Introduction
Pancreatic cancer is a highly malignant carcinoma
characterized by a dismal patient prognosis; therefore, the
development of more effective treatments is urgently required.
Gemcitabine (GEM) has been used worldwide as a key agent for
chemotherapy for the treatment of pancreatic cancer after it was
reported that it significantly prolonged overall survival in a
phase III trial compared with 5-fluorouracil (5-FU) (1). S-1 (TS-1; Taiho Pharmaceutical Co.,
Ltd., Tokyo, Japan), an oral fluoropyrimidine developed in Japan,
consists of tegafur (FT), 5-chloro-2,4-dihydroxypyridine (CDHP),
and oteracil potassium (Oxo) at a molar ratio of 1:0.4:1 (2). FT is a prodrug of 5-FU and is
catabolized by dihydropyrimidine dehydrogenase (DPD) (3,4). CDHP
competitively inhibits DPD, and thereby maintains effective
concentrations of 5-FU in both plasma and tumor tissues (5). Oxo inhibits phosphorylation of 5-FU in
the gastrointestinal tract, thereby reducing the gastrointestinal
toxicity of 5-FU (6). The antitumor
effect of S-1 has already been demonstrated in a single agent or
combination regimen against a variety of solid tumors, such as
gastric cancer, colorectal cancer and non-small cell lung cancer
(7–11). Regarding pancreatic cancer, a
randomized phase III trial of GEM and S-1 monotherapy and GEM + S-1
combination (GS) therapy for pancreatic cancer with locally
advanced or distant metastases (GEST) has shown that S-1 was not
inferior to GEM. The GEM therapy group had a median overall
survival (OS) of 8.8 months and the S-1 therapy group had a median
OS of 9.7 months [hazard ratio (HR) 0.96] (12). Although the superiority of GS
therapy (10.1 months median OS) to GEM therapy was not verified, GS
significantly prolonged the progression-free survival of secondary
endpoints compared with GEM and in a subgroup analysis, GS
significantly prolonged the OS compared with GEM in locally
advanced pancreatic cancer. For adjuvant chemotherapy for
pancreatic cancer after resection, a phase III trial of GEM and S-1
therapy has shown that the median OS at the time of the 5-year
follow-up between April 2007 and January 2016 was 46.5 months in
the S-1 group and 25.5 months in the GEM group, and the HR of the
S-1 group vs. the GEM group was 0.57 (0.44–0.72), indicating S-1
therapy was superior to GEM therapy (P<0.0001; log-rank test)
(13). On the basis of these
trials, GEM and S-1 are recognized as key drugs for pancreatic
cancer in Japan.
Our previous research using pancreatic cancer cells
showed that a combination of S-1 and GEM is more effective than
either drug alone, both in vitro and in vivo.
Moreover, a combination of S-1 and GEM induced a greater degree of
S-phase arrest than either agent alone (14,15).
Mechanistic studies showed that both drugs led to increased
phosphorylation of checkpoint kinase 1 (Chk1). Chk1 is activated
when DNA is damaged, when it stops the cell cycle and catalyzes DNA
repair (16). Therefore, it is
expected that after the induction of DNA damage with antitumor
drugs in combination with a Chk1 inhibitor, DNA damage checkpoints
are inactivated and eventually cause cell death in a more potent
manner. Several Chk1 inhibitors have been developed and clinical
trials have been conducted for both monotherapy or combination
treatments with antitumor drugs (17–22).
Prexasertib (LY2606368) is one of the inhibitors of Chk1 and
checkpoint kinase 2 (Chk2) and inhibits Chk1 more strongly than
Chk2 by causing DNA double-strand breaks and apoptosis when used as
a single agent (23).
The aim of the present study was to investigate the
combined effect of the Chk1 inhibitor prexasertib with GEM, S-1,
and GS on the pancreatic cancer cell line SUIT-2. Moreover, we
conducted further mechanistic analysis of the combined effects.
Materials and methods
Cell cultures
The human pancreatic cancer cell line SUIT-2 was
obtained from the Japanese Collection of Research Bioresources
(Osaka, Japan). SUIT-2 cells were grown in RPMI-1640 medium
(FUJIFILM Wako Pure Chemical Corp., Osaka, Japan) supplemented with
10% fetal bovine serum (FBS), penicillin, and streptomycin.
Antitumor agents
GEM, 5-FU, and CDHP were purchased from Tokyo
Chemical Industry Co., Ltd. (Tokyo, Japan), and prexasertib was
purchased from Selleck Biotech (Osaka, Japan).
Antibodies
Monoclonal antibodies against phosphorylated-Chk1
(Ser296) (product #90178, 1:1,000 dilution), phosphorylated-Chk1
(Ser317) (product #12302, 1:1,000 dilution), phosphorylated-Chk1
(Ser345) (product #2348, 1:1,000 dilution), Chk1 (product #2360,
1:1,000 dilution), γH2AX (Ser139) (product #9718, 1:1,000
dilution), H2AX (product #2595, 1:1,000 dilution), caspase 9
(product #9508, 1:1,000 dilution), caspase 3 (product #9665,
1:1,000 dilution), cleaved PARP (Asp214) (product #5625, 1:1,000
dilution), Mcl-1 (product #94296, 1:1,000 dilution), Bcl-2 (product
#2870, 1:1,000 dilution), Bcl-xL (product #2764, 1:1,000 dilution)
and Bax (product #2772, 1:1,000 dilution) were purchased from Cell
Signaling Technology. The monoclonal antibody against β-actin was
purchased from Sigma-Aldrich (Merck KGaA, 1:5,000 dilution).
Purified mouse anti-cytochrome c (cat. no. 556433, 1:1,000
dilution) was purchased from BD Biosciences.
Cell proliferation assay
Cells were seeded in 96-well plates at a density of
1×103 cells/well. After 24 h, the culture medium was
replaced with 0.2 ml of fresh medium containing GEM, S-1, and/or
prexasertib at each concentration. After a further 72 h, 20 µl of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
reagent (Sigma-Aldrich; Merck KGaA) 0.5% in phosphate buffered
saline (PBS) was added to each well. The plate was then incubated
for 4 h, and 0.1 ml of dimethyl sulfoxide (Kanto Chemical Co. Inc.,
Tokyo, Japan) was added to each well to dissolve the formazan
crystals. Absorbance was then measured in each well using a
microplate reader at a wavelength of 620 nm. Inhibitory
concentration (IC50) values were then calculated. A
classical isobologram was used to evaluate the synergistic effects
of GEM + prexasertib and S-1 + prexasertib (24). The combination index (CI) was
calculated using CalcuSyn (Biosoft, Cambridge, UK) and synergy
level classifications were determined. A CI <1 indicates
synergy, a CI=1 indicates additive effects, and a CI >1
indicates antagonistic effects. All experiments were repeated at
least three times.
Analysis of cell death
Cells were seeded on 6-well plates
(6.0×104 cells/well) in RPMI-1640 medium supplemented
with 10% FBS. After 24 h, the culture medium was replaced with 2.0
ml of fresh medium containing GEM (0.3 ng/ml), S-1 (0.2 µg/ml),
and/or prexasertib (5 nM). After a further 72 h, mono- and
oligonucleosomes in the cytoplasmic fraction were measured by the
Cell Death Detection ELISA kit (Roche Diagnostics, Basel,
Switzerland; cat. no. 1544675) according to the manufacturer's
instructions. Floating and attached cells were collected and
homogenized in 500 µl of incubation buffer. The wells of a 96-well
plate were coated with antihistone antibodies at room temperature
for 1 h and incubated with the lysates, horseradish
peroxidase-conjugated anti-DNA antibodies, and the substrate, and
the absorbance was read at 405 nm. Furthermore, after 72 h of
culture on a 6-well plate under the same conditions, apoptotic
cells were assessed by examining their nuclear morphology under a
fluorescence microscope after staining with 1.2 mM Hoechst 33342
for 5 min.
Western blot analysis
Cells (3.0×105) were seeded in dishes and
cultured for 24 h. The medium was replaced by fresh medium
containing the drugs (GEM 0.3 ng/ml, S-1 0.2 µg/ml, or prexasertib
10 nM) and the cells were incubated for the indicated times (24,
48, and 72 h). Cells were rinsed with PBS and scraped into cell
lysis buffer M (FUJIFILM Wako Pure Chemical Corp.) dissolved in
complete protease inhibitor cocktail (Roche) and PhosSTOP
phosphatase inhibitor cocktail (Roche). After incubation on ice for
20 min, cell lysates were obtained by centrifugation at 15,000 × g
for 15 min at 4°C. Protein concentrations were determined and equal
amounts (30 µg) of total protein were separated on 7.5–12% sodium
dodecyl sulfate polyacrylamide gels at a constant current of 30 mA.
Separated proteins were then transferred to Immobilon
polyvinylidene difluoride membranes (Millipore) at 150 mA. The
membranes were blocked with 1% Difco™ skim milk (BD Biosciences)
and hybridized overnight at 4°C with various primary antibodies.
Membranes were then probed with a horseradish peroxidase-conjugated
anti-rabbit or anti-mouse antibody (Dako Denmark A/S, Glostrup,
Denmark; cat. nos. P0448 or P0260) and chemiluminescence was
developed using a SuperSignal West Pico Chemiluminescent Substrate
(Thermo Fisher Scientific, Inc.). The band intensities of Bcl-2 and
β-actin were analyzed using Image J 1.41 (National Institutes of
Health, Bethesda, MD, USA).
Analysis of the release of cytochrome
c
Cells (3.0×105) were seeded in dishes and
cultured for 2 h. The medium was replaced with fresh medium
containing the drugs (GEM 0.3 ng/ml, S-1 0.2 µg/ml, or prexasertib
10 nM) and the cells were incubated for the indicated time (24, 48,
and 72 h). Cells were washed with PBS and treated with 0.05%
trypsin, and then resuspended in 100 µl of ice-cold digitonin lysis
buffer (0.01% digitonin in PBS). After 5 min on ice, the cells were
centrifuged at 14,000 × g for 5 min, and the supernatant was
subjected to western blot analysis with cytochrome
c-specific antibody.
Small interfering RNAs and
transfection of cells
Silencer® Select Validated siRNA (small
interfering RNA) against Chk1 and Silencer® Select
Negative Control siRNA were purchased from Thermo Fisher
Scientific, Inc. The day before siRNA transfection, the cells
(1×105) were seeded into 6-well plates and incubated
overnight at 37°C without antibiotics. Cells were then treated with
siRNA (final concentration of 25 nM) in RPMI-1640 medium in the
presence of the DharmaFECT transfection reagent according to the
manufacturer's instructions (GE Healthcare Japan, Tokyo, Japan).
After incubation for 24 h at 37°C, the medium containing the
mixture of DharmaFECT and siRNA was replaced by RPMI-1640 medium
containing 10% FBS and cells were incubated for a further 24 h with
or without the drugs (GEM 0.8 ng/ml, S-1 0.5 µg/ml).
Statistical analysis
Data from the MTT assays, ELISA and western blot
analyses are expressed as the mean ± standard deviation.
Differences between the groups were examined for statistical
significance using analysis of variance (ANOVA) followed by
Fisher's protected least significant difference analysis. A P-value
of <0.05 was considered statistically significant. Statistical
analysis was performed using a BellCurve created in Excel for
Windows (Social Survey Research Information Co., Ltd., Tokyo,
Japan).
Results
Combinations of prexasertib and
antitumor drugs (GEM or S-1) exhibit a synergistic
antiproliferative effect on SUIT-2 cells
An MTT assay was used to investigate whether
prexasertib and an antitumor drug (GEM or S-1) exhibit synergistic
effects on the inhibition of cell growth in SUIT-2 cells. S-1 is a
combination drug comprising three compounds, but in our study, we
omitted the Oxo component (Oxo is used to reduce the incidence of
gastrointestinal side effects) and instead used a combination of
5-FU and CDHP at a ratio of 1:2 based on the blood concentration
ratio of the combination in humans (25). First, we calculated the
IC50 values for each drug. The IC50 values
for prexasertib, GEM, and S-1 were 30.8±6.04 nM, 0.642±0.048 ng/ml
and 0.506±0.219 µg/ml, respectively. Simultaneous administration of
various concentrations of prexasertib and an antitumor drug (GEM or
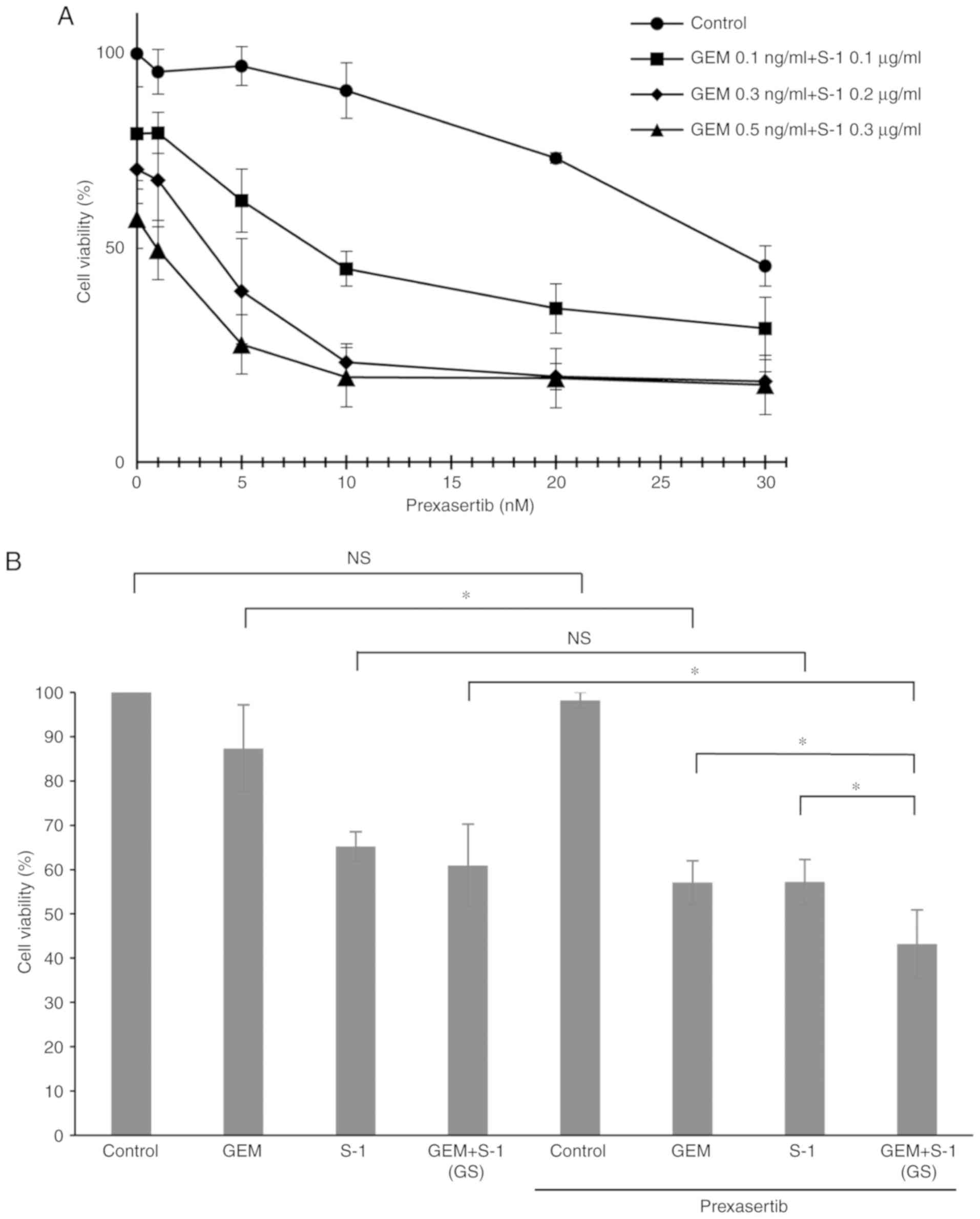
S-1) resulted in greater inhibition of SUIT-2 cell growth (Fig. 1A and B). The IC50 values
of prexasertib + GEM and prexasertib + S-1 were connected using a
dotted line in a classical isobologram to evaluate any possible
synergistic effects (Fig. 1C and
D). The data revealed that each of these drugs had a
synergistic effect, and a combination of prexasertib and GEM had a
stronger synergistic effect than prexasertib and S-1 in the SUIT-2
cells. The combination index also demonstrated that the combined
effect of prexasertib + GEM and prexasertib + S-1 on cell viability
was synergistic (CI <1) for many concentration combinations
(Table I). We further investigated
the effect of the combination of all three drugs [prexasertib + GEM
+ S-1 (GS)]. Prexasertib + GS showed significantly greater
inhibition of cell proliferation than prexasertib + GEM or
prexasertib + S-1 (P<0.05) (Fig. 2A
and B).
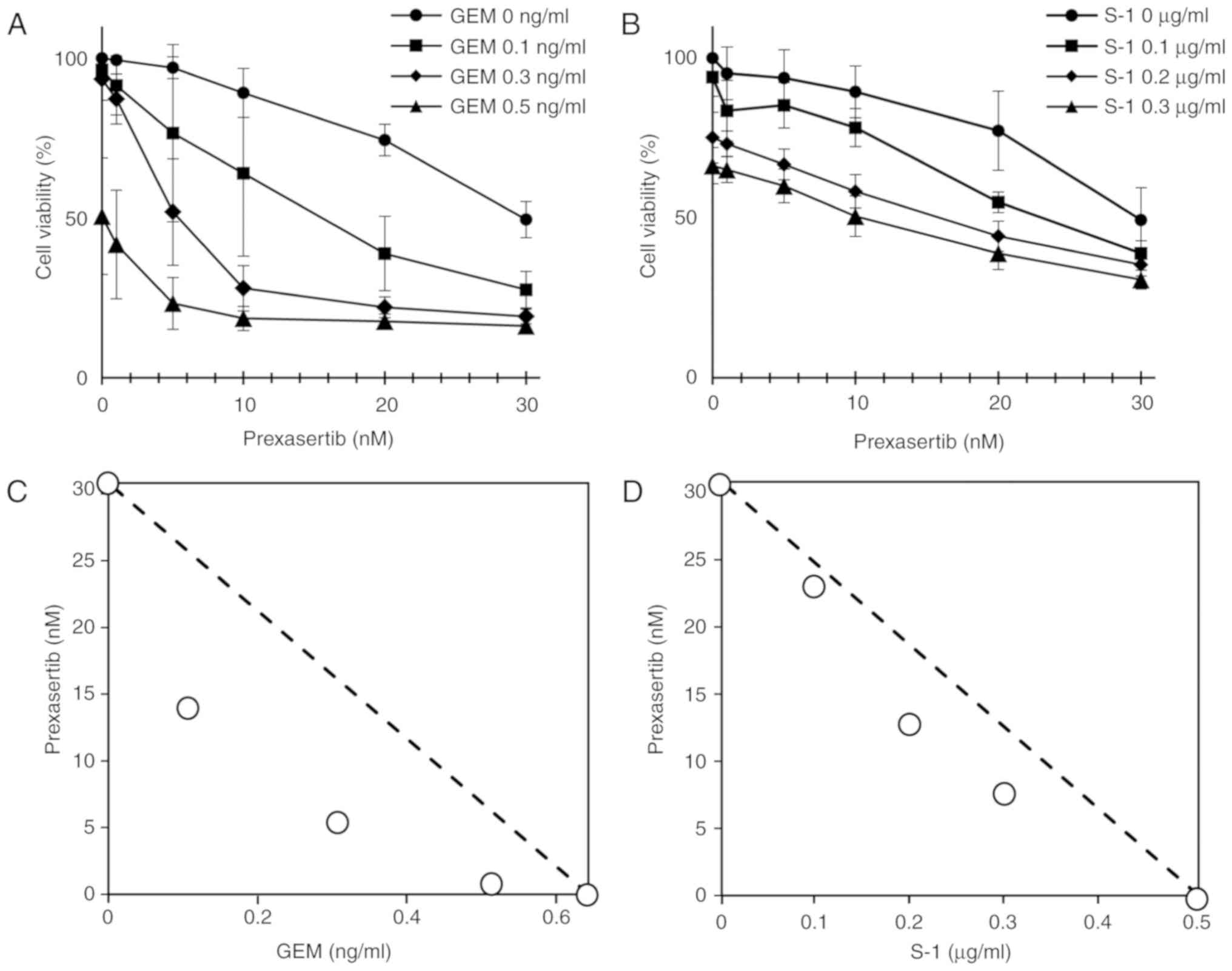 | Figure 1.Prexasertib enhanced the antitumor
effect of (A) GEM and (B) S-1. SUIT-2 cells were treated with a
combination of prexasertib (0, 1, 5, 10, 20, or 30 nM) and GEM (0,
0.1, 0.3, or 0.5 ng/ml), or S-1 (0, 0.1, 0.2, or 0.3 µg/ml) for 72
h. Values shown are the mean ± standard deviation (SD) from at
least 3 independent experiments. A classical isobologram shows the
combined effect of (C) prexasertib + GEM and (D) prexasertib +S-1
in SUIT-2 cells. The IC50 values for prexasertib (30.8
nM) and GEM (0.642 ng/ml), as well as those for prexasertib and S-1
(0.506 µg/ml), are connected with a dotted line to distinguish the
area of synergism (under the line) and the area of antagonism
(above the line). Plots on the dotted line indicate an additive
effect. Both prexasertib + GEM and prexasertib + S-1 showed a
synergistic effect. GEM, gemcitabine. |
 | Table I.Combination indexes (CI) calculated
for the combination of prexasertib + GEM or prexasertib + S-1 (CI
<1 indicates synergy). |
Table I.
Combination indexes (CI) calculated
for the combination of prexasertib + GEM or prexasertib + S-1 (CI
<1 indicates synergy).
|
| Prexasertib
(nM) |
|---|
|
|
|
|---|
|
| 10 | 20 | 30 |
|---|
| GEM (ng/ml) |
|
|
|
|
0.1 | 0.488a | 0.564a | 0.679a |
|
0.3 | 0.456a | 0.584a | 0.723a |
|
0.5 | 0.495a | 0.653a | 0.793a |
| S-1 (µg/ml) |
|
|
|
|
0.1 | 3.016 | 0.807a | 0.749a |
|
0.2 | 1.007 | 0.702a | 0.753a |
|
0.3 | 0.820a | 0.661a | 0.716a |
Induction of apoptosis by prexasertib
+ [GEM + S-1 (GS)]
We also investigated apoptotic cell death in SUIT-2
cells using a Cell Death Detection ELISA kit (Roche). This kit
includes mouse monoclonal antibodies directed against DNA and
histones and allows the specific determination of mono- and
oligonucleosomes in the cytoplasmic fraction of cell lysates.
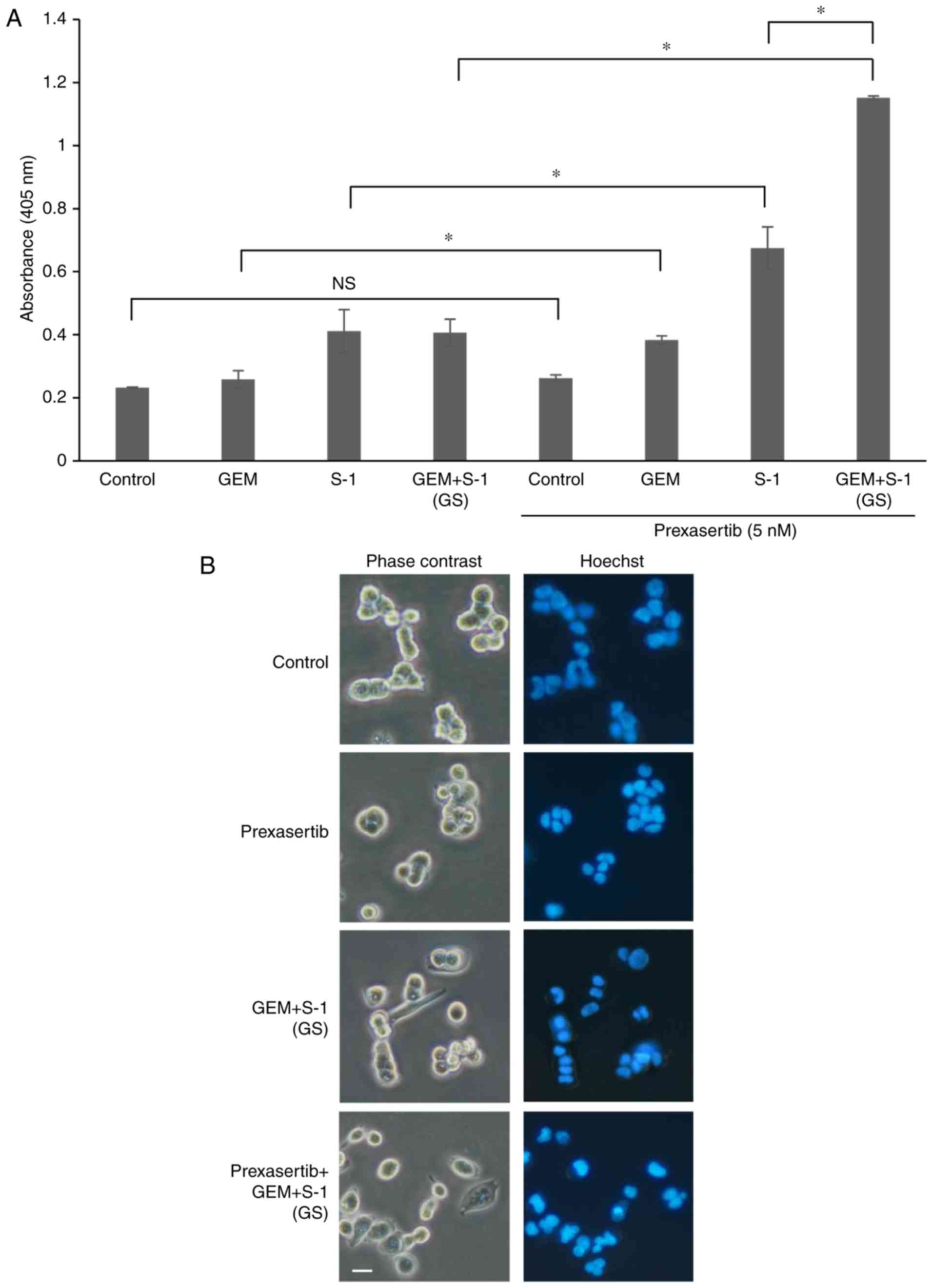
Assays using the ELISA kit confirmed that the treatment of SUIT-2
cells with prexasertib at 5 nM + GS significantly increased the
extent of apoptotic cell death compared with the other groups
(P<0.05) (Fig. 3A). In addition,
the absorbance for prexasertib at 10 nM with GS was 2.26±0.14,
which was an approximately 2-fold increase compared to prexasertib
at 5 nM with GS. Fig. 3B shows
morphological changes in SUIT-2 cells treated with several drugs
for 72 h. The cells treated with prexasertib + GS developed
apoptotic features including nuclear fragmentation.
Effect of prexasertib + GS on Chk1
signaling and apoptotic pathways
We performed western blotting to measure the change
in signals from apoptosis due to the combination of prexasertib +
GS. We used prexasertib at 10 nM, which induced apoptosis in
combination with GS more efficiently than at 5 nM in our pilot
experiments. To examine the phosphorylation of the DNA
damage-induced cell cycle checkpoint protein Chk1, western blotting
was used to assess the levels of phosphorylated Chk1 (pS296, pS317,
and pS345) in the SUIT-2 cells (Fig.
4A). Prexasertib suppressed the phosphorylation of Chk1 pS296,
which is an autophosphorylation site and most important for the
activation of Chk1, at 24 h. Prexasertib + GS increased the
phosphorylation of Chk1 pS317 and Chk1 pS345, which are DNA damage
markers. The expression of γH2AX, which is also a DNA damage
marker, was also induced to a higher degree following prexasertib +
GS treatment. However, the expression of total H2AX did not
correlate with the expression of γH2AX. We investigated the
apoptotic mechanism induced by prexasertib + GS. The expression
levels of cleaved caspase 3 and cleaved PARP were increased at 48 h
(Fig. 4B). We also investigated the
effect of prexasertib + GS on the Bcl-2 family. Cleaved Mcl-1 was
increased after 48 h of prexasertib + GS treatment and Bcl-2 was
markedly decreased at 24 h; this effect was prolonged until after
72 h (Fig. 4C). We next examined
the effects of prexasertib + GS on the release of cytochrome
c from mitochondria in SUIT-2 cells. When the cells were
exposed to prexasertib + GS, a significant increase in the release
of cytochrome c was observed within 48 h (Fig. 4D). These results suggest a
correlation between the release of cytochrome c and the
induction of apoptosis by prexasertib + GS. The expression of Bcl-2
at 72 h was significantly decreased by prexasertib + GS (P<0.05)
(Fig. 4E).
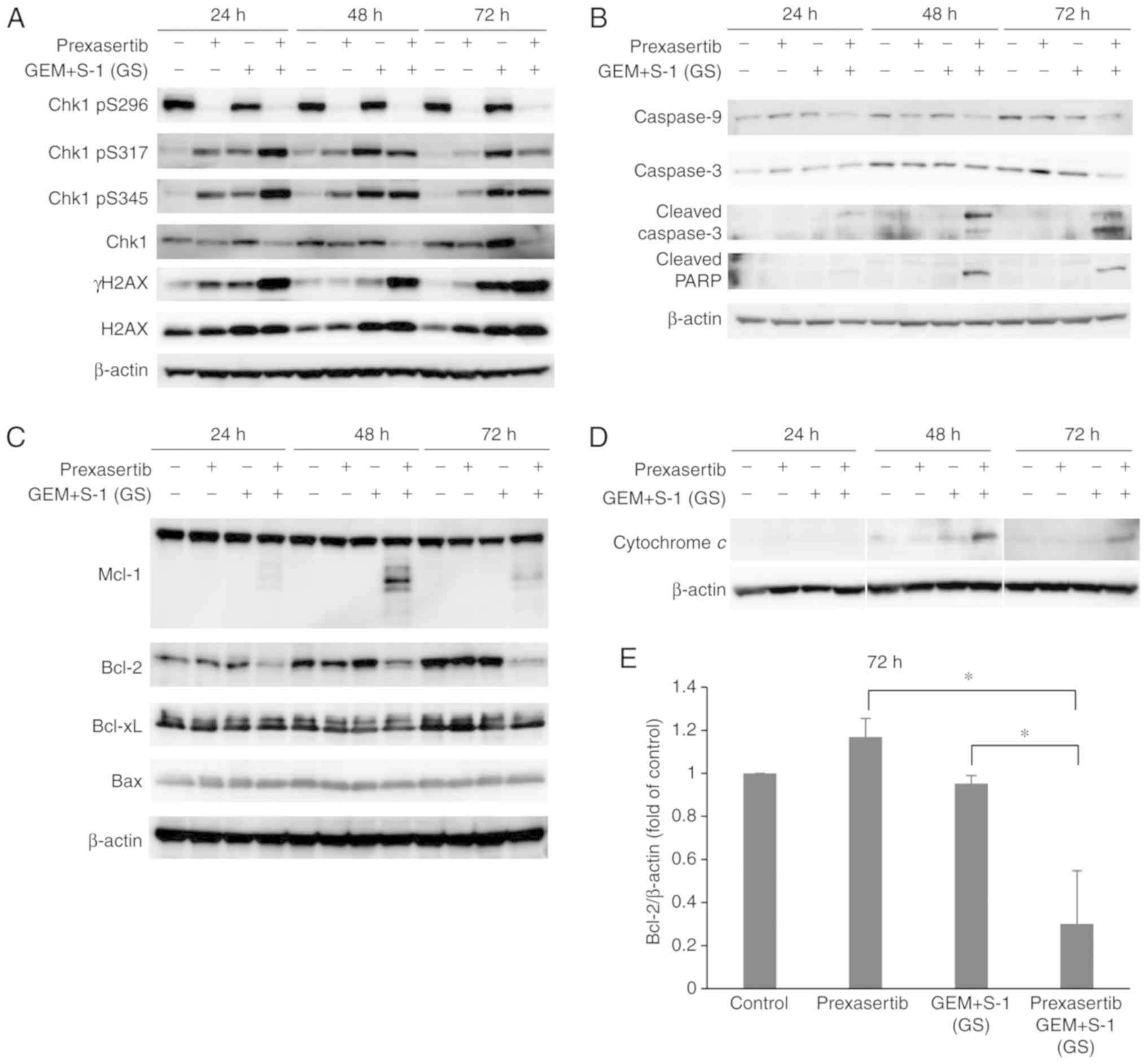 | Figure 4.(A) Effect of prexasertib, GEM + S-1
and prexasertib + GEM + S-1 on Chk1 (pS296, pS317, pS345), Chk1,
γH2AX, and H2AX levels in SUIT-2 cells. (B and C) Western blot
analysis revealed the levels of caspase 9, caspase 3, and PARP and
Bcl-2 family. (D) Release of cytochrome c from mitochondria
was detected. SUIT-2 cells were treated for 24, 48, and 72 h either
without (control) or with prexasertib, GEM + S-1, or prexasertib +
GEM + S-1. (E) Level of expression of Bcl-2 at 72 h normalized to
that of β-actin (Bcl-2/β-actin) and reported as the fold change
compared with the control value. The relative band intensities of
Bcl-2 and β-actin were quantified using densitometric analysis.
Values are expressed as the mean ± SD of three independent
experiments. *P<0.05 indicates a significant difference. GEM,
gemcitabine; GS, GEM + S-1 combined treatment; Chk1, checkpoint
kinase 1; PARP, poly(ADP-ribose) polymerase. |
Effects of Chk1-specific siRNA on
combination treatment with GEM and S-1
It is important to investigate whether prexasertib
enhances the combination of GEM and S-1 activity through Chk1
inhibition. We used siRNA to selectively deplete Chk1 in the cells
to investigate Chk1 inhibition and its contribution to the effect
of GEM and S-1 combined treatment (GS). After transfection, the
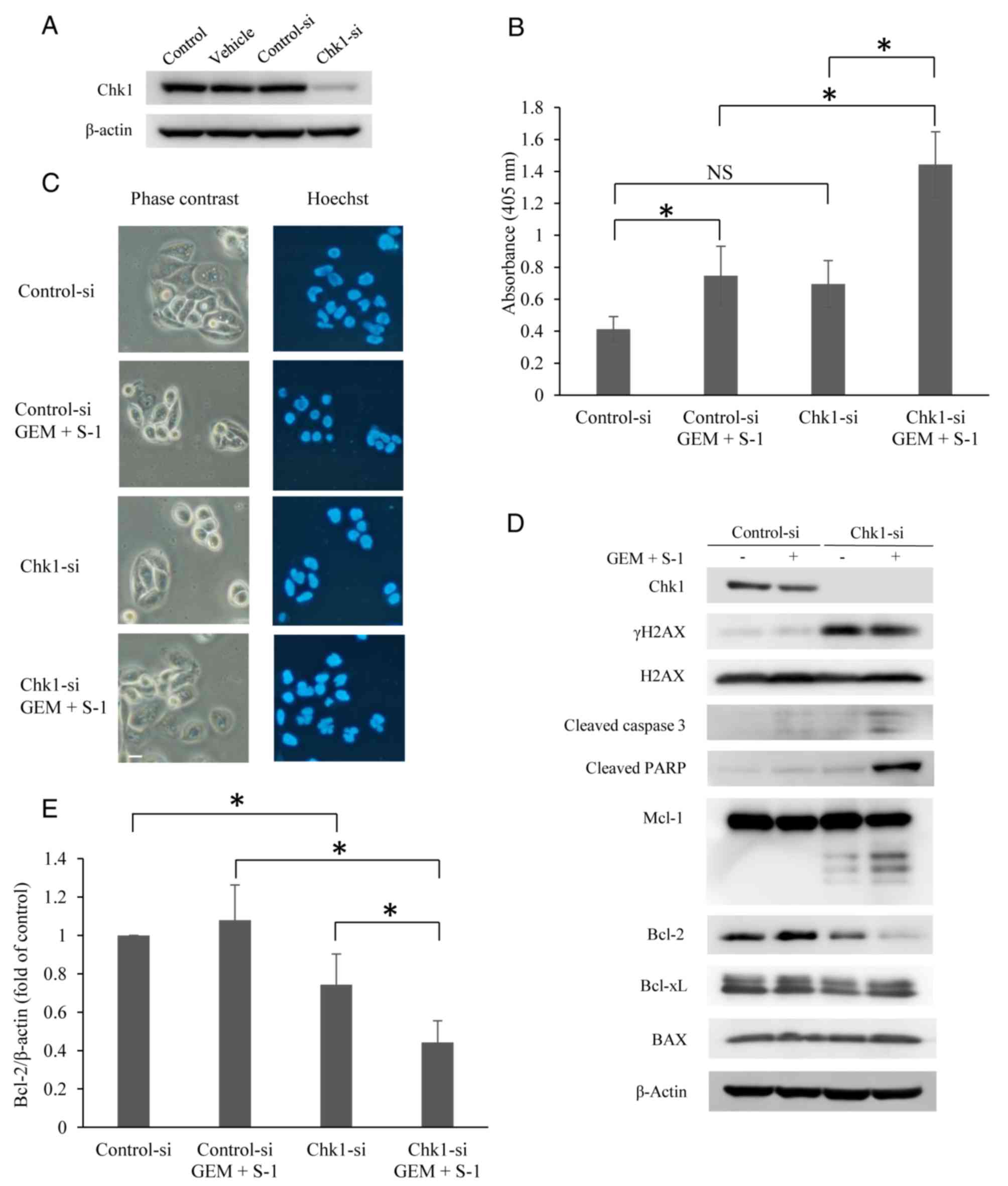
levels of expression of Chk1 were analyzed by western blotting
(Fig. 5A). As shown in Fig. 5A, treatment of SUIT-2 cells with
Chk1 siRNA (Chk1-si) significantly suppressed the expression of
Chk1 compared with cells treated with non-silencing siRNA
(Control-si) or with vehicle alone. Compared with the Control-si
(nonspecific siRNA)-transfected cells treated with GS, the Chk1-si
cells treated with GS showed significantly higher levels of
apoptosis at 24 h (P<0.05) (Fig.
5B). Only the Chk1-si cells treated with GS developed obvious
apoptotic features (Fig. 5C),
including nuclear fragmentation. When we examined the mechanism of
apoptosis in the Chk1-si transfected SUIT-2 cells after GS
treatment, the expression of Bcl-2 was decreased after 24 h
(Fig. 5D). Knockdown of Chk1 using
siRNA showed almost the same apoptotic signaling pathway as
prexasertib when used in combination with GS (Fig. 4B and C). However, the fact that
γH2AX was not induced in the Control-si-treated GS group is thought
to be due to differences in several experimental conditions. The
expression of Bcl-2 was also significantly decreased by Chk1-si +
GS (P<0.05) (Fig. 5E).
Discussion
In the present study, the results revealed that
combinations of prexasertib and antitumor drugs (GEM or S-1) have a
synergistic antiproliferative effect and a combination of
prexasertib and GS (GEM and S-1) was the most effective in SUIT-2
cells. Furthermore, the combination of all three drugs (prexasertib
+ GS) induced apoptotic cell death through the suppression of
Bcl-2.
DNA checkpoints are activated by DNA damage, and
serve to arrest the cell cycle and catalyze DNA repair. DNA
checkpoints are controlled by two major signaling pathways, namely,
the ATM/Chk2/p53 pathway and the ATR/Chk1/Cdc25A pathway (26). In many cancers, the ATM/Chk2/p53
pathway is impaired; therefore, Chk1 inhibitors are expected to
selectively induce mitotic cell death in cancer cells (26). Preclinical studies in solid tumor
and blood cancer models have shown that prexasertib as a single
agent or in combination with other agents has antitumor activity
both in vitro and in vivo (23,27–30).
Dysregulation of Chk1-dependent replication and checkpoint control
by prexasertib is thought to be an extremely efficient approach to
the induction of apoptosis in cancer cells. We previously showed
that both GEM and S-1 led to the increased phosphorylation of Chk1
(15). Therefore, the effects of
both drugs can be enhanced by combining them with an effective Chk1
inhibitor. The results showed a synergistic effect for each drug.
Although prexasertib + GS increased the phosphorylation of
Chk1-S317 and Chk1-S345, which are DNA damage markers, prexasertib
strongly suppressed the phosphorylation of Chk1-S296, which is the
most important autophosphorylation site for the activation of Chk1
(26). Previous reports have also
shown that prexasertib suppresses the autophosphorylation of
Chk1-S296 (23,27,29).
Although there have been several reports that show
that GEM in combination with other Chk1 inhibitors has a strong
antitumor effect (20,31,32),
this study is the first to show that the combined effect with S-1,
which is an oral fluorinated pyrimidine drug, led to high Chk1
inhibition and efficiently induced apoptosis. It has also been
reported that the Chk1 pathway is involved in the resistance
mechanism of 5-FU (33). In
addition, our results showed that prexasertib + GS had a stronger
effect than prexasertib + GEM or prexasertib + S-1, and this may be
due to greater induction of Chk1 phosphorylation by GS than by GEM
or S-1 alone.
Few studies have conducted a close examination of
the mechanism of apoptosis via combinations of a Chk1 inhibitor and
antitumor drugs. Therefore, we examined the Bcl-2 family, which has
been a target of drug discovery in recent years. Western blot
analysis showed that prexasertib + GS suppressed Bcl-2. Bcl-2 is
located in mitochondria and controls apoptosis by inhibiting the
release of cytochrome c (34). The selective Bcl-2 inhibitor
venetoclax (ABT-199) has already shown a strong effect on
refractory chronic lymphocytic leukemia in combination with
rituximab in a phase III trial (35). It has been reported that Bcl-2
expression is intimately involved in the induction of apoptosis in
pancreatic cancer cells (36). In
human colon cancer cell lines, it has been reported that the
sensitivity of anticancer agents depends on the expression level of
Bcl-2 (37); thus, the detailed
mechanism and effective combinations with existing antitumor drugs
must be investigated. We have not clarified the detailed
relationship between the Chk1 and Bcl-2 mechanisms in the
synergistic effect of prexasertib + GS; therefore, it is necessary
to investigate the correlation. In addition, the combination of low
concentrations of the three agents caused apoptosis efficiently;
therefore, we did not investigate the mechanism for the two-drug
combinations (prexasertib + GEM or prexasertib + S-1). We plan to
investigate the two-drug combinations (prexasertib + GEM or
prexasertib + S-1) in future research. Similarly, only one cell
line was used in the present study, and therefore multiple
pancreatic cancer cell lines must be utilized in future
research.
Furthermore, we showed that prexasertib + GS
increased the levels of cleaved Mcl-1. Mcl-1 is a member of the
Bcl-2 family and has an antiapoptotic effect. It has been reported
that Mcl-1 cleaved by caspase 3 in non-small cell lung cancer cells
treated with antitumor drugs triggers apoptosis and the cleavage by
caspase partially impairs the antiapoptotic activity of Mcl-1
(38). Drug research targeting
Mcl-1 has been progressing in recent years, but the detailed
mechanisms of Mcl-1 during apoptosis induced by antitumor drugs are
still unknown and further investigation is required.
The standard regimens currently used worldwide as
chemotherapy for metastatic pancreatic cancer are FOLFIRINOX or GEM
+ nab-paclitaxel (39,40). However, FOLFIRINOX frequently
induces grade 3 adverse events and GEM + nab-PTX results in a high
frequency of peripheral neuropathy and neutropenia. Thus, even
though both regimens have antitumor effects in pancreatic cancer,
they can be administered only to patients with a good performance
status due to their high toxicity. In Japan, GS is used for
perioperative chemotherapy or in the elderly for pancreatic cancer
(41,42). Regarding cholangiocarcinoma, GS has
been proven not to be inferior to GEM and cisplatin (GC) therapy,
which are worldwide standard treatments (43). Because GC has severe side effects,
such as vomiting and acute kidney injury, a regimen of either GC or
GS can be selected according to the patient's situation. In
addition to pancreatic cancer, the combined effect of prexasertib +
GS in cholangiocarcinoma must be examined.
Several Chk1 inhibitors have been developed, and the
results of several clinical trials have been reported (18–22,44,45).
However, none of the drugs can be used in clinical settings for
various reasons, such as poor efficacy and high toxicity.
Prexasertib is currently under development and shows single-agent
activity against ovarian cancer and squamous cell carcinoma
(46,47). However, it frequently leads to grade
4 neutropenia. Therefore, it is desirable to combine existing
antitumor drugs and prexasertib to create regimens that have fewer
side effects via using lower doses.
Prexasertib enhanced the antitumor effect of GEM +
S-1 through the induction of apoptosis in pancreatic cancer cells
with downregulation of anti-apoptotic Bcl-2 protein. Prexasertib
could be a useful agent to enhance the effect of GEM or fluorinated
pyrimidine drugs, which are standard drugs for the treatment of
pancreatic cancer.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YMo designed the study, performed the experiments,
analyzed the data, and wrote the manuscript. KT coordinated the
experiments and the writing of the manuscript. OT supervised the
research and experiments. AT coordinated the data analysis and the
writing of the manuscript. KW, MH, and TH collected and reviewed
the data and coordinated the writing of the manuscript. YMa
coordinated the scientific research and the writing of the
manuscript. All authors read and approved the final manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declared that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
GEM
|
gemcitabine
|
|
GS
|
GEM + S-1
|
|
Chk1
|
checkpoint kinase 1
|
|
5-FU
|
5-fluorouracil
|
|
FT
|
tegafur
|
|
CDHP
|
5-chloro-2,4-dihydroxypyridine
|
|
Oxo
|
oteracil potassium
|
|
DPD
|
dihydropyrimidine dehydrogenase
|
|
OS
|
overall survival
|
|
HR
|
hazard ratio
|
|
Chk2
|
checkpoint kinase 2
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
PBS
|
phosphate-buffered saline
|
|
siRNA
|
small interfering RNA
|
|
GC
|
GEM and cisplatin
|
References
|
1
|
Burris HA III, Moore MJ, Andersen J, Green
MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo
AM, Tarassoff P, et al: Improvements in survival and clinical
benefit with gemcitabine as first-line therapy for patients with
advanced pancreas cancer: A randomized trial. J Clin Oncol.
15:2403–2413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shirasaka T, Shimamato Y, Ohshimo H,
Yamaguchi M, Kato T, Yonekura K and Fukushima M: Development of a
novel form of an oral 5-fluorouracil derivative (S-1) directed to
the potentiation of the tumor selective cytotoxicity of
5-fluorouracil by two biochemical modulators. Anticancer Drugs.
7:548–557. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tatsumi K, Fukushima M, Shirasaka T and
Fujii S: Inhibitory effects of pyrimidine, barbituric acid and
pyridine derivatives on 5-fluorouracil degradation in rat liver
extracts. Jpn J Cancer Res. 78:748–755. 1987.PubMed/NCBI
|
|
4
|
Fukui Y, Oka T, Nagayama S, Danenberg PV,
Danenberg KD and Fukushima M: Thymidylate synthase,
dihydropyrimidine dehydrogenase, orotate phosphoribosyltransferase
mRNA and protein expression levels in solid tumors in large scale
population analysis. Int J Mol Med. 22:709–716. 2008.PubMed/NCBI
|
|
5
|
Shirasaka T, Nakano K, Takechi T, Satake
H, Uchida J, Fujioka A, Saito H, Okabe H, Oyama K, Takeda S, et al:
Antitumor activity of 1 M tegafur-0.4 M
5-chloro-2,4-dihydroxypyridine-1 M potassium oxonate (S-1) against
human colon carcinoma orthotopically implanted into nude rats.
Cancer Res. 56:2602–2606. 1996.PubMed/NCBI
|
|
6
|
Shirasaka T, Shimamoto Y and Fukushima M:
Inhibition by oxonic acid of gastrointestinal toxicity of
5-fluorouracil without loss of its antitumor activity in rats.
Cancer Res. 53:4004–4009. 1993.PubMed/NCBI
|
|
7
|
Sasako M, Sakuramoto S, Katai H, Kinoshita
T, Furukawa H, Yamaguchi T, Nashimoto A, Fujii M, Nakajima T and
Ohashi Y: Five-year outcomes of a randomized phase III trial
comparing adjuvant chemotherapy with S-1 versus surgery alone in
stage II or III gastric cancer. J Clin Oncol. 29:4387–4393. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Koizumi W, Narahara H, Hara T, Takagane A,
Akiya T, Takagi M, Miyashita K, Nishizaki T, Kobayashi O, Takiyama
W, et al: S-1 plus cisplatin versus S-1 alone for first-line
treatment of advanced gastric cancer (SPIRITS trial): A phase III
trial. Lancet Oncol. 9:215–221. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ohtsu A, Baba H, Sakata Y, Mitachi Y,
Horikoshi N, Sugimachi K and Taguchi T: Phase II study of S-1, a
novel oral fluorophyrimidine derivative, in patients with
metastatic colorectal carcinoma. S-1 cooperative colorectal
carcinoma study group. Br J Cancer. 83:141–145. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Okamoto I, Yoshioka H, Morita S, Ando M,
Takeda K, Seto T, Yamamoto N, Saka H, Asami K, Hirashima T, et al:
Phase III trial comparing oral S-1 plus carboplatin with paclitaxel
plus carboplatin in chemotherapy-naïve patients with advanced
non-small-cell lung cancer: Results of a west Japan oncology group
study. J Clin Oncol. 28:5240–5246. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kubota K, Sakai H, Katakami N, Nishio M,
Inoue A, Okamoto H, Isobe H, Kunitoh H, Takiguchi Y, Kobayashi K,
et al: A randomized phase III trial of oral S-1 plus cisplatin
versus docetaxel plus cisplatin in Japanese patients with advanced
non-small-cell lung cancer: TCOG0701 CATS trial. Ann Oncol.
26:1401–1408. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ueno H, Ioka T, Ikeda M, Ohkawa S,
Yanagimoto H, Boku N, Fukutomi A, Sugimori K, Baba H, Yamao K, et
al: Randomized phase III study of gemcitabine plus S-1, S-1 alone,
or gemcitabine alone in patients with locally advanced and
metastatic pancreatic cancer in Japan and Taiwan: GEST study. J
Clin Oncol. 31:1640–1648. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Uesaka K, Boku N, Fukutomi A, Okamura Y,
Konishi M, Matsumoto I, Kaneoka Y, Shimizu Y, Nakamori S, Sakamoto
H, et al: Adjuvant chemotherapy of S-1 versus gemcitabine for
resected pancreatic cancer: A phase 3, open-label, randomised,
non-inferiority trial (JASPAC 01). Lancet. 388:248–257. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yoshizawa J, Takizawa A, Takeuchi O,
Hiraku O, Sasaki K, Morimoto Y, Atsuda K, Inoue G, Suzuki Y,
Asanuma F and Yamada Y: Experimental study of combination therapy
with S-1 against pancreatic cancer. Cancer Chemother Pharmacol.
64:1211–1219. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Morimoto Y, Takeuchi O, Takizawa A,
Yoneyama H, Asanuma F, Suzuki Y, Atsuda K and Yamada Y: Effect of a
combination of S-1 and gemcitabine on cell cycle regulation in
pancreatic cancer cell lines. Anticancer Drugs. 23:505–514. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jackson SP and Bartek J: The DNA-damage
response in human biology and disease. Nature. 461:1071–1078. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dent P, Tang Y, Yacoub A, Dai Y, Fisher PB
and Grant S: CHK1 inhibitors in combination chemotherapy: Thinking
beyond the cell cycle. Mol Interv. 11:133–140. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li T, Christensen SD, Frankel PH, Margolin
KA, Agarwala SS, Luu T, Mack PC, Lara PN Jr and Gandara DR: A phase
II study of cell cycle inhibitor UCN-01 in patients with metastatic
melanoma: A California cancer consortium trial. Invest New Drugs.
30:741–748. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Webster JA, Tibes R, Morris L, Blackford
AL, Litzow M, Patnaik M, Rosner GL, Gojo I, Kinders R, Wang L, et
al: Randomized phase II trial of cytosine arabinoside with and
without the CHK1 inhibitor MK-8776 in relapsed and refractory acute
myeloid leukemia. Leuk Res. 61:108–116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sausville E, Lorusso P, Carducci M, Carter
J, Quinn MF, Malburg L, Azad N, Cosgrove D, Knight R, Barker P, et
al: Phase I dose-escalation study of AZD7762, a checkpoint kinase
inhibitor, in combination with gemcitabine in US patients with
advanced solid tumors. Cancer Chemother Pharmacol. 73:539–549.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wehler T, Thomas M, Schumann C,
Bosch-Barrera J, Viñolas Segarra N, Dickgreber NJ, Dalhoff K,
Sebastian M, Corral Jaime J, Alonso M, et al: A randomized, phase 2
evaluation of the CHK1 inhibitor, LY2603618, administered in
combination with pemetrexed and cisplatin in patients with advanced
nonsquamous non-small cell lung cancer. Lung Cancer. 108:212–216.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Karp JE, Thomas BM, Greer JM, Sorge C,
Gore SD, Pratz KW, Smith BD, Flatten KS, Peterson K, Schneider P,
et al: Phase I and pharmacologic trial of cytosine arabinoside with
the selective checkpoint 1 inhibitor Sch 900776 in refractory acute
leukemias. Clin Cancer Res. 18:6723–6731. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
King C, Diaz HB, McNeely S, Barnard D,
Dempsey J, Blosser W, Beckmann R, Barda D and Marshall MS:
LY2606368 causes replication catastrophe and antitumor effects
through CHK1-dependent mechanisms. Mol Cancer Ther. 14:2004–2013.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wagenpfeil S, Treiber U and Lehmer A:
Statistical analysis of combined dose effects for experiments with
two agents. Artif Intell Med. 37:65–71. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hirata K, Horikoshi N, Aiba K, Okazaki M,
Denno R, Sasaki K, Nakano Y, Ishizuka H, Yamada Y, Uno S, et al:
Pharmacokinetic study of S-1, a novel oral fluorouracil antitumor
drug. Clin Cancer Res. 5:2000–2005. 1999.PubMed/NCBI
|
|
26
|
Goto H, Izawa I, Li P and Inagaki M: Novel
regulation of checkpoint kinase 1: Is checkpoint kinase 1 a good
candidate for anti-cancer therapy? Cancer Sci. 103:1195–1200. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ghelli Luserna Di Rorà A, Iacobucci I,
Imbrogno E, Papayannidis C, Derenzini E, Ferrari A, Guadagnuolo V,
Robustelli V, Parisi S, Sartor C, et al: Prexasertib, a Chk1/Chk2
inhibitor, increases the effectiveness of conventional therapy in
B-/T- cell progenitor acute lymphoblastic leukemia. Oncotarget.
7:53377–53391. 2016.PubMed/NCBI
|
|
28
|
Zeng L, Beggs RR, Cooper TS, Weaver AN and
Yang ES: Combining Chk1/2 inhibition with Cetuximab and radiation
enhances in vitro and in vivo cytotoxicity in head and neck
squamous cell carcinoma. Mol Cancer Ther. 16:591–600. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lowery CD, VanWye AB, Dowless M, Blosser
W, Falcon BL, Stewart J, Stephens J, Beckmann RP, Bence Lin A and
Stancato LF: The checkpoint kinase 1 inhibitor prexasertib induces
regression of preclinical models of human neuroblastoma. Clin
Cancer Res. 23:4354–4363. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Brill E, Yokoyama T, Nair J, Yu M, Ahn YR
and Lee JM: Prexasertib, a cell cycle checkpoint kinases 1 and 2
inhibitor, increases in vitro toxicity of PARP inhibition by
preventing Rad51 foci formation in BRCA wild type high-grade serous
ovarian cancer. Oncotarget. 8:111026–111040. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Barnard D, Diaz HB, Burke T, Donoho G,
Beckmann R, Jones B, Barda D, King C and Marshall M: LY2603618, a
selective CHK1 inhibitor, enhances the anti-tumor effect of
gemcitabine in xenograft tumor models. Invest New Drugs. 34:49–60.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Montano R, Thompson R, Chung I, Hou H,
Khan N and Eastman A: Sensitization of human cancer cells to
gemcitabine by the Chk1 inhibitor MK-8776: Cell cycle perturbation
and impact of administration schedule in vitro and in vivo. BMC
Cancer. 13:6042013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fujinaka Y, Matsuoka K, Iimori M, Tuul M,
Sakasai R, Yoshinaga K, Saeki H, Morita M, Kakeji Y, Gillespie DA,
et al: ATR-Chk1 signaling pathway and homologous recombinational
repair protect cells from 5-fluorouracil cytotoxicity. DNA Repair
(Amst). 11:247–258. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: Release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Seymour JF, Kipps TJ, Eichhorst B, Hillmen
P, D'Rozario J, Assouline S, Owen C, Gerecitano J, Robak T, De la
Serna J, et al: Venetoclax-Rituximab in relapsed or refractory
chronic lymphocytic leukemia. N Engl J Med. 378:1107–1120. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dong J, Zhao YP, Zhou L, Zhang TP and Chen
G: Bcl-2 upregulation induced by miR-21 via a direct interaction is
associated with apoptosis and chemoresistance in MIA PaCa-2
pancreatic cancer cells. Arch Med Res. 42:8–14. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hakata S, Terashima J, Shimoyama Y, Okada
K, Fujioka S, Ito E, Habano W and Ozawa S: Differential
sensitization of two human colon cancer cell lines to the antitumor
effects of irinotecan combined with 5-aza-2′-deoxycytidine. Oncol
Lett. 15:4641–4648. 2018.PubMed/NCBI
|
|
38
|
Wang T, Yang Z, Zhang Y, Zhang X, Wang L,
Zhang S and Jia L: Caspase cleavage of Mcl-1 impairs its
anti-apoptotic activity and proteasomal degradation in non-small
lung cancer cells. Apoptosis. 23:54–64. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Conroy T, Desseigne F, Ychou M, Bouché O,
Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de
la Fouchardière C, et al: FOLFIRINOX versus gemcitabine for
metastatic pancreatic cancer. N Engl J Med. 364:1817–1825. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Von Hoff DD, Ervin T, Arena FP, Chiorean
EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et
al: Increased survival in pancreatic cancer with nab-paclitaxel
plus gemcitabine. N Engl J Med. 369:1691–1703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Motoi F, Ishida K, Fujishima F, Ottomo S,
Oikawa M, Okada T, Shimamura H, Takemura S, Ono F, Akada M, et al:
Neoadjuvant chemotherapy with gemcitabine and S-1 for resectable
and borderline pancreatic ductal adenocarcinoma: Results from a
prospective multi-institutional phase 2 trial. Ann Surg Oncol.
20:3794–3801. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ishii H, Yamashita N, Ueno M, Ohkawa S,
Saito AM and Sekimoto M: A randomised controlled trial of
gemcitabine hydrochloride plus S-1 combination therapy versus
gemcitabine hydrochloride therapy alone in pancreatic cancer
patients aged ≥75 years: A study protocol for an open-label
randomised feasibility study. BMJ Open Gastroenterol.
5:e0001872018.PubMed/NCBI
|
|
43
|
Mizusawa J, Morizane C, Okusaka T,
Katayama H, Ishii H, Fukuda H and Furuse J; Hepatobiliary and
Pancreatic Oncology Group of the Japan Clinical Oncology Group, :
Randomized phase III study of gemcitabine plus S-1 versus
gemcitabine plus cisplatin in advanced biliary tract cancer: Japan
clinical oncology group study (JCOG1113, FUGA-BT). J Clin Oncol.
46:385–388. 2016.
|
|
44
|
Scagliotti G, Kang JH, Smith D, Rosenberg
R, Park K, Kim SW, Su WC, Boyd TE, Richards DA, Novello S, et al:
Phase II evaluation of LY2603618, a first-generation CHK1
inhibitor, in combination with pemetrexed in patients with advanced
or metastatic non-small cell lung cancer. Invest New Drugs.
34:625–635. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Laquente B, Lopez-Martin J, Richards D,
Illerhaus G, Chang DZ, Kim G, Stella P, Richel D, Szcylik C,
Cascinu S, et al: A phase II study to evaluate LY2603618 in
combination with gemcitabine in pancreatic cancer patients. BMC
Cancer. 17:1372017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lee JM, Nair J, Zimmer A, Lipkowitz S,
Annunziata CM, Merino MJ, Swisher EM, Harrell MI, Trepel JB, Lee
MJ, et al: Prexasertib, a cell cycle checkpoint kinase 1 and 2
inhibitor, in BRCA wild-type recurrent high-grade serous ovarian
cancer: A first-in-class proof-of-concept phase 2 study. Lancet
Oncol. 19:207–215. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hong DS, Moore K, Patel M, Grant SC,
Burris HA III, William WN Jr, Jones S, Meric-Bernstam F, Infante J,
Golden L, et al: Evaluation of prexasertib, a checkpoint kinase 1
inhibitor, in a phase Ib study of patients with squamous cell
carcinoma. Clin Cancer Res. 24:3263–3272. 2018. View Article : Google Scholar : PubMed/NCBI
|