholamine
hormones including adrenaline and norepinephrine, and catecholamine
hormones are usually secreted under stressful conditions (19). Recent studies have revealed that β-AR
signaling is also involved in tumorigenesis and metastasis
(20–23). For example, it has been documented
that the activation of β-AR signaling can promote hepatocellular
carcinoma cell growth in vitro and in vivo (24). Either the use of β-AR antagonists or
the genetic deletion of β2- and β3-AR in stromal cells has been
shown to suppress the early metastasis of prostate cancer (25). β-AR blockers have been used for
treating hypertension for decades, and they have exhibited a
notable antitumor efficacy in clinical trials (26,27).
However, the role of the β-AR pathway in the tumorigenesis and
progression of NB, which is a type of neural system tumor, is not
clear, and the underlying regulatory mechanisms have not been well
studied. Thus, it is necessary to investigate the role of β-AR
signaling in NB and the novel mechanisms underlying the
β-AR-mediated regulation of NB.
Autophagy is an evolutionarily conserved catabolic
process by which cells capture, deliver, degrade and recycle
damaged protein aggregates and organelles to sustain metabolic
homeostasis; it helps the cells survive stressful conditions such
as nutrient deprivation (28–30). There are three main types of
autophagy: Macro-autophagy, micro-autophagy, and chaperone-mediated
autophagy (31). Hereafter, the term
‘autophagy’ refers to macro-autophagy. A normal, basal level of
autophagy is important for the timely clearance of unfavorable
cellular components to prevent the buildup of toxic materials
(32,33), while the deregulation of autophagy is
closely associated with multiple diseases including cancer,
inflammation, and neurodegenerative diseases (34,35). The
role of autophagy in tumors is really complex and
context-dependent. Autophagy can promote or suppress tumorigenesis
and the aggressive progression of tumors based on the tumor
microenvironment. For instance, the ablation of Atg7+/+
tumor cells in a KRASG12D mutation-driven lung cancer
mouse model has been shown to be accompanied by the accumulation of
defective mitochondria and cell growth arrest, resulting in the
inhibition of tumorigenesis (36). On
the other hand, autophagy defects can lead to genomic instability,
activation of DNA damage response, and oxidative stress, which can
contribute to tumorigenesis and tumor progression (37). In NB, it has been reported that the
targeted inhibition of the autophagy-related protein unc-51-like
autophagy kinase 1 (ULK1) can promote NB cell apoptosis and
suppress NB cell growth (38). It is
known that autophagy is usually activated upon starvation; however,
whether autophagy can be stimulated under stressful conditions in
NB has not yet been reported. Thus, in the present study, we aimed
to investigate the novel underlying regulatory mechanisms of
autophagy in NB.
Importantly, we used clinical tumor samples and
cellular assays to ascertain the role of β-AR signaling and
autophagy in NB, hoping to uncover a novel regulatory mechanism
involving the β-AR pathway and autophagy, and provide potential
therapeutic targets for treating NB.
Materials and methods
Reagents and antibodies
Isoprenaline (ISO, I5627), formoterol (product no.
F9552), ICI118,551 (product no. I127), atenolol (product no. A7655)
and 3-methyladenine (3-MA, product no. M9281) were procured from
Sigma-Aldrich/Merck. SBI-0206965 was procured from Selleck
Chemicals. Antibodies for β1-AR (product no. ab3442), β2-AR
(product no. ab182136), N-MYC (product no. ab24193), and Beclin-1
(product no. ab109631 for IHC assay) were obtained from Abcam.
Antibodies for LC3B (product no. 3868), beclin-1 (product no.
3495), CREB (product no. 48H2), p-CREB (product no. 87G3), p-ULK1
(product no. 5869S) and ULK1 (product no. 8054T) were obtained from
Cell Signaling Technology. LC3-II (cat. no. A11923) for IHC
staining was purchased from Abclonal. ULK1 for the IHC assay was
obtained from Zen BioScience.
Clinical samples and
immunohistochemistry (IHC) assay
The clinical samples of ganglioneuroma (GN),
ganglioneuroblastoma (GNB, nodular and intermixed), and NB were
obtained from the Guangzhou Women and Children's Medical Center
(26, 34 and 71 samples, respectively). The date range of sample
collection was from January 2003 to September 2015. These samples
were used for constructing tissue microarrays (TMAs) by the
Shanghai Outdo Biotech Company. The clinical information of these
samples is documented in Table I. For
the IHC staining of the TMAs, the clinical tumor samples were fixed
and then embedded in paraffin. The tissue slides were treated with
endogenous peroxidase blocking solution and normal goat serum to
block nonspecific background interactions. Then, the tissues were
incubated with anti-β1-AR (dilution 1:200), -β2-AR (dilution
1:200), -ULK1 (dilution 1:200), -beclin-1 (dilution 1:200) and
-LC3-II (dilution 1:200) antibodies at 4°C overnight. After
incubation with the primary antibodies, the slides were incubated
with horseradish peroxidase-labeled anti-rabbit or mouse secondary
antibodies for 30 min at room temperature; they were then stained
with the chromogenic substrate diaminobenzidine (DAB; Dako; Agilent
Technologies, Inc.), and counterstained with hematoxylin. The
evaluation of tissue microarray scores was carried out by two
independent researchers based on the positive staining intensity
and the percentage of positively stained cells. The IHC results
were scored as 0 (negative), 1–2 (weakly positive), 3–4 (moderately
positive), 5–6 (strongly positive), and high expression of tumor
cells were defined as scores >3.
 | Table I.Clinical information of the GN, GNB
and NB samples. |
Table I.
Clinical information of the GN, GNB
and NB samples.
| Tumor | Age | Sex | N-MYC status | INSS | Total no. |
|---|
| NB | <18 months
(43) | Male (43) | Amplified (8) | I (29) | 71 |
|
| >18 months
(28) | Female (28) | Non-amplified
(37) | II (15) |
|
|
|
|
|
| III (10) |
|
|
|
|
|
| IV (13) |
|
| GNB | <18 months
(5) | Male (16) | Amplified (2) | I (11) | 34 |
|
| >18 months
(29) | Female (18) | Non-amplified
(17) | II (9) |
|
|
|
|
|
| III (4) |
|
|
|
|
|
| IV (7) |
|
| GN | <18 months
- | Male (10) | Amplified - | I (16) | 26 |
|
| >18 months
(26) | Female (16) | Non-amplified
(3) | II (5) |
|
|
|
|
|
| III - |
|
|
|
|
|
| IV - |
|
This study was approved by the Human Ethics
Committee of the Affiliated Guangzhou Women and Children's Medical
Center, Zhongshan School of Medicine. All patients gave informed
consent before participation in the present study.
Cell culture
NB cells including SK-N-AS, SK-N-SH, SHEP, SK-N-BE2,
SK-N-BE2c and Kelly was used as cell models. Among these cell lines
SK-N-AS, SK-N-SH and SHEP were N-MYC non-amplified cells, while
SK-N-BE2, SK-N-BE2c and Kelly were N-MYC amplified cells. These
cell lines were a kind gift from Professor Li Bo, Sun Yat-Sen
Medical School. These cells were cultured in high-glucose
Dulbecco's modified Eagle's medium (DMEM; Corning, Inc.)
supplemented with 10% heat-inactivated fetal bovine serum (FBS;
Gibco; Thermo Fisher Scientific, Inc.) at 37°C and 5%
CO2 conditions in a humidified incubator.
CCK-8 assay
For the CCK-8 assay, SK-N-BE2 and Kelly cells were
seeded into 96-well plates at densities of 3×103 and
5×103 cells per well, respectively. These cells were
treated with selective and non-selective β-AR agonists and
antagonists including isoprenaline, ICI118,551 and atenolol for the
indicated time periods. Then, the absorbance of the samples was
detected at 450 nm according to the manufacturer's
instructions.
Western blot assay
Proteins from NB cells were harvested using lysis
buffer (glycerol, 0.5 M Tris-HCl, 10% SDS, ddH2O), and
the protein concentrations were detected using the BCA kit. Then,
the protein samples were treated with 10X loading buffer (0.2%
bromophenol blue, β-mercaptoethanol) and 30 µg protein was
separated on 10 and 15% SDS-PAGE gels; the resulting protein bands
were transferred onto PVDF membranes. The membranes were blocked
with 5% skimmed milk for 1 h at room temperature. Then, they were
incubated with the indicated antibodies anti-β1-AR (dilution
1:1,000), -β2-AR (dilution 1:1,000), -ULK1 (dilution 1:1,000),
-p-ULK1 (dilution 1:1,000), CREB (dilution 1:1,000), p-CREB
(dilution 1:1,000), -LC3-II (dilution 1:1,000), N-MYC (dilution
1:1,000) and β-actin (dilution 1:6,000) at 4°C overnight with
shaking, followed by incubation with horseradish peroxidase
(HRP)-labeled secondary antibodies (dilution 1:3,000) for 2 h at
room temperature. The enhanced chemiluminescent (ECL) kit (P90720;
Millipore) was used to visualize the western blot results, and
ImageJ (×64; National Institutes of Health, Bethesda, MD, USA)
software was applied to quantify the densitometry.
EdU assay and autophagic flux
detection
For EdU staining, a total number of 2×105
SK-N-BE2 and Kelly cells were mounted onto coverslips in 6-well
plates. After being cultured for 24 h, the cells were treated with
the indicated reagents for another 24 h. Then, they were incubated
with EdU reagents (Nanjing KeyGen Biotech Co., Ltd.) for 2 h,
followed by subsequent staining according to the manufacturer's
instructions. The images were captured using a fluorescence
microscope at a magnification of ×100 (Olympus BX63; Olympus Corp.,
Tokyo, Japan). A recombinant adenovirus expression vector
containing tandem RFP-GFP-LC3 (Hanbio Biotechnology Co.) was used
to assess the change of autophagic flux. The MOI was 100, and the
cells were incubated with the adenoviruses for 24 h before the
subsequent treatments. The images were obtained using a confocal
microscope at a magnification of ×630 (LSM 780; Carl Zeiss).
Transfection with small interfering
RNA (siRNA)
siRNAs were used to knock down the expression of
β2-AR and CREB. The siRNAs were synthesized by Guangzhou
RiboBio Co., Ltd. (Guangzhou, China). For interfering with the
functions of β2-AR and CREB, SK-N-BE2 cells were seeded into 6-well
plates at a density of 5×105 cells per well, and
transfected with 100 nM of the respective siRNAs using the
transfection reagent RNAiMAX (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's instructions. After
culturing the cells for 48 h, the interference efficiency was
verified by western blotting; the cells were subjected to the
subsequent treatments based on the above methods. The siRNA
sequences of β2-AR and CREB used in our experiments are as follows:
β2-AR (CATCCTCCTAAATTGGATA) and CREB (siRNA1, CGTAGAAAGAAGAAAGAAT;
siRNA2, GCCACAGATTGCCACATTA; and siRNA3, GCAATACAGCTGGCTAACA).
Statistical analysis
Student's t-test and one-way ANOVA with post hoc
contrasts by a Newman-Keuls test were used to assess the
differences between two or three groups by means of GraphPad Prism
5 software (GraphPad Software, Inc.). P-values <0.05 were
considered statistically significant.
Results
Expression of β1- and β2-AR is
significantly increased in NB
To investigate the role of β-AR in NB, we first
detected the expression of β-AR in the clinical tumor samples. The
expression of β-adrenergic receptors was detected in samples of
three peripheral neuroblastic tumors (PNTs) including GN, GNB
(nodular and intermixed) and NB. Among these subtypes, NB is the
most malignant and consistently shows an unfavorable prognosis and
low survival rate; the other two types of tumors consistently
exhibit a good prognosis compared to NB (9). β-AR includes β1-AR, β2-AR and β3-AR. We
mainly tested the expression of β1-AR and β2-AR in our study. IHC
results using the TMA samples revealed that the levels of β1-AR and
β2-AR were significantly upregulated in the NB samples, compared to
the levels noted in the GN and GNB samples (Fig. 1A). The representative images of IHC
staining scores of clinical NB samples are shown in Fig. 1B. We further evaluated the expression
of β1-AR and β2-AR in different clinical stages of NB, but there
was no notable correlation between the β1-AR and β2-AR expression
in the different clinical stages of NB (Fig. 1C). This result suggests that the β-AR
pathway may play a crucial role in the tumorigenesis of NB.
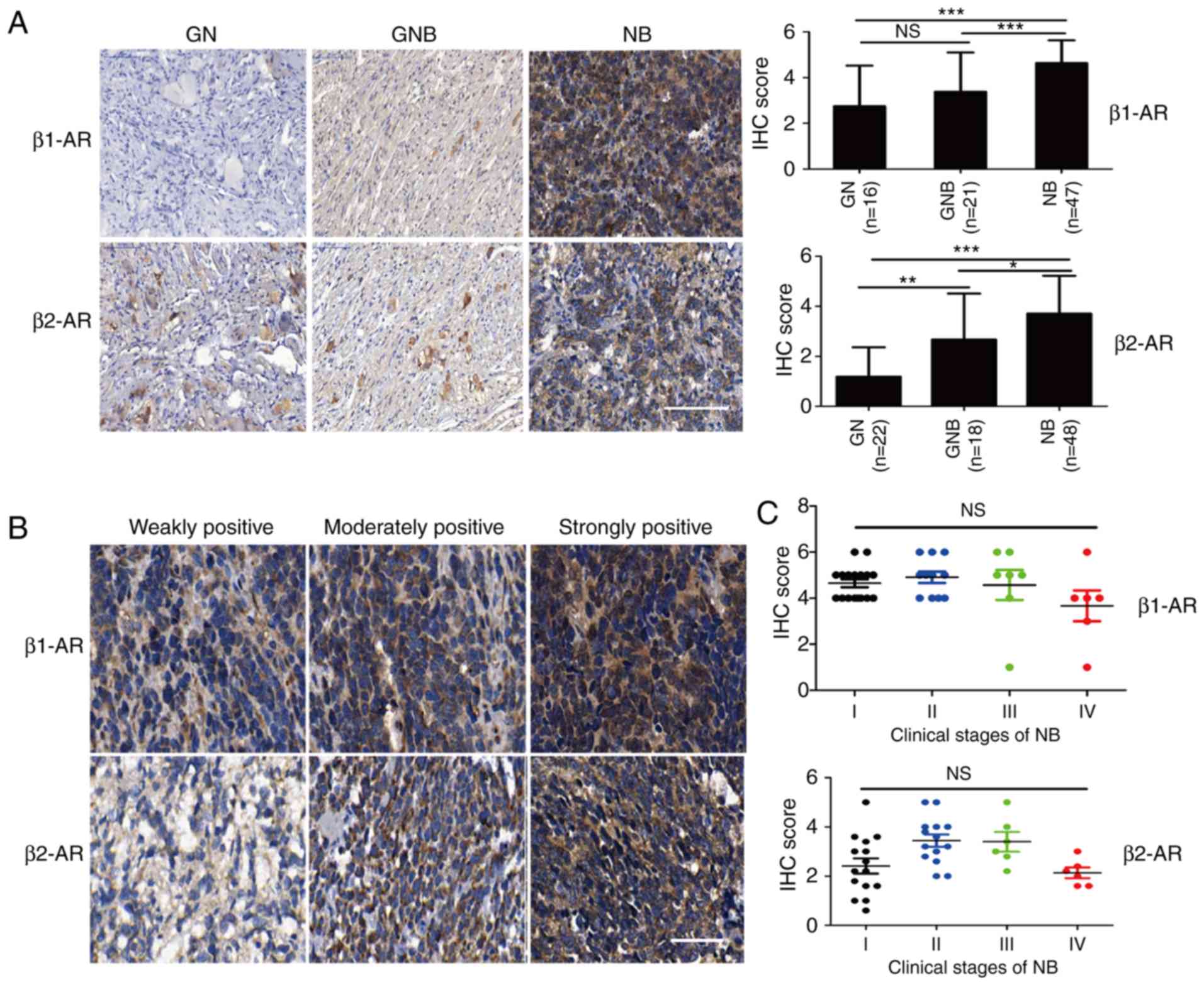 | Figure 1.Expression of β2-AR is significantly
increased in the neuroblastoma tissues. (A) IHC staining was used
to assess the levels of β1-AR and β2-AR in tissue microarrays, and
the IHC scores were calculated. Scale bar, 100 µm. (B)
Representative images of IHC staining scores are shown. Scale bar,
50 µm. (C) Correlation between the β1-AR and β2-AR levels in NB of
different clinical stages is shown. Student's unpaired t-test was
used to assess the difference between two groups. All values are
expressed as the mean ± SEM. The experiment was repeated three
times. *P<0.05, **P<0.01, ***P<0.001. n.s., not
significant. IHC, immunohistochemistry; NB, neuroblastoma; GN,
ganglioneuroma; GNB, ganglioneuroblastoma; β-AR, β-adrenergic
receptor. |
Activation of β2-AR signaling promotes
NB cell proliferation
To further investigate the role of β-AR signaling
pathway in NB cell growth, the expression of β1-AR, β2-AR and N-MYC
was detected in six different NB cell lines. The amplification
status of N-MYC is an important prognostic marker for NB. The
expression levels of β1-AR and β2-AR in six NB cell lines are shown
in Fig. 2A. We chose two cell lines
SK-N-BE2 and Kelly which express high level of β2-AR as cell
models. Treatment with the non-selective β-AR agonist isoprenaline
(ISO) increased the viability of the NB SK-N-BE2 and Kelly cell
lines (Fig. 2B and C). In order to
demonstrate which β-AR is responsible for the promotive effect of
β-AR activation on cell survival, we utilized specific antagonists
of β1-AR (atenolol) and β2-AR (ICI118,551) to ascertain the role of
the β-AR pathway in NB cell growth. The results showed that
treatment with the β1-AR antagonist atenolol did not affect NB cell
survival, while treatment with the β2-AR antagonist ICI118,551
suppressed the cell viability in a concentration-dependent manner
(Fig. 2D). These findings demonstrate
that the increased proliferation of NB cells induced by β-AR
activation occurs through β2-AR signaling. However, the mechanisms
underlying the promotive role of the β2-AR pathway on NB cell
proliferation have not yet been clarified.
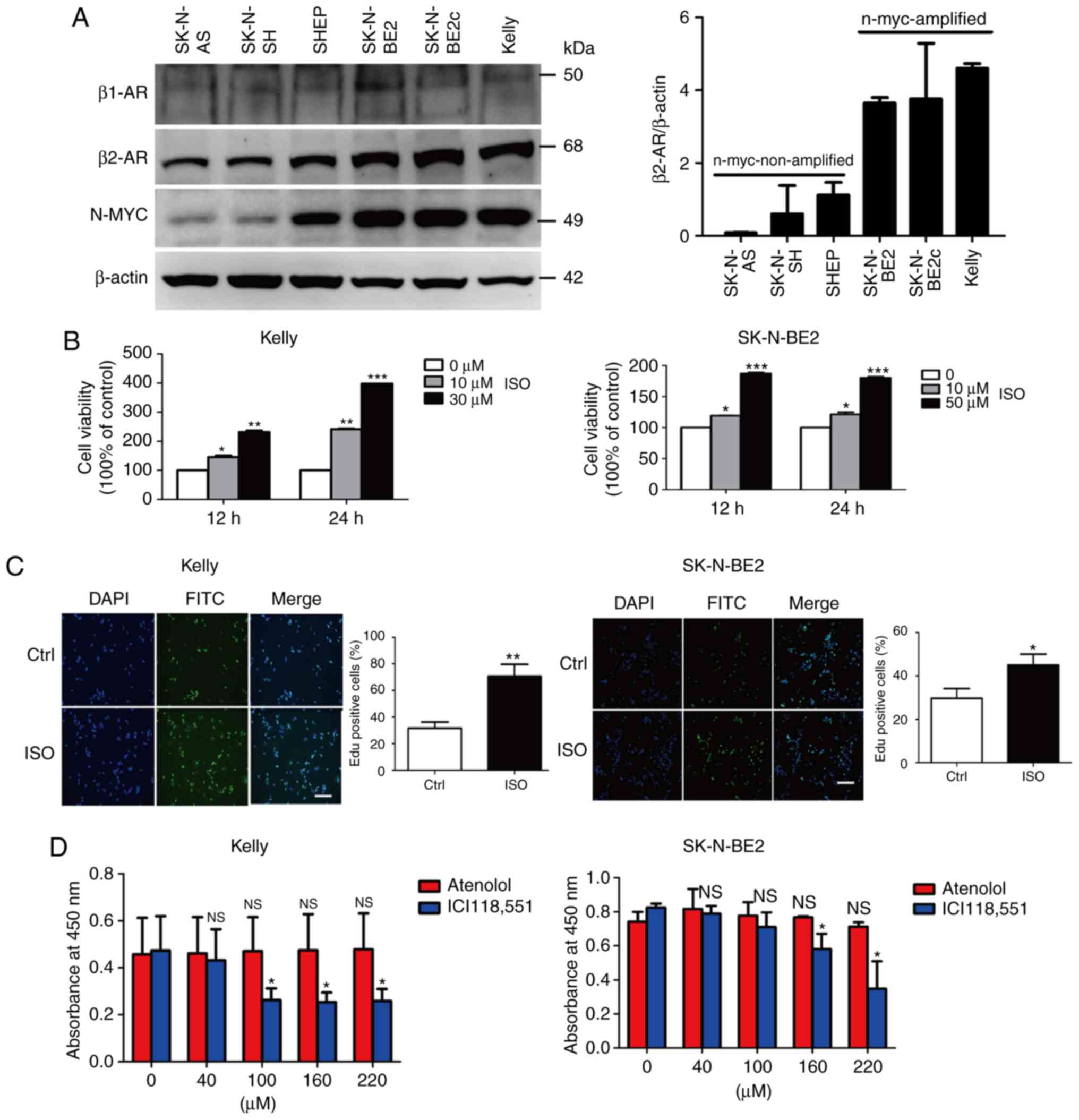 | Figure 2.Stimulation of NB cell lines with ISO
promotes their proliferation. (A) The protein expression of β1-AR,
β2-AR and N-MYC was evaluated in six types of NB cells using
western blot assay. (B) SK-N-BE2 cells were treated with 10 and 50
µM ISO, and Kelly cells were treated with 10 and 30 µM ISO for 12
and 24 h; CCK-8 assay was used to analyze the cell viability. (C)
SK-N-BE2 and Kelly cells were treated with 10 µM ISO, and EdU assay
was performed to test the proliferation of the ISO-treated cells.
(D) NB cells were treated with different concentrations of atenolol
or ICI118,551 for 24 h, and then cell viability was detected via
the CCK-8 assay. Scale bar, 100 µm. Data are expressed as the mean
± SEM of three experiments. Student's unpaired t-test was used to
assess the difference between two groups. Multigroup comparisons of
the means were carried out by one-way analysis of variance (ANOVA)
test with post hoc contrasts by a Newman-Keuls test. *P<0.05,
**P<0.01, ***P<0.001. β-AR, β-adrenergic receptor; ISO,
agonist isoprenaline; MYCN, MYCN proto-oncogene, BHLH transcription
factor; NB, neuroblastoma. |
Expression of autophagy markers is
significantly increased in NB
Considerable research has demonstrated the critical
role of the autophagy pathway in the initiation and progression of
tumors, and autophagy plays a dual role in tumorigenesis and
metastasis. To study the role of autophagy in NB, we assessed the
expression of the important autophagy markers ULK1, beclin-1 and
microtubule-associated protein 1 light chain 3-II (LC3-II) in the
clinical tumor samples. ULK1 is a serine/threonine-protein kinase,
and the activation of ULK1 can initiate autophagy via the
phosphorylation of beclin-1 and the subsequent enhancement of the
formation and activity of the autophagic complex
Atg14/beclin-1/VPS34 (39). LC3-II is
located in the autophagosome and promotes the fusion of
autophagosomes with lysosomes, leading to the formation of
autolysosomes. The IHC results showed that the levels of ULK1,
beclin-1 and LC3-II were significantly increased in the clinical
samples of NB, compared to these levels in the GN and GNB samples
(Fig. 3). Clinical correlation
analysis revealed that the expression of ULK1, beclin-1 and LC3-II
correlated positively with that of β2-AR, but not β1-AR (Tables II and III), implying that there is a crosstalk
between the β2-AR pathway and autophagy in NB. Thus, we further
investigated the role of β2-AR signaling in regulating autophagy in
NB cells.
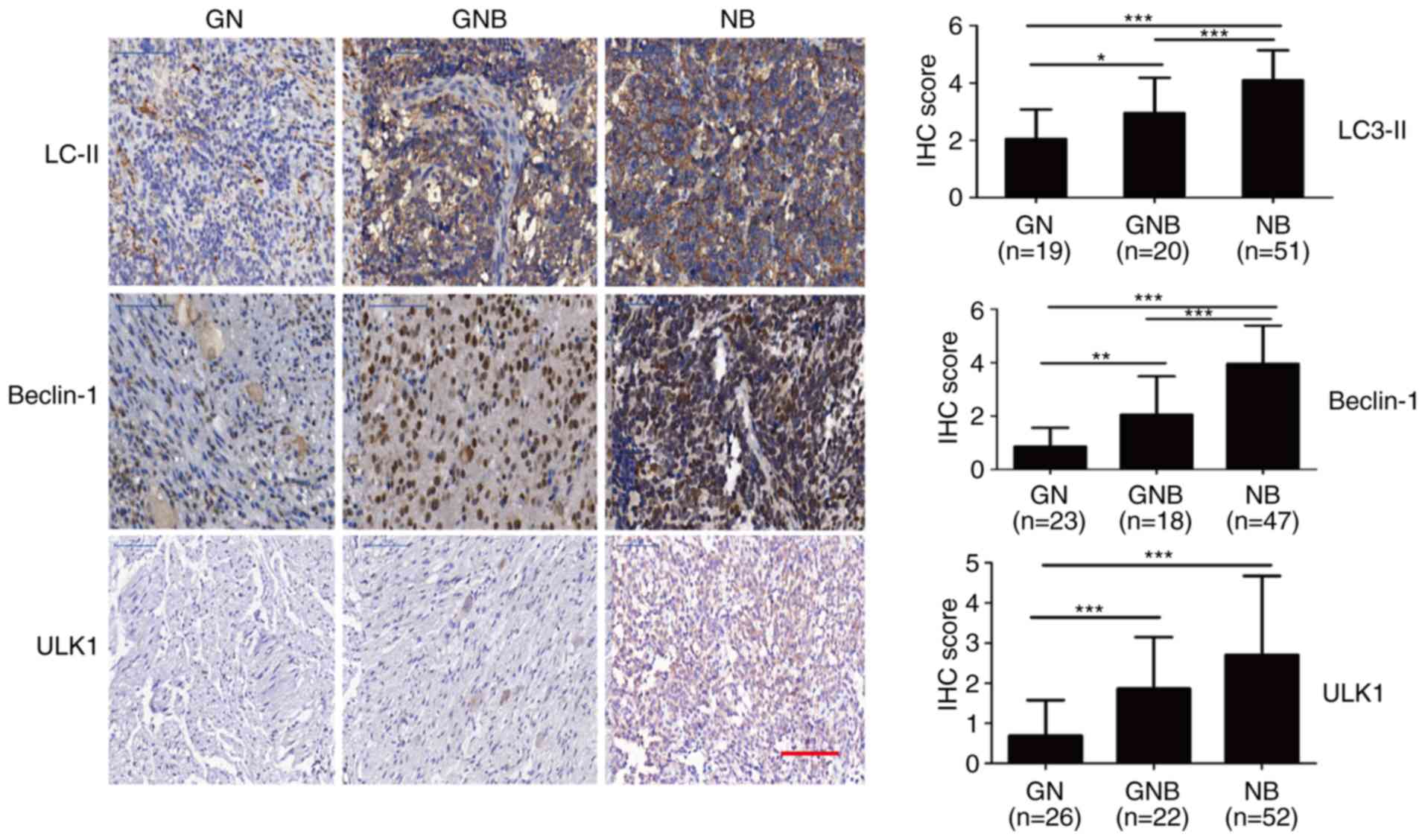 | Figure 3.β2-AR expression is positively
correlated with autophagy markers in NB. The expression levels of
the autophagy markers ULK1, beclin-1 and LC3-II were markedly
elevated in the NB samples, compared to the levels noted in the GN
and GNB samples. Scale bar, 100 µm. Student's unpaired t-test was
used to assess the difference between two groups. All values are
expressed as the mean ± SEM. The experiment was repeated three
times. *P<0.05, **P<0.01, ***P<0.001. IHC,
immunohistochemistry; NB, neuroblastoma; GN, ganglioneuroma; GNB,
ganglioneuroblastoma; β-AR, β-adrenergic receptor; ULK1,
unc-51-like autophagy kinase 1; LC3-II, microtubule-associated
protein 1 light chain 3-II. |
| Table II.Correlation of β1-AR with autophagic
markers. |
Table II.
Correlation of β1-AR with autophagic
markers.
|
| β1-AR |
|---|
|
|
|
|---|
|
| Mean ± SD | r | P-value |
|---|
| LC3-II | 0.88±4.15 | 0.31 | 0.05 |
| Beclin-1 | 4.07±0.34 | 0.24 | 0.14 |
| ULK1 | 3.28±1.77 | 0.24 | 0.14 |
| Table III.Correlation of β2-AR with autophagic
markers. |
Table III.
Correlation of β2-AR with autophagic
markers.
|
| β1-AR |
|---|
|
|
|
|---|
|
| Mean ± SD | r | P-value |
|---|
| LC3-II | 4.19±0.86 | 0.47 | 0.001 |
| Beclin-1 | 4.21±1.23 | 0.40 | 0.01 |
| ULK1 | 3.27±1.70 | 0.36 | 0.03 |
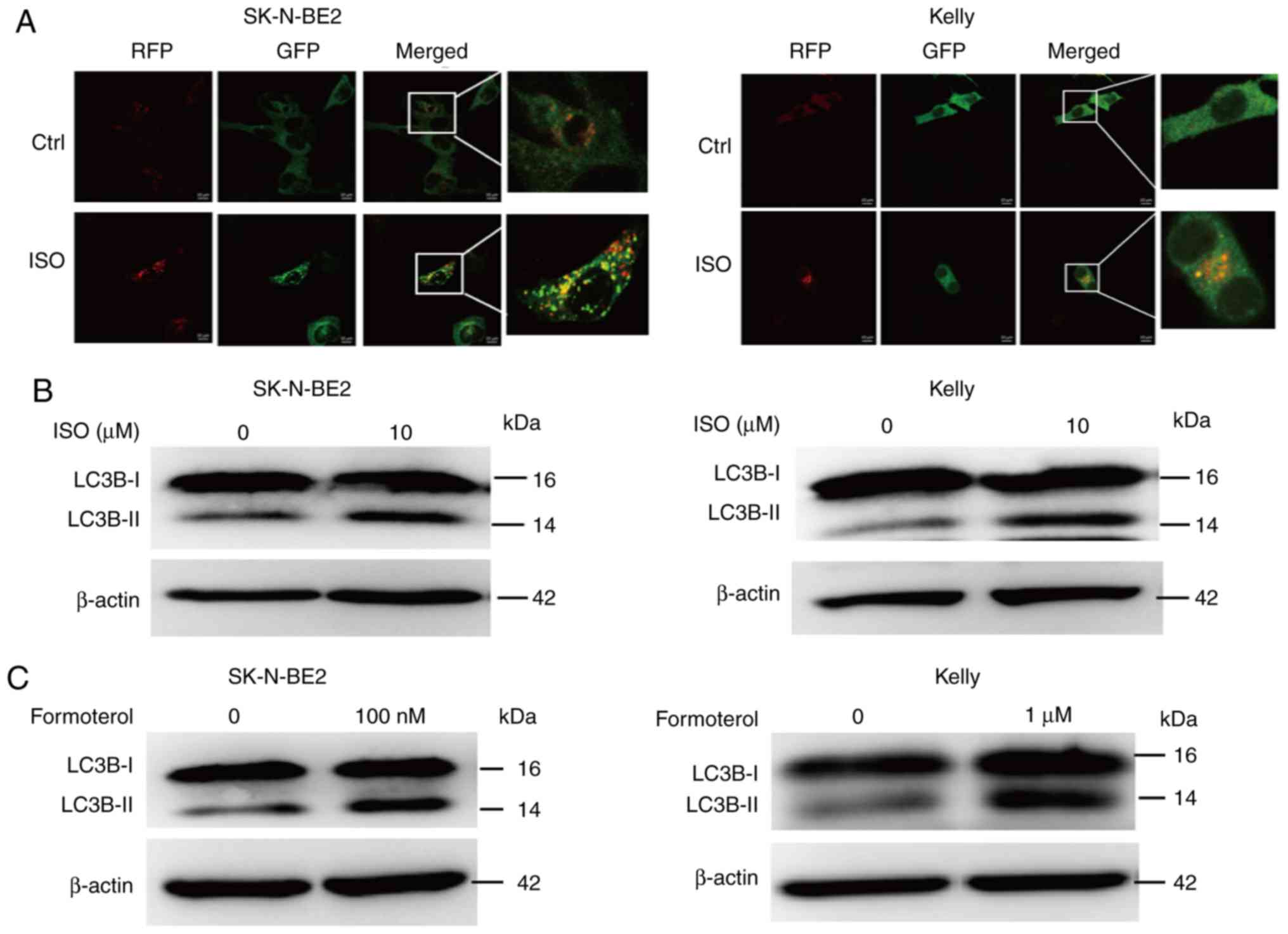
β2-AR signaling activates the
autophagy pathway in NB cells
To explore the role of β-AR in autophagy, NB cells
were stimulated with the β-AR agonist ISO, and then expression of
the autophagy marker LC3 was detected. Upon its synthesis, LC3 is
immediately cleaved by Atg4 and is converted to its cytoplasmic
form LC3-I. During autophagy activation, cytoplasmic LC3-I is
conjugated with phosphatidylethanolamine (PE) by the action of Atg3
and Atg7, and is then translocated into the membranes of
autophagosomes, turning into autophagic LC3-II; thus, it promotes
the elongation and maturation of autophagosomes. Then, mature
autophagosomes fuse with lysosomes to form autolysosomes, degrading
the components within themselves. LC3-II has always been used as a
molecular marker for the activation of autophagy. Firstly, we used
GFP-RFP-labeled adenovirus expressing LC3 to evaluate the change of
autophagic flux. During the late stage of the autophagy process,
the formation of autolysosomes causes a decrease in pH, which
results in the quenching of GFP, as GFP is sensitive to acidic
conditions. The results showed that ISO treatment improved the
accumulation of yellow (autophagosomes) and red (autolysosomes)
vacuoles (Fig. 4A). The western
blotting results suggested that the treatment of NB cells with ISO
promoted the expression of LC3B-II in the SK-N-BE2 and Kelly cell
lines (Fig. 4B), revealing that the
activation of β-AR signaling promotes the initiation of autophagy.
Treating NB cells with the specific β2-AR agonist formoterol also
promoted the expression of LC3B-II (Fig.
4C). These results suggest that the β2-AR pathway promotes the
activation of autophagy in NB cells.
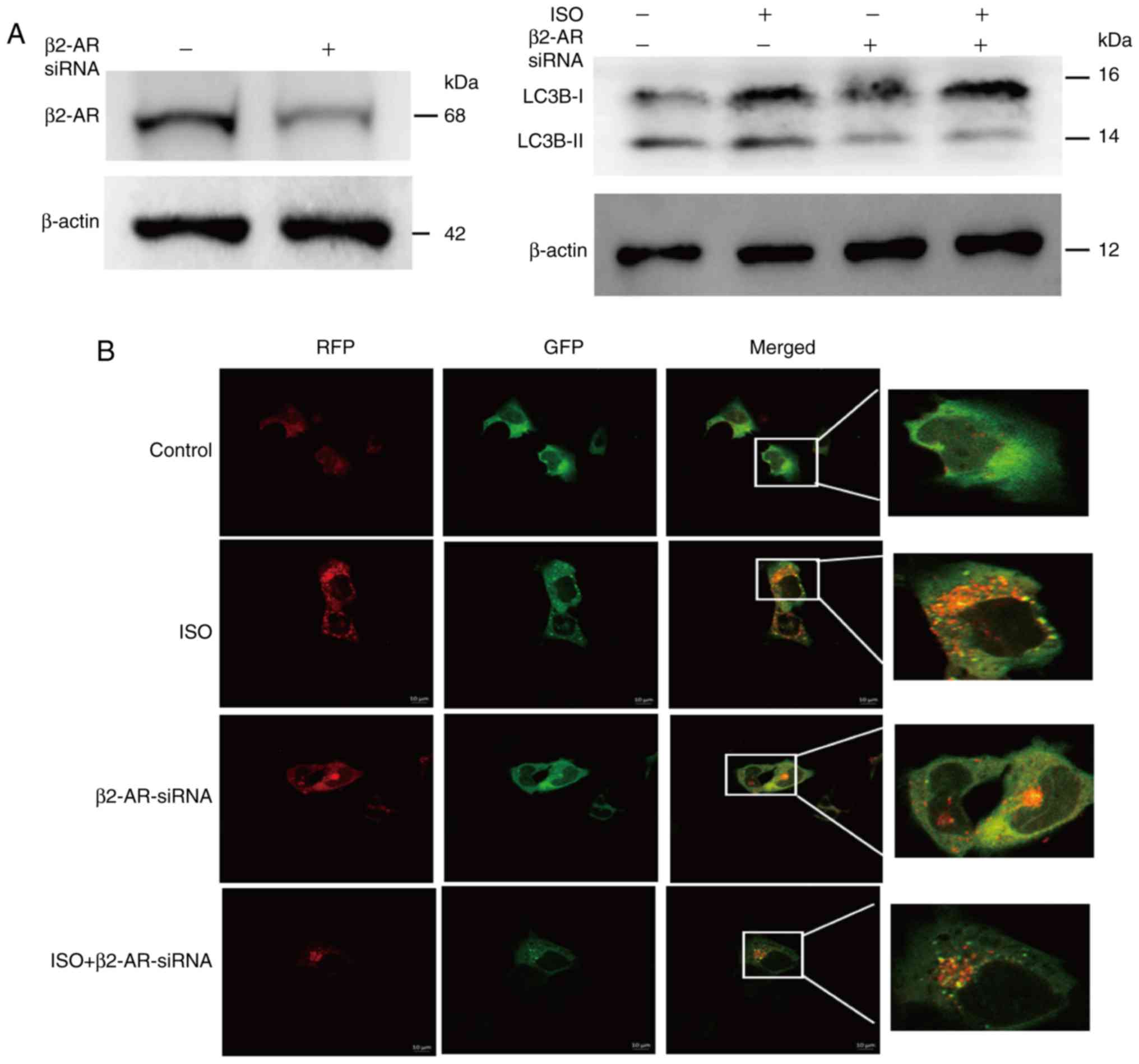
Knockdown of β2-AR expression inhibits
autophagy activation
To further investigate the role of β2-AR on the
autophagy of NB cells, we performed a genetic loss-of-function
assay to determine the ISO-induced effects of β2-AR on NB cell
autophagy. We transfected NB cells with β2-AR-specific siRNA to
inhibit the expression of β2-AR and non-specific scrambled siRNA as
a control. The western blot analysis showed that the knockdown of
β2-AR suppressed the ISO-induced expression of LC3B-II (Fig. 5A). The results of the
GFP-RFP-LC3-labeled adenovirus assay showed that inhibition of
β2-AR expression decreased the ISO-induced formation of yellow
(autophagosomes) and red (autolysosomes) vacuoles (Fig. 5B), suggesting that β-AR signaling
activates autophagy via the β2-AR pathway.
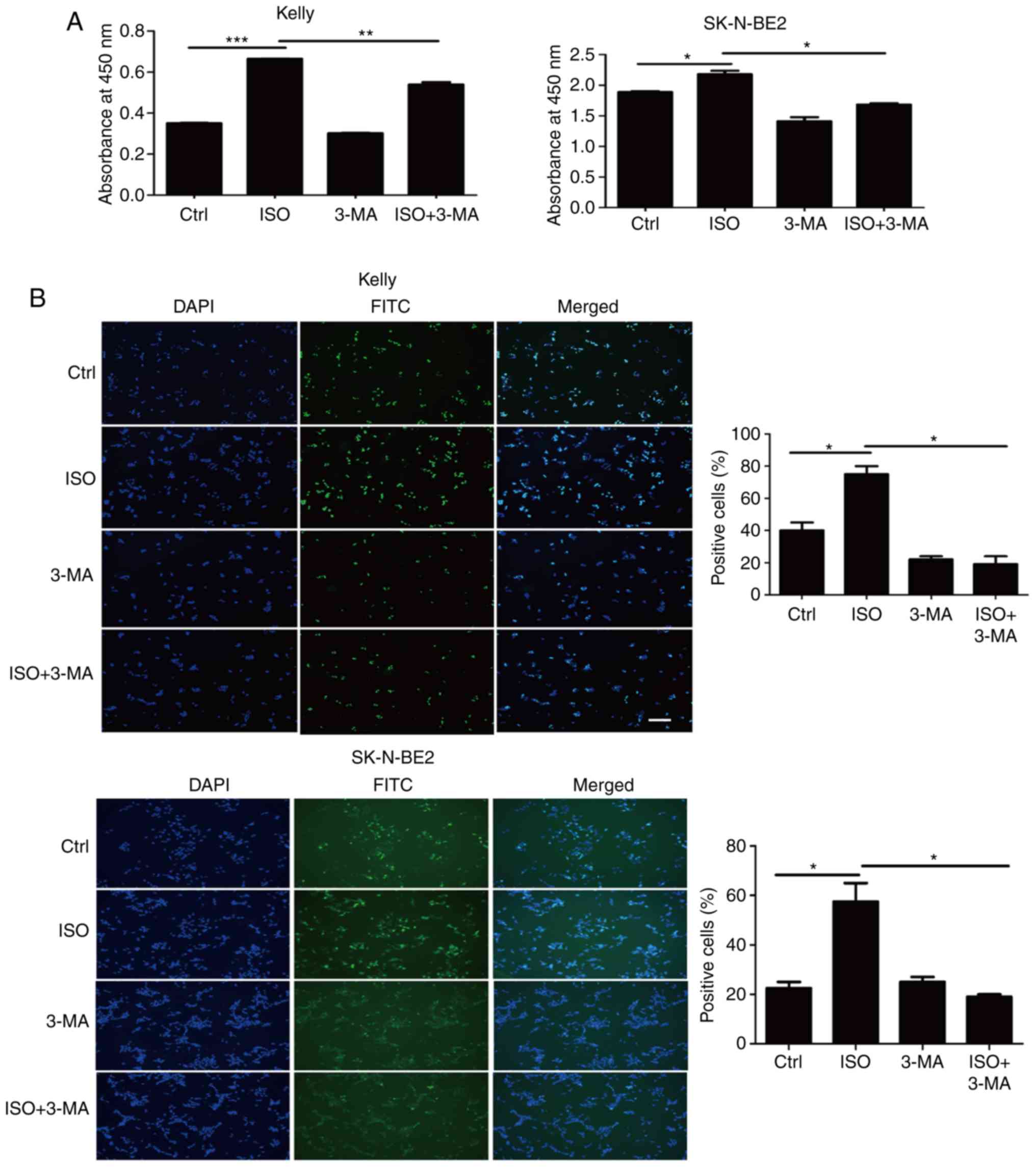
Inhibition of autophagy suppresses NB
cell growth
Our cellular functional assays demonstrated that
stimulation of NB cells with ISO promoted cell growth and activated
autophagy. Further clinical analysis revealed that there was a
positive correlation between β2-AR and the autophagy markers ULK1,
beclin-1, and LC3-II in the clinical samples of NB. Thus, we
proposed that the promotive effect of β2-AR on NB cell
proliferation may depend on autophagy. To verify this hypothesis,
we inhibited autophagy using 3-MA, which is a pharmacological
reagent commonly used to block the formation of the
beclin-1/Atg14/VPS34 complex, leading to the prevention of
autophagy activation. Our results of the CCK-8 and EdU assays
showed that treatment with 3-MA abolished the ISO-induced
enhancement of NB proliferation in the Kelly and SK-N-BE2 cell
lines (Fig. 6A and B). These results
suggest that the β2-AR signaling pathway promotes NB cell growth by
activating the autophagy pathway.
β2-AR activates autophagy via
CREB-regulated ULK1 expression
In order to further investigate the molecular
mechanism of β-AR signaling-stimulated autophagy, we investigated
the role of transcriptional factor cAMP-response element binding
protein (CREB) in regulating autophagy. CREB is a downstream
regulator of β-AR signaling. It has been reported that the
activation of CREB under starvation conditions could directly
target autophagy-related gene ulk1 to induce autophagy in
hepatic cells (40). As CREB is a
downstream factor of β-AR signaling, thus, we hypothesized that
CREB may also regulate ISO-activated autophagy in neuroblastoma.
Above all, we aimed to investigate the regulatory role of CREB in
ULK1 expression, which is important to clarify the regulatory
mechanism of CREB in autophagy of NB cells. To verify this
hypothesis, first of all, we detected the level of p-CREB in
clinical samples of GN, GNB and NB; the phosphorylation of CREB was
notably upregulated in the NB samples, compared to that noted in
the GN and GNB samples (Fig. 7A).
Treatment of NB cells with β-AR agonist ISO promoted the
phosphorylation of CREB and the autophagy-related protein ULK1 in
NB SK-N-BE2 cells (Fig. 7B). Then, we
used siRNA to inhibit the expression of CREB; the results of the
CREB knockdown assay revealed that CREB knockdown abolished the
ISO-induced enhancement of ULK1 and LC3B-II expression (Fig. 7C). As ULK1 expression is downregulated
after CREB knockdown, we hypothesized that CREB activates autophagy
by targeting ULK1. Further study demonstrated that use of the
specific ULK1 inhibitor SBI-0206965 inhibited the ISO-induced
expression of LC3B-II (Fig. 7D),
suggesting that ISO induces autophagy through CREB-mediated ULK1
expression. These results showed that β-AR activated autophagy via
CREB-regulated ULK1 expression.
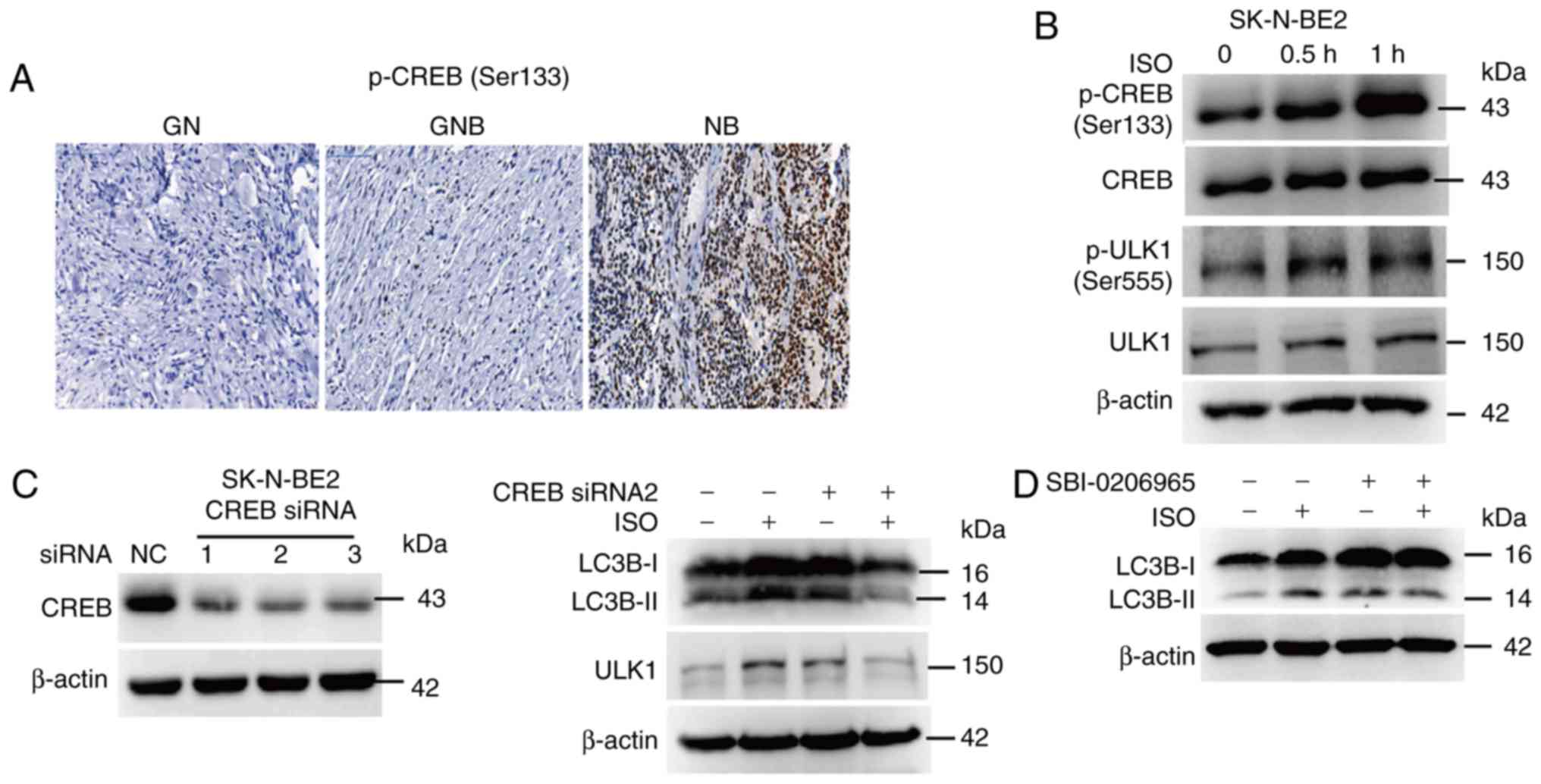 | Figure 7.β-AR signaling activates autophagy
via the CREB pathway in NB. (A) Expression of p-CREB was detected
by IHC in the clinical samples of GN, GNB and NB; scale bar, 100
µm. (B) SK-N-BE2 cells were treated with 10 µM ISO for 0.5 and 1 h;
western blotting was performed to detect p-CREB and p-ULK1
expression. (C) SK-N-BE2 cells were transfected with 100 nM CREB
siRNA for 24 h and then stimulated with 10 µM ISO for 24 h; western
blotting assay was performed to detect the expression of ULK1 and
LC3B-I and -II. (D) SK-N-BE2 cells were pretreated with 10 µM
SBI-0206965 for 1 h and then stimulated with 10 µM ISO for 24 h;
the expression of LC3B-I and -II was detected by western blotting
analysis. Experiments were replicated thrice and representative
images are shown. IHC, immunohistochemistry; β-AR, β-adrenergic
receptor; ISO, isoprenaline; NB, neuroblastoma; GN, ganglioneuroma;
GNB, ganglioneuroblastoma; CREB, cAMP-response element binding
protein; LC3-II, microtubule-associated protein 1 light chain
3-II. |
Discussion
Most importantly, the present study revealed that
β-AR signaling is aberrantly activated in NB, and the expression of
β1-AR and β2-AR is significantly enhanced in clinical samples of
NB, compared to the case for those of GN and GNB. Cellular
functional assays showed that β2-AR is the key β-adrenergic
receptor responsible for NB cell growth. Clinical correlation
analysis revealed that β2-AR expression was positively associated
with the autophagy markers ULK1, beclin-1 and LC3-II. Further
analyses of the underlying mechanisms uncovered that activation of
β2-AR stimulated the autophagy cascade, and that the promotive
effect of the β2-AR pathway on NB cell growth was autophagy
dependent. These findings broaden our understanding of the role of
autophagy and β2-AR in NB, and clarify the involvement of a novel
regulatory pathway of β2-AR-activated autophagy in NB cell
growth.
The initiation and aggressive progression of cancer
are closely dependent on the tumor microenvironment, in which tumor
cells can interact with other cells including myofibroblast cells,
macrophages and endothelial cells, to facilitate their growth and
distant dissemination (41). Many
studies have reported that nerves also play an important role in
modulating tumor cell survival and metastasis. For example, Magnon
et al (25) found that
pharmacological or surgical sympathectomy and genetic ablation of
stromal cell β2-AR and β3-AR prevented early-stage development of
prostate cancer cells in a mouse model, suggesting that the
microenvironmental nerves play a pivotal role in the initiation and
distant dissemination of prostate cancer. Apart from prostate
cancer, β2-AR signaling can enhance breast cancer cell invasion
(42). These findings revealed a
promotive role of β-AR signaling in tumors. In our study, we
demonstrated that the β-AR pathway plays a pro-survival role in NB
cells, as the expression of β1- and β2-AR was notably upregulated
in NB specimens, compared to that in the samples of the less
malignant tumors GN and GNB. Further functional studies suggested
that β2-AR is the key receptor responsible for NB cell growth.
Clinical stage analysis revealed that although the β1- and β2-AR
expression levels were increased in the NB samples, they were not
correlated with the different clinical stages of NB, indicating
that β-AR signaling is important for the tumorigenesis of NB, but
may not be crucial for the metastasis of NB. There may be other
signaling pathways regulating NB metastasis, which needs further
investigations.
The role of autophagy in tumors is context-dependent
and complex, and it is involved in the mediation of cancer stem
cell viability, resistance to anoikis, drug resistance, and tumor
growth and metastasis (43).
Autophagy can promote or suppress tumor growth and progression in
different types of cancer and microenvironmental settings. For
instance, Yang et al demonstrated that blocking autophagy
inhibited pancreatic ductal adenocarcinoma (PDAC) cell growth in an
autochthonous mouse model, and that autophagy plays an important
role in maintaining pancreatic tumor growth (44). In another study concerning
non-small-cell lung cancer, Cai et al reported that CK1α
suppressed non-small-cell lung cancer growth by stabilizing PTEN
and inducing autophagy and activating the tumor-suppressive
PTEN/AKT/FOXO3a/Atg7 axis (45),
suggesting an inhibitory role of autophagy in non-small-cell lung
cancer. In the present study, we demonstrated that autophagy plays
a promotive role in NB cell growth, as evidenced by the increased
expression of the important autophagy markers ULK1, beclin-1 and
LC3-II in clinical samples of NB, compared to that noted in GN and
GNB. This suggests that autophagy activation is crucial for the
tumorigenesis of NB. Further clinical correlation analysis revealed
that the expression of ULK1, beclin-1 and LC3-II is positively
correlated with the expression of β2-AR in clinical samples of NB,
suggesting that β2-AR signaling may promote NB cell growth by
activating the autophagy pathway. Cellular assays demonstrated that
β2-AR promotes NB cell survival by stimulating the autophagy
pathway, revealing that autophagy plays an important role in
β2-AR-mediated NB cell growth. Autophagy is also involved in tumor
metastasis; however, in our study, we only investigated the effect
of autophagy on NB cell growth, and not its role in NB cell
invasion and metastasis. Hence, further studies are needed to
evaluate the role of autophagy in the different clinical stages and
metastasis of NB.
Our research on the related underlying mechanisms
revealed that β2-AR signaling activates autophagy via the
downstream transcriptional factor CREB. As illustrated in our
study, ISO promotes the phosphorylation of CREB, which leads us to
speculate that β-AR activates autophagy via the CREB pathway. We
found that siRNA-mediated knockdown of CREB abolished the
ISO-induced accumulation of ULK1 and LC3-II. As ULK1 is a crucial
upstream kinase that can initiate the autophagy pathway, we
proposed that ISO activates autophagy through ULK1. Treatment of NB
cells with the specific ULK1 inhibitor SBI-0206965 inhibited the
ISO-induced enhancement of LC3B-II expression, suggesting that ISO
induces autophagy via ULK1. In conclusion, our findings
demonstrated the presence of a new regulatory mechanism underlying
the involvement of β2-AR and the autophagy cascade in NB cell
growth.
In conclusion, the present study demonstrated that
the β2-AR pathway plays an important role in the progression of NB,
and β2-AR signaling enhances NB cell proliferation via the
activation of autophagy. Our findings clarify the existence of a
novel crosstalk between autophagy and β2-AR in the regulation of NB
cell growth, and provide a novel therapeutic strategy for treating
NB more effectively by specifically targeting β2-AR and
autophagy.
Acknowledgements
We thank Professor Bo Li for providing the
neuroblastoma cell lines.
Funding
The present study was funded by the National Nature
Science Foundation of China (grant nos. 81572342, 81770808,
81872165, 81570871, 81570764, 81600641 and 81602199); the National
Key Sci-Tech Special Project of China (grant nos. 2013ZX09102-053
and 2015GKS-355); the Key Project of Nature Science Foundation of
Guangdong Province, China (grant nos. 2015A030311043 and
2016A030311035); the Guangdong Natural Science Fund (grant nos.
2014A020212023, 2014A030313073, 2015A030313029 and 2015A030313103);
the Guangdong Science Technology Project (grant nos. 2017A020215075
and 2015B090903063).
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
PJ and TY analyzed and interpreted the IHC staining
results of the NB tissue microarray. JD, MH, JX and CL designed and
drafted the manuscript. WQ, TZ, ZY, GG and XY finally approved the
version to be published. All authors read and approved the final
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
This study was approved by the Human Ethics
Committee of the Affiliated Guangzhou Women and Children's Medical
Center, Zhongshan School of Medicine. All patients gave informed
consent before participation in this study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
References
|
1
|
Brodeur GM: Neuroblastoma: Biological
insights into a clinical enigma. Nat Rev Cancer. 3:203–216. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Johnsen JI, Dyberg C, Fransson S and
Wickström M: Molecular mechanisms and therapeutic targets in
neuroblastoma. Pharmacol Res. 131:164–176. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Johnsen JI, Kogner P, Albihn A and
Henriksson MA: Embryonal neural tumours and cell death. Apoptosis.
14:424–438. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ratner N, Brodeur GM, Dale RC and Schor
NF: The ‘neuro’ of neuroblastoma: Neuroblastoma as a
neurodevelopmental disorder. Ann Neurol. 80:13–23. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Davidoff AM: Neuroblastoma. Semin Pediatr
Surg. 21:2–14. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Matthay KK, Maris JM, Schleiermacher G,
Nakagawara A, Mackall CL, Diller L and Weiss WA: Neuroblastoma. Nat
Rev Dis Primers. 2:160782016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen AM, Trout AT and Towbin AJ: A review
of neuroblastoma image-defined risk factors on magnetic resonance
imaging. Pediatr Radiol. 48:1337–1347. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cohn SL, Pearson AD, London WB, Monclair
T, Ambros PF, Brodeur GM, Faldum A, Hero B, Iehara T, Machin D, et
al: The international neuroblastoma risk group (INRG)
classification system: An INRG task force report. J Clin Oncol.
27:289–297. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Luksch R, Castellani MR, Collini P, De
Bernardi B, Conte M, Gambini C, Gandola L, Garaventa A, Biasoni D,
Podda M, et al: Neuroblastoma (peripheral neuroblastic tumours).
Crit Rev Oncol Hematol. 107:163–181. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gonzalez Malagon SG and Liu KJ: ALK and
GSK3: Shared features of neuroblastoma and neural crest cells. J
Exp Neurosci. 12:11790695187924992018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Westermark UK, Wilhelm M, Frenzel A and
Henriksson MA: The MYCN oncogene and differentiation in
neuroblastoma. Semin Cancer Biol. 21:256–266. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bell E, Chen L, Liu T, Marshall GM, Lunec
J and Tweddle DA: MYCN oncoprotein targets and their therapeutic
potential. Cancer Lett. 293:144–157. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cruickshank JM: The role of beta-blockers
in the treatment of hypertension. Adv Exp Med Biol. 956:149–166.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tank AW and Lee Wong D: Peripheral and
central effects of circulating catecholamines. Compr Physiol.
5:1–15. 2015.PubMed/NCBI
|
|
15
|
Lukewich MK, Rogers RC and Lomax AE:
Divergent neuroendocrine responses to localized and systemic
inflammation. Semin Immunol. 26:402–408. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Padro CJ and Sanders VM: Neuroendocrine
regulation of inflammation. Semin Immunol. 26:357–368. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shakola F, Suri P and Ruggiu M: Splicing
regulation of pro-inflammatory cytokines and chemokines: At the
interface of the neuroendocrine and immune systems. Biomolecules.
5:2073–2100. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lan H, Hoos LM, Liu L, Tetzloff G, Hu W,
Abbondanzo SJ, Vassileva G, Gustafson EL, Hedrick JA and Davis HR:
Lack of FFAR1/GPR40 does not protect mice from high-fat
diet-induced metabolic disease. Diabetes. 57:2999–3006. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim-Fuchs C, Le CP, Pimentel MA,
Shackleford D, Ferrari D, Angst E, Hollande F and Sloan EK: Chronic
stress accelerates pancreatic cancer growth and invasion: A
critical role for beta-adrenergic signaling in the pancreatic
microenvironment. Brain Behav Immun. 40:40–47. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Payne JK: State of the science: Stress,
inflammation, and cancer. Oncol Nurs Forum. 41:533–540. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dal Monte M, Casini G, Filippi L, Nicchia
GP, Svelto M and Bagnoli P: Functional involvement of
beta3-adrenergic receptors in melanoma growth and vascularization.
J Mol Med (Berl). 91:1407–1419. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen H, Liu D, Yang Z, Sun L, Deng Q, Yang
S, Qian L, Guo L, Yu M, Hu M, et al: Adrenergic signaling promotes
angiogenesis through endothelial cell-tumor cell crosstalk. Endocr
Relat Cancer. 21:783–795. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen D, Xing W, Hong J, Wang M, Huang Y,
Zhu C, Yuan Y and Zeng W: The beta2-adrenergic receptor is a
potential prognostic biomarker for human hepatocellular carcinoma
after curative resection. Ann Surg Oncol. 19:3556–3565. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu FQ, Fang T, Yu LX, Lv GS, Lv HW, Liang
D, Li T, Wang CZ, Tan YX, Ding J, et al: ADRB2 signaling promotes
HCC progression and sorafenib resistance by inhibiting autophagic
degradation of HIF1α. J Hepatol. 65:314–324. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Magnon C, Hall SJ, Lin J, Xue X, Gerber L,
Freedland SJ and Frenette PS: Autonomic nerve development
contributes to prostate cancer progression. Science.
341:12363612013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Barron TI, Connolly RM, Sharp L, Bennett K
and Visvanathan K: Beta blockers and breast cancer mortality: A
population-based study. J Clin Oncol. 29:2635–2644. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Grytli HH, Fagerland MW, Fossa SD and
Tasken KA: Association between use of beta-blockers and prostate
cancer-specific survival: A cohort study of 3561 prostate cancer
patients with high-risk or metastatic disease. Eur Urol.
65:635–641. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Efeyan A, Comb WC and Sabatini DM:
Nutrient-sensing mechanisms and pathways. Nature. 517:302–310.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang X, Overholtzer M and Thompson CB:
Autophagy in cellular metabolism and cancer. J Clin Invest.
125:47–54. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wirawan E, Vanden Berghe T, Lippens S,
Agostinis P and Vandenabeele P: Autophagy: For better or for worse.
Cell Res. 22:43–61. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Scrivo A, Bourdenx M, Pampliega O and
Cuervo AM: Selective autophagy as a potential therapeutic target
for neurodegenerative disorders. Lancet Neurol. 17:802–815. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim KH and Lee MS: Autophagy - a key
player in cellular and body metabolism. Nat Rev Endocrinol.
10:322–337. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
White E: The role for autophagy in cancer.
J Clin Invest. 125:42–46. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Menzies FM, Fleming A, Caricasole A, Bento
CF, Andrews SP, Ashkenazi A, Fullgrabe J, Jackson A, Jimenez
Sanchez M, Karabiyik C, et al: Autophagy and neurodegeneration:
Pathogenic mechanisms and therapeutic opportunities. Neuron.
93:1015–1034. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mizushima N: A(beta) generation in
autophagic vacuoles. J Cell Biol. 171:15–17. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guo JY, Karsli-Uzunbas G, Mathew R, Aisner
SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, et
al: Autophagy suppresses progression of K-ras-induced lung tumors
to oncocytomas and maintains lipid homeostasis. Genes Dev.
27:1447–1461. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mathew R, Karp CM, Beaudoin B, Vuong N,
Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al:
Autophagy suppresses tumorigenesis through elimination of p62.
Cell. 137:1062–1075. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dower CM, Bhat N, Gebru MT, Chen L, Wills
CA, Miller BA and Wang HG: Targeted inhibition of ULK1 promotes
apoptosis and suppresses tumor growth and metastasis in
neuroblastoma. Mol Cancer Ther. 17:2365–2376. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Russell RC, Tian Y, Yuan H, Park HW, Chang
YY, Kim J, Kim H, Neufeld TP, Dillin A and Guan KL: ULK1 induces
autophagy by phosphorylating Beclin-1 and activating VPS34 lipid
kinase. Nat Cell Biol. 15:741–750. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar
S, Sun X, Yoon G, Kang Y, Zhong W, et al: Transcriptional
regulation of autophagy by an FXR-CREB axis. Nature. 516:108–111.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mierke CT: The fundamental role of
mechanical properties in the progression of cancer disease and
inflammation. Rep Prog Phy. 77:0766022014. View Article : Google Scholar
|
|
42
|
Creed SJ, Le CP, Hassan M, Pon CK, Albold
S, Chan KT, Berginski ME, Huang Z, Bear JE, Lane JR, et al:
β2-adrenoceptor signaling regulates invadopodia formation to
enhance tumor cell invasion. Breast Cancer Res. 17:1452015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mowers EE, Sharifi MN and Macleod KF:
Autophagy in cancer metastasis. Oncogene. 36:1619–1630. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang A, Herter-Sprie G, Zhang H, Lin EY,
Biancur D, Wang X, Deng J, Hai J, Yang S, Wong KK, et al: Autophagy
sustains pancreatic cancer growth through both cell-autonomous and
nonautonomous mechanisms. Cancer Discov. 8:276–287. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cai J, Li R, Xu X, Zhang L, Lian R, Fang
L, Huang Y, Feng X, Liu X, Li X, et al: CK1α suppresses lung tumour
growth by stabilizing PTEN and inducing autophagy. Nat cell Biol.
20:465–478. 2018. View Article : Google Scholar : PubMed/NCBI
|