Introduction
Chromatin regulators play an important role in
DNA-templated processes including transcription, DNA replication
and repair, and are strongly associated with tumorigenesis
(1–3).
We previously identified a bromodomain-containing, AAA+ ATPase
protein ANCCA (AAA+ nuclear coregulatory cancer-associated protein,
also known as ATPase family AAA domain containing 2 or ATAD2), as a
nuclear coactivator for estrogen and androgen receptors (4,5). ANCCA
regulates estrogen- or androgen-induced expression of genes
involved in proliferation and survival of cancer cells. Further
studies demonstrated that ANCCA/ATAD2 is overexpressed in different
types of human cancers including breast, lung, liver, gastric,
cervical and endometrial cancer, and that its overexpression in
TNBC and other cancers is strongly correlated with poor prognosis
(6–14). Mechanistically, ANCCA/ATAD2 likely
functions by facilitating the assembly of histone-modifying protein
complexes at target gene chromatin loci of E2F, Myc and nuclear
receptor-regulated genes (4,5,8,15,16).
Notably, it was demonstrated that ANCCA/ATAD2 may also be involved
in DNA replication through its interactions with acetylated
histones (17), indicating that the
bromodomain protein may play a role in DNA/chromatin-based
processes beyond transcriptional regulation. Recently, a drug
discovery campaign by pharmaceutical companies has led to the
identification of several small-molecule probes against its
bromodomain (18,19), paving the way to development of
ANCCA-targeting drugs for treatment of many types of cancer.
DNA damages and consequent aberrant repair are
fundamental genetic processes that are often derailed in cell
transformation and tumorigenesis (20). On the other hand, anti-cancer
radiation and chemotherapy yield clinical benefits by causing
genomic instability and DNA damages in cancer cells. Thus,
identification of key pathways and regulators for DNA repair in
cancer cells, especially those evoked by anti-cancer therapies, can
provide important information for enhancing the therapy benefit in
cancer treatment. In mammalian cells, there are at least five major
mechanisms to repair different types of DNA damage (21). These are base excision repair (BER),
mismatch repair (MMR), nucleotide excision repair (NER), and
double-strand break repair (DSB), which includes homologous
recombination (HR) and non-homologous end joining (NHEJ). Although
gene transcription and DNA repair are two seemingly separate
processes, accumulating evidence has indicated that they are highly
coordinated in response to genotoxic stress conditions (22). For example, transcriptional regulation
of core factors in BER, NER and MMR facilitates DNA repair, whereas
DNA repair factors often monitor the transcription process to
maintain fidelity and genome integrity. Recent studies also
indicate that transcription factors such as E2F1 can be recruited
onto the DNA damage foci where they can directly regulate the DNA
repair process (23). One emerging,
critical mechanism underlying transcription factors-mediated local
DNA repair may be related to their functions to alter chromatin
structure and facilitate the repair efficacy (22), suggesting that chromatin regulators
with chromatin-remodeling activity could be the extended DNA repair
machinery components. In fact, numerous chromatin-modifying
proteins and remodeling complexes have been shown to play crucial
roles in DNA damage response and repair by facilitating the local
chromatin structure change and nucleosome dynamics (24). For example, the recruitment of a
lysine methyltransferase (KMT) MMSET/NSD2 to the DSB sites mediates
H4K20 methylation which, in turn, facilitates the formation of
γH2AX-MDC1-53BP1 complex and repair of the damaged DNA (25). Conversely, although bromodomain
protein BRD4 in its full-length may promote DNA repair through
regulation of the expression of DNA repair genes (26), its shorter isoform can recruit the
condensin II chromatin remodeling complex to acetylated histones at
the damaged site to inhibit the DNA damage response (27).
In the present study, the possibility that ANCCA is
an important responder of cancer cells to genotoxic stress
condition was examined. It was revealed that ANCCA protein
expression was strongly induced by DNA-damaging, anticancer agents
and that increased ANCCA upregulated the expression and activation
of key DNA damage response and repair factors including BRCA1.
Moreover, it was revealed that ANCCA silencing sensitized TNBC
cells to carboplatin. Collectively, these results provide the first
evidence indicating that the bromodomain protein ANCCA is an
important mediator of DNA damage response and repair in cancer
cells and that therapeutics targeting ANCCA hold promise in
enhancing chemotherapy efficacy in TNBC.
Materials and methods
Cell culture, siRNA transfections and
drug treatment
MDA-MB-468 and H1299 cells were grown in RPMI-1640
medium or DMEM respectively, supplemented with 10% fetal bovine
serum (Hyclone; GE Healthcare Life Sciences), at 37°C in 5%
CO2 incubators. They were purchased from ATCC and
recently authenticated using short tandem repeat profiling and used
for experiments within 3 to 8 passages after thawing. They were
also frequently tested to ensure the absence of mycoplasma. For
siRNA transfection, the cells were transfected with Dharmafect 1
(GE Healthcare Dharmacon, Inc.) according to the manufacturer's
protocol. The sequences for the siRNAs were ANCCA#6,
GCUACUGUUUACUAUCAGGCU; ANCCA#7, CAAGCUGCUAAGCCUCCUAUAUU; ANCCA#58,
GUAGGAUUAGAAGUCGUUAUA; ATM, GCGCCUGAUUCGAGAUCCU; ATR,
AACCUCCGUGAUGUUGCUUGA; control targeting the luciferase gene,
CTTACGCTGAGTACTTCGA. For chemotherapy drug treatments, cells were
plated at equal densities, treated with different concentrations of
drugs and harvested for western blotting (WB) at different
time-points as indicated in the experiments.
WB and qRT-PCR
Protein samples were prepared by lysing cells in
modified RIPA buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2 mM
EGTA, 5 mM EDTA, 1% Triton X-100, 0.1% SDS, 1% deoxycholate, 10 mM
NaF, 0.5 mM Na3VO4, and 10% glycerol). Protein concentrations were
measured using Bio-Rad DC Protein Assay kit. Lysates (50–100 µg)
were separated on a 10% SDS-PAGE gel and transferred to a PVDF
membrane. The membrane was then blocked with 5% milk in 1X TBST
buffer at room temperature for 1 h with shaking and, after washing
in TBST, incubated with different specific primary antibodies
overnight at 4°C. After washing in TBST, the membrane was incubated
with appropriate secondary antibody at room temperature for 1 h.
Visualization/detection of proteins on the membrane was performed
using enhanced chemiluminescence (ECL) WB Reagents from GE
Healthcare (RPN2106) followed by exposure to X-ray film. The
antibody for ANCCA was generated and purified as previously
described (6). Other primary
antibodies were obtained from Cell Signaling Technology, Inc.
(BRAC1, cat. no. 9010; pBRCA1-S1524, cat. no. 9009; pATR-S428, cat.
no. 2853; ATR, cat. no. 2790; pATM-S1981, cat. no. 4526; ATM, cat.
no. 2873; pChk1-S345, cat. no. 2348; Chk1, cat. no. 2360;
pChk2-T68, cat. no. 2661; and GAPDH, cat. no. 2118) or from Santa
Cruz Biotechnology, Inc. (53BP1, cat. no. sc-22760; Rad51, cat. no.
sc-8349; E2F1, cat. no. sc-251; β-actin, cat. no. sc-47778).
Antibodies were used at 1:1,000 dilutions, except antibody against
beta-actin which was used at 1;2,000 dilutions. qRT-PCR was
performed as described with primers reported previously (6).
Immunofluorescence (IF)
For IF, after washing with PBS, the cells were fixed
by 4% formaldehyde in PBS for 10 min at 4°C. After fixation, the
slides were rinsed with PBS. Cells were permeabilized for 10 min at
room temperature (RT) with 0.1% Triton X-100 in PBS and blocked
with 5% (FBS) in PBS (blocking solution) for 30 min at RT. After 2
h of incubation with primary antibodies diluted in the blocking
solution and being rinsed three times in PBS, slides were incubated
for 1 h with the appropriate secondary antibodies diluted 1:1,000
in blocking solution. The following antibodies and their dilutions
were used: γH2AX (cat. no. 05-636; 1:200; EMD Millipore), 53BP1
(cat. no. sc-22760; 1:500; Santa Cruz Biotechnology, Inc.), and
ANCCA (1:200; homemade described as aforementioned). Following
rinses (four times in PBS), slides were mounted with Vectashield
mounting medium (Vector Laboratories, Inc.) and sealed with clear
nail polish. Images were acquired using a Zeiss LSM510 scanning
confocal microscope or Olympus BX61 at the same exposure
settings.
Chromatin immunoprecipitation (ChIP)
assay
ChIP was performed essentially as previously
described (28) with the following
modifications. MDA-MB-468 cells were treated with 1 µM carboplatin
for 12 h and harvested for ChIP. The crude chromatin solutions were
first cleared with Protein A beads (Invitrogen; Thermo Fisher
Scientific, Inc.) that were pre-coated with pre-immune serum for 2
h at 4°C. The pre-cleared supernatants were then incubated with
indicated antibodies at 4°C overnight prior to precipitation with
Protein A beads that had been pre-blocked with BSA and sonicated
salmon sperm DNA. ChIP DNA was analyzed by real-time PCR with
SYBR-Green (Takara Bio.) on a PCR machine. Enrichment of genomic
DNA was presented as the percentage of recovery relative to the
input. The primers for BRCA1 promoter were forward,
5′-CGACTGCTTTGGACAATAGGTAGCG-3′ and reverse,
5′-GAGTAGAGGCTAGAGGGCAGGCAC-3′.
Colony formation assay
MDA-MB-468 cells were transfected with indicated
siRNAs. Forty-eight hours after transfection, the cells were
treated with chemotherapy drugs for 2 h. For MDA-MB-468 cells, 1.0,
2.5, 5.0 or 10 µM carboplatin were used to obtain a curve. Cells
were then trypsinized and plated. After re-plating, the cells were
incubated for 10–12 days with medium changed every 3 days. The
cells in the plates were then fixed with 10% formaldehyde in PBS,
and stained with 0.5% crystal violet. The cells were incubated for
30 min and the stain was removed. The number of cell colonies were
counted using ImageJ (version 1.46; National Institutes of
Health).
Homologous recombination assay
Homologous recombination (HR) was assessed using a
direct repeat green fluorescent protein (DR-GFP) assay as
previously described (29,30). Briefly, H1299-DR-GFP cells with an
integrated DR-GFP construct were transiently transfected with
pCMV3×nlsI-SceI along with different siRNAs separately
including control, ANCCA, or RAD51 siRNA. At 72 h after
transfection, the cells were trypsinized and assessed for GFP
expression with a BD flow cytometer. The results were analyzed by
FlowJo software (version 9.4.9; FlowJo LLC).
Statistical analysis
Results were reported as the means ± standard
deviations. According to the number of groups and variances, data
were analyzed with unpaired Student's t-test or one-way
ANOVA with Tukey's multiple comparison test used for the post hoc
test (GraphPad Prism; version 5.0; GraphPad Software, Inc.). Any
difference was considered as significant if the probability
<0.05 (P<0.05).
Results
ANCCA protein expression is induced by
DNA-damaging drugs and ionizing radiation (IR)
Cellular DNA damage response and repair (DDR)
involve induction of expression and/or activities of many proteins
including the chromatin regulators. Our inspection of ANCCA amino
sequences revealed that ANCCA protein contains at its C-terminus,
five (S/T)Q sites potentially phosphorylated by ATM/ATR kinases
(Fig. 1A). Notably, two of the sites,
S1277 and S1302, were demonstrated in a proteomics study to be
phosphorylated upon UV-induced DNA damage (31). One common effect of ATM/ATR
phosphorylation is to stabilize the substrate protein. To examine
whether ANCCA protein level could be altered upon DNA damage,
MDA-MB-468 cells, a TNBC cell line, were treated with several
DNA-damaging agents and then subjected to immunoblotting analysis.
Results revealed in Fig. 1B
demonstrated that drugs that cause single and/or double DNA strand
breaks, such as cisplatin, carboplatin, doxorubicin (Doxo) and
mitomycin C (MMC), induced ANCCA protein expression as early as 6 h
after the drug treatment (Fig. 1B).
The induction persisted for at least 24 h. However, paclitaxel
(Pacl), which inhibits spindle function, and methotrexate (MTX),
which inhibits the metabolism of folic acid and thus the synthesis
of macromolecules, did not induce sustained ANCCA expression.
MDA-MB-468 cells express a GOF mutant (R273H) form of p53. To
examine whether p53 plays a role in the ANCCA induction, a p53-null
lung cancer cell line H1299 was treated with the same drugs.
Notably, similar results (Fig. 1C)
were observed, indicating that cancer cell p53 status is not a
crucial factor in the induction. Since IR can cause immediate and
direct DNA breaks, H1299 cells were also exposed to IR and ANCCA
expression was examined. As early as 2 h after IR exposure, the
induction of ANCCA was detected (Fig.
1D).
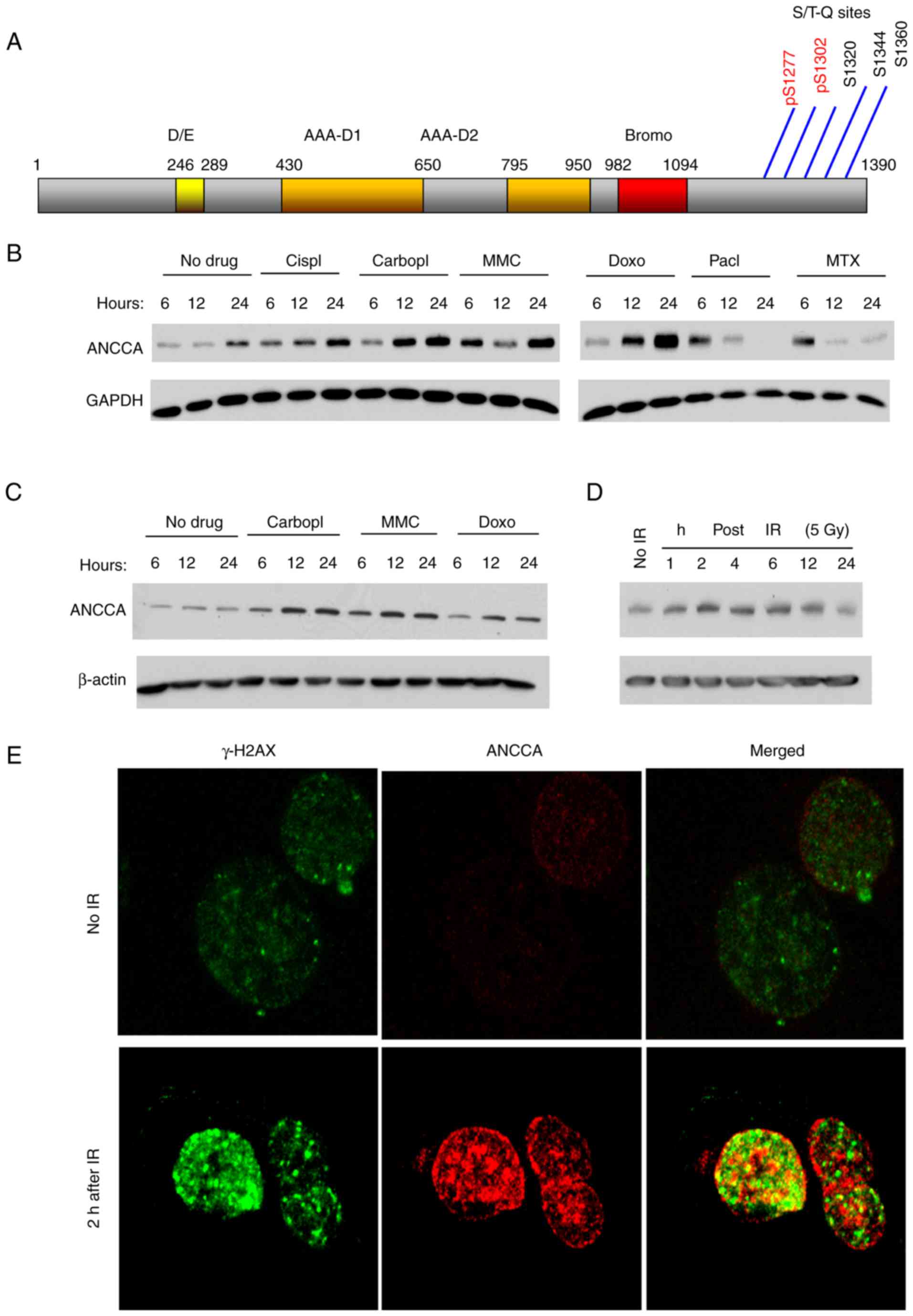 | Figure 1.ANCCA is induced in cancer cells upon
DNA damage. (A) Schematic diagram of ANCCA/ATAD2 protein with
indicated functional domains and potential ATM or ATR
phosphorylation sites. (B) MDA-MB-468 cells were treated with 10 µM
Cispl, 20 µM Carbopl, 1.0 µM MMC, 0.1 µM Doxo, 0.1 µM Pacl, 1.0 µM
MTX or vehicle for indicated time-points. Cells were then harvested
for immunoblotting. (C) H1299 cells were treated with the indicated
chemo-drugs as in A before being harvested for immunoblotting. (D)
H1299 cells were exposed to IR at a dose of 5 Gy and harvested at
indicated time-points after IR before being harvested for
immunoblotting. (E) H1299 cells were exposed to IR at a dose of 5
Gy for 2 h before being harvested for immune-staining with rH2A.x
(Alexa 488-labeled) or ANCCA antibody. ANCCA, AAA+ nuclear
coregulatory cancer-associated protein; Cispl, cisplatin; Carbopl,
carboplatin; MMC, mitomycin C; Doxo, doxorubincin; Pacl,
paclitaxel; MTX, methotrexate; IR, ionizing radiation. |
It is well known that formation of γ-H2AX
(phosphorylated H2AX) foci at the DNA damage sites is one of the
earliest and most important events in DNA damage response (DDR). To
examine whether ANCCA is associated with the DNA damage foci, cells
were exposed to IR and stained with γ-H2AX and ANCCA antibodies for
confocal IF analysis. The IF analysis revealed that similar to
γ-H2AX, ANCCA could also form punctate structures with an increased
intensity after IR treatment (Fig.
1E). However, the majority of ANCCA punctates did not
co-localize with γ-H2AX foci. Only a small fraction of them
displayed potential co-localization which were revealed as yellow
punctates under the confocal microscope (Fig. 1E). Collectively, the results indicated
that, upon DNA damage, ANCCA protein was induced and redistributed
in the nucleus of cancer cells.
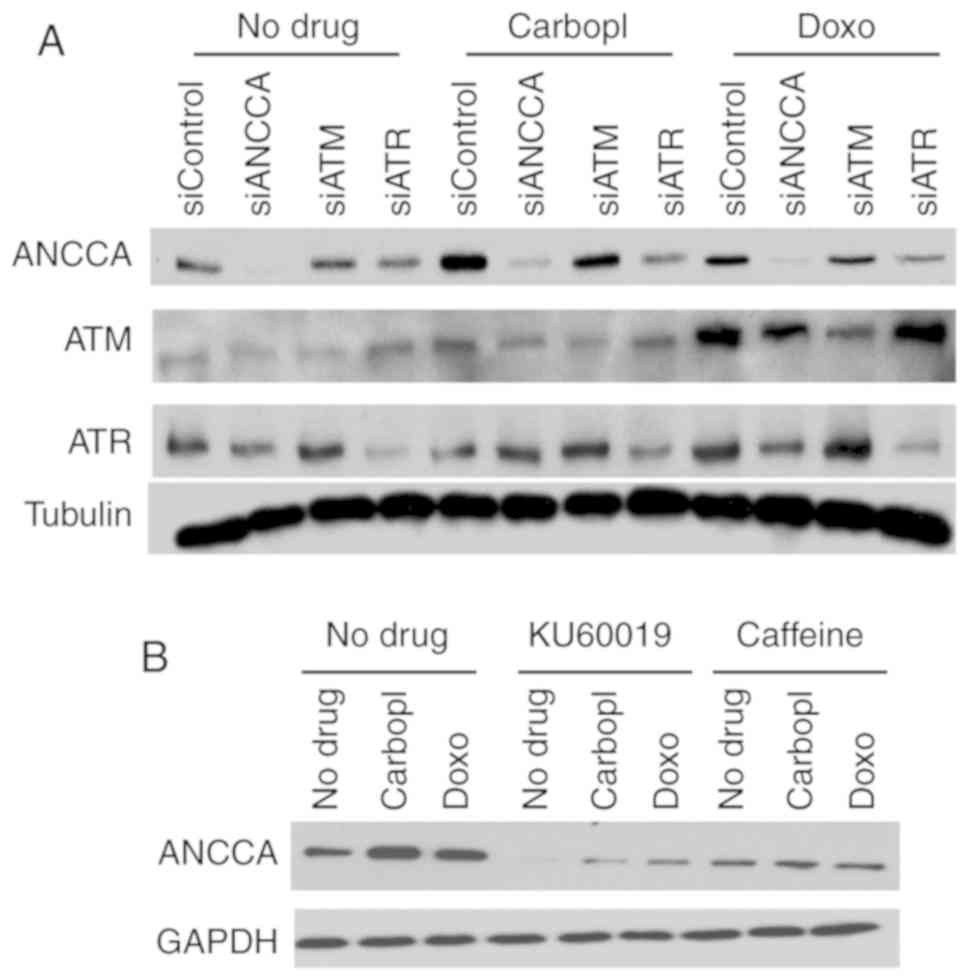
ANCCA induction is mediated by ATM and
ATR
ATM and ATR kinases are central upstream regulators
of DDR. To investigate whether DNA damage-induced ANCCA expression
is regulated by the ATM/ATR pathway, ATM or ATR were knocked down
before treatment with drugs. Results in Fig. 2A revealed that in comparison to
siControl, silencing of ATM in MDA-MB-468 cells markedly decreased
ANCCA induction by carboplatin or doxorubicin. Likewise, in
comparison to the siControl, knockdown of ATR also effectively
mitigated the induction (Fig. 2A).
However, in regularly growing cells without drug treatment, ATM or
ATR knockdown did not affect ANCCA expression in the Western
blotting. Furthermore, treatment of the cells with caffeine (a
general ATM and ATR inhibitor) or KU60019 (a specific inhibitor of
ATM) almost completely eliminated carboplatin or
doxorubicin-induced ANCCA upregulation (Fig. 2B). These results strongly indicated
that DNA-damaging drug-induced ANCCA protein induction was mediated
by the key DDR regulators ATM and ATR.
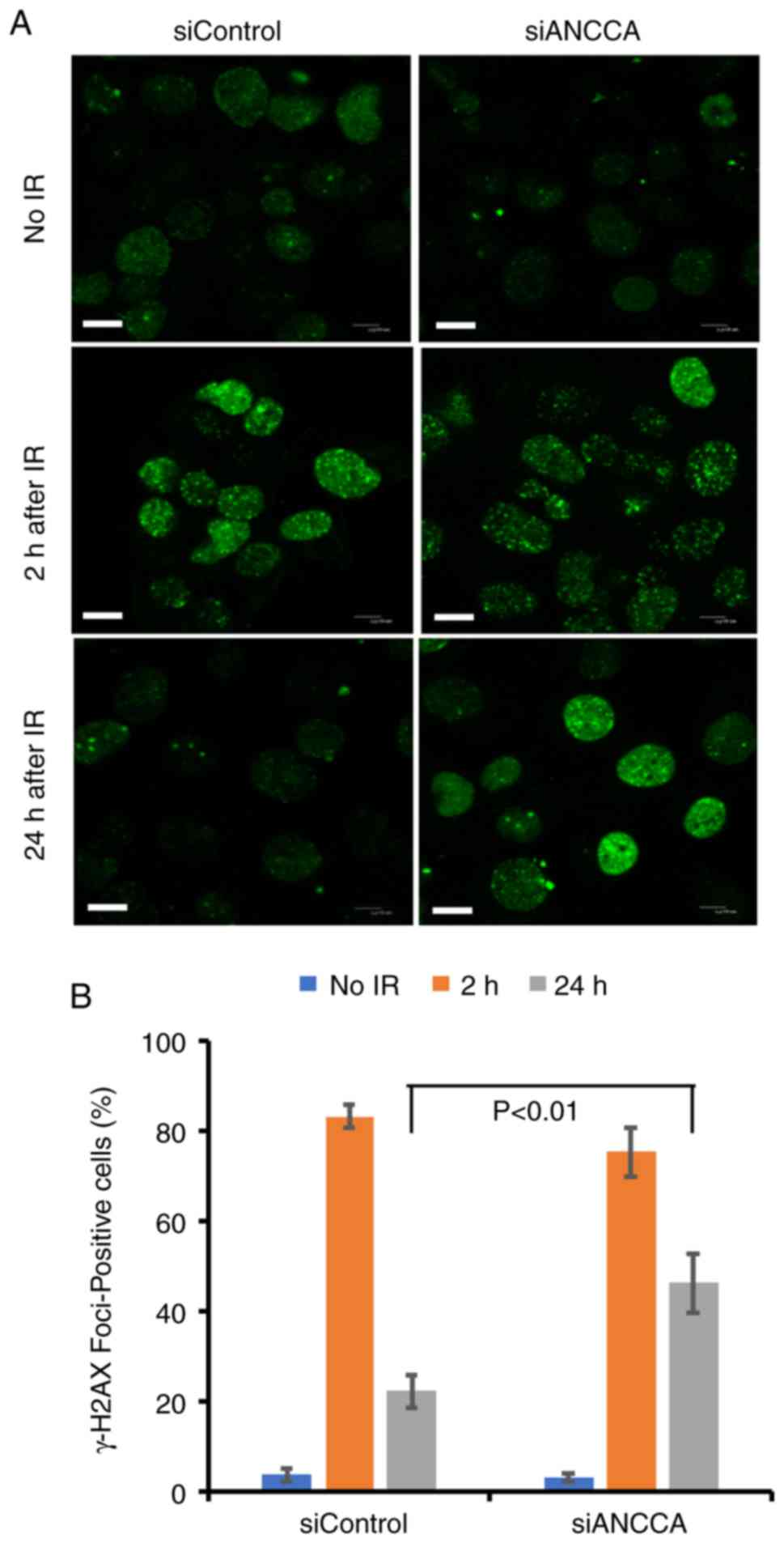
ANCCA silencing does not affect DNA
damage foci formation but delays their dissolution
It was next examined whether ANCCA silencing
influenced γ-H2AX foci formation or their dissolution. The
dissolution occurs during or after the completion of the repair.
Immunofluorescence analysis revealed that γ-H2AX foci were rapidly
generated 2 h after radiation in both the ANCCA-silenced group and
the control group (Fig. 3A and B),
indicating that ANCCA does not play a critical role in the
formation of γ-H2AX foci. Notably, when the dissolution of γ-H2AX
foci was analyzed, a significant delay in the ANCCA-silenced group
was detected, with foci remaining ~2-fold higher than the control
group at 24 h after IR. In addition, the foci formation of another
important DDR protein 53BP1 was also analyzed following exposure to
DNA-damaging agent MMC. At 48 h after MMC treatment, the
ANCCA-silenced group still had a higher percentage of
53BP1-positive cells than the control group (data not shown). These
results indicated that although ANCCA may not play an essential
role in the initial step of DDR, it may be involved in the timely
completion of the repair process of DNA damage.
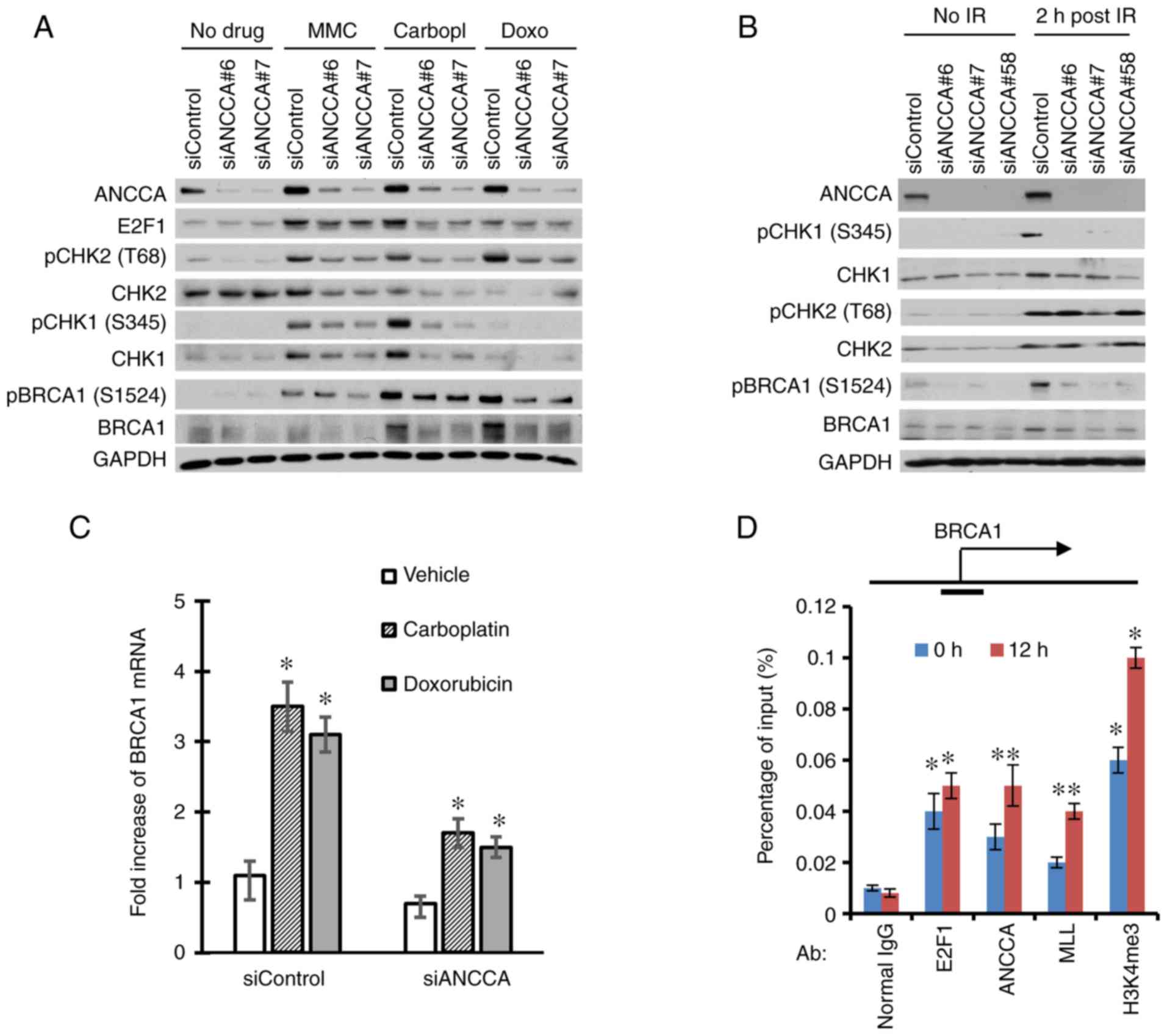
ANCCA controls CHK1 and CHK2 signaling
and the expression of BRCA1
To explore the possible function of ANCCA in DNA
damage response and repair, ANCCA was silenced by siRNA and the
effect on signaling and the expression of major DDR proteins was
examined. As revealed in Fig. 4A,
consistent with previous studies, treatment of cells with the
genotoxic drugs (i.e., MMC, carboplatin and doxorubicin) induced a
DNA damage response with strong activation of CHK1 and CHK2 kinases
as indicated by their markedly increased phosphorylation and the
increased phosphorylation of the BRCA1 protein. Notably, ANCCA
depletion strongly diminished the activation of Chk1 and Chk2 as
well as the activation of BRCA1. Similar effects were also observed
in cells treated with IR (Fig. 4B).
Notably, ANCCA silencing also decreased the total protein level of
BRCA1 elevated by carboplatin and doxorubicin or IR (Fig. 4A and B). It has been revealed that
effective DNA damage response and repair necessitates
transcriptional upregulation of genes involved in DDR including
BRCA1 (32,33). Consistent with previous studies,
carboplatin and doxorubicin treatment significantly increased BRCA
mRNA expression. However, the increase was largely diminished by
ANCCA knockdown (Fig. 4C). Since
ANCCA is a chromatin-bound protein and has been revealed to
function as a coactivator of transcription factors such as ER, AR
or E2F1, it was further investigated whether ANCCA could regulate
BRCA1 gene expression directly. In fact, ChIP analysis revealed
that ANCCA was recruited to BRCA1 gene promoter, and that
carboplatin treatment significantly enhanced the ANCCA recruitment
(Fig. 4D). Notably, the treatment
also increased the accumulation of transcriptionally active histone
mark H3K4me3 and the H3K4 methyltransferase MLL at BRCA1 promoter
(Fig. 4D), indicating that ANCCA may
mediate the chemotherapy drug induction of BRCA1 expression through
recruitment of MLL1 and H3K4 methylation of local chromatin.
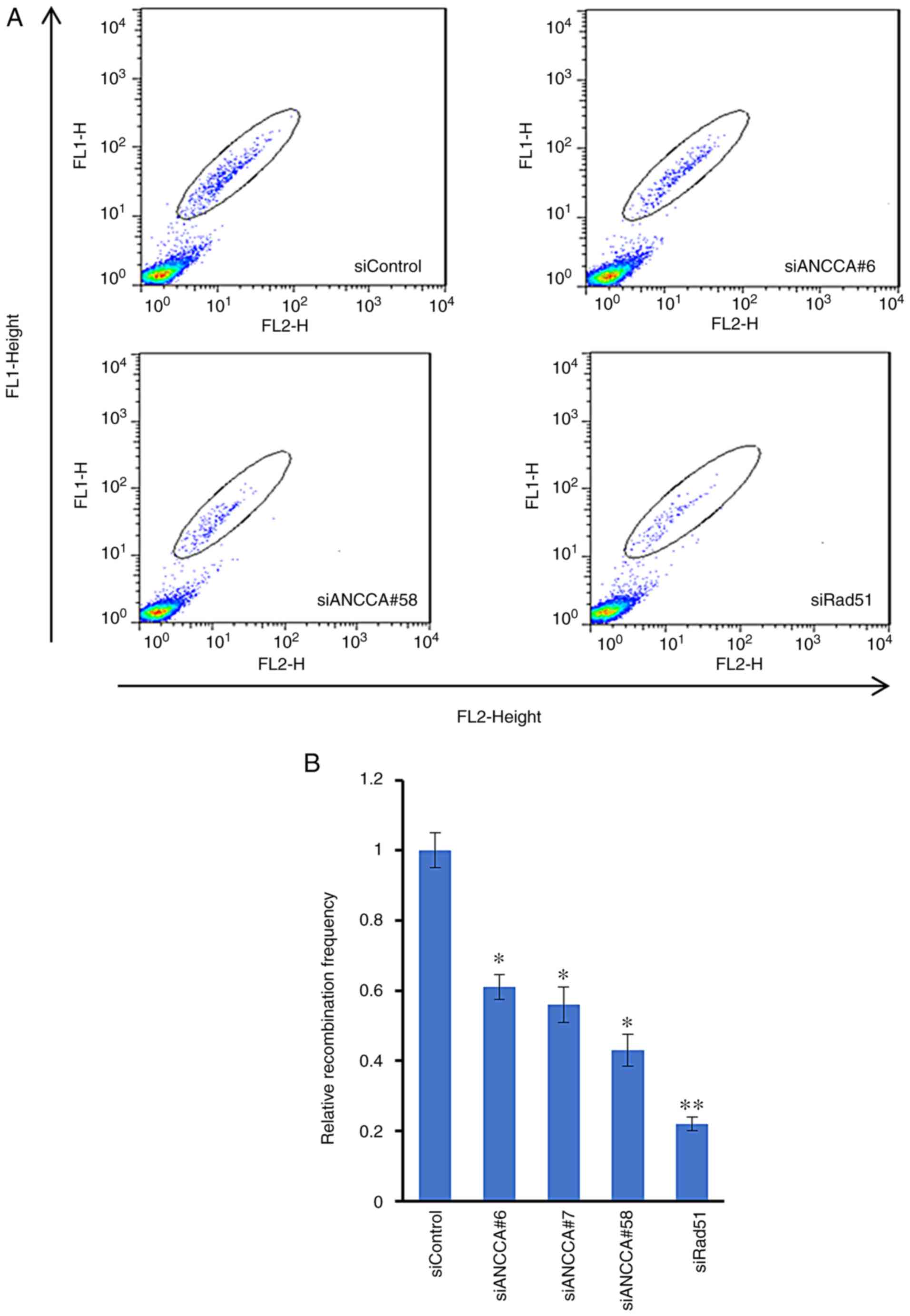
ANCCA knockdown affects homologous
recombination
BRCA1 is important in homologous recombination (HR),
one of the major pathways for DNA repair and cell survival. Having
determined that ANCCA controls the expression and activation of
BRAC1, it was next examined whether ANCCA plays a role in
modulating HR. A DR-GFP assay was thus performed. H1299-DR-GFP
cells were co-transfected with I-SceI endonuclease
expression plasmid and either control siRNA, ANCCA siRNA or Rad51
siRNA. Cells were harvested at 72 h after transfection to allow
sufficient recombination to occur. Similar to the HR inhibition
effect (to ~22% of control) by depletion of Rad51, the key
component of HR, knockdown of ANCCA also significantly reduced HR
repair efficiency to ~43 to 60% by the different siRNAs, indicating
an important role of ANCCA in the HR repair pathway (Fig. 5).
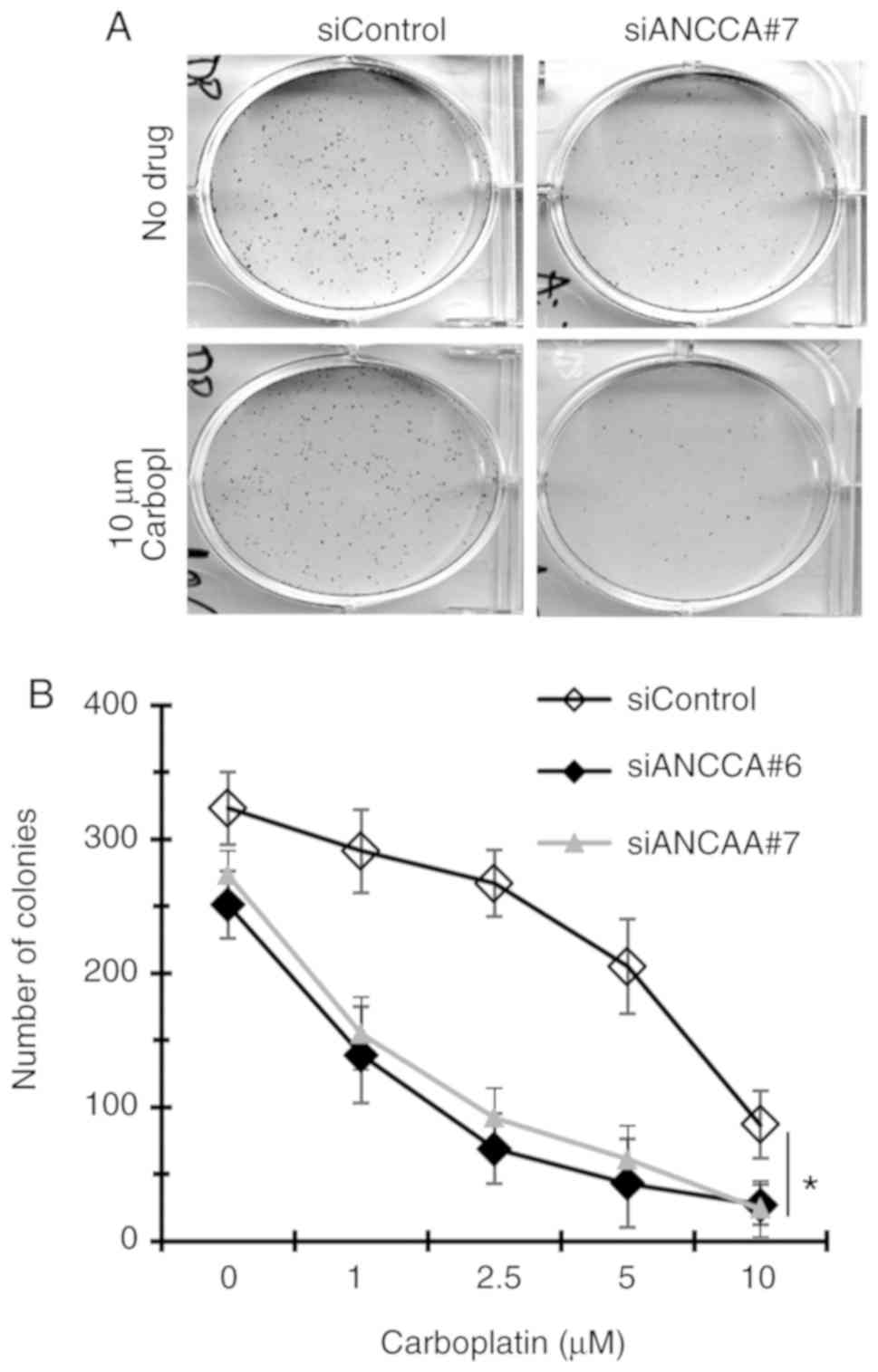
ANCCA knockdown sensitizes TNBC cells
to DNA-damaging drugs
Given the important functions of ANCCA in DNA damage
response and repair, whether ANCCA silencing affects the efficacy
of DNA-damaging drugs was next examined. Thus, colony formation
assays were performed with MDA-MB-468 TNBC cells treated by a
combination of ANCCA siRNA and carboplatin. Results in Fig. 6 revealed that when compared to the
control siRNA, ANCCA knockdown alone markedly decreased the number
of colonies (from 323 in the siControl to 251 in siANCCA#6), which
is consistent with our previous findings that ANCCA plays a
positive role in promoting breast cancer cell proliferation and
survival (6). Notably, when cells
were also treated with different concentrations of carboplatin,
more marked decreases of colony numbers in cells with ANCCA
knockdown were observed than in cells with siRNA-control. For
instance, at 1 µM carboplatin, colonies of si-ANCCA-6 cells
decreased ~45% (from 251 in vehicle-treated to 139 in 1 µM
carboplatin-treated) whereas colonies of siRNA-control cells
decreased only ~10% (from 323 to 291). Likewise, 2.5 µM carboplatin
treatment of siANCCA#6 cells caused >75% reduction in colony
numbers whereas it took 10 µM of the drug to cause a similar
effect. Therefore, ANCCA silencing could strongly sensitize cancer
cells to killing by DNA-damaging drugs such as carboplatin.
Discussion
Genotoxic stress or DNA damage elicits cellular DDR
responses that are complex and integrated to ensure the survival of
both normal and cancer cells. In the context of cancer cells,
identification of major cellular DDR factors can be of value in
providing new strategies in enhancing the efficacy of
chemotherapies. In the present study, several lines of evidence
were provided that ANCCA, which has a bromodomain that associates
with acetylated histones and functions in transcriptional
regulation (7,15), is also involved in DNA damage repair.
First, it was revealed that ANCCA protein was strongly induced in
response to various DNA-damaging agents, including chemotherapeutic
drugs and IR, which requires the activity of DNA damage-activated
kinases (ATM and ATR). Secondly, it was demonstrated that ANCCA
knockdown significantly delayed γ-H2AX and 53BP1 foci dissolution.
Thirdly, it was revealed that ANCCA depletion inhibited the
expression and activation of numerous DNA damage response factors,
including Chk1, Chk2 and BRCA1. Finally, it was demonstrated that
ANCCA knockdown also severely impaired the HR-dependent repair
efficiency.
Unlike many DDR factors, ANCCA is abundantly
expressed in cells and tumors of many types of cancers. Yet, it was
revealed that its protein level was strongly induced by DNA damage
agents and that this induction could be readily observed within 2 h
of IR or 6 h of chemo-drug treatment. Further analysis indicated
that the ANCCA induction does not appear to involve transcriptional
activation (data not shown). Multiple putative ATM/ATR-mediated
phosphorylation sites can be identified at the C-terminus of ANCCA.
A previous proteomics study revealed that at least two of the
putative ATM/ATR phosphorylation sites at the C-terminus of ANCCA
(S1277 and S1302) could be phosphorylated by UV-induced DNA damage
(31). Although the exact role of the
phosphorylation events is unclear, it is tempting to speculate that
the phosphorylation at either or both of the sites by ATM/ATR in
response to genotoxic stress upregulates the stability of ANCCA
protein. In support of this hypothesis, it was revealed that
suppression of the expression or function of ATM and ATR
effectively diminished ANCCA induction.
The exact role and functional mechanisms of ANCCA
involvement in DDR are unclear at this point. One important role
revealed in this study is its direct control of BRCA1. As one of
the core factors of HR, BRCA1 expression regulation in response to
DNA damage has been revealed to be dynamic and sometimes
context-dependent (32,33). Several transcriptional activating and
repressing complexes containing factors such as E2F1, Rb and BRCA1
itself were revealed to be important in control of BRCA1 gene
transcription. Effective assembly and disassembly of the complexes
at BRCA1 gene regulatory enhancer and promoter will likely be part
of the important regulatory events that underlie the dynamics of
its regulation in response to DNA damage. Given the AAA type of
ATPase possessed by ANCCA, it can be envisaged that through its
physical association with acetylated histones via its bromodomain
and with DNA-binding factors such as E2Fs, ANCCA can play a pivotal
role in facilitating the assembly and/or disassembly of the
regulatory complexes. The present ChIP data lend some support to
this model. Notably, a recent study demonstrated that overexpressed
ATAD2/ANCCA could increase the expression of PLK4, a
serine/threonine kinase, which plays important functions in
tumorigenesis and radiation resistance in glioblastoma models
(34), which is in line with the
notion that ANCCA is a major mediator of DNA damage-based
therapeutic resistance. To date, the exact mechanism(s) of how
ANCCA depletion led to the diminished activation of Chk1 and Chk2
is unclear. One possibility is that ANCCA depletion causes
decreased expression of BRCA1, which in turn leads to diminished
Chk1 activation. As well established, BRCA1 can regulate the
expression and activation of Chk1. Alternatively, ANCCA depletion
inhibited ATM protein expression as revealed in Fig. 2A, which in turn diminished Chk1 and
Chk2 activation.
In addition to the transcriptional regulation of DNA
repair genes, it was also observed that ANCCA depletion not only
reduced the expression of Chk1 and Chk2 and BRCA1 but also their
phosphorylation (and hence activation). These results raise the
possibility that DNA damage-induced ANCCA may also participate in
some of the DNA damage signaling and/or repair processes. One major
feature of genotoxic insults is a change of histone acetylation
landscape at the damaged chromatin (26). One recent proteomics study revealed
that, in addition to proteins of chromatin remodeling complexes as
anticipated, ANCCA is also associated with proteins and enzymes of
DNA replication and repair (35). The
latter includes Top2a, PARP1, and BLM. Considering that ANCCA has a
bromodomain that associates with the acetylated H3 and H4, it is
thus possible that enhanced histone acetylations at the damaged
loci guide ANCCA recruitment. Once recruited, ANCCA can assist the
repair by facilitating assembly and/or loading of repair complexes
through its physical association with the complexes and its ATPase
activity. This hypothetical recruitment mode is consistent with our
IF data in Fig. 1E and our failure to
detect ANCCA association with γ-H2AX in co-IP experiments (data not
shown). Future studies are required to examine the model with
small-molecule inhibitors specifically targeting ANCCA bromodomain
or its ATPase, once the inhibitors become available for use in cell
cultures or animal models. Further studies with the inhibitors can
also be conducted to examine whether targeting ANCCA in combination
with different chemotherapeutics (e.g., Topo-II inhibitors such as
doxorubicin and DNA crosslinking agents such as carboplatin) can
elicit different therapeutic efficacy.
Our previous studies demonstrated that ANCCA
overexpression may serve as a poor prognostic marker for TNBC and
that it is crucial for proliferation and survival of cancer cells
(6). In addition, ANCCA was also
revealed to control the expression of B-Myb, histone
methyltransferase EZH2 and an Rb-E2F core program for
proliferation, as well as a subset of mitotic kinesins and survival
genes (6,15,36).
Collectively with our findings of the functions of ANCCA in DNA
damage repair for TNBC cells, our studies revealed that ANCCA can
be a valuable target for the treatment of TNBC. In particular,
inhibition of ANCCA may resensitize tumors of advanced TNBC to
chemotherapy or radiation. Moreover, findings of this study also
provide rationale for determination of whether ANCCA overexpression
represents a prognostic factor for early relapse from certain
chemotherapies or radiation therapy.
Acknowledgements
We would like to thank Dr R.G Bristow for
H1299-DR-GFP cells and Mr. Neeraj Lal for technical help.
Funding
The present study was supported by the NIH grants
R01CA113860 and R01CA224900 (HWC) and R01CA213830 (JJL). NPA was a
trainee of an NIH T32 training grant.
Availability of data and materials
The materials used in this study are available from
the corresponding author upon reasonable request. All data analyzed
in this study are included in this article.
Authors' contributions
ZD, NPA, JJL and HWC designed the experiments. ZD,
NPA, CZC, MF, JW and JS performed the experiments and analyzed the
data. ZD, CZC, JS, JJL and HWC wrote the manuscript. JJL and HWC
edited the manuscript. All authors read and approved the final
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Liu B, Yip R and Zhou Z: Chromatin
remodeling, DNA damage repair and aging. Curr Genomics. 13:533–547.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Soria G, Polo SE and Almouzni G: Prime,
repair, restore: The active role of chromatin in the DNA damage
response. Mol Cell. 46:722–734. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Price BD and D'Andrea AD: Chromatin
remodeling at DNA double-strand breaks. Cell. 152:1344–1354. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zou JX, Guo L, Revenko AS, Tepper CG, Gemo
AT, Kung HJ and Chen HW: Androgen-induced coactivator ANCCA
mediates specific androgen receptor signaling in prostate cancer.
Cancer Res. 69:3339–3346. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zou JX, Revenko AS, Li LB, Gemo AT and
Chen HW: ANCCA, an estrogen-regulated AAA+ ATPase coactivator for
ERalpha, is required for coregulator occupancy and chromatin
modification. Proc Natl Acad Sci USA. 104:18067–18072. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kalashnikova EV, Revenko AS, Gemo AT,
Andrews NP, Tepper CG, Zou JX, Cardiff RD, Borowsky AD and Chen HW:
ANCCA/ATAD2 overexpression identifies breast cancer patients with
poor prognosis, acting to drive proliferation and survival of
triple-negative cells through control of B-Myb and EZH2. Cancer
Res. 70:9402–9412. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Caron C, Lestrat C, Marsal S, Escoffier E,
Curtet S, Virolle V, Barbry P, Debernardi A, Brambilla C, Brambilla
E, et al: Functional characterization of ATAD2 as a new
cancer/testis factor and a predictor of poor prognosis in breast
and lung cancers. Oncogene. 29:5171–5181. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ciro M, Prosperini E, Quarto M, Grazini U,
Walfridsson J, McBlane F, Nucifero P, Pacchiana G, Capra M,
Christensen J and Helin K: ATAD2 is a novel cofactor for MYC,
overexpressed and amplified in aggressive tumors. Cancer Res.
69:8491–8498. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang M, Zhang C, Du W, Yang X and Chen Z:
ATAD2 is overexpressed in gastric cancer and serves as an
independent poor prognostic biomarker. Clin Transl Oncol.
18:776–781. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang J, Huang J, Luo L, Chen Z, Guo Y and
Guo L: Significance of PRO2000/ANCCA expression, a novel
proliferation-associated protein in hepatocellular carcinoma.
Cancer Cell Int. 14:332014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Krakstad C, Tangen IL, Hoivik EA, Halle
MK, Berg A, Werner HM, Ræder MB, Kusonmano K, Zou JX, Øyan AM, et
al: ATAD2 overexpression links to enrichment of B-MYB-translational
signatures and development of aggressive endometrial carcinoma.
Oncotarget. 6:28440–28452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hwang HW, Ha SY, Bang H and Park CK: ATAD2
as a poor prognostic marker for hepatocellular carcinoma after
curative resection. Cancer Res Treat. 47:853–861. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shang P, Meng F, Liu Y and Chen X:
Overexpression of ANCCA/ATAD2 in endometrial carcinoma and its
correlation with tumor progression and poor prognosis. Tumour Biol.
36:4479–4485. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zheng L, Li T, Zhang Y, Guo Y, Yao J, Dou
L and Guo K: Oncogene ATAD2 promotes cell proliferation, invasion
and migration in cervical cancer. Oncol Rep. 33:2337–2344. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Revenko AS, Kalashnikova EV, Gemo AT, Zou
JX and Chen HW: Chromatin loading of E2F-MLL complex by
cancer-associated coregulator ANCCA via reading a specific histone
mark. Mol Cell Biol. 30:5260–5272. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang J, Yang J, Lei Y, Gao H, Wei T, Luo
L, Zhang F, Chen H, Zeng Q and Guo L: An
ANCCA/PRO2000-miR-520a-E2F2 regulatory loop as a driving force for
the development of hepatocellular carcinoma. Oncogenesis.
5:e2292016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Koo SJ, Fernández-Montalván AE, Badock V,
Ott CJ, Holton SJ, von Ahsen O, Toedling J, Vittori S, Bradner JE
and Gorjánácz M: ATAD2 is an epigenetic reader of newly synthesized
histone marks during DNA replication. Oncotarget. 7:70323–70335.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bamborough P, Chung CW, Furze RC, Grandi
P, Michon AM, Watson RJ, Mitchell DJ, Barnett H, Prinjha RK, Rau C,
et al: Aiming to miss a moving target: Bromo and extra terminal
domain (BET) selectivity in constrained ATAD2 inhibitors. J Med
Chem. 61:8321–8336. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fernández-Montalván AE, Berger M, Kuropka
B, Koo SJ, Badock V, Weiske J, Puetter V, Holton SJ, Stöckigt D,
Ter Laak A, et al: Isoform-selective ATAD2 chemical probe with
novel chemical structure and unusual mode of action. ACS Chem Biol.
12:2730–2736. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lord CJ and Ashworth A: The DNA damage
response and cancer therapy. Nature. 481:287–294. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Altieri F, Grillo C, Maceroni M and
Chichiarelli S: DNA damage and repair: From molecular mechanisms to
health implications. Antioxid Redox Signal. 10:891–937. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fong YW, Cattoglio C and Tjian R: The
intertwined roles of transcription and repair proteins. Mol Cell.
52:291–302. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Biswas AK and Johnson DG: Transcriptional
and nontranscriptional functions of E2F1 in response to DNA damage.
Cancer Res. 72:13–17. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hauer MH and Gasser SM: Chromatin and
nucleosome dynamics in DNA damage and repair. Genes Dev.
31:2204–2221. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pei H, Zhang L, Luo K, Qin Y, Chesi M, Fei
F, Bergsagel PL, Wang L, You Z and Lou Z: MMSET regulates histone
H4K20 methylation and 53BP1 accumulation at DNA damage sites.
Nature. 470:124–128. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li X, Baek G, Ramanand SG, Sharp A, Gao Y,
Yuan W, Welti J, Rodrigues DN, Dolling D, Figueiredo I, et al: BRD4
promotes DNA repair and mediates the formation of TMPRSS2-ERG gene
rearrangements in prostate cancer. Cell Rep. 22:796–808. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Floyd SR, Pacold ME, Huang Q, Clarke SM,
Lam FC, Cannell IG, Bryson BD, Rameseder J, Lee MJ, Blake EJ, et
al: The bromodomain protein Brd4 insulates chromatin from DNA
damage signalling. Nature. 498:246–250. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang J, Zou JX, Xue X, Cai D, Zhang Y,
Duan Z, Xiang Q, Yang JC, Louie MC, Borowsky AD, et al: ROR-γ
drives androgen receptor expression and represents a therapeutic
target in castration-resistant prostate cancer. Nat Med.
22:488–496. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chan N, Koritzinsky M, Zhao H, Bindra R,
Glazer PM, Powell S, Belmaaza A, Wouters B and Bristow RG: Chronic
hypoxia decreases synthesis of homologous recombination proteins to
offset chemoresistance and radioresistance. Cancer Res. 68:605–614.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Luoto KR, Meng AX, Wasylishen AR, Zhao H,
Coackley CL, Penn LZ and Bristow RG: Tumor cell kill by c-MYC
depletion: Role of MYC-regulated genes that control DNA
double-strand break repair. Cancer Res. 70:8748–8759. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Boeing S, Williamson L, Encheva V, Gori I,
Saunders RE, Instrell R, Aygün O, Rodriguez-Martinez M, Weems JC,
Kelly GP, et al: Multiomic analysis of the UV-induced DNA damage
response. Cell Rep. 15:1597–1610. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Christmann M and Kaina B: Transcriptional
regulation of human DNA repair genes following genotoxic stress:
Trigger mechanisms, inducible responses and genotoxic adaptation.
Nucleic Acids Res. 41:8403–8420. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
De Siervi A, De Luca P, Byun JS, Di LJ,
Fufa T, Haggerty CM, Vazquez E, Moiola C, Longo DL and Gardner K:
Transcriptional autoregulation by BRCA1. Cancer Res. 70:532–542.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang J, Zuo J, Wang M, Ma X, Gao K, Bai X,
Wang N, Xie W and Liu H: Pololike kinase 4 promotes tumorigenesis
and induces resistance to radiotherapy in glioblastoma. Oncol Rep.
41:2159–2167. 2019.PubMed/NCBI
|
|
35
|
Morozumi Y, Boussouar F, Tan M, Chaikuad
A, Jamshidikia M, Colak G, He H, Nie L, Petosa C, de Dieuleveult M,
et al: Atad2 is a generalist facilitator of chromatin dynamics in
embryonic stem cells. J Mol Cell Biol. 8:349–362. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zou JX, Duan Z, Wang J, Sokolov A, Xu J,
Chen CZ, Li JJ and Chen HW: Kinesin family deregulation coordinated
by bromodomain protein ANCCA and histone methyltransferase MLL for
breast cancer cell growth, survival, and tamoxifen resistance. Mol
Cancer Res. 12:539–549. 2014. View Article : Google Scholar : PubMed/NCBI
|