Introduction
Ewing sarcomas (ES) are aggressive mesenchymal
tumors, which most often occur in children, adolescents and young
adults. They are the second most prevalent malignant bone tumors
(1). The exact genesis of ES is still
unclear. Currently, mesenchymal stem cells (MSC) are considered as
the most probable cells of origin (2). Various gene translocations, which define
the disease, are discussed as a trigger for ES formation. The most
common rearrangement is the reciprocal t(11; 22)(q24; q12)
translocation, which occurs in approximately 85% of all cases and
fuses the Ewing sarcoma breakpoint region (EWS) 1 gene with the
Friend leukemia integration (FLI) 1 gene (1).
The EWS-FLI1 oncoprotein acts as a transcription
factor and promotes, among others, the expression of glioma
associated oncogene family 1 (GLI1) (3) and insulin-like growth factor 1 (IGF1)
mRNA, while numerous additional targets are repressed by EWS-FLI1
(1). Hence, overexpression of GLI1
protein triggered by EWS-FLI1 may be assumed, although this has not
been confirmed for the ES cell lines A673, RD-ES and SK-N-MC, which
all express the EWS-FLI1 fusion protein (4), while GLI1 protein expression is commonly
reduced compared to that in MSC (5).
GLI1 expression in the ES cell line TC-71 has been previously shown
at the mRNA and protein levels (3).
In TC-71 cells, arsenic trioxide (ATO) was found to have no effect
on GLI1 protein expression but inhibited the transcriptional
activity of GLI1 proven by reduced expression of target genes
(6). The authors attributed the
ATO-dependent apoptosis induction in ES cell lines (determined by
TUNEL assay) at least partially to GLI1 inhibition (6).
Fusion protein-positive ES form genomic stable
tumors with a few additional somatic mutations mostly affecting p16
and p53 function. Yet, EWS-FLI1 expression is heterogeneous leading
phenotypic plasticity and chemotherapy increases mutation numbers,
which has been linked to poor outcome (4,7,8). Acquired multidrug resistance is a common
feature of ES, which have been shown to express both multidrug
resistance-associated protein 1 (MRP1) and multidrug resistance
protein 1 (MDR1), also known as P-glycoprotein 1 (PGP1) (9–11). Both,
the SK-N-MC and TC-71 (also EWS-FLI1-positive) cell lines were
established after chemotherapy from a metastatic site (SK-N-MC) or
local recurrence (TC-71) (8). In
contrast, RD-ES cells originate from a primary tumor (12). The A673 cell line has been established
without further documentation of the site of origin. Neither for
RD-ES nor for A673 is a treatment history available.
For ES, the actual ESMO-PaedCan-EURACAN guideline
recommends multimodal chemotherapy including doxorubicin,
cyclophosphamide, ifosfamide, vincristine, dactinomycin and
etoposide (Eto) combined with radiation therapy (13). Eto acts as a topoisomerase-2-inhibitor
and clinically achievable doses in the plasma of ES patients are
approximately 10 µg/ml (8). Patients
suffering from recurrent or metastatic ES have a poor prognosis.
Previously achieved cumulative doses of chemotherapeutic agents
afford the individualization of second line treatments which may
also contain Eto (13).
Topoisomerase-2 inhibition by Eto causes DNA damage, cell cycle
arrest and apoptosis via p53-dependent and -independent pathways
(14). Activation of the intrinsic
mitochondrial apoptosis pathway by Eto has been shown to be
dependent on active glycogen synthase kinase-3 (GSK-3) (15–17).
ATO has been established for the treatment of acute
promyelocytic leukemia (APL) (18).
Moreover, it has been shown to induce cell death of different solid
tumor cells in vitro at concentrations of 0.1–5 µM, which
can also be obtained in the plasma of patients (5,19–21). ATO has the property of binding and
inhibiting the GLI1 protein directly (22) but also targets many additional
pathways implicated in proliferation, metabolism and cell death
(23). Apoptosis induction by ATO is
based on several interconnected mechanisms: First, inhibition of
mitogenic signaling pathways including the Hedgehog (Hh) and Akt
pathways (22,24,25);
second, enhanced production of reactive oxygen species (ROS)
(26,27); third, induction of endoplasmic
reticulum stress and suppression of proteasome activity (24,27);
fourth, mitochondrial disruption and subsequent activation of the
intrinsic apoptotic pathway (26–28), which
may be facilitated by suppression of Bcl-2 expression (26), fifth, stabilization of securin and
cyclin B directly inhibiting mitotic progression, which leads to
centrosome fragmentation dependent apoptosis preferentially
affecting rapidly growing cancer cells compared to differentiated
resting cells (29). In human
epidermoid carcinoma cells high doses of ATO (20 µM) have been
shown to induce ERK phosphorylation dependent GSK-3 inhibition
leading to increased p21 expression as well as apoptosis (30). In human rhabdomyosarcoma cell lines 1
µM ATO had a minor effect on GSK-3β serine 9 phosphorylation. Yet,
it significantly increased lithium chloride-induced GSK-3β
inhibition and apoptosis (31).
GSK-3β has been shown to act either in a
pro-apoptotic or anti-apoptotic manner dependent on the pathway.
The mitochondrial intrinsic apoptotic pathway is activated by
GSK-3β, whereas the death receptor-mediated extrinsic apoptotic
signaling pathway is suppressed (32). Since both Eto and ATO are capable of
activating the intrinsic mitochondrial apoptosis pathway (15,27), it
can be assumed that active GSK-3β is required for apoptosis
induction by both drugs.
The combined application of ATO and Eto on ES cell
lines has been shown to effectively reduce proliferation and
viability concomitant with cell death induction (5). The present study investigated the effect
of different chronological orders of low dose ATO and Eto
administration on apoptosis induction in ES cell lines and explored
the effect of both drugs on inhibitory GSK-3β phosphorylation as
well as the ATO impact on multidrug transporter gene MRP1 and MDR1
expression.
Materials and methods
Reagents
ATO (Trisenox, Pharmacy of the University Hospital
Tuebingen, Germany) was dissolved in purified water, and Eto
(Selleckchem, Munich, Germany) was dissolved in dimethyl sulfoxide.
For cell culture treatment stock solutions were further diluted in
culture medium.
Cell lines and culture
RD-ES and A673 cells were obtained from CLS Cell
Lines Service GmbH (Eppelheim, Germany). SK-N-MC (ATCC HTB-10) were
purchased from the American Type Culture Collection (ATCC,
Manassas, VA, USA). TC-71 cells were purchased from DSMZ
(Braunschweig, Germany). RD-ES and SK-N-MC cells were maintained in
RPMI-1640 medium with L-glutamine (Gibco, Life Technologies; Thermo
Fisher Scientific, Inc.) supplemented with 15% fetal calf serum
(FCS) (Biochrom, Berlin, Germany). A673 cells were cultivated in
Dulbecco's modified Eagle's medium (DMEM) with GlutaMAX, 4.5 g/l
D-glucose (Gibco, Life Technologies; Thermo Fisher Scientific,
Inc.) supplemented with 10% FCS. TC-71 cells were maintained in
Iscove's modified Dulbecco's medium (IMDM), supplemented with 10%
FCS.
Bone marrow-derived MSC were isolated at the
University Hospital Tuebingen after written informed consent of the
patients (approved by the ethics committee of the medical faculty,
project no. 401/2013 BO2), propagated as previously described
(33) and confirmed to represent
multi-lineage differentiation potential toward chondrocytes,
adipocytes and osteocytes (data not shown).
All cell lines were purchased from ATCC, CLS or
DSMZ. Cryo-stocks were made and low passage cultures were used from
these stocks. Cell line authentication was not performed as these
cells derived from established cell banks and were submitted to
in-house quality controls.
All cells were cultivated at 37°C in a humidified
atmosphere containing 5% CO2 and were regularly tested
for absence of mycoplasma contamination by PCR (AppliChem,
Darmstadt, Germany).
RNA isolation and RT-qPCR
RNA was isolated using the innuPREP RNA Mini Kit
(Analytik Jena AG, Jena, Germany). One µg of RNA was reversely
transcribed using the innuSCRIPT reverse transcriptase (Analytik
Jena AG). cDNA (50 ng) was analyzed in duplicate reactions by
quantitative RT-PCR (qRT-PCR) using gene-specific primers and the
SYBR Select Master Mix for CFX (Thermo Fisher Scientific, Inc.) in
a total volume of 10 µl. QRT-PCR was carried out in a CFX96
Real-Time PCR instrument (Bio-Rad Laboratories, Inc., Munich,
Germany) and was analyzed using the CFX Manager™ software (Bio-Rad
Laboratories, Inc.). Relative expression levels were calculated as
fold change compared to MSC or basal expression using the ΔΔCt
(2−ΔΔCt) method with TATA box binding protein (TBP) as a
reference gene (34). The qRT-PCR
primers were used as previously published: GLI1 (35), MDR1 (36), MRP1 (37) and TBP (38).
Cytotoxicity assay
Cell Titer 96® AQueous One Solution Cell
Proliferation assay (Promega, Mannheim, Germany) (MTS) was used to
measure cell viability via redox enzyme activity, according to the
protocol provided by the manufacturer. TC-71 (0.3×104
cells/well) or RD-ES (0.5×104 cells/well, Fig. S1) cells were grown in transparent
F-bottom 96-well plates. Twenty-four hours after seeding, the cells
were incubated in the presence of ATO or Eto as indicated for
another 96 h at 37°C in a humidified atmosphere of 5%
CO2 in air. At the end of the incubation period, MTS
reagent was added to the wells, and the plate was incubated for 1.5
h protected from light. Absorbance was recorded at 490 nm with a
reference wavelength of 630 nm using an EL 800 reader (BioTek,
Winooski, VT, USA).
IC50 determination
The half maximal inhibitory concentration
(IC50) values of ATO and Eto were determined for TC-71
cells by nonlinear regression using GraphPad Prism V8.0 software
(GraphPad Software, Inc., La Jolla, CA, USA).
Caspase assay
Apo-ONE® Homogeneous Caspase-3/7 Assay
(Promega) was used according to the protocol provided by the
manufacturer. A673 (0.5×104 cells/well), RD-ES
(0.25×104 cells/well), TC-71 (0.15×104
cells/well) and MSC (0.5×104 cells/well) were grown in
black 96-well plates with opaque F-bottom. Twenty-four hours after
seeding, the cells were incubated in the presence of ATO, Eto or
combinations as indicated for another 96 h at 37°C in a humidified
atmosphere of 5% CO2 in air. Excess primary
drug-solution was removed prior to secondary drug application. At
the end of the incubation period, the Z-DEVD-R110 substrate was
added to the wells, and the plate was incubated for 1 h protected
from light. Fluorescence was recorded at an excitation wavelength
of 485 nm and emission wavelength of 528 nm with a Wallac 1420
Victor2 multilabel counter (Perkin Elmer, Waltham, MA, USA).
Western blot analysis
A673, RD-ES, SK-N-MC, TC-71 cells or MSC
(2.5×105 cells/well) were incubated in 12-well plates
for 48 h. For analysis, the cells were washed with PBS and lysed in
protein lysis buffer (40 mM Tris/HCl pH 7.4, 300 mM NaCl, 2 mM
EDTA, 20% glycerol, 2% Triton X-100) supplemented with proteinase
inhibitor at 4°C. Insoluble material was removed by centrifugation.
The protein concentration in the supernatant was determined by
Bradford protein assay. An amount 40 µg of the protein samples was
separated by 10% SDS-PAGE and transferred to a hydrophobic
polyvinylidene difluoride (PVDF) membrane (Immobilon-P; Merck KGaA,
Darmstadt, Germany). After blocking with 5% powdered milk (Carl
Roth, Karlsruhe, Germany) in TBS-T, the membranes were incubated
with primary antibodies: GLI1 rabbit pAb #2553, β-tubulin (9F3)
rabbit mAb #2128 (both at a dilution 1:1,000) (Cell Signaling
Technology, Leiden, Netherlands) with gentle shaking overnight at
4°C according to the manufacturer's protocols. Membranes were
washed three times with TBS-T. Secondary antibody (horseradish
peroxidase-conjugated anti-rabbit pAb, dilution 1:10,000, Jackson
Immuno Research, West Grove, PA, USA) was added for 2 h, and the
membranes were washed another three times with TBS-T. Proteins were
detected using ECL Western Blotting Substrate (Thermo Fisher
Scientific, Inc.) with membranes exposed to Amersham Hyperfilm ECL
(GE Healthcare, Pittsburgh, PA, USA). A pre-stained protein ladder
(PageRuler Plus; Thermo Fisher Scientific, Inc.) was used for
determination of molecular weights. ImageJ Software version 1.51s
(National Institutes of Health, Bethesda, MD, USA) was utilized for
western blot quantification.
ELISA
A673 (0.5×106 cells/well), RD-ES
(0.5×106 cells/well) and TC-71 (0.2×105
cells/well) cells were incubated with the drug concentrations
indicated for 48 h in 6-well plates and the GSK3 beta
(Total/Phospho) InstantOne™ ELISA kit (cat. no. 85-86173-11; Thermo
Fisher Scientific, Inc.) was carried out according to the
manufacturer's instructions.
Statistical analysis
All statistical tests were performed using GraphPad
Prism V8.0 software and statistical differences were analyzed by
two-way ANOVA with P≤0.05, P≤0.01 and P≤0.001 considered as
indicative of statistical significance. Multiple comparisons
between groups were performed using Tukey's test.
Results
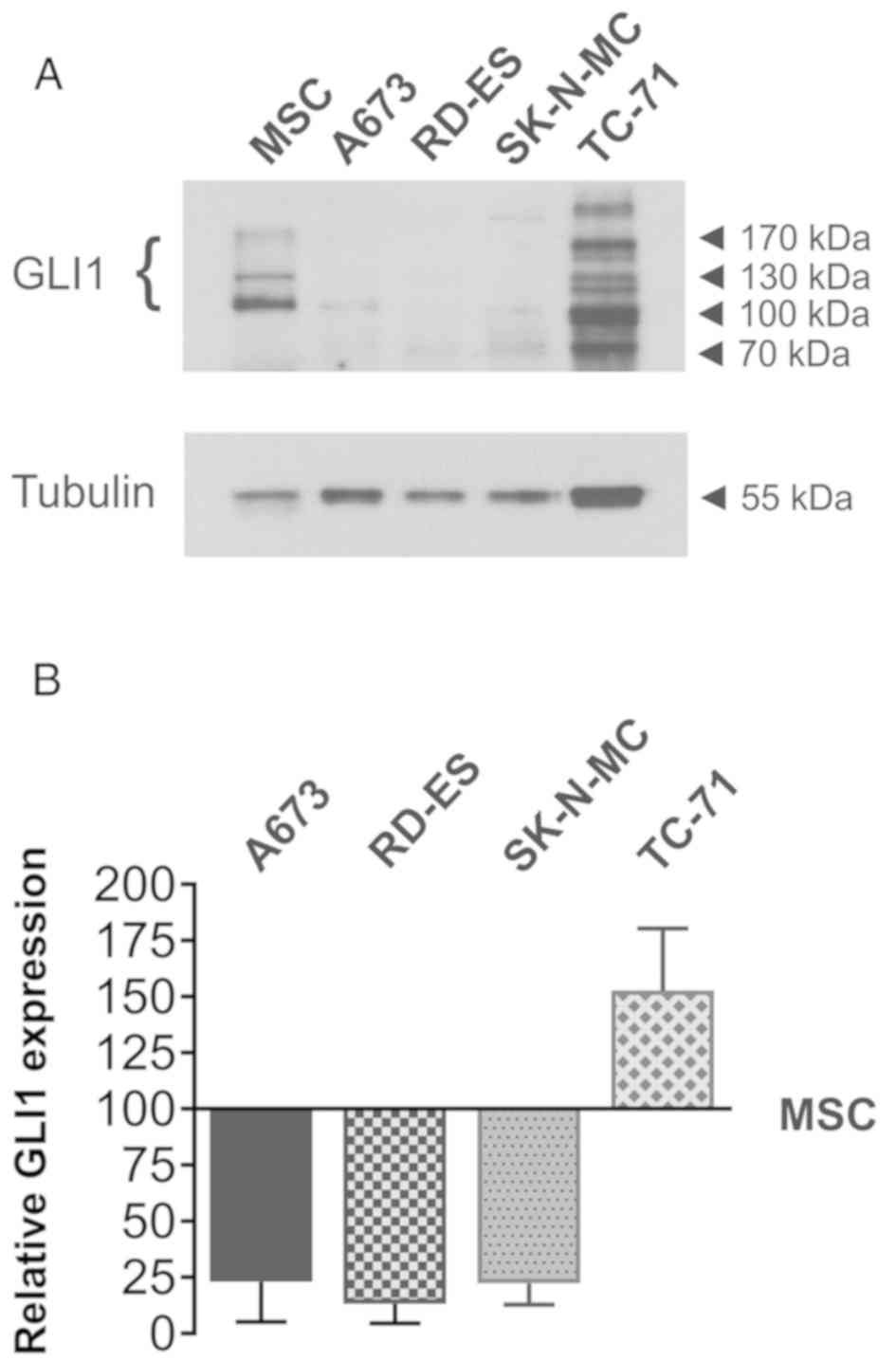
GLI1 protein expression in ES cell
lines
GLI1 protein expression in A673, RD-ES and SK-N-MC
cells in comparison to MSC was analyzed in a previous publication
(5). For a better comparison of GLI1
protein expression in TC-71 cells, western blotting was repeated
including the previously analysed ES cell lines and MSC isolated
from a different patient (Fig. 1A and
B). All GLI1 splice variants with molecular masses between 100
and 160 kDa (39) were quantified and
normalized to tubulin expression. The graph shows the full length
GLI1 protein expression relative to MSC (Fig. 1B). As previously shown (5), GLI1 protein abundance was reduced in the
A673, RD-ES and SK-N-MC cells when compared to MSC, whereas TC-71
cells showed an increased GLI1 protein expression. Compared to the
MSC used in (5), the GLI1 protein
expression difference was more pronounced in the actual
experiments.
Viability reduction by ATO and
etoposide in TC-71 cells
MTS viability assays were performed to determine the
IC50 values of ATO and Eto in the TC-71 cell line
(Table I). The IC50 value
for ATO was 2.12 µM, whereas the half maximal inhibitory
concentration of Eto was determined as 0.3 µM, which was comparable
to the drug sensitivity of the other ES cell lines (5).
 | Table I.ATO and Eto sensitivity of the TC-71
cell line. |
Table I.
ATO and Eto sensitivity of the TC-71
cell line.
| Inhibitor | Cell line | IC50
(µM) |
|---|
| ATO | TC-71 | 2.12 |
| Eto | TC-71 | 0.3 |
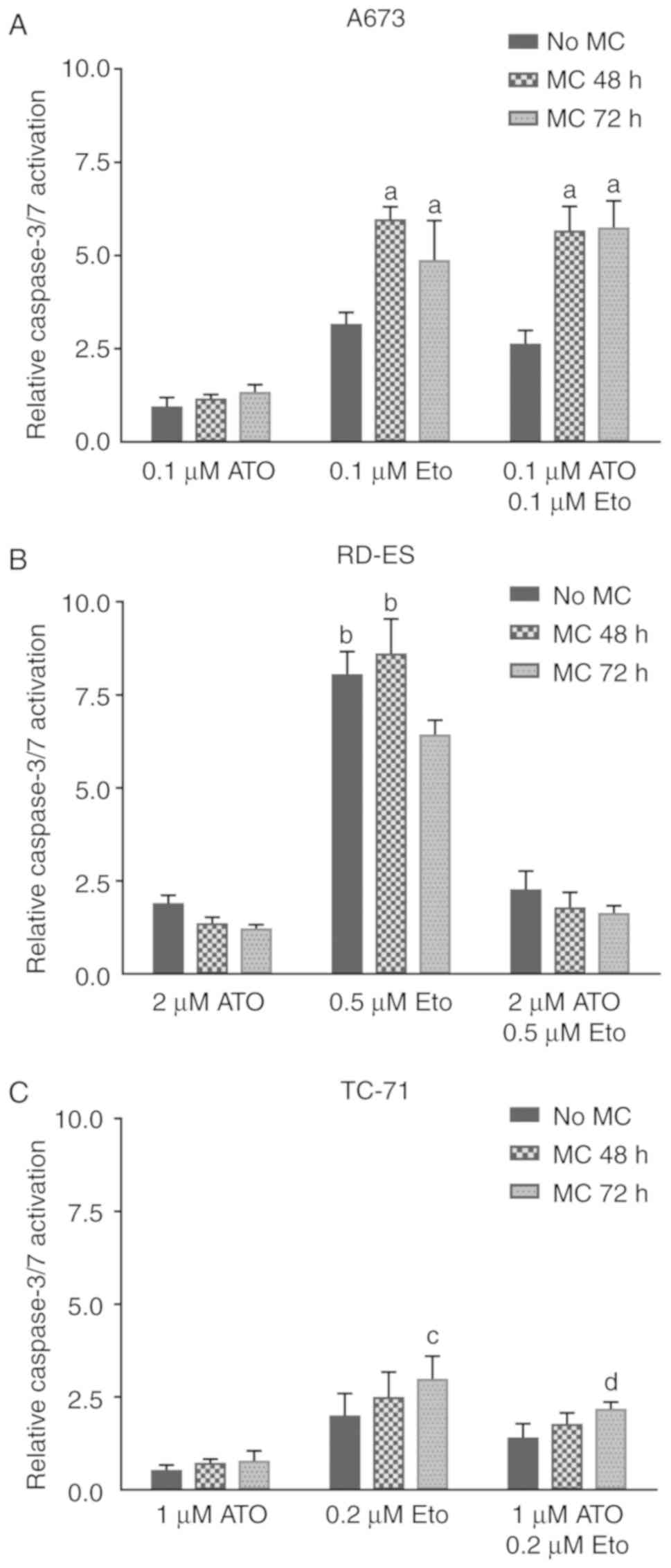
Influence of the temporal order of
repeated drug treatment on caspase activation in ES cell lines
To determine the effect of ATO, Eto and combinations
of both drugs on caspase-dependent apoptosis a caspase-3/7 activity
assay was performed (Fig. 2A-C).
A673, RD-ES and TC-71 cells were either incubated for 96 h without
medium change (MC) or fresh drug solution was added after 48 or 72
h, respectively. Drug doses applied were set individually per cell
line as low as possible depending on previous dose finding tests
and IC50 values. The ATO doses applied to the A673,
RD-ES and TC-71 cells led to a maximal 2-fold increase in
caspase-3/7 activation with only slight differences between the
treatment sequences (Fig. 2A-C). The
general ability of ATO to induce caspase-3 cleavage was previously
shown in A673 and SK-N-MC cells applying higher doses, whereas 5 µM
also ATO induced only minor caspase-3 cleavage in RD-ES cells
(5). Compared to ATO, the Eto doses
applied in the actual experiments induced more pronounced relative
caspase-3/7 activation, which was also influenced by treatment
chronology. In A673 cells (Fig. 2A),
fresh drug application after 48 or 72 h was both favorable compared
to a single Eto treatment (P≤0.001). In contrast, in RD-ES cells
(Fig. 2B) caspase activation could
not be further enhanced by fresh drug application after 48 h,
whereas a medium change providing fresh Eto after 72 h
significantly reduced the detected caspase-3/7 activity (P≤0.001).
In TC-71 cells (Fig. 2C), a second
Eto application induced higher caspase activity, which was
significant for the medium change after 72 h compared to the
incubation without medium change (P≤0.01). In A673 cells the
combined application of ATO and Eto resembled the trend of Eto
treatment (Fig. 2A) with both the
repeated drug application after 48 or 72 h inducing higher caspase
activity compared to the single drug application (P≤0.001). On the
contrary, in RD-ES cells (Fig. 2B)
the combined application of ATO and Eto in any chronology
restricted the caspase activity to a level observed for ATO alone.
In TC-71 cells (Fig. 2C), the
combined drug application resembled the Eto treatment with a second
drug application inducing elevated caspase activity, which was
significant for the medium change after 72 h compared to the
incubation without medium change (P≤0.05). To summarize, in the
A673 and TC-71 cells caspase activity after the combination
treatment was dominated by the Eto effect, whereas in RD-ES cells
ATO application suppressed the pro-apoptotic Eto impact.
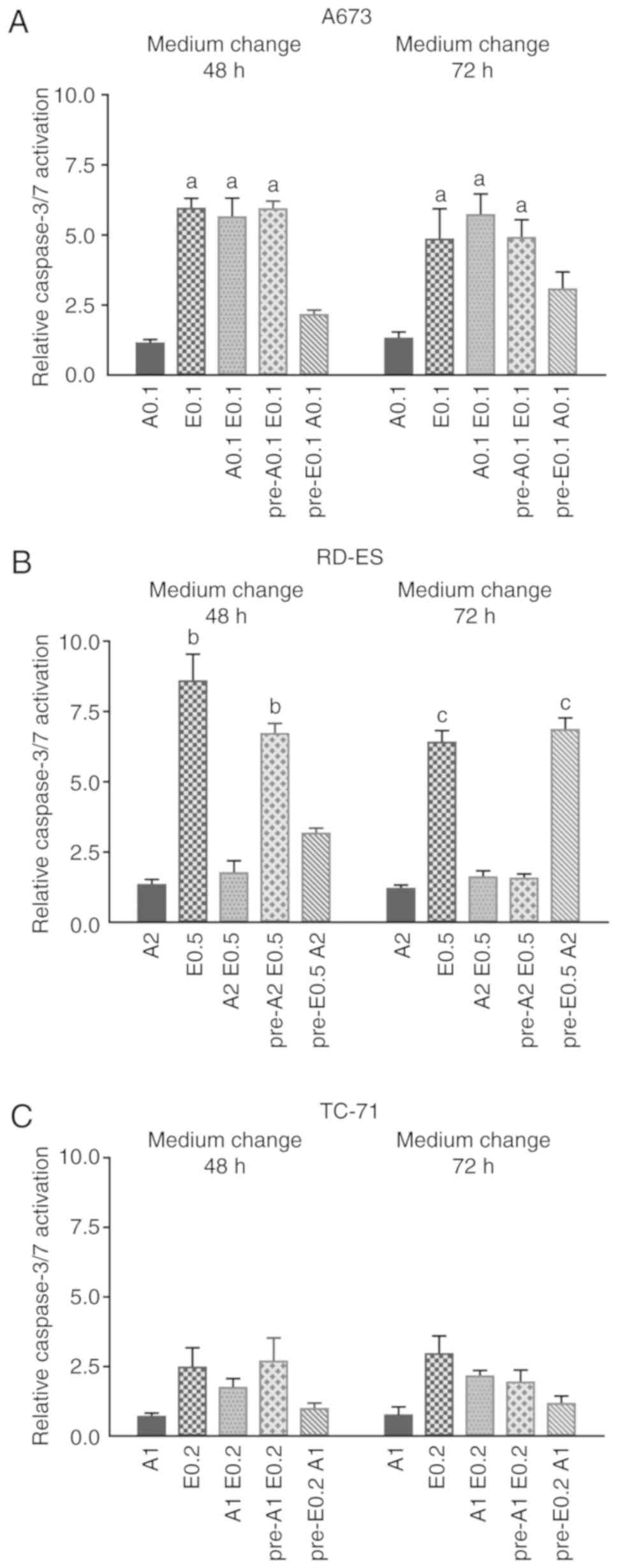
Influence of pre-treatment chronology
on caspase activation in ES cell lines
To further address the interdependence of ATO and
Eto effects on caspase activation in the ES cell lines (A673, RD-ES
and TC-71) consecutive ATO and Eto treatment was compared to single
or combined drug application applying a medium change after 48 or
72 h (Fig. 3A-C). In A673 cells
(Fig. 3A), the treatment with Eto,
the combined treatment with ATO and Eto and the pre-treatment with
ATO followed by Eto incubation all led to comparable caspase-3/7
activation, regardless of when the medium was changed, which was
significantly higher compared to Eto pre-treatment followed by ATO
(P≤0.001). In addition, in the case of pre-Eto incubation in A673
cells, a later medium change applying ATO after 72 h increased
caspase activity compared to the ATO application 48 h after initial
Eto treatment. In RD-ES cells (Fig.
3B), pure Eto incubation and pre-ATO treatment followed by Eto
after 48 h, both were significantly more effective compared to the
combined treatment and pre-Eto application followed by ATO after 48
h. Remarkably, when the medium was changed after 72 h, pre-Eto
treatment followed by ATO showed a similar effect on caspase
activation compared to Eto alone, which was both significantly
higher compared to the combined treatment and pre-ATO application.
Additionally, RD-ES viability as determined by MTS assay (Fig. S1) was significantly less compromised
by pre-ATO treatment followed by Eto compared to all other
treatments, whereas combined treatment led to the most significant
viability reduction. In TC-71 cells (Fig.
3C), the applied doses of ATO and Eto caused no significant
differences in caspase activation. Although, similar to A673 cells,
the treatment with Eto, the combined treatment with ATO and Eto and
the pre-treatment with ATO followed by Eto incubation were all
slightly more effective compared to pre-Eto incubation followed by
ATO. In summary, a distinct caspase activation was observed after
Eto treatment in all 3 ES cell lines examined, whereas individual
ATO treatment sequences cell line-specifically suppressed caspase
activation by Eto.
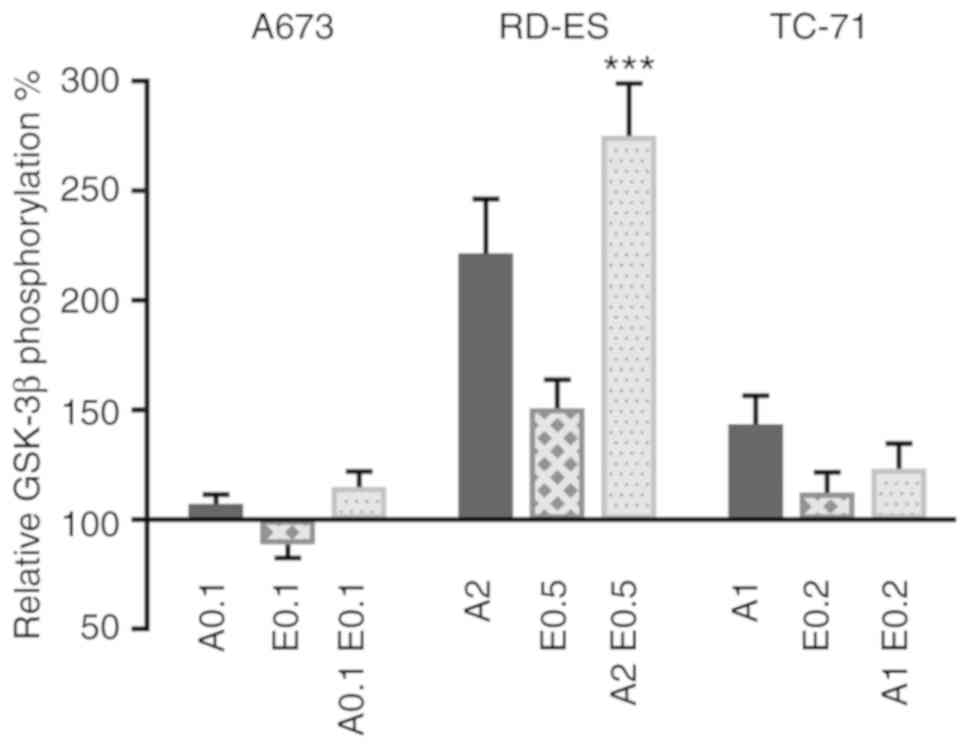
ATO and Eto affect GSK-3β
phosphorylation in ES cells
GSK-3β is implicated in various signaling pathways
affecting metabolism, cell survival and apoptosis (40). Yet, the impact of ATO and Eto on
inhibitory GSK-3β serine 9 phosphorylation in ES cells has not been
investigated to date. To examine the individual and combined Eto
and ATO effects in the ES cell lines (A673, RD-ES and TC-71) both
drugs were applied for 48 h, followed by an ELISA assay to
determine total GSK-3β and phosphorylated GSK-3β protein levels
(Fig. 4). In the A673 cells, the ATO
and Eto doses applied barely changed the relative GSK-3β
phosphorylation. In contrast, in the RD-ES cells especially the
combined application of ATO and Eto increased the relative GSK-3β
phosphorylation (P≤0.001 compared to single treatment). ATO alone
also considerably induced relative GSK-3β phosphorylation in the
RD-ES cells. In the TC-71 cells, ATO slightly increased relative
GSK-3β phosphorylation, whereas the combination treatment had less
impact. This indicates that the applied ATO drug doses specifically
affect the inhibitory GSK-3β phosphorylation in RD-ES cells, where
the pronounced GSK-3β phosphorylation after combination treatment
coincides with restricted caspase activity.
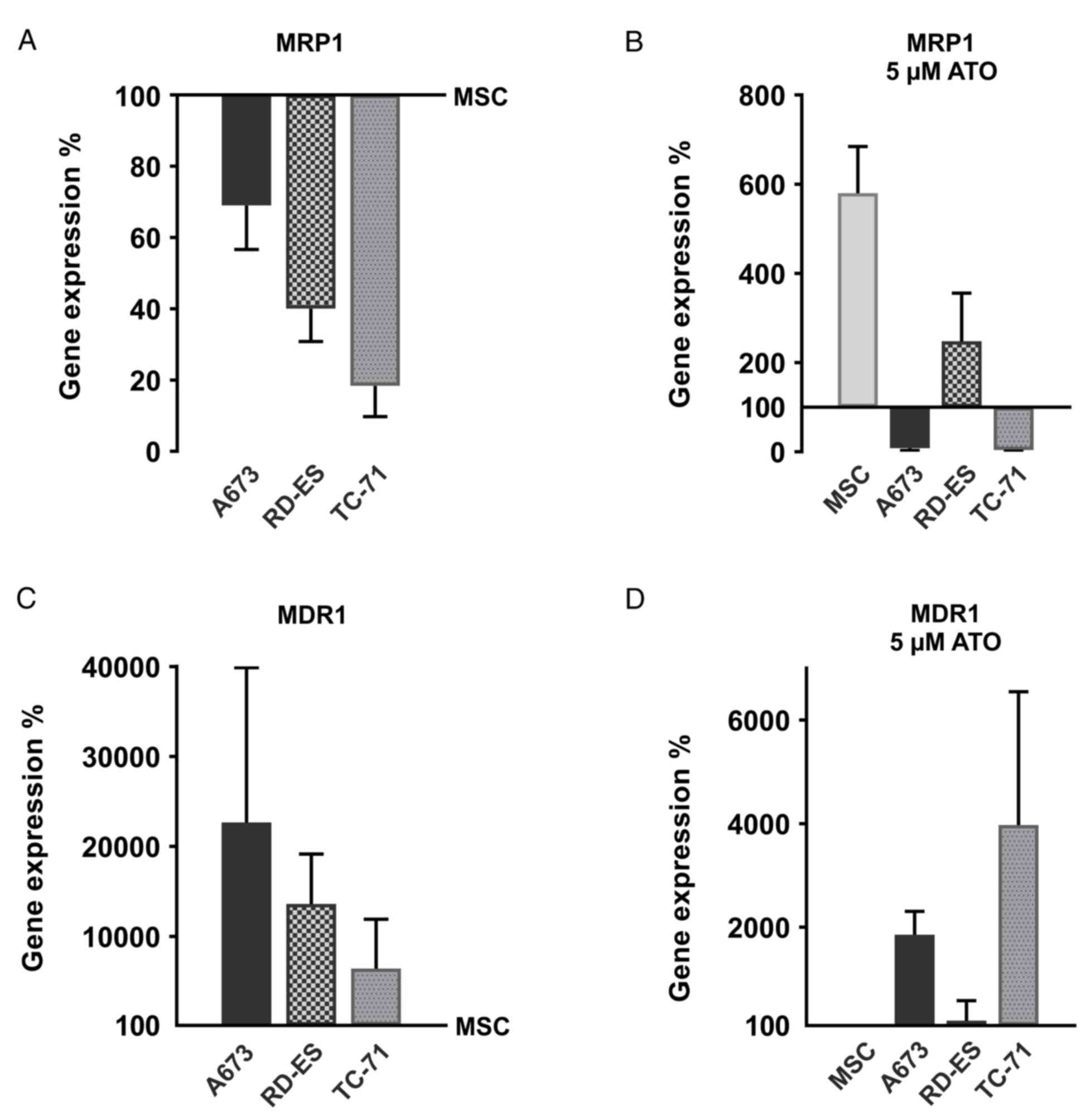
Inherent and ATO-dependent multidrug
transporter gene expression in ES cells
MRP1 and MDR1 expression have been previously shown
in ES samples and cell lines (9,10). Both
multidrug transporters are implicated in acquired Eto resistance of
various cancer cells (41). To
provide an impression of relative MRP1 and MDR1 mRNA abundance,
qRT-PCR was performed in the ES cell lines (A673, RD-ES and TC-71)
compared to the MSC (Fig. 5A and C).
In addition, the ATO impact on MRP1 and MDR1 mRNA expression in ES
cell lines and MSC was examined (Fig. 5B
and D). Indeed, MRP1 mRNA expression was abundant in MSC,
whereas all ES cell lines showed a distinct, yet, compared to MSC.
reduced MRP1 mRNA expression (Fig.
5A). After 96 h treatment with 5 µM ATO, the MRP1 mRNA
expression increased approximately 6-fold in the MSC. In addition,
the RD-ES cells showed an enhanced MRP1 mRNA expression, whereas
ATO reduced MRP1 mRNA abundance in the A673 and TC-71 cells,
actually increasing the expression differences between MSC and the
two ES cell lines. In contrast, basic MDR1 mRNA expression was more
pronounced in the ES cell lines compared to MSC, where MDR1
expression was at the detection limit of the qRT-PCR. In
particular, A673 cells showed robust MDR1 expression, which was
still enhanced by ATO treatment. Moreover in the TC-71 cells, ATO
increased MDR1 expression, whereas MDR1 remained nearly unchanged
in the MSC and RD-ES cells. Therefore, ES cell lines exhibit an
obvious surplus of MDR1 mRNA compared to MSC, which was still
enhanced by ATO in the A673 and TC-71 cells.
Discussion
ES are characterized by a poor differentiation
status with mesenchymal progenitor cell features (42), limited cell adhesion (43) and a heterogeneous EWS-FLI1 protein
expression promoting phenotypic diversity (4). These features contribute to
aggressiveness, early metastasis and acquired treatment resistance
of ES (1,8) necessitating new treatment strategies
which consider the specific nature of ES.
As previously shown, the single and combined
application of ATO and Eto are capable of impairing metabolic
activity, as assessed by MTS viability assay, and to induce
significant cell death, as determined by eFluor 450 incorporation,
in the ES cell lines A673, RD-ES and SK-N-MC, which was clearly
increased after combined treatment compared to individual treatment
in the RD-ES and SK-N-MC cells. In addition, western blot analysis
indicated induction of poly(ADP-ribose) polymerase 1 (PARP1) and
caspase 3 cleavage upon Eto treatment of A673, RD-ES and SK-N-MC
cells. Notably, apoptosis induction by ATO, manifested as caspase-3
and PARP1 cleavage, was only observed in A673 and SK-N-MC cells,
indicating the existence of additional caspase-3-independent cell
death mechanisms at least in RD-ES cells (5). Indeed, ATO has been shown to activate
several cell death pathways in human tumor cells including
ROS-induced PARP1-dependent or caspase-3-dependent mitochondrial
apoptosis but also necrosis (21,28,44,45).
In contrast, Eto mainly induced caspase-3-dependent apoptotic cell
death in ES cells (5,12).
In the present study, the ATO and Eto doses applied
on RD-ES cells were consistent with previously published doses. In
A673 cells, the ATO dose was maintained whereas Eto was reduced to
one-tenth of the previous dose which was still efficient to induce
caspase activation (5). Using low
dose ATO and Eto, the intensity of caspase-3/7 activation was
mainly determined by the Eto doses in A673 and TC-71 cells, whereas
in RD-ES cells ATO application actively suppressed Eto-induced
apoptosis dependent on the treatment chronology. Notably, in RD-ES
cells a late medium change applying ATO after 72 h of Eto
incubation could no more suspend apoptosis induction, whereas a
48-h Eto incubation followed by 48 h ATO largely suppressed
apoptosis. On the other hand, a 48-h ATO pre-incubation was not
able to suppress subsequent Eto-dependent apoptosis, whereas only
24 h Eto application after a 72-h ATO pre-incubation was not
sufficient to obtain a high caspase activation rate. Considering
viability reduction in RD-ES cells, a strong decline could be
observed upon combined treatment, which is consistent with previous
results (5). Yet, ATO pre-incubation
largely prevented viability reduction by Eto. In the experiments,
the applied ATO and Eto doses were set individually dependent on
previous dose finding tests and IC50 values. Since A673
cells exhibited the highest sensitivity, the low ATO dose applied
may not interfere with Eto-dependent apoptosis to the same extent
as observed in the other cell lines. TC-71 cells, although being
highly Eto sensitive in viability assays, showed only comparably
low caspase activation rates upon Eto treatment with a tendency of
apoptosis reduction by concomitant or subsequent ATO treatment.
Based on previous and actual observations, augmented cell death of
RD-ES cells ascertained upon combination treatment with both drugs
(5) must be triggered by a
caspase-3/7-independent pathway since caspase activation is
impaired by concomitant ATO treatment. Taking the example of these
three individual ES cell lines a remarkable heterogeneity of ES
response to ATO and Eto treatment becomes apparent, complicating
the prediction of treatment success.
Indeed, ATO affects various signaling pathways with
low doses up to 0.5 µM actually inducing differentiation in APL
cells (18), whereas higher doses up
to 5 µM commonly induce cell death (5,19,21,28,44,45).
ATO directly acts on the Hh pathway by binding and inhibiting GLI
transcription factors (46). Yet, of
all the ES cell lines tested, only TC-71 cells showed a marked GLI1
protein expression, which was increased compared to the MSC.
However, this did not coincide with an enhanced caspase-3/7
activation by ATO treatment, excluding inhibition of GLI1-dependent
transcription as a crucial trigger for caspase-dependent apoptosis
induction in ES cells. Since GSK-3β serine 9 phosphorylation
interferes with apoptosis induction in different cancer cell lines
(32), physiological ATO and Eto
doses were tested in ES cells concerning their impact on inhibitory
GSK-3β serine 9 phosphorylation. Actually, ATO and especially the
combination of Eto and ATO largely induced inhibitory GSK-3β
phosphorylation in RD-ES cells, coinciding with suppression of
apoptosis in this cell line, whereas GSK-3β phosphorylation in A673
and TC-71 cells was only marginally affected by the ATO and Eto
doses applied. Therefore, the mechanism of apoptosis suppression in
RD-ES cells may depend on ATO-mediated GSK-3β inhibition and
subsequent suppression of mitochondrial apoptosis (47).
Multidrug resistance is common in recurrent and
metastatic ES (8,11). MRP1 protein expression has been shown
in A673, RD-ES and SK-N-MC cells (10). Both, MRP1 and MDR1 can actively
exclude Eto from human cells (41).
In addition, MRP1 and potentially also MDR1 transport arsenic out
of human cells (48,49). Interestingly, ATO itself has been
shown to induce multidrug resistance via upregulation of MRP1
expression (50), while it suppresses
GLI1-dependent MRP1 and MDR1 transcription (51). Multidrug transporter expression by MSC
is an established feature (52),
which may contribute to observed drug tolerance (5). To elucidate the basal and ATO-dependent
MRP1 and MDR1 expression in A673, RD-ES and TC-71 cells compared to
MSC, qRT-PCR was performed, showing that MSC indeed express higher
MRP1 mRNA levels compared to the ES cell lines A673, RD-ES and
TC-71, while MDR1 mRNA expression was clearly more abundant in the
ES cell lines. ATO treatment had a diverse effect on MRP1 and MDR1
expression. While MRP1 mRNA expression increased upon ATO
incubation in MSC and RD-ES, it was attenuated in A673 and TC-71
cells. Yet, MDR1 mRNA expression was enhanced by ATO in A673 and
TC-71 cells. To summarize, ATO reinforces the differences of MRP1
and MDR1 mRNA expression in MSC compared to A673 and TC-71 ES
cells. Nevertheless, further experiments are needed to clarify the
impact of ATO as well as Eto and ATO combination treatment on MRP1
and MDR1 protein expression and activity in ES cell lines to
determine a possible interference with drug resistance.
New therapeutic approaches for ES targeting receptor
tyrosine kinases, the EWS-FLI1 fusion protein and immunotherapies
using T cells and NK cells are currently being evaluated in
clinical trials (53). Yet, usually
only a small subset of patients responds to the individual
therapies underlining the intricate heterogeneity of ES (54). Combining ATO and Eto remains a
promising approach for cell death induction in ES, although
individual ES characteristics potentially impairing the efficacy
must be considered.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JEL and RR carried out the experiments. JEL, RR,
KAB, SBS, RH and FT collected and analyzed the data. KAB and FT
designed the experiments. KAB and JEL wrote, improved and finalized
the manuscript. All authors read and approved the final manuscript
and agree to be accountable for all aspects of the research in
ensuring that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Ethics approval was provided by The Ethics Committee
of the Medical Faculty of the University Hospital Tuebingen,
project no. 401/2013 BO2. Bone marrow-derived MSC were isolated at
the University Hospital Tuebingen after written informed consent of
the patients.
Patient consent for publication
Not applicable.
Authors' information
Julia E. Lenz, ORCID 0000-0001-5890-3489. Frank
Traub, ORCID 0000-0002-6400-3257.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ATO
|
arsenic trioxide
|
|
ES
|
Ewing sarcoma(s)
|
|
EWS
|
Ewing sarcoma breakpoint region
|
|
Eto
|
etoposide
|
|
FLI1
|
Friend leukemia integration 1
|
|
GLI1
|
glioma associated oncogene family
1
|
|
GSK3-β
|
glycogen synthase kinase-3β
|
|
MDR1
|
multidrug resistance protein 1
|
|
MRP1
|
multidrug resistance-associated
protein 1
|
|
MSC
|
mesenchymal stem cells
|
References
|
1
|
Ross KA, Smyth NA, Murawski CD and Kennedy
JG: The biology of Ewing sarcoma. ISRN Oncol.
2013:7597252013.PubMed/NCBI
|
|
2
|
Tirode F, Laud-Duval K, Prieur A, Delorme
B, Charbord P and Delattre O: Mesenchymal stem cell features of
Ewing tumors. Cancer Cell. 11:421–429. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Beauchamp E, Bulut G, Abaan O, Chen K,
Merchant A, Matsui W, Endo Y, Rubin JS, Toretsky J and Uren A: GLI1
is a direct transcriptional target of EWS-FLI1 oncoprotein. J Biol
Chem. 284:9074–9082. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Franzetti GA, Laud-Duval K, van der Ent W,
Brisac A, Irondelle M, Aubert S, Dirksen U, Bouvier C, de Pinieux
G, Snaar-Jagalska E, et al: Cell-to-cell heterogeneity of
EWSR1-FLI1 activity determines proliferation/migration choices in
Ewing sarcoma cells. Oncogene. 36:3505–3514. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Boehme KA, Nitsch J, Riester R,
Handgretinger R, Schleicher SB, Kluba T and Traub F: Arsenic
trioxide potentiates the effectiveness of etoposide in Ewing
sarcomas. Int J Oncol. 49:2135–2146. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Beauchamp EM, Ringer L, Bulut G, Sajwan
KP, Hall MD, Lee YC, Peaceman D, Ozdemirli M, Rodriguez O,
Macdonald TJ, et al: Arsenic trioxide inhibits human cancer cell
growth and tumor development in mice by blocking hedgehog/GLI
pathway. J Clin Invest. 121:148–160. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sand LG, Szuhai K and Hogendoorn PC:
Sequencing overview of Ewing sarcoma: A journey across genomic,
epigenomic and transcriptomic landscapes. Int J Mol Sci.
16:16176–16215. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
May WA, Grigoryan RS, Keshelava N, Cabral
DJ, Christensen LL, Jenabi J, Ji L, Triche TJ, Lawlor ER and
Reynolds CP: Characterization and drug resistance patterns of
Ewing's sarcoma family tumor cell lines. PLoS One. 8:e800602013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Oda Y, Dockhorn-Dworniczak B, Jürgens H
and Roessner A: Expression of multidrug resistance-associated
protein gene in Ewing's sarcoma and malignant peripheral
neuroectodermal tumor of bone. J Cancer Res Clin Oncol.
123:237–239. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roundhill EA and Burchill SA: Detection
and characterisation of multi-drug resistance protein 1 (MRP-1) in
human mitochondria. Br J Cancer. 106:1224–1233. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Roessner A, Ueda Y, Bockhorn-Dworniczak B,
Blasius S, Peters A, Wuisman P, Ritter J, Paulussen M, Jürgens H
and Böcker W: Prognostic implication of immunodetection of P
glycoprotein in Ewing's sarcoma. J Cancer Res Clin Oncol.
119:185–189. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mauz-Körholz C, Kachel M, Harms-Schirra B,
Klein-Vehne A, Tunn PU and Körholz D: Drug-induced caspase-3
activation in a Ewing tumor cell line and primary Ewing tumor
cells. Anticancer Res. 24:145–149. 2004.PubMed/NCBI
|
|
13
|
Casali PG, Bielack S, Abecassis N, Aro HT,
Bauer S, Biagini R, Bonvalot S, Boukovinas I, Bovee JVMG, Brennan
B, et al: ESMO Guidelines Committee, paedcan and ERN EURACAN: Bone
sarcomas: ESMO-PaedCan-EURACAN clinical practice guidelines for
diagnosis, treatment and follow-up. Ann Oncol. 29 (Suppl
4):iv79–iv95. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Clifford B, Beljin M, Stark GR and Taylor
WR: G2 arrest in response to topoisomerase II inhibitors: The role
of p53. Cancer Res. 63:4074–4081. 2003.PubMed/NCBI
|
|
15
|
Lin CF, Tsai CC, Huang WC, Wang YC, Tseng
PC, Tsai TT and Chen CL: Glycogen synthase kinase-3β and caspase-2
mediate ceramide- and etoposide-induced apoptosis by regulating the
lysosomal-mitochondrial axis. PLoS One. 11:e01454602016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee KI, Su CC, Yang CY, Hung DZ, Lin CT,
Lu TH, Liu SH and Huang CF: Etoposide induces pancreatic β-cells
cytotoxicity via the JNK/ERK/GSK-3 signaling-mediated
mitochondria-dependent apoptosis pathway. Toxicol In Vitro.
36:142–152. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ngok-Ngam P, Watcharasit P, Thiantanawat A
and Satayavivad J: Pharmacological inhibition of GSK3 attenuates
DNA damage-induced apoptosis via reduction of p53 mitochondrial
translocation and Bax oligomerization in neuroblastoma SH-SY5Y
cells. Cell Mol Biol Lett. 18:58–74. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lengfelder E, Hofmann WK and Nowak D:
Impact of arsenic trioxide in the treatment of acute promyelocytic
leukemia. Leukemia. 26:433–442. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Boehme KA, Zaborski JJ, Riester R,
Schweiss SK, Hopp U, Traub F, Kluba T, Handgretinger R and
Schleicher SB: Targeting hedgehog signalling by arsenic trioxide
reduces cell growth and induces apoptosis in rhabdomyosarcoma. Int
J Oncol. 48:801–812. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Au WY, Tam S, Fong BM and Kwong YL:
Determinants of cerebrospinal fluid arsenic concentration in
patients with acute promyelocytic leukemia on oral arsenic trioxide
therapy. Blood. 112:3587–3590. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kang YH, Yi MJ, Kim MJ, Park MT, Bae S,
Kang CM, Cho CK, Park IC, Park MJ, Rhee CH, et al:
Caspase-independent cell death by arsenic trioxide in human
cervical cancer cells: Reactive oxygen species-mediated
poly(ADP-ribose) polymerase-1 activation signals apoptosis-inducing
factor release from mitochondria. Cancer Res. 64:8960–8967. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Beauchamp EM and Uren A: A new era for an
ancient drug: Arsenic trioxide and Hedgehog signaling. Vitam Horm.
88:333–354. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dawood M, Hamdoun S and Efferth T:
Multifactorial modes of action of arsenic trioxide in cancer cells
as analyzed by classical and network pharmacology. Front Pharmacol.
9:1432018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chiu HW, Tseng YC, Hsu YH, Lin YF, Foo NP,
Guo HR and Wang YJ: Arsenic trioxide induces programmed cell death
through stimulation of ER stress and inhibition of the
ubiquitin-proteasome system in human sarcoma cells. Cancer Lett.
356:762–772. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kozono S, Lin YM, Seo HS, Pinch B, Lian X,
Qiu C, Herbert MK, Chen CH, Tan L, Gao ZJ, et al: Arsenic targets
Pin1 and cooperates with retinoic acid to inhibit cancer-driving
pathways and tumor-initiating cells. Nat Commun. 9:30692018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jiang L, Wang L, Chen L, Cai GH, Ren QY,
Chen JZ, Shi HJ and Xie YH: As2O3 induces apoptosis in human
hepatocellular carcinoma HepG2 cells through a ROS-mediated
mitochondrial pathway and activation of caspases. Int J Clin Exp
Med. 8:2190–2196. 2015.PubMed/NCBI
|
|
27
|
Yen YP, Tsai KS, Chen YW, Huang CF, Yang
RS and Liu SH: Arsenic induces apoptosis in myoblasts through a
reactive oxygen species-induced endoplasmic reticulum stress and
mitochondrial dysfunction pathway. Arch Toxicol. 86:923–933. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kumar S, Yedjou CG and Tchounwou PB:
Arsenic trioxide induces oxidative stress, DNA damage, and
mitochondrial pathway of apoptosis in human leukemia (HL-60) cells.
J Exp Clin Cancer Res. 33:422014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
States JC: Disruption of mitotic
progression by arsenic. Biol Trace Elem Res. 166:34–40. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang HS, Liu ZM and Cheng YL: Involvement
of glycogen synthase kinase-3β in arsenic trioxide-induced p21
expression. Toxicol Sci. 121:101–109. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schleicher SB, Zaborski JJ, Riester R,
Zenkner N, Handgretinger R, Kluba T, Traub F and Boehme KA:
Combined application of arsenic trioxide and lithium chloride
augments viability reduction and apoptosis induction in human
rhabdomyosarcoma cell lines. PLoS One. 12:e01788572017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Beurel E and Jope RS: The paradoxical pro-
and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic
apoptosis signaling pathways. Prog Neurobiol. 79:173–189. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Battula VL, Treml S, Bareiss PM, Gieseke
F, Roelofs H, de Zwart P, Müller I, Schewe B, Skutella T, Fibbe WE,
et al: Isolation of functionally distinct mesenchymal stem cell
subsets using antibodies against CD56, CD271, and mesenchymal stem
cell antigen-1. Haematologica. 94:173–184. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Laurendeau I, Ferrer M, Garrido D, D'Haene
N, Ciavarelli P, Basso A, Vidaud M, Bieche I, Salmon I and Szijan
I: Gene expression profiling of the hedgehog signaling pathway in
human meningiomas. Mol Med. 16:262–270. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xie J, Li DW, Chen XW, Wang F and Dong P:
Expression and significance of hypoxia-inducible factor-1α and
MDR1/P- glycoprotein in laryngeal carcinoma tissue and hypoxic
Hep-2 cells. Oncol Lett. 6:232–238. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
König J, Hartel M, Nies AT, Martignoni ME,
Guo J, Büchler MW, Friess H and Keppler D: Expression and
localization of human multidrug resistance protein (ABCC) family
members in pancreatic carcinoma. Int J Cancer. 115:359–367. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Benaitreau D, Dieudonné MN, Dos Santos E,
Leneveu MC, Mazancourt PD and Pecquery R: Antiproliferative effects
of adiponectin on human trophoblastic cell lines JEG-3 and BeWo.
Biol Reprod. 80:1107–1114. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Amable L, Gavin E, Kudo K, Meng E, Rocconi
RP, Shevde LA and Reed E: GLI1 upregulates C-JUN through a specific
130-kDa isoform. Int J Oncol. 44:655–661. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
McCubrey JA, Steelman LS, Bertrand FE,
Davis NM, Sokolosky M, Abrams SL, Montalto G, D'Assoro AB, Libra M,
Nicoletti F, et al: GSK-3 as potential target for therapeutic
intervention in cancer. Oncotarget. 5:2881–2911. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gottesman MM: Mechanisms of cancer drug
resistance. Annu Rev Med. 53:615–627. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kauer M, Ban J, Kofler R, Walker B, Davis
S, Meltzer P and Kovar H: A molecular function map of Ewing's
sarcoma. PLoS One. 4:e54152009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chaturvedi A, Hoffman LM, Jensen CC, Lin
YC, Grossmann AH, Randall RL, Lessnick SL, Welm AL and Beckerle MC:
Molecular dissection of the mechanism by which EWS/FLI expression
compromises actin cytoskeletal integrity and cell adhesion in Ewing
sarcoma. Mol Biol Cell. 25:2695–2709. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
McCafferty-Grad J, Bahlis NJ, Krett N,
Aguilar TM, Reis I, Lee KP and Boise LH: Arsenic trioxide uses
caspase-dependent and caspase-independent death pathways in myeloma
cells. Mol Cancer Ther. 2:1155–1164. 2003.PubMed/NCBI
|
|
45
|
Scholz C, Wieder T, Stärck L, Essmann F,
Schulze-Osthoff K, Dörken B and Daniel PT: Arsenic trioxide
triggers a regulated form of caspase-independent necrotic cell
death via the mitochondrial death pathway. Oncogene. 24:1904–1913.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Raju GP: Arsenic: A potentially useful
poison for hedgehog-driven cancers. J Clin Invest. 121:14–16. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yang K, Chen Z, Gao J, Shi W, Li L, Jiang
S, Hu H, Liu Z, Xu D and Wu L: The Key roles of GSK-3β in
regulating mitochondrial activity. Cell Physiol Biochem.
44:1445–1459. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shukalek CB, Swanlund DP, Rousseau RK,
Weigl KE, Marensi V, Cole SP and Leslie EM: Arsenic triglutathione
[As(GS)3] transport by multidrug resistance protein 1 (MRP1/ABCC1)
is selectively modified by phosphorylation of Tyr920/Ser921 and
glycosylation of Asn19/Asn23. Mol Pharmacol. 90:127–139. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Maciaszczyk-Dziubinska E, Wawrzycka D and
Wysocki R: Arsenic and antimony transporters in eukaryotes. Int J
Mol Sci. 13:3527–3548. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Seo T, Urasaki Y and Ueda T: Establishment
of an arsenic trioxide-resistant human leukemia cell line that
shows multidrug resistance. Int J Hematol. 85:26–31. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Santisteban M: ABC transporters as
molecular effectors of pancreatic oncogenic pathways: The
Hedgehog-GLI model. J Gastrointest Cancer. 41:153–158. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Erdei Z, Lőrincz R, Szebényi K, Péntek A,
Varga N, Likó I, Várady G, Szakács G, Orbán TI, Sarkadi B and Apáti
A: Expression pattern of the human ABC transporters in pluripotent
embryonic stem cells and in their derivatives. Cytometry B Clin
Cytom. 86:299–310. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Brown HK, Schiavone K, Gouin F, Heymann MF
and Heymann D: Biology of bone sarcomas and new therapeutic
developments. Calcif Tissue Int. 102:174–195. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yu H, Ge Y, Guo L and Huang L: Potential
approaches to the treatment of Ewing's sarcoma. Oncotarget.
8:5523–5539. 2017.PubMed/NCBI
|