Introduction
During tumor development, oncogenic cells are under
the surveillance of the host immune system. This leads to the
selection of cells with adaptations that confer a survival
advantage (1). The reduced expression
of surface major histocompatibility complex class I (MHC-I)
molecules is one of the most frequent mechanisms of evasion from
immune reactions in different human tumors, ranging from 15% in
renal carcinoma to 93% in lung cancer and >75% in most types of
epithelial-derived tumors (2). The
majority of MHC-I aberrations are reversible and are often
associated with defects in the antigen-processing machinery (APM).
In that case, reduced MHC-I expression is usually caused by
epigenetic silencing of the genes coding for MHC-I heavy chains or
APM components (3) and may be
restored by cytokines [e.g., interferon (IFN)-γ or tumor-necrosis
factor (TNF)-α]. Mutations or chromosomal aberrations that affect
genes encoding MHC-I heavy chains, β-2 microglobulin, proteins
regulating MHC-I expression, or APM components, are responsible for
the irreversible changes in surface MHC-I expression detected in
approximately one-third of human tumors (4). An analysis of genomic datasets generated
from thousands of solid tumors including samples of 18 tumor types
revealed an association of immune cytolytic activity based on
granzyme A and perforin expression with mutations in the invariant
MHC-I chain (β-2 microglobulin) and MHC-I (HLA) loci (5), further confirming that reduced
production of MHC-I molecules is an important mechanism of tumor
immune evasion. However, despite the frequency and clinical
importance of MHC-I downregulation, overcoming this escape
mechanism by cancer immunotherapy has not been sufficiently
investigated.
The efficacy of cancer immunotherapy may be enhanced
by a combination of different immunotherapeutic approaches that
include activation of both adaptive and innate immunity and
inhibition of immunosuppressive mechanisms (6,7). Such
combinations have achieved a notable antitumor effect in
preclinical models (8,9). However, although some mechanisms
contributing to the antitumor effect are MHC-I-independent, the
association of MHC-I expression on tumor cells with treatment
efficacy has not been investigated. In our previous study, we
examined combined immunotherapy of tumors induced in mice by the
TC-1/A9 cells characterized by reversible MHC-I downregulation
(10), and found that the combination
of DNA immunization with either α-galactosylceramide (GalCer) or
the synthetic oligodeoxynucleotide ODN1826, carrying
immunostimulatory CpG motifs, induced temporary tumor regression.
CD8+ T cells, IFN-γ, and NK1.1+ cells were
involved in this response. For ODN1826, antitumor activity of
M1-polarized macrophages was also suggested.
In the present study, TC-1/dB2m cells with a
deactivated β-2 microglobulin gene (B2m) were developed as a
model of tumor cells with irreversible MHC-I downregulation in
order to examine tumor growth, immune cell infiltration and
sensitivity to immunotherapy by DNA vaccination combined with
ODN1826 injection.
Materials and methods
Animals
A total of 250 female C57BL/6NCrl mice
(7–8-weeks-old and weighing 17–22 g) were obtained from Charles
River Laboratories to be used in animal experiments after at least
2 weeks of acclimatization. The mice were housed (n=5 per cage) and
maintained under specific pathogen-free conditions and a 12/12-h
light/dark cycle in a temperature-controlled room (20–24°C) with a
relative humidity of 50–60%. The animals had access to food and
water ad libitum. All animal handling procedures complied to
the guidelines for the proper treatment of laboratory animals at
the Czech Center for Phenogenomics (BIOCEV).
Cell lines
TC-1 tumor cells (Cellosaurus ID: CVCL_4699; kindly
provided by T.-C. Wu, Johns Hopkins University) were prepared by
transformation of C57BL/6 mouse primary lung cells with human
papillomavirus (HPV) 16 E6/E7 oncogenes and activated
H-ras (11). TC-1/A9 cells
with reversibly downregulated MHC-I expression were derived from
TC-1 cells as described previously (12). The cells were grown in high-glucose
Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich; Merck
KGaA) supplemented with 10% fetal bovine serum (Biosera), 2 mM
L-glutamine, 100 U/ml penicillin and 100 µg/ml
streptomycin.
Plasmids
The pBSC (13) and
pBSC/PADRE.E7GGG (14) plasmids were
used in immunization experiments. The pBSC/PADRE.E7GGG plasmid
contains the HPV16 E7 oncogene with three point mutations in
the pRb-binding site (E7GGG) (13)
and the helper Pan HLA-DR reactive epitope (PADRE) designed in
silico (15).
Deactivation of B2m with the
CRISPR/Cas9 system
The deactivation of the B2m gene was
performed with the GeneArt CRISPR Nuclease Vector Kit (Thermo
Fisher Scientific, Inc.). The target site
5′-CCGAGCCCAAGACCGTCTAC-3′ located in exon 2 was designed using an
online software (http://crispr.mit.edu/) and cloned into the CRISPR
nuclease vector as the corresponding annealed oligonucleotides
synthetized by Integrated DNA Technologies. The resultant plasmid
was multiplied in Escherichia coli XL-1 Blue cells, isolated
by the NucleoSpin Plasmid Kit (Macherey-Nagel), and verified by
sequencing with the BigDye Terminator v3.1 Cycle Sequencing Kit
(Applied Biosystems; Thermo Fisher Scientific, Inc.). This plasmid
was transfected into TC-1 cells by Lipofectamine 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.). The cells carrying the transfected
vector were selected by magnetic beads (Dynabeads FlowComp Human
CD4; Thermo Fisher Scientific, Inc.) based on the human CD4
reporter gene encoded by the vector. Clones were prepared from
isolated cells and analyzed by flow cytometry for MHC-I and β-2
microglobulin surface expression. The resultant clone with the
deactivated B2m gene was designated as TC-1/dB2m.
Treatment with IFN-γ
Cells were stimulated with 200 U/ml mouse
recombinant IFN-γ (PeproTech, Inc.) for 48 h.
Flow cytometry
Cells grown in tissue culture were harvested with
trypsin, washed with PBS, and stained with the following monoclonal
antibodies diluted in FACS buffer (2% fetal bovine serum and 0.03%
sodium azide in PBS) at 4°C for 30 min: FITC-labeled mouse anti-B2m
(clone S19.8; 1:100 dilution; Santa Cruz Biotechnology, Inc.),
FITC-labeled mouse anti-mouse H-2Kb (clone CTKb; 1:200
dilution; BD Pharmingen; BD Biosciences), FITC-labeled mouse
anti-mouse H-2Db (clone 28-14-8; 1:400 dilution; BD
Pharmingen; BD Biosciences), or PE-labeled rat anti-mouse CD1d
(clone 1B1; 1:100 dilution; BD Pharmingen; BD Biosciences).
Subsequently, the cells were washed twice and measured on an
LSRFortessa flow cytometer (BD Biosciences). The results were
analyzed using FlowJo software v10.5.3 (BD Biosciences).
For analysis of tumor-infiltrating cells,
single-cell suspensions were prepared from tumors with a longest
diameter of 5–10 mm by using the gentleMACS Octo Dissociator
(Miltenyi Biotec, GmbH), as described previously (16). The obtained cells were stained with
two panels of fluorescence-labeled antibodies (Table I) to identify several subpopulations
of lymphoid and myeloid cells (gating strategy in Figs. S1 and S2, respectively). Viability staining was
performed with Fixable Viability Dye eFluor 455UV (eBioscience;
Thermo Fisher Scientific, Inc.) in PBS, prior to surface staining.
To detect the nuclear Foxp3 transcription factor, the cells were
treated with Fixation/Permeabilization Concentrate (eBioscience;
Thermo Fisher Scientific, Inc.) diluted 1:3 with
Fixation/Permeabilization Diluent (eBioscience; Thermo Fisher
Scientific, Inc.). Fixation and permeabilization were followed by a
washing step with permeabilization buffer (eBioscience; Thermo
Fisher Scientific, Inc.) and Foxp3 staining.
 | Table I.Antibodies used for flow
cytometry. |
Table I.
Antibodies used for flow
cytometry.
| Antigen | Conjugate | Clone | Source | Staining | Panels |
|---|
| CD11b | BV421 | M1/70 | BioLegend | Surface |
| a |
| CD11c | APC-Cy7 | N418 | BioLegend | Surface |
| a |
| CD25 | APC | PC61.5 | eBiosciences | Surface | a |
|
| CD3 | APC-Cy7 | 145-2C11 | BioLegend | Surface | a |
|
| CD317 | APC | 927 | BioLegend | Surface |
| a |
| CD4 | BV510 | RM4-5 | BioLegend | Surface | a |
|
| CD45 | Alexa Fluor
700 | 30-F11 | BioLegend | Surface | a | a |
| CD8 | FITC | 53-6.7 | BD Pharmingen | Surface | a |
|
| F4/80 | BV510 | BM8 | BioLegend | Surface |
| a |
| Foxp3 | PE | FJK-16s | eBiosciences | Nuclear | a |
|
| Ly6C | BV786 | HK1.4 | BioLegend | Surface |
| a |
| Ly6G | FITC | 1A8 | BioLegend | Surface |
| a |
| MHC-II | PE-Cy7 | 114.15.2 | BioLegend | Surface |
| a |
| NK1.1 | BV650 | PK136 | BioLegend | Surface | a |
|
| PD-1 |
PE-Cy7/PEb | 29F.1A12 | BioLegend | Surface | a | b |
| PD-L1 | BV650 | 10F.9G2 | BioLegend | Surface |
| a |
| TCR γ/δ | BV605 | GL3 | BioLegend | Surface | a |
|
In vitro cell proliferation assay
Approximately half a million live cells were seeded
into three 10-cm dishes. The cells were counted after 24, 48 or 72
h (one dish at each interval) using a hemocytometer. Proliferation
was evaluated by non-linear regression for exponential growth.
Calculations were performed using Prism 8 software (GraphPad
Software, Inc.).
Preparation of gene gun
cartridges
Plasmid DNA was coated onto 1-µm gold
particles (Bio-Rad Laboratories. Inc.) according to the
manufacturer's recommendations. Each cartridge contained 1
µg DNA coated onto 0.5 mg of gold particles (13).
Oncogenicity of TC-1/dB2m cells
Counts of 3×104, 1×105 or
3×105 TC-1/dB2m cells suspended in 0.15 ml PBS were
subcutaneously (s.c.) inoculated into the backs of mice (n=5 per
group) under anesthesia with ketamine (100 mg/kg) and xylazine (16
mg/kg). Tumor growth was measured three times per week, and tumor
size was calculated using the formula (height × length × width)
π/6. To determine the effect of B2m deactivation on the
metastatic capacity of the TC-1-derived cells, the mice were s.c.
injected with 3×105 TC-1/dB2m cells. When the size of
the tumors reached 2 cm in any of the measured dimensions, the mice
were sacrificed by cervical dislocation and dissected. The lungs
were inspected for macrometastases, stained with hematoxylin and
eosin, and examined under a light microscope in the Czech Center
for Phenogenomics to detect micrometastases.
Immunization experiments
The mice were immunized using a gene gun (Bio-Rad
Laboratories, Inc.) three times with two shots each delivering 1
µg of plasmid DNA. The DNA was applied into the shaven skin
of the abdomen at a discharge pressure of 400 psi. In preventive
immunization experiments, mice (n=5 per group) were first immunized
with the pBSC/PADRE.E7GGG plasmid at 1-week interval. The pBSC
plasmid was used as a negative control. One week after the last
immunization, 3×105 TC-1 or TC-1/dB2m cells or
3×104 TC-1/A9 cells were s.c. inoculated into the backs
of the mice. In the combined immunotherapy experiments,
3×105 TC-1/dB2m cells were s.c. injected and DNA
immunization was performed after 3, 6 and 10 days. DNA vaccination
was combined with an intraperitoneal (i.p.) injection of 50
µg ODN1826 (Generi Biotech) or 2 µg GalCer (Abcam) diluted
in 200 µl PBS. These immunostimulants were injected in three
or five doses, as indicated in Fig.
3. Control mice received PBS.
In vivo depletion experiments
Different subpopulations of immune cells were
depleted with the following antibodies (Bio X Cell) injected i.p.
in a volume of 200 µl of PBS: 100 µg anti-CD4 (clone
GK1.5), 100 µg anti-CD8 (clone 2.43), or 100 µg
anti-NK1.1 (clone PK136). These antibodies were applied 2 days
before and after inoculation of tumor cells (3×104 TC-1
cells or 3×105 TC-1/dB2m cells), and then at 3–4-day
intervals for 5 weeks. Moreover, 1 mg carrageenan IV
(Sigma-Aldrich; Merck KGaA) dissolved in 200 µl PBS was
inoculated on the same days to deplete macrophages. For
neutralization of IFN-γ, 300 µg anti-IFN-γ (clone P4-6A2;
Bio X Cell) was injected 2 days prior and 5, 12, 19 and 26 days
after tumor cell inoculation.
In immunotherapeutic experiments, antibodies and
carrageenan were administered from the 7th day onwards after
inoculation of tumor cells.
Statistical analysis
Cell proliferation and tumor growth were evaluated
by two-way analysis of variance (ANOVA) and the Sidak multiple
comparisons test. Intergroup comparisons of flow cytometry data
were made by one-way ANOVA and Dunnett's multiple comparisons test.
Calculations were performed using GraphPad Prism 8 (GraphPad
Software, Inc.), and the results were considered statistically
significant at P<0.05.
Results
In vitro characterization of the TC-1
clone with B2m gene deactivation
To abrogate MHC-I expression on TC-1 tumor cells,
the B2m gene was deactivated by the CRISPR/Cas9 system and
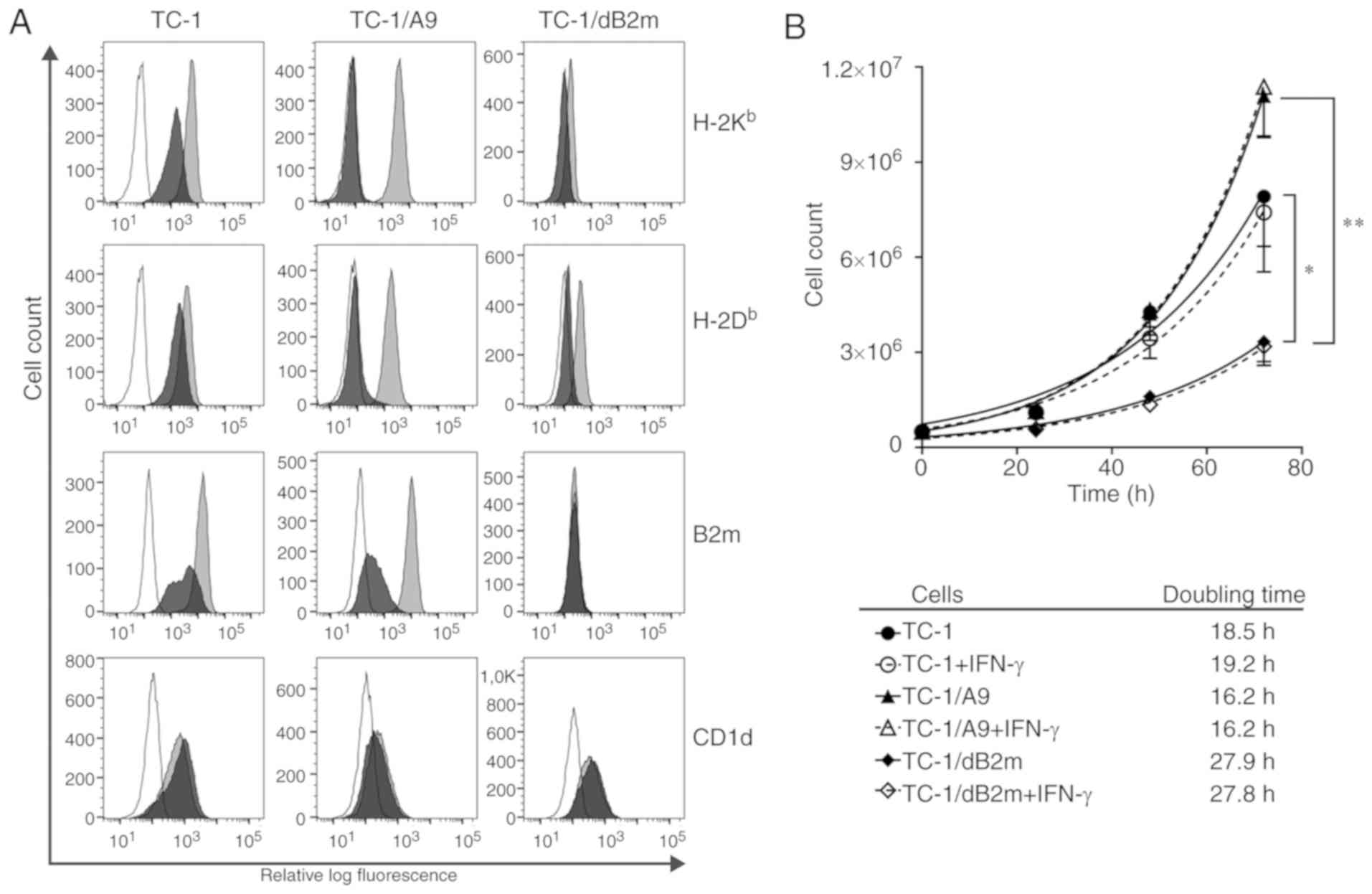
the TC-1/dB2m cell line was derived. These cells did not express
β-2 microglobulin or MHC-I heavy chains on their surface (Fig. 1A). Following stimulation with IFN-γ,
β-2 microglobulin was still absent on TC-1/dB2m cells, but weak
MHC-I expression was induced, particularly for H-2Db
molecules.
As β-2 microglobulin also forms a complex with the
CD1d molecules expressed on the cell surface and CD1d expression
has been demonstrated on TC-1 and TC-1/A9 cells (17), CD1d expression was detected on
TC-1/dB2m cells. B2m deactivation did not prevent surface
expression of the CD1d molecules (Fig.
1A).
Next, the proliferation rates of TC-1/dB2m, TC-1 and
TC-1/A9 cells were compared. The doubling time of TC-1/dB2m cells
was significantly increased by ~9 and 11 h in comparison with TC-1
and TC-1/A9 cells, respectively (Fig.
1B). Incubation with IFN-γ slightly reduced TC-1 cell
proliferation, but did not affect TC-1/A9 and TC-1/dB2m cells.
In summary, deactivation of the B2m gene in
the TC-1/dB2m cell line resulted in abrogation of β-2 microglobulin
production and downregulation of surface MHC-I expression, which
was associated with reduced proliferation rate.
Deactivation of the B2m gene alters
oncogenicity/immunogenicity of tumor cells
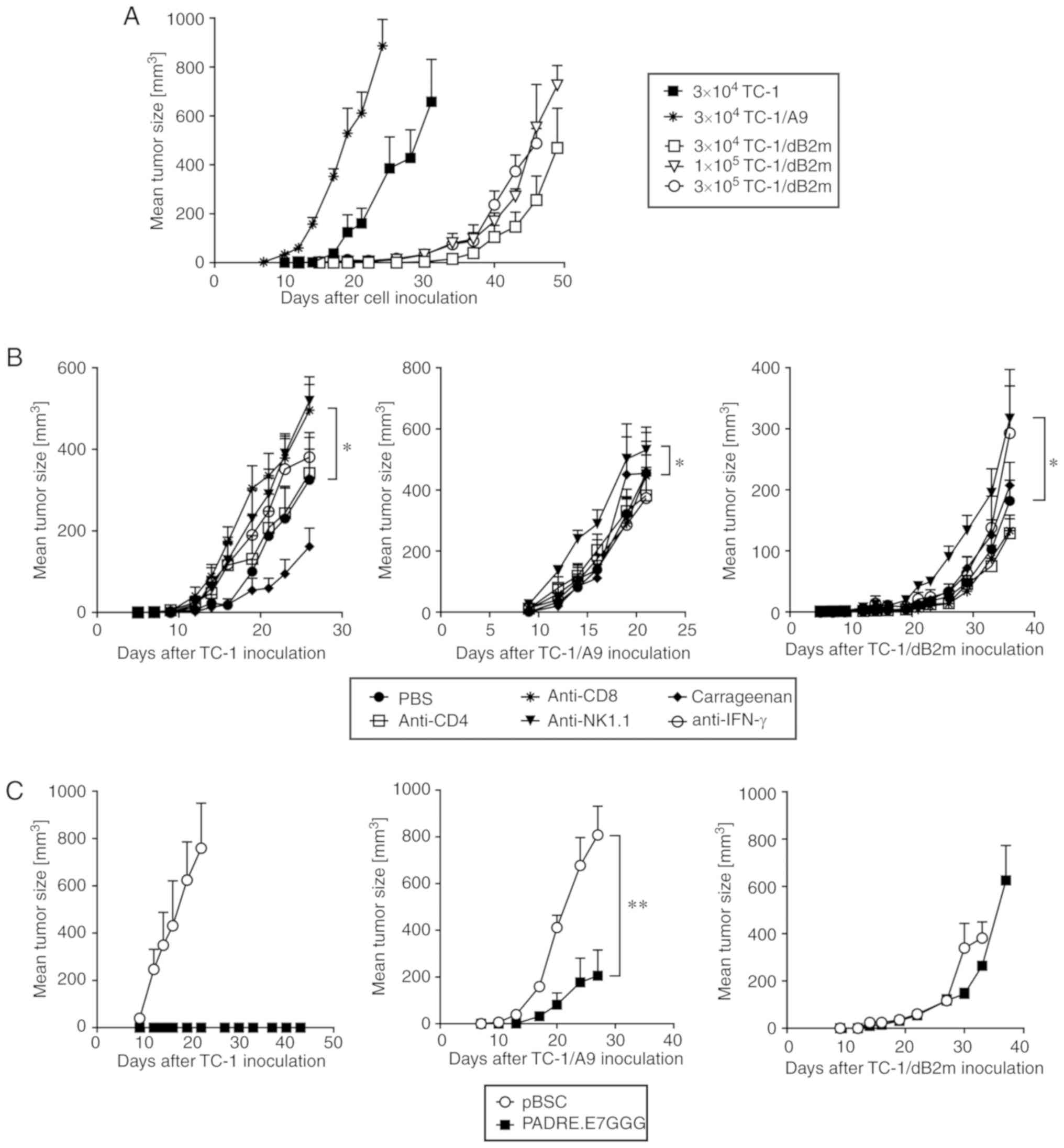
To induce tumor formation, mice were inoculated s.c.
with 3×104 TC-1 or TC-1/A9 cells. As a pilot experiment
demonstrated delayed growth of tumors induced by this number of
TC-1/dB2m cells, we also tested inoculation using higher numbers:
1×105 and 3×105 TC-1/dB2m cells (Fig. 2A). However, even for the highest
TC-1/dB2m cell number (3×105), tumor growth was delayed
by ~20 days compared with tumors induced by 3×104 TC-1
cells. Moreover, tumors developing from TC-1/dB2m cells were more
elongated in comparison with TC-1-induced tumors, particularly at
the early growth phase. Spontaneous lung metastasis formation was
not observed after s.c. induced tumors.
To identify the immune cells that could inhibit the
growth of TC-1/dB2m-induced tumors, some subpopulations were
depleted in vivo by monoclonal antibodies (Fig. 2B). For TC-1-induced tumors,
CD8+ and NK1.1+ cells were found to
contribute to the reduction of tumor growth, and macrophages
supported this growth. NK1.1+ cells also inhibited
TC-1/A9 and TC-1/dB2m tumors, but there was no involvement of
CD8+ cells or macrophages. In all types of tumors, the
depletion of CD4+ cells or neutralization of IFN-γ did
not significantly affect tumor growth (the antitumor effect of
IFN-γ was only apparent during the initial phase of the growth of
TC-1 tumors).
Next, the sensitivity of TC-1/dB2m-induced tumors to
adaptive immunity activated against the HPV16 E7 oncoprotein was
evaluated. While TC-1 tumors are highly sensitive to therapeutic
DNA immunization by the PADRE.E7GGG vaccine (14), the sensitivity of TC-1/A9 tumors to
DNA vaccination is low (10). After
more efficient preventive immunization with the PADRE.E7GGG gene,
the growth of TC-1/A9-induced tumors was significantly reduced, but
the TC-1/dB2m tumors were resistant to DNA vaccination (Fig. 2C). The development of control TC-1
tumors was inhibited in all mice.
Collectively, these findings indicate that
deactivation of the B2m gene resulted in delayed tumor
growth and resistance to adaptive immunity.
TC-1/dB2m cells are slightly sensitive
to combined immunotherapy
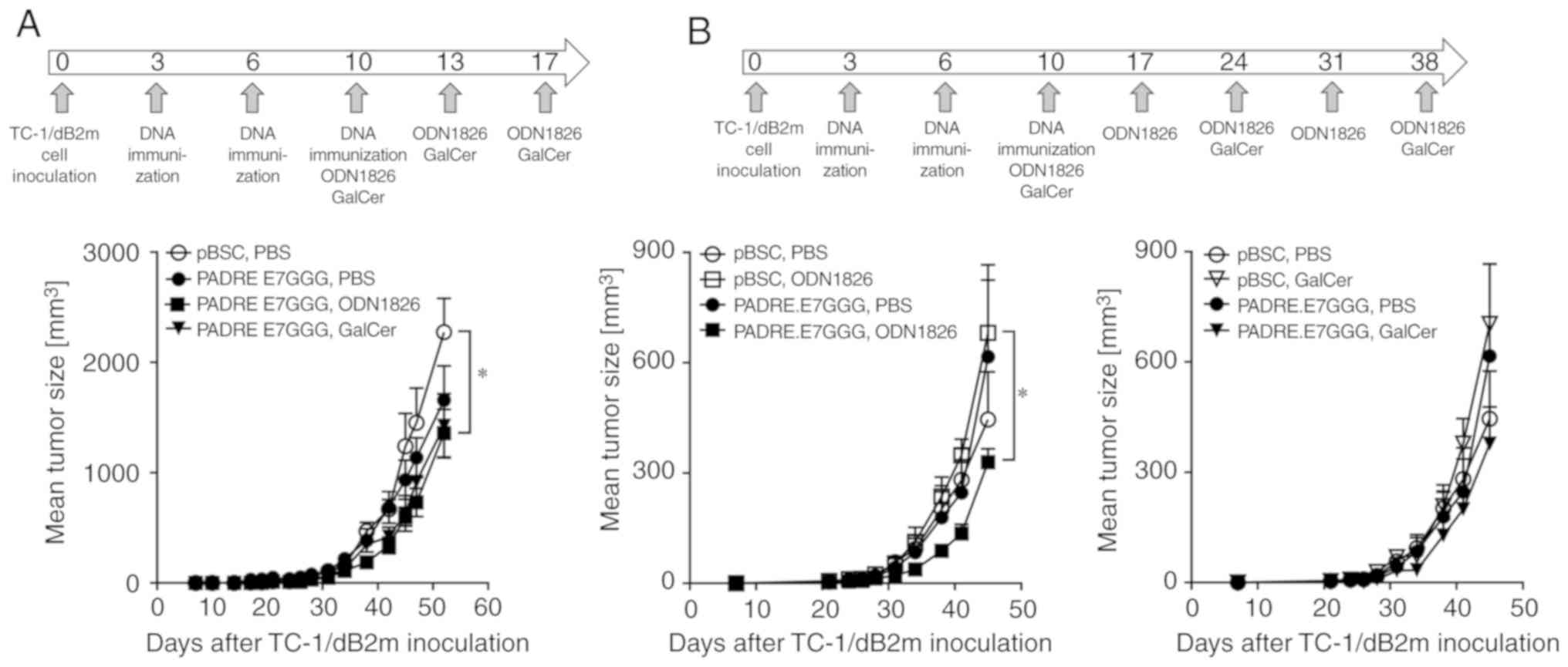
As combinations of DNA vaccination with i.p.
injection of ODN1826 or GalCer reduced tumor growth of TC-1/A9
cells (10), these immunotherapies
were also examined against TC-1/dB2m cells. However, only a weak
antitumor effect was observed (Fig.
3A). Due to the delayed growth of TC-1/dB2m tumor, and in an
attempt to enhance antitumor immunity, the interval between the
injections of immunostimulatory drugs was prolonged and the number
of ODN1826 doses was increased from three to five. Although these
modifications only exerted a weak effect, a significant reduction
of tumor growth was achieved by ODN1826 combined with DNA
immunization (Fig. 3B). This
experiment also demonstrated that a combination of DNA vaccination
with ODN1826 or GalCer was necessary for the antitumor response, as
either therapy alone did not result in tumor reduction. Thus,
repeated experiments suggested a weak inhibition of tumor
development following combined immunotherapy, and this effect was
more obvious for ODN1826.
NK1.1+ cells mainly
contribute to the antitumor effect of combined immunotherapy
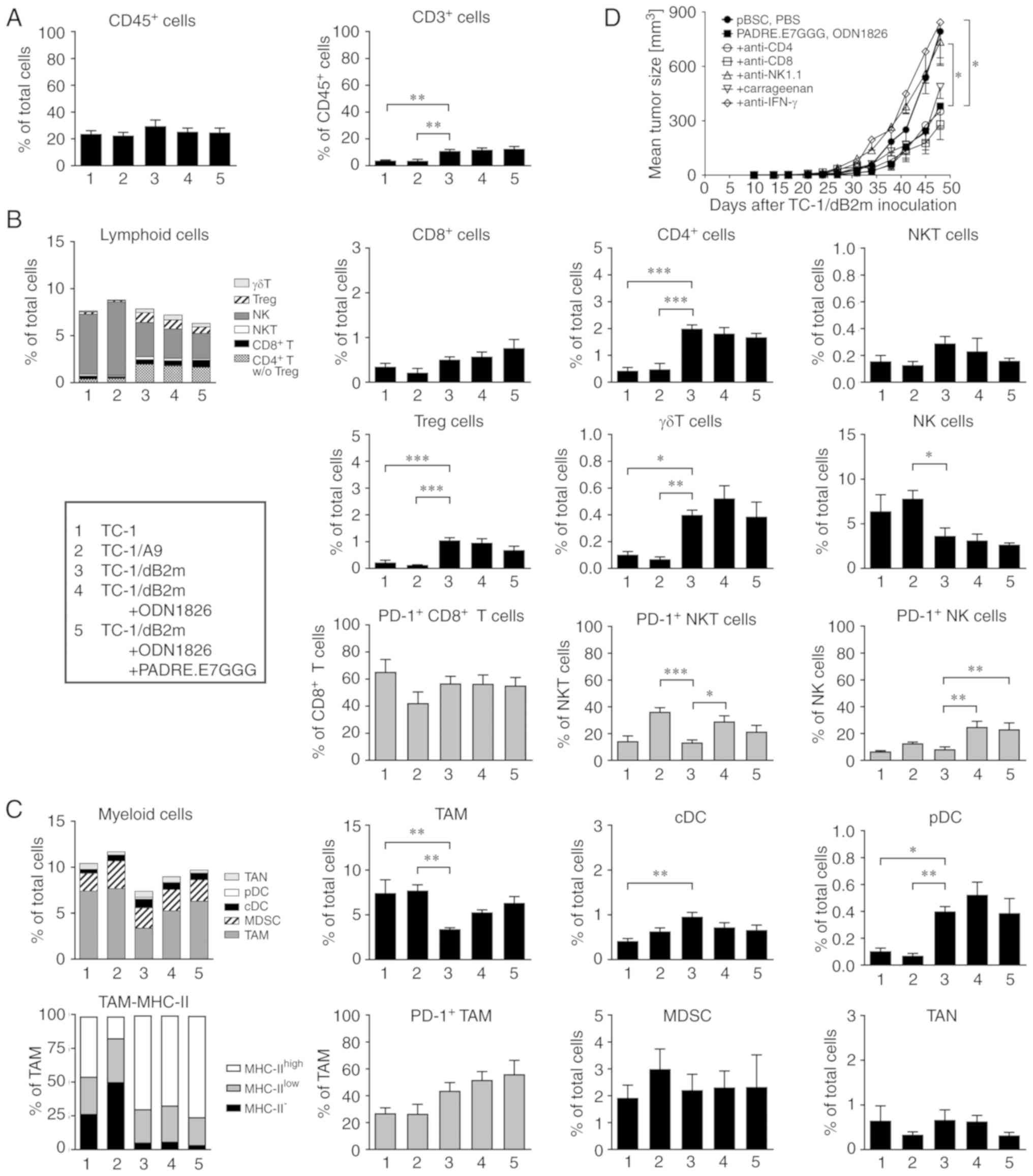
In order to identify the immune cells involved in
the antitumor response to combined therapy against TC-1/dB2m
tumors, tumor-infiltrating cells were first analyzed by flow
cytometry using two panels of monoclonal antibodies. In this
experiment, tumor-infiltrating cells in TC-1- and TC-1/A9-induced
tumors were also compared. The numbers of CD45+ cells in
the tumors were comparable for all three cell lines examined, and
did not change after therapy of TC-1/dB2m-induced tumors. In tumors
that developed from TC-1/dB2m cells, CD3+ cells
comprised a significantly higher proportion of CD45+
cells (~11%) that was not altered following immunotherapy (Fig. 4A).
Among lymphoid cells (Fig.
4B), NK cells (CD3−NK1.1+) were
predominant in all types of tumors, but their proportion was
significantly lower in TC-1/dB2m compared with that in TC-1/A9
tumors (accounting for 13 and 30% of CD45+ cells,
respectively). On the contrary, the proportion of CD4+ T
cells and γδ T cells was significantly higher in TC-1/dB2m tumors
compared with those in TC-1/A9 and TC-1 tumors. Regulatory T cells
(Treg; CD4+CD25+Foxp3+) were
partially responsible for the increase in CD4+ T cells.
The numbers of NKT cells
(CD3+TCRγ/δ−NK1.1+) were also
higher in TC-1/dB2m tumors, but this difference was not
significant. After immunotherapy of TC-1/dB2m tumors, the
proportion of any lymphoid subpopulation was not significantly
altered. Significantly enhanced PD-1 expression was only observed
on NK and NKT cells.
Tumor-associated macrophages (TAMs;
CD11b+Ly6G−Ly6C−F4/80+)
comprised a major subpopulation of myeloid cells in all types of
tumors (Fig. 4C). In TC-1/dB2m
tumors, their numbers were significantly lower compared with those
in TC-1 and TC-1/A9 tumors, but they expressed a higher level of
MHC-II molecules that are considered a marker of M1-polarized
macrophages (18,19). Following immunotherapy of TC-1/dB2m
tumors, the numbers of macrophages and their PD-1 expression were
slightly enhanced. In TC-1/dB2m tumors generated in non-treated
mice, the populations of dendritic cells, both conventional (cDC;
CD11c+Ly6G−Ly6C−F4/80−MHC-II+)
and plasmacytoid (pDC;
CD11c+CD11b−Ly6G−Ly6C+F4/80−MHC-II+CD317+),
were significantly higher compared with those in TC-1 and TC-1/A9
tumors and were not altered after immunotherapy. The numbers of
myeloid-derived suppressor cells
(CD11c+CD11b+Ly6G−Ly6Chigh)
and tumor-associated neutrophils
(CD11b+Ly6GhighLy6Clow) were
comparable in all types of tumors.
Next, we examined cells involved in antitumor
immunity by in vivo depletion. In mice treated with
immunotherapy (DNA vaccination plus injection of ODN1826), tumor
growth was significantly enhanced only after elimination of
NK1.1+ cells (Fig. 4D).
Neutralization of IFN-γ suggested that this cytokine played a
crucial role in the antitumor effect. After depletion of
CD8+ cells, tumor growth was comparable to that in mice
treated with anti-NK1.1+ until day 27 after inoculation
of tumor cells. Subsequently, the growth of tumors was similarly
reduced in animals with depleted CD8+ cells and
administered immunotherapy alone.
In summary, combined immunotherapy of
TC-1/dB2m-induced tumors did not result in significantly increased
immune cell infiltration. NK1.1+ cells and IFN-γ
contributed to the weak antitumor effect. Enhanced expression of
the PD-1 receptor on NK and NKT cells suggests that both types of
cells may be involved in this effect.
Discussion
The production of β-2 microglobulin is often
abrogated by genetic alterations in human tumors (20). As these modifications lead to
irreversible downregulation of surface MHC-I expression that is
associated with resistance to CD8+ cytotoxic T
lymphocytes (CTLs), they enable evasion of adaptive immune
responses generated during tumor development or induced by
immunotherapy. Such resistance has also been demonstrated for a
blockade of the PD-1 immune checkpoint by a monoclonal antibody
(21). Therefore, the development of
relevant tumor models is necessary for studies of experimental
cancer immunotherapy that may result in enhancement of clinical
trial efficacy.
The CRISPR/Cas9 system has been recently used for
the deactivation of the B2m gene in two mouse tumor cell
lines, namely melanoma B16F10 and breast cancer EO-771 cells
(22). In the present study, the
B2m gene was deactivated in the mouse TC-1 cell line, which
is often used to examine various cancer therapies. B2m
deactivation in TC-1/dB2m cells was associated with loss of surface
MHC-I expression, but when inducibility by IFN-γ was tested, a
slight restoration of MHC-I expression on the cell surface,
particularly of molecules from the D locus, was observed. As β-2
microglobulin expression remained negative, it was hypothesized
that β-2 microglobulin-free MHC-I heavy chains were displayed on
the cells. Such molecules, particularly H-2Db, have been
reported for both β-2 microglobulin-negative and -positive mouse
cells (23–25); however, the role of IFN-γ stimulation
was not described in these studies. In a human neuroblastoma cell
line producing β-2 microglobulin, the expression of β-2
microglobulin-free MHC-I molecules was enhanced upon
differentiation with either retinoic acid or serum starvation.
Incubation with IFN-γ increased the surface expression of MHC-I
heterodimers, but not of β-2 microglobulin-free MHC-I molecules
(26). In concordance with the
published results (27), the present
study demonstrated that B2m deactivation did not prevent
CD1d surface expression.
Deactivation of the B2m gene significantly
reduced the proliferation of TC-1/dB2m cells and the growth of
TC-1/dB2m-induced tumors. These results correspond to the findings
that β-2 microglobulin promotes cell proliferation, migration, and
invasion in different tumor types (28,29). A key
role in this β-2 microglobulin-mediated signaling was attributed to
its binding with hemochromatosis (HFE) protein, a non-classical
MHC-I molecule that regulates iron concentration in cells.
Following formation of the β-2 microglobulin/HFE complex, iron
influx is inhibited and numerous intracellular pathways are
affected (30).
A reduced proliferation rate may contribute to the
delay in tumor growth, but does not appear to be a crucial factor
responsible for the substantially decreased oncogenicity of
TC-1/dB2m cells. As in vivo depletion was associated with
partial restoration of tumor growth after application of
NK1.1-specific antibody, enhanced sensitivity to elimination by NK
cells may be more important. Das et al (22) observed a similar reduction of
oncogenicity following B2m deactivation in two mouse tumor
cell lines and also suggested a role of NK cells in this
phenomenon.
The effect of MHC-I downregulation after B2m
deactivation was also manifested by the loss of sensitivity to
depletion of CD8+ cells and to adaptive immunity induced
by DNA immunization and mediated by CTLs. As similar effects were
observed for TC-1/A9 cells, where a combination of DNA immunization
and an adjuvant (ODN1826 or GalCer) significantly reduced tumor
growth (10), the efficacy of this
combined immunotherapy was also examined against TC-1/dB2m tumors.
However, only the combination of DNA vaccination and ODN1826
injection significantly inhibited tumor growth, and this effect was
less notable compared with that against TC-1/A9-induced tumors.
Flow cytometric analysis of tumor-infiltrating
immune cells and in vivo depletion after immunotherapy
revealed several marked differences between TC-1/dB2m and TC-1/A9
tumors: i) TC-1/dB2m tumors contained more pDCs, CD4+ T,
Treg, and γδ T cells, but fewer TAMs and NK cells. However, this
observation should be interpreted with caution, as infiltration of
tumors with immune cells is a dynamic process with specific
kinetics of individual subpopulations (8,31–33), which hampers a direct comparison among
tumors induced by different cells. Although we strived to analyze
tumors of similar size, the composition of the cell infiltrate may
be affected by the markedly different growth of TC-1-, TC-1/A9- and
TC-1/dB2m-induced tumors. ii) Following combined immunotherapy,
none of the examined subpopulations of infiltrating immune cells
was increased in the TC-1/dB2m tumors, while in the TC-1/A9 tumors,
most subpopulations were increased, particularly CD8+ T
cells (10). iii)
MHC-IIhigh TAMs were predominant in TC-1/dB2m tumors,
even without immunotherapy (while in the TC-1/A9 tumors, they were
predominant only after immunotherapy). Reduced numbers of M2 TAMs
were mainly responsible for this effect. Movahedi et al
reported similar results for the 4T1 cell line (18). Progressing tumors induced by these
cells accumulated MHC-IIhigh TAMs, in contrast to tumors
induced by 3LL or TS/A cells, and these TAMs remained M1 polarized.
Therefore, it was concluded that the proportions of TAM subsets
were tumor-dependent. iv) For TC-1/A9 tumors, NK1.1+
cells, CD8+ cells and TAMs cooperated in the antitumor
response, but only NK1.1+ cells were significantly
implicated in the delay of TC-1/dB2m tumor growth. Enhanced PD-1
expression on both NK and NKT cells after immunotherapy suggested
activation of both cell types by treatment and their possible
involvement in antitumor immunity.
By using in vivo depletion of
CD8+, CD4+ and NK1.1+ cells,
studies comparing immunotherapy against TC-1 cells and TC-1 clones
with reversible MHC-I downregulation demonstrated that only
CD8+ T cells were necessary for the antitumor effect
against TC-1 cells, and that both CD8+ and
NK1.1+ cells may be involved in the inhibition of tumors
induced by cells with reversibly downregulated MHC-I expression
(34,35), which was confirmed in our previous
study with TC-1/A9 cells (10). This
study extended these observations for the TC-1 clone with
irreversible MHC-I downregulation, demonstrating that
NK1.1+ cells were the most important for the antitumor
effect stimulated by immunotherapy against TC-1/dB2m cells. For
TC-1-induced tumors, two other studies demonstrated the cooperation
of CD8+ T cells with other immune cells in the antitumor
response after immunotherapy, but suggested that CD8+ T
cells were not the main cytotoxic cells eliminating
MHC-I-proficient TC-1 cells (8,36). Our
study with TC-1/A9 cells, which are deficient in MHC-I expression,
also demonstrated a role of CD8+ T cells activated by
DNA vaccination in the antitumor response (10). However, in the present study, despite
the fact that DNA vaccination was necessary for the induction of
the antitumor effect by combined immunotherapy, the level of
CD8+ T cells in the tumors was not increased by
treatment, and the function of CD8+ T cells was not
proven by in vivo depletion. The only observation suggesting
the involvement of CD8+ T cells was derived from the
in vivo depletion experiment, where augmented tumor growth
was recorded at the initial phase of tumor development (up to day
27) following application of anti-CD8. As tumor infiltration by
immune cells is a dynamic process (8), this fact should be confirmed by further
studies investigating a more efficient immunotherapy against
TC-1/dB2m cells, which may also help elucidate the role of
CD8+ T cells and other immune cells in this tumor
model.
As combined immunotherapy only weakly inhibited the
growth of TC-1/dB2m tumors and did not affect infiltration of these
tumors by immune cells, immunosuppressive mechanisms most likely
prevail in the microenvironment of early-stage tumors. This
condition resembles human MHC-I-negative tumors that are often
devoid of immune cells in the tumor parenchyma and contain
unfunctional immune cells in the tumor stroma (37,38).
Therefore, for successful immunotherapy of such tumors, activation
of adaptive and innate immunity should be accompanied by
appropriate inhibition of immunosuppression and recovery of MHC-I
expression (4,39,40).
The mechanisms of immune reactions against
TC-1/dB2m-induced tumors were not completely elucidated in the
present study, and will be the subject of further analyses. The
present results suggest that, following immunotherapy,
NK1.1+ cells were the major cell type exhibiting
antitumor activity against TC-1/dB2m tumors. The possible
involvement of both NK and NKT cells was indicated by enhanced PD-1
expression, which suggested the activation and subsequent
inactivation of these cells. The antitumor effect of NKT cells,
which constitute a minor subpopulation of NK1.1+ cells,
was also suggested by immunotherapy with GalCer, as this adjuvant
activates NKT cells. However, NK cells are likely the main effector
cells, as they can eliminate tumor cells with downregulated MHC-I
expression. Unfortunately, they can also contribute to tumor immune
evasion (41). The antitumor effect
of ODN1826 may not be dependent on adaptive immunity (42); however, DNA immunization was necessary
for the reduced growth of TC-1/dB2m tumors. The role of
CD8+ T cells in antitumor reactions was not clearly
confirmed by in vivo depletion, but this experiment
indicated the effect of these cells during early tumor growth. At a
later stage of tumor development, CD8+ T cells are
likely inactivated by the immunosuppressive tumor microenvironment
(43).
In conclusion, deactivation of the B2m gene
led to the creation of the TC-1/dB2m cell line, which is
characterized by irreversible MHC-I downregulation, and a reduced
proliferation rate and tumor growth. These cells displayed loss of
sensitivity to DNA immunization and, in comparison to the TC-1/A9
cells with reversible MHC-I downregulation, they responded more
weakly to combined immunotherapy consisting of DNA vaccination and
either ODN1826 or GalCer injection. Moreover, infiltration of
TC-1/dB2m tumors with immune cells was not enhanced after
immunotherapy, and only NK1.1+ cells were confirmed to
contribute to the antitumor effect. In a set with TC-1 and TC-1/A9
cells, the TC-1/dB2m cell line may be utilized for enhancement of
cancer immunotherapy with a potentially high clinical benefit, as
human tumors are heterogeneous in terms of MHC-I expression, which
enables evasion of the immune response and confers resistance to
therapy.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Pavlina Vesela,
Kristyna Klecakova and Nela Vaclavikova for technical assistance.
We also acknowledge the Imaging Methods Core Facility at BIOCEV for
their support with obtaining the flow cytometry data presented in
this article.
Funding
The present study was funded by the Czech Science
Foundation (grant nos. GA16-04477S and GA19-00816S); the European
Regional Development Fund (grant nos.
CZ.02.1.01/0.0/0.0/16_019/0000785, CZ.1.05/1.1.00/02.0109,
CZ.1.05/2.1.00/19.0400 and CZ.1.05/2.1.00/19.0395); the Ministry of
Education, Youth and Sports of the Czech Republic (grant nos.
LQ1604 and LM2015040); and Charles University (grant no.
SVV-2017-260426).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
MS conceived and designed the study. KL, AG, IP, JV
and MS performed the experiments. MS, IP, KL and JV analyzed and
interpreted the data. KL, MS and IP wrote the manuscript. All
authors have read and approved the final version of this manuscript
for publication.
Ethics approval and consent to
participate
All animal experimental procedures were performed
in compliance with Directive 2010/63/EU, and animal protocols were
approved by the Sectoral Expert Committee of the Czech Academy of
Sciences for Approval of Projects of Experiments on Animals
(reference no. 46/2016, 16 May 2016).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Khong HT and Restifo NP: Natural selection
of tumor variants in the generation of ‘tumor escape’ phenotypes.
Nat Immunol. 3:999–1005. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Garrido F, Ruiz-Cabello F and Aptsiauri N:
Rejection versus escape: The tumor MHC dilemma. Cancer Immunol
Immunother. 66:259–271. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reinis M: Immunotherapy of MHC class
I-deficient tumors. Future Oncol. 6:1577–1589. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Garrido F, Aptsiauri N, Doorduijn EM,
Garcia Lora AM and van Hall T: The urgent need to recover MHC class
I in cancers for effective immunotherapy. Curr Opin Immunol.
39:44–51. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rooney MS, Shukla SA, Wu CJ, Getz G and
Hacohen N: Molecular and genetic properties of tumors associated
with local immune cytolytic activity. Cell. 160:48–61. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Smyth MJ, Ngiow SF, Ribas A and Teng MW:
Combination cancer immunotherapies tailored to the tumour
microenvironment. Nat Rev Clin Oncol. 13:143–158. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zappasodi R, Merghoub T and Wolchok JD:
Emerging concepts for immune checkpoint blockade-based combination
therapies. Cancer Cell. 33:581–598. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thoreau M, Penny HL, Tan K, Regnier F,
Weiss JM, Lee B, Johannes L, Dransart E, Le Bon A, Abastado JP, et
al: Vaccine-induced tumor regression requires a dynamic cooperation
between T cells and myeloid cells at the tumor site. Oncotarget.
6:27832–27846. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moynihan KD, Opel CF, Szeto GL, Tzeng A,
Zhu EF, Engreitz JM, Williams RT, Rakhra K, Zhang MH, Rothschilds
AM, et al: Eradication of large established tumors in mice by
combination immunotherapy that engages innate and adaptive immune
responses. Nat Med. 22:1402–1410. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Grzelak A, Polakova I, Smahelova J,
Vackova J, Pekarcikova L, Tachezy R and Smahel M: Experimental
combined immunotherapy of tumours with major histocompatibility
complex class I downregulation. Int J Mol Sci. 19:E36932018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin KY, Guarnieri FG, Staveley-O'Carroll
KF, Levitsky HI, August JT, Pardoll DM and Wu TC: Treatment of
established tumors with a novel vaccine that enhances major
histocompatibility class II presentation of tumor antigen. Cancer
Res. 56:21–26. 1996.PubMed/NCBI
|
|
12
|
Smahel M, Sima P, Ludvikova V, Marinov I,
Pokorna D and Vonka V: Immunisation with modified HPV16 E7 genes
against mouse oncogenic TC-1 cell sublines with downregulated
expression of MHC class I molecules. Vaccine. 21:1125–1136. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Smahel M, Sima P, Ludvikova V and Vonka V:
Modified HPV16 E7 genes as DNA vaccine against E7-containing
oncogenic cells. Virology. 281:231–238. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Smahel M, Polakova I, Duskova M, Ludvikova
V and Kastankova I: The effect of helper epitopes and cellular
localization of an antigen on the outcome of gene gun DNA
immunization. Gene Ther. 21:225–232. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Alexander J, Sidney J, Southwood S,
Ruppert J, Oseroff C, Maewal A, Snoke K, Serra HM, Kubo RT and
Sette A: Development of high potency universal DR-restricted helper
epitopes by modification of high affinity DR-blocking peptides.
Immunity. 1:751–761. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kaštánková I, Poláková I, Dušková M and
Šmahel M: Combined cancer immunotherapy against aurora kinase A. J
Immunother. 39:160–170. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Simova J, Indrova M, Bieblová J, Mikyskova
R, Bubeník J and Reinis M: Therapy for minimal residual tumor
disease: β-galactosylceramide inhibits the growth of recurrent
HPV16-associated neoplasms after surgery and chemotherapy. Int J
Cancer. 126:2997–3004. 2010.PubMed/NCBI
|
|
18
|
Movahedi K, Laoui D, Gysemans C, Baeten M,
Stange G, Van den Bossche J, Mack M, Pipeleers D, In't Veld P, De
Baetselier P and Van Ginderachter JA: Different tumor
microenvironments contain functionally distinct subsets of
macrophages derived from Ly6C(high) monocytes. Cancer Res.
70:5728–5739. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mills CD and Ley K: M1 and M2 macrophages:
The chicken and the egg of immunity. J Innate Immun. 6:716–726.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bernal M, Ruiz-Cabello F, Concha A,
Paschen A and Garrido F: Implication of the β2-microglobulin gene
in the generation of tumor escape phenotypes. Cancer Immunol
Immunother. 61:1359–1371. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zaretsky JM, Garcia-Diaz A, Shin DS,
Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY,
Abril-Rodriguez G, Sandoval S, Barthly L, et al: Mutations
associated with acquired resistance to PD-1 blockade in melanoma. N
Engl J Med. 375:819–829. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Das K, Eisel D, Lenkl C, Goyal A,
Diederichs S, Dickes E, Osen W and Eichmüller SB: Generation of
murine tumor cell lines deficient in MHC molecule surface
expression using the CRISPR/Cas9 system. PLoS One. 12:e01740772017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Potter TA, Boyer C, Verhulst AM, Golstein
P and Rajan TV: Expression of H-2Db on the cell surface in the
absence of detectable beta 2 microglobulin. J Exp Med. 160:317–322.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Allen H, Fraser J, Flyer D, Calvin S and
Flavell R: Beta 2-microglobulin is not required for cell surface
expression of the murine class I histocompatibility antigen H-2Db
or of a truncated H-2Db. Proc Natl Acad Sci USA. 83:7447–7451.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bix M and Raulet D: Functionally conformed
free class I heavy chains exist on the surface of beta 2
microglobulin negative cells. J Exp Med. 176:829–834. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Marozzi A, Meneveri R, Bunone G, De Santis
C, Lopalco L, Beretta A, Agresti A, Siccardi AG, Della Valle G and
Ginelli E: Expression of β2m-free HLA class I heavy chains in
neuroblastoma cell lines. Scand J Immunol. 37:661–667. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim HS, Garcia J, Exley M, Johnson KW,
Balk SP and Blumberg RS: Biochemical characterization of CD1d
expression in the absence of beta2-microglobulin. J Biol Chem.
274:9289–9295. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang WC, Wu D, Xie Z, Zhau HE, Nomura T,
Zayzafoon M, Pohl J, Hsieh CL, Weitzmann MN, Farach-Carson MC and
Chung LW: Beta2-Microglobulin is a signaling and growth-promoting
factor for human prostate cancer bone metastasis. Cancer Res.
66:9108–9116. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nomura T, Huang WC, Zhau HE, Wu D, Xie Z,
Mimata H, Zayzafoon M, Young AN, Marshall FF, Weitzmann MN and
Chung LW: Beta2-microglobulin promotes the growth of human renal
cell carcinoma through the activation of the protein cinase A,
Cyclic AMP–responsive element-binding protein, and vascular
endothelial growth factor axis. Clin Cancer Res. 12:7294–7305.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nomura T, Huang WC, Zhau HE, Josson S,
Mimata H and Chung LW: β2-Microglobulin-mediated signaling as a
target for cancer therapy. Anticancer Agents Med Chem. 14:343–352.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sneed RA, Stevenson AP and Stewart CC:
Quantitation of the host cell infiltration kinetics of the
nonimmunogenic colon 26 tumor by multiparameter flow cytometry. J
Leukoc Biol. 46:547–555. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kennedy BC, Maier LM, D'Amico R, Mandigo
CE, Fontana EJ, Waziri A, Assanah MC, Canoll P, Anderson RC,
Anderson DE and Bruce JN: Dynamics of central and peripheral
immunomodulation in a murine glioma model. BMC Immunol. 10:112009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bindea G, Mlecnik B, Tosolini M,
Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T,
Lafontaine L, Berger A, et al: Spatiotemporal dynamics of
intratumoral immune cells reveal the immune landscape in human
cancer. Immunity. 39:782–795. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cheng WF, Hung CF, Lin KY, Ling M, Juang
J, He L, Lin CT and Wu TC: CD8 + T cells, NK cells and IFN-gamma
are important for control of tumor with downregulated MHC class I
expression by DNA vaccination. Gene Ther. 10:1311–1320. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Indrová M, Símová J, Bieblová J, Bubeník J
and Reinis M: NK1.1+ cells are important for the development of
protective immunity against MHC I-deficient, HPV16-associated
tumours. Oncol Rep. 25:281–288. 2011.PubMed/NCBI
|
|
36
|
van der Sluis TC, Sluijter M, van Duikeren
S, West BL, Melief CJ, Arens R, van der Burg SH and van Hall T:
Therapeutic peptide vaccine-induced CD8 T cells strongly modulate
intratumoral macrophages required for tumor regression. Cancer
Immunol Res. 3:1042–1051. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Perea F, Bernal M, Sánchez-Palencia A,
Carretero J, Torres C, Bayarri C, Gómez-Morales M, Garrido F and
Ruiz-Cabello F: The absence of HLA class I expression in non-small
cell lung cancer correlates with the tumor tissue structure and the
pattern of T cell infiltration. Int J Cancer. 140:888–899. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Aptsiauri N, Ruiz-Cabello F and Garrido F:
The transition from HLA-I positive to HLA-I negative primary
tumors: The road to escape from T-cell responses. Curr Opin
Immunol. 51:123–132. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ugurel S, Spassova I, Wohlfarth J, Drusio
C, Cherouny A, Melior A, Sucker A, Zimmer L, Ritter C, Schadendorf
D and Becker JC: MHC class-I downregulation in PD-1/PD-L1 inhibitor
refractory Merkel cell carcinoma and its potential reversal by
histone deacetylase inhibition: A case series. Cancer Immunol
Immunother. 68:983–990. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang S, Kohli K, Black RG, Yao L,
Spadinger SM, He Q, Pillarisetty VG, Cranmer LD, Van Tine BA, Yee
C, et al: Systemic interferon-γ increases MHC class I expression
and T-cell infiltration in cold tumors: Results of a phase 0
clinical trial. Cancer Immunol Res. 7:1237–1243. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Di Vito C, Mikulak J, Zaghi E, Pesce S,
Marcenaro E and Mavilio D: NK cells to cure cancer. Semin Immunol.
41:1012722019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wright SE, Rewers-Felkins KA, Chowdhury
NI, Ahmed J and Srivastava SK: Prevention of human adenocarcinoma
with CpG-ODN in a mouse model. Oncol Lett. 4:1061–1063. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Maimela NR, Liu S and Zhang Y: Fates of
CD8+ T cells in tumor microenvironment. Comput Struct Biotechnol J.
17:1–13. 2018. View Article : Google Scholar : PubMed/NCBI
|