Introduction
Malignant tumors are one of the most important
causes of death worldwide. In most countries, including China, lung
cancer has the highest morbidity and mortality and is still
increasing yearly. Smoking, diet, air pollution and genetic factors
are all related to the occurrence of lung cancer (1–3).
According to histopathology, lung cancer can be divided into
non-small cell lung cancer (NSCLC) and small-cell lung cancer
(SCLC), of which NSCLC accounts for ~85% of all lung cancer
(4). Despite the continuous
improvement of medical advancements, the diagnosis and treatment of
NSCLC has not progressed significantly over the past few decades,
and the overall 5-year survival rate remains at ~15% (5,6). Since
there are no specific clinical symptoms in the early stage of lung
cancer, most lung cancer patients are already in the advanced stage
at the time of diagnosis, which means that tumor cells have already
metastasized or have the characteristics of invasion and metastasis
(7). Therefore, identifying key
factors and elucidating their molecular mechanisms in invasion and
metastasis of NSCLC are particularly important for reducing patient
mortality and improving the quality of life of patients.
The invasion and metastasis of malignant tumors, is
an extremely complicated process coordinated by multiple steps and
factors. Studies have revealed that epithelial-mesenchymal
transition (EMT) plays an important role in lung cancer invasion
and metastasis, occurring in the early stage of lung cancer
invasion and metastasis (8). EMT
refers to the biological process by which polar epithelial cells
are transformed into mesenchymal cells under certain specific
physiological or pathological conditions. EMT is one of the most
important mechanisms for malignant tumor cells to acquire the
ability of migration and invasion, and plays a vital part in the
occurrence and development of cancer (9). In the process of EMT, the epithelial
cells are less exposed to surrounding cells and matrix, and cell
adhesion is weakened. The epithelial cells obtain the ability of
higher migration, invasion, anti-apoptosis and degradation of the
extracellular matrix, exhibiting strong mesenchymal characteristics
and transforming into mesenchymal cells. In addition, the related
gene expression profile of the cells changes (10,11).
The morphology of the cells changes significantly after EMT. For
example, the cells become narrow and long, and form pseudopods. The
intercellular gaps significantly increase, and the cells become
more mobile. EMT is the result of the coordination of many factors,
involving the effects of transcription factors, microRNA
regulation, the TGF-signaling pathway as well as other chemical
signaling pathways.
Transforming growth factor-β (TGF-β) is a
multifunctional cytokine that triggers diverse cellular processes
including tissue fibrosis, growth arrest and EMT (12). Previous studies have revealed that
TGF-β can promote EMT progression by activating kinase-dependent
signaling processes and is currently the classical inducing factor
for the induction of EMT in lung cancer cells (10,13).
The EMT process induced by TGF-β can be expressed as changes in the
expression of some molecular markers, such as a loss of the
epithelial phenotype indicated by downregulation of E-cadherin and
the gain of the mesenchymal phenotype indicated by upregulation of
N-cadherin and vimentin (14).
TGF-β inhibits epithelial cell proliferation in the early stage of
cancer, but promotes tumor growth and metastasis in the later
stage. Studies have revealed that patients with higher TGF-β levels
have a poor prognosis (15).
Therefore, it was hypothesized that inhibition of TGF-β or its
receptors can suppress the activation of the EMT pathway, thereby
impairing the ability of tumor cell invasion and metastasis. TGF-β
mainly exerts its biological function through the SMAD protein
family. Specifically, binding of TGF-β to its receptor leads to
phosphorylation of SMAD2/3, which binds to SMAD4 and enters the
nucleus, where the SMAD transcription complex regulates the
expression of specific target genes (16,17).
Previous studies have reported that SMAD3 plays a key role in the
occurrence and development of various cancers (18,19),
and that SMAD3 can promote the invasion and metastasis of tumor
cells through its mediated EMT (20,21).
Loss or lack of SMAD3 will impede the EMT process and may alleviate
epithelial deterioration (18,19).
In addition, studies have revealed that silencing of SMAD3 can
inhibit the migration and invasion of nasopharyngeal carcinoma
cells (22), and the downregulated
expression of N-cadherin can also be detected (14); miR-140 inhibited the migration and
invasion of colorectal cancer cells by targeting SMAD3 (23). These data indicated that SMAD3 plays
an essential role in TGF-β-induced EMT progression, however, in
this process, the regulatory mechanisms of miRNA are still
unclear.
miRNAs are a class of endogenous non-coding RNA with
a wide distribution and a length of 19–25 nucleotides. They mainly
form partial complementary sequences by binding to the 3′-UTR,
5′-UTR or coding region of a specific target gene, thereby
affecting the transcriptional stability and post-transcriptional
translation process of the target gene (24). The expression of ~30–50% of
protein-coding genes in humans may be regulated by targeted miRNAs
(25). Therefore, miRNAs play an
important role in the fine regulation of a variety of biological
processes, including cell proliferation, differentiation,
apoptosis, migration and tumor formation (26,27).
In addition, miRNAs must also regulate TGF-signaling by targeting
the expression of key members in the TGF- pathway (28), known as TGF-β pathway-associated
miRNAs. Several studies have revealed that miR-203 can inhibit the
cell invasion of NSCLC and nasopharyngeal carcinoma, and is
frequently downregulated in NSCLC (26,29,30).
Ding et al revealed that miR-203 plays an important role in
TGF-β-induced EMT progression and is downregulated in highly
metastatic breast cancer cells (9).
These studies indicated that miR-203 may regulate the process of
EMT in NSCLC by regulating the TGF-β signaling pathway, and the
mechanism of miR-203 in this process remains to be further
elucidated.
In the present study, miR-203 was transfected into
NSCLC cells to verify the hypothesis that SMAD3 is a target gene
for miR-203, and miR-203 regulates the hypothesis that SMAD3
inhibits TGF-β-induced EMT and tumor invasion and metastasis. The
present results clarified that miR-203 in NSCLC cell line can
suppress the expression of SMAD3, affect the TGF-β-induced EMT
process, inhibit the invasion and metastasis of tumor cells, and
provide a new experimental basis for the diagnosis and treatment of
NSCLC.
Materials and methods
Human tissue samples
Fresh NSCLC tissue samples from 10 patients (32–61
years old) and their corresponding paracancerous samples were
collected in the study (n=10). The patients were diagnosed with
NSCLC based on pathology and did not receive any chemotherapy
and/or radiotherapy before surgery. There were 6 males and 4
females with an average age of 48.70±11.25 years. All of the
specimens were examined and evaluated by two independent
pathologists. Clinicopathological data were collected from the
patient medical records and are presented in Table I. All patients provided their
written informed consent and ethics approval was obtained from the
Ethics Committees of the First Affiliated Hospital of Wenzhou
Medical University (2017063).
 | Table I.Clinicopathological characteristics
of the NSCLC patients. |
Table I.
Clinicopathological characteristics
of the NSCLC patients.
|
Characteristics | Total (10, %) |
|---|
| Sex |
|
|
Male | 6 (60.0) |
|
Female | 4 (40.0) |
| Age (years) |
|
|
<40 | 4 (40.0) |
|
≥40 | 6 (60.0) |
| Histologic
type |
|
|
ADC | 6 (60.0) |
|
SQCC | 4 (40.0) |
| Lymph node
metastasis |
|
|
Yes | 3 (30.0) |
| No | 7 (70.0) |
| TNM stage |
|
|
I–II | 7 (70.0) |
|
III–IV | 3 (30.0) |
Cell lines and cell cultures
Human NSCLC cell line H226 cells (Institute of Cell
Sciences, Chinese Academy of Sciences) were cultured in modified
RPMI-1640 medium (Hyclone, GE Healthcare Life Sciences),
supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.) and a mixture of antibiotics (penicillin,
Sigma-Aldrich; streptomycin, Invitrogen), and the cells were
incubated in a humidified 5% CO2 incubator at 37°C.
Real time-qPCR (RT-qPCR)
RNA, according to the manufacturer's protocol, was
extracted from the cells using the RNAiso Plus kit (Thermo Fisher
Scientific, Inc.). Synthesis of cDNA with reverse transcriptase was
performed using the M-MLV First Strand kit (Invitrogen; Thermo
Fisher Scientific, Inc.). The primer sequences used for RT-qPCR
were as follows: miR-203-F, ACACTCCAGCTGGGAGTGGTTCTTAACAGTTC and
miR-203-R, TGGTGTCGTGGAGTCG; SMAD3-F, TGGACGCAGGTTCTCCAAAC and
SMAD3-R, CCGGCTCGCAGTAGGTAAC; SMAD2-F, ATCTTGCCATTCACTCCGCC and
SMAD2-R, CTGTTCTCCACCACCTGCTC; E-cadherin-F, ATTTTTCCCTCGACACCCGAT
and E-cadherin-R, TCCCAGGCGTAGACCAAGA; N-cadherin-F,
TGCGGTACAGTGTAACTGGG and N-cadherin-R, GAAACCGGGCTATCTGCTCG;
vimentin-F, AGTCCACTGAGTACCGGAGAC and vimentin-R,
CATTTCACGCATCTGGCGTTC; Snail-F, ACTGCAACAAGGAATACCTCAG and Snail-R,
GCACTGGTACTTCTTGACATCTG; U6-F, CTCGCTTCGGCAGCACA and U6-R,
AACGCTTCACGAATTTGCGT; GAPDH-F, CTGGGCTACACTGAGCACC and GAPDH-R,
AAGTGGTCGTTGAGGGCAATG. The cycling parameters (31) were as follows: One cycle at 94°C for
5 min, 35 cycles of a denaturing step at 94°C for 30 sec, an
annealing step at 55°C for 30 sec, an extension step at 72°C for 1
min and, lastly, one cycle of an additional extension at 72°C for
10 min. PCR products were analyzed by 3% (w/v) agarose gel
electrophoresis. All reactions were carried out in triplicate using
SYBR-Green on the ABI StepOnePlus Real-time PCR instrument (Applied
Biosystems; Thermo Fisher Scientific, Inc.), using standard cycling
parameters. Standard SYBR-Green PCR conditions were used, with an
annealing temperature at 59°C and 40 cycles. The 2−ΔΔCq
method (32) was used to calculate
the relative expression of miR-203, SMAD3, SMAD2, E-cadherin,
N-cadherin, vimentin and Snail mRNA. As an internal control, mRNAs
of U6 and GAPDH were measured under the same reaction conditions.
All samples were tested in triplicate.
Immunohistochemistry
The paraffin-embedded tissue pieces were cut into
4-µm-thick sections, mounted on the slides and heated. The sections
were dewaxed in xylene, and rehydrated in a gradient of ethanol
through a series of alcohol gradient solutions. Digestion with 3
mol/l urea for 30 min was performed to expose target antigens in
the tissue. After antigen addition to the citrate solution, the
sections were treated with 3% H2O2 solution
for 10 min, light was avoided, and then the sections were treated
with 5% bovine serum albumin (BSA) for 30 min. The sections were
then incubated with the monoclonal rabbit anti-human Smad3 antibody
(dilution 1:100; product code ab40854; Abcam), placed in a
refrigerator at 4°C overnight, and reacted for 45 min in a 37°C
water temperature chamber the next day. Visualization of antibody
binding was performed using DAB staining. The nuclei were stained
with hematoxylin for 90 sec, then soaked in hydrochloric acid
alcohol differentiation solution for 7 sec, and the gelatin was
sealed after dehydration. The results of immunostaining were
independently assessed by two pathologists.
The IHC score was based on staining intensity and
percentage of positive cells, as follows: The intensity score was 0
(no staining), 1 (weak staining), 2 (moderate staining) and 3
(strong staining); the proportion score was 0 (<5% positive
cells), 1 (6–25% positive cells), 2 (26–50% positive cells), 3
(51–75% positive cells) and 4 (>75% positive cells). The final
staining fraction was obtained by multiplying strength and
proportion fraction: 0 (negative), + (1–4), ++
(5–8) and +++ (9–12). For
statistical analysis, negative or positive final staining scores
were combined into the low-expression group, while ++ or +++ final
staining scores were combined into the high-expression group.
All the tissue points on the tissue chip were
positively scored with the same criteria, specifically, the product
of the staining intensity of the target cells and the percentage of
positive cells. Positive cells were distinguished from background
or non-specific staining. The staining intensity was scored
according to the staining characteristics of the target cells: 0
for non-staining, 1 for light yellow, 2 for brownish yellow and 3
for brown.
Western blot analysis
Protein markers used in western blot analysis were
as follows: p-SMAD2 (cat. no. ARG55037), SMAD2 (cat. no. ARG54942),
p-SMAD3 (cat. no. ARG51797), SMAD3 (cat. no. ARG53570), SMAD4 (cat.
no. ARG54741, all from arigo Biolaboratories) and GAPDH (product
code ab8245; Abcam). After H226 cells were treated with ice-cold
RIPA buffer (Beyotime Institute of Biotechnology, Inc.), the
supernatant was collected by centrifugation at 14000 × g. The
concentration of protein was determined by the BCA method. The
total protein concentration of the extracted sample was adjusted to
2 µg/ml. Total protein (20 µg), was transferred to the
polyvinylidene fluoride (PVDF) membrane by 10% SDS-PAGE gel
electrophoresis, and was blocked with 5% non-fat milk for 1 h at
room temperature, and then incubated overnight at 4°C after
treatment with a primary antibody (SMAD2, 1:1,000; SMAD3, 1:1,000;
SMAD4, 1:1,000; p-SMAD2, 1:1,000; p-SMAD3, 1:1,000; GAPDH,
1:10,000). After washing away the excess primary antibody, TBST was
used to dilute the corresponding HRP-labeled secondary antibody, so
that the PVDF membrane could be immersed in the secondary antibody
incubation solution (goat anti-mouse IgG-HRP, cat. no. sc-2058,
dilution 1:3,000; goat anti-rabbit IgG-HRP, cat. no. sc-2301,
dilution 1:3,000; Santa Cruz Biotechnology, Inc.), and incubation
followed for 2 h at 37°C on a shaking table. Next, the PVDF
membrane was washed 5–6 times with TBST, and the color was
developed by ECL chemiluminescence (Beyotime Institute of
Biotechnology). The gel images were obtained with an Alpha Gel
imager (Alpha Innotech Co.), and absorbance analysis was performed
using Quantity One v4.62 software (Bio-Rad Laboratories). The
expression level of the target protein was expressed by the
absorbance ratio between its absorbance and the corresponding
GAPDH. The experiment was repeated 3 times.
Cell transfection
H226 cells were seeded at 2×105
cells/well in 6-well plates and transfected with miR-203 mimics,
miR-203 inhibitors, and scrambled controls (miR-NC) (Shanghai
GenePharma CO., Ltd.) using Invitrogen™ Lipofectamine™ 2000 (Thermo
Fisher Scientific, Inc.). The sequences of these miRNAs were:
miR-203 mimics, 5′-TGCTTTGGCCACTGACTGTCC-3′; miR-203 inhibitors,
5′-ACGAAACCGGTGACTGACAGG-3′; miR-NC, 5′-TCGCCACATGATCGCCTAAGT-3′.
The expression levels of all transfected genes were confirmed with
RT-PCR. Cells were transfected with appropriate miRNA, siRNA
oligonucleotides and plasmids using Lipofectamine 2000 reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) following the
manufacturer's instructions. The medium was replenished 6 h after
transfection.
Transwell assays
For the invasion assay, the upper Transwell chamber
(Corning, Inc.) was coated with 50 ml of 20 mg/ml Matrigel for
filtering. The invasive chamber (Corning, Inc.) with an 8-µm pore
polycarbonate membrane was pretreated for 2 h in serum-free DMEM
(Gibco; Thermo Fisher Scientific, Inc.) medium at 37°C, 5%
CO2. After the removal of the medium, 0.25%
trypsin-digested monolayer cells were added in an amount of 1 ml/25
cm2 of surface area. The cells were resuspended and
100–200 µl were used for counting after pouring out the excess
trypsin. The passaged cells were digested with 0.25% trypsin,
washed with PBS, and added to the culture solution to prepare a
suspension of the cells to be tested, and cell density was counted
and calculated. H226 cells were plated into 6-well plates, and
transfected with miR-NC, miR-203 mimics, and miR-203 inhibitors 12
h later. After 12 h of transfection, the cells in the 6-well plate
were re-plated into the Transwell chamber with 1.5×104
cells/well. After transfection for 18 h, the cells in the upper
chamber were replaced after the cells were attached, and the cells
were treated with TGF (10 ng/ml). Cell status was observed after
treatment with TGF for 48 h, cell samples were collected, and cells
on the lower surface of the chamber were fixed with 4%
paraformaldehyde for 30 min. After washing 3 times with PBS, the
samples were stained with crystal violet (0.5 mM) dye for 30 min,
and observed and photographed under the microscope. For the
migration assay, a procedure similar to invasion assay was
performed using the migration chamber without Matrigel.
Dual luciferase assay
When the cell density in the cell culture flask
reached 70–80%, the H226 cells were digested with trypsin. Cells
were collected by centrifugation at 40 × g, and then the cells were
placed in the 24-well plate to make the density reach ~60% after
resuspended. After mixture, the cells continued to be cultured in
an incubator at 37°C. Plasmid transfection was performed using the
TurboFect™ transfection kit (Fermentas, Inc.), and fresh medium was
replaced 12 h after transfection. After 72 h of transfection, the
cells were washed twice with PBS, and 250 µl of 1X PLB lysis buffer
(Thermo Fisher Scientific, Inc.) was then added to each well. LAR
II reagent (100 µl) and lysate (20 µl) were added into a 96-well
plate, and then 100 µl of Stop & Glo substrate (Promega Corp.)
was added within 10 sec to measure the luciferase activity. After
exporting the data, the results were analyzed and plotted using
GraphPad Prism 5 software (GraphPad Software, Inc.).
Statistical analysis
Statistical analysis was conducted by SPSS 17.0
(SPSS, Inc.) statistical software. Measured data were compared
using t-tests. The count data were analyzed by Chi-square test. The
rank-sum test was employed to compare the clinical grade data sets.
The course of disease and score were statistically expressed as the
mean ± standard deviation. Student's t-test analysis was used for
comparison between groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
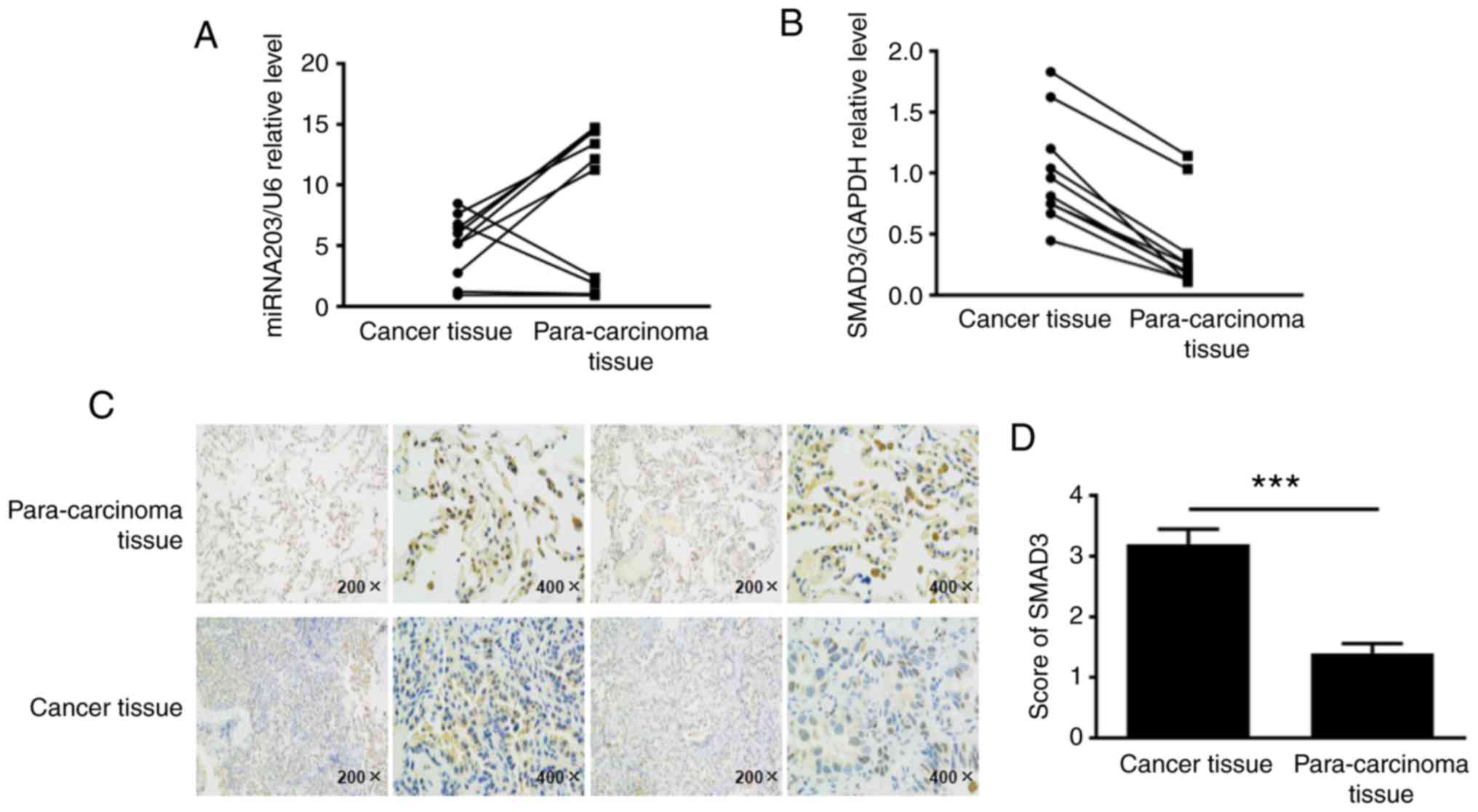
Expression of miR-203 and SMAD3 in
human NSCLC tissues
To elucidate the expression levels of miR-203 and
SMAD3 in non-small cell lung cancer tissues, ~10 fresh NSCLC tissue
samples and their corresponding paracancerous samples were
collected, total RNA was extracted, and the expression levels of
miR-203 and SMAD3 mRNA were detected by RT-PCR. The results
revealed that miR-203 RNA levels were decreased in human NSCLC
tissues compared with paracancerous tissues, while SMAD3 mRNA
levels were upregulated (Fig. 1A and
B). Tissue paraffin-embedded sections were used to detect the
expression of SMAD3 protein by immunohistochemistry, according to
the cell positive ratio, the score was 0 for ~0–5%, 1 for ~6–25%, 2
for ~26–50%, 3 for ~51–75%, and 4 for >75% (Fig. 1C and D). Collectively, these
findings indicated that the SMAD3 protein was highly expressed in
cancer tissues (P<0.001).
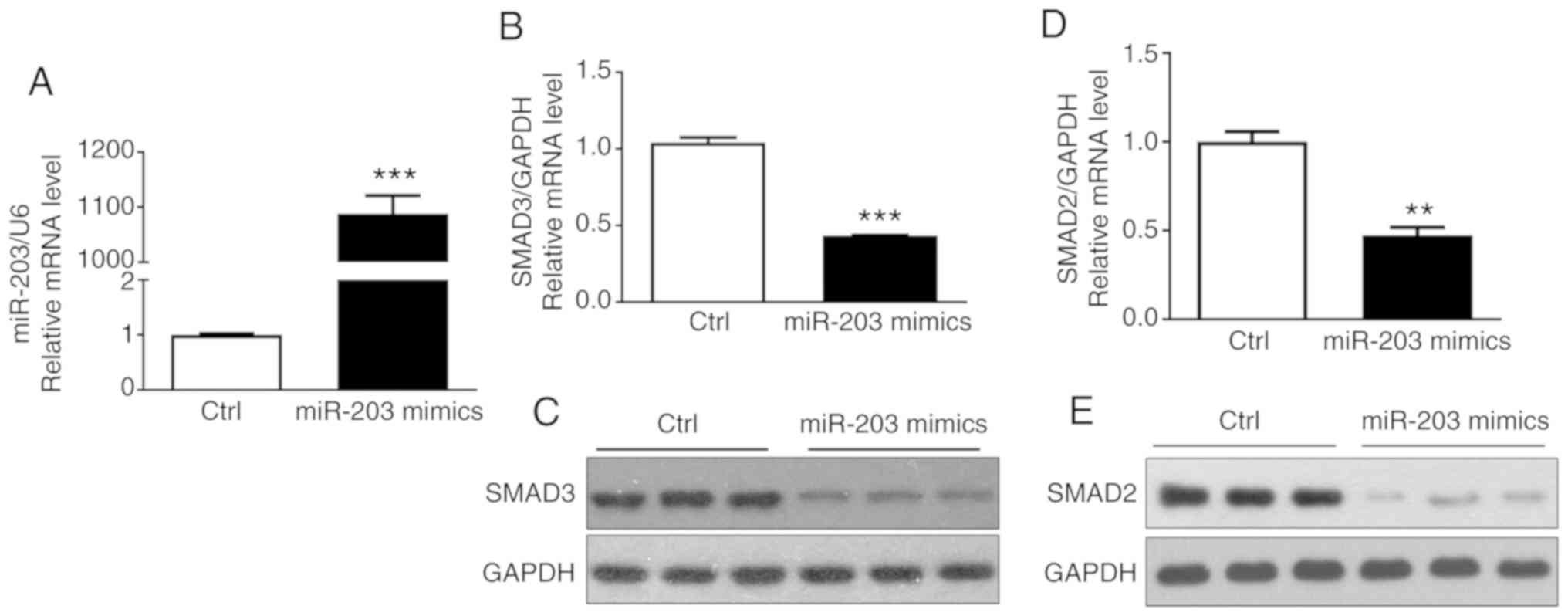
miR-203 affects the expression of
SMAD3 in NSCLC cell lines
To determine that miR-203 affected the expression of
SMAD3 in NSCLC cells, miR-203 mimics were transfected into H226
cells, and the transfection efficiency of miR-203 and the
expression of SMAD3 and SMAD2 mRNA were then detected by RT-PCR.
The results indicated that miR-203 mRNA expression was
significantly increased (P<0.05) (Fig. 2A), while the mRNA expression of
SMAD3 and SMAD2 was significantly decreased (P<0.05) (Fig. 2B and D). In addition, the western
blotting assay confirmed that the protein expression of SMAD3 and
SMAD2 was also significantly reduced (Fig. 2C and E), consistent with the
aforementioned results. These results demonstrated that miR-203
inhibited the expression of SMAD3 and SMAD2.
Effect of miR-203 on the TGF-β-induced
EMT process
In order to clarify the role of miR-203 in
TGF-β-induced EMT, miR-203 mimics and miR-203 inhibitor were
synthesized according to miR-203 sequence, and then they were
transiently transfected into the H226 cell line, respectively. In
addition, the random mimics fragment miR-NC was also synthesized as
a control. The RT-PCR results revealed that the miR-203 mRNA
expression was significantly increased after the miR-203 mimics
were transfected (P<0.001) (Fig.
3B), while it was significantly decreased after the miR-203
inhibitor was transfected (P<0.01) (Fig. 3C). The experimental groups were
divided into a control group, TGF-β group, TGF-β+miR-NC group,
TGF-β+miR-203 mimics group, and TGF-β+miR-203 inhibitor group. The
changes in cell morphology of H226 cells induced by TGF-β were
first identified, and it was revealed that the intercellular gap
was significantly increased. When cells were transfected with
miR-203 mimics, the intercellular gap was significantly reduced.
After the inhibition of miR-203, the intercellular space was
significantly larger compared with the TGF-β+miR-NC group (Fig. 3A). RT-PCR was used to detect the
mRNA expression of several related factors during EMT. The results
revealed that the mRNA expression of the epithelial marker
E-cadherin was decreased after TGF-β induction, while the
expression of mesenchymal markers Snail, N-cadherin and vimentin
was upregulated, indicating significant differences (P<0.05).
However, transfection of miR-203 mimics significantly reversed the
aforementioned effects. Compared with the TGF-β+miR-NC group, the
mRNA levels of E-cadherin in the TGF-β+miR-203 mimics group was
significantly increased (P<0.05), whereas the mRNA levels of
Snail N-cadherin and vimentin were significantly decreased
(P<0.05). Conversely, after the transfection of miR-203
inhibitor, the mRNA level of E-cadherin in the TGF-β+miR-203
inhibitor group was significantly decreased (P<0.05), while the
mRNA levels of Snail, N-cadherin and vimentin were significantly
increased (P<0.05) (Fig. 3D-G).
Western blotting was used to detect the expression of p-SMAD2,
SMAD2, p-SMAD3, SMAD3 and SMAD4 in the TGF-β pathway under
different TGF-β stimulation conditions. These results indicated
that in the presence of TGF-β, the protein expression of SMAD3 and
p-SMAD3 were significantly reduced after transfection with miR-203
mimics compared with the TGF-β-miR-NC group (P<0.05). Notably,
after transfection with miR-203 inhibitor, there was no significant
difference in SMAD3, while p-SMAD3 expression was significantly
upregulated compared with the TGF-β-miR-NC group (P<0.05)
(Fig. 3H and I). Collectively,
these findings indicated that miR-203 may inhibit TGF-β-induced EMT
progression by blocking SMAD3 phosphorylation.
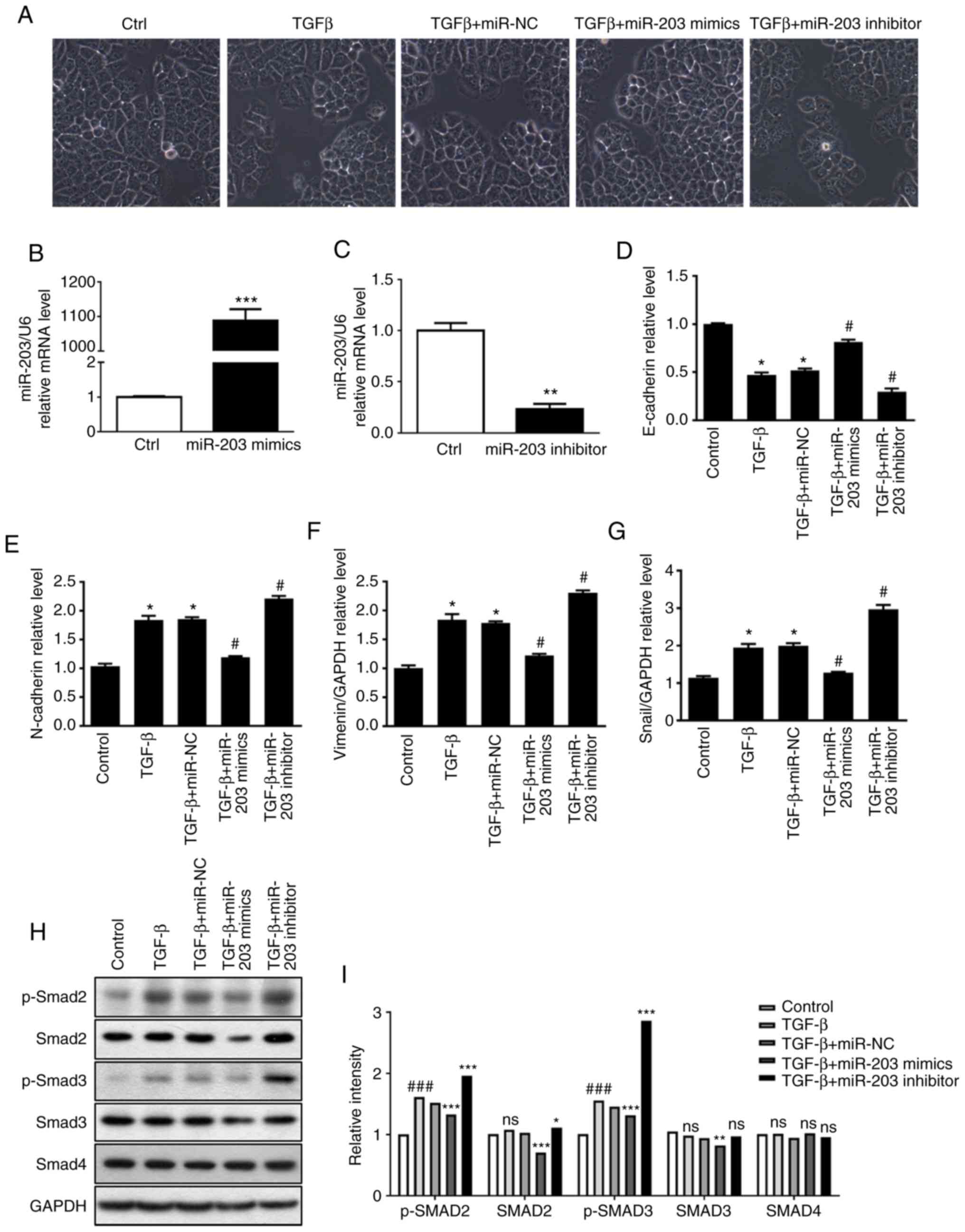 | Figure 3.(A) The effect of miR-203 expression
on H226 cell morphology and EMT phenomenon. RT-qPCR was used to
detect (B) the transfection efficiency of miR-203 mimics, (C) the
transfection efficiency of miR-203 inhibitor, the expression of (D)
E-cadherin, (E) N-cadherin, (F) vimentin, (G) Snail mRNA during
EMT. (H and I) Protein expression of p-SMAD2, SMAD2, p-SMAD3, SMAD3
and SMAD4. *P<0.5, **P<0.01, ***P<0.001, compared with the
control group. #P<0.5, ###P<0.01,
compared with the TGF-β+miR-NC group. EMT, epithelia-mesechymal
transition; TGF-β, transforming growth factor. |
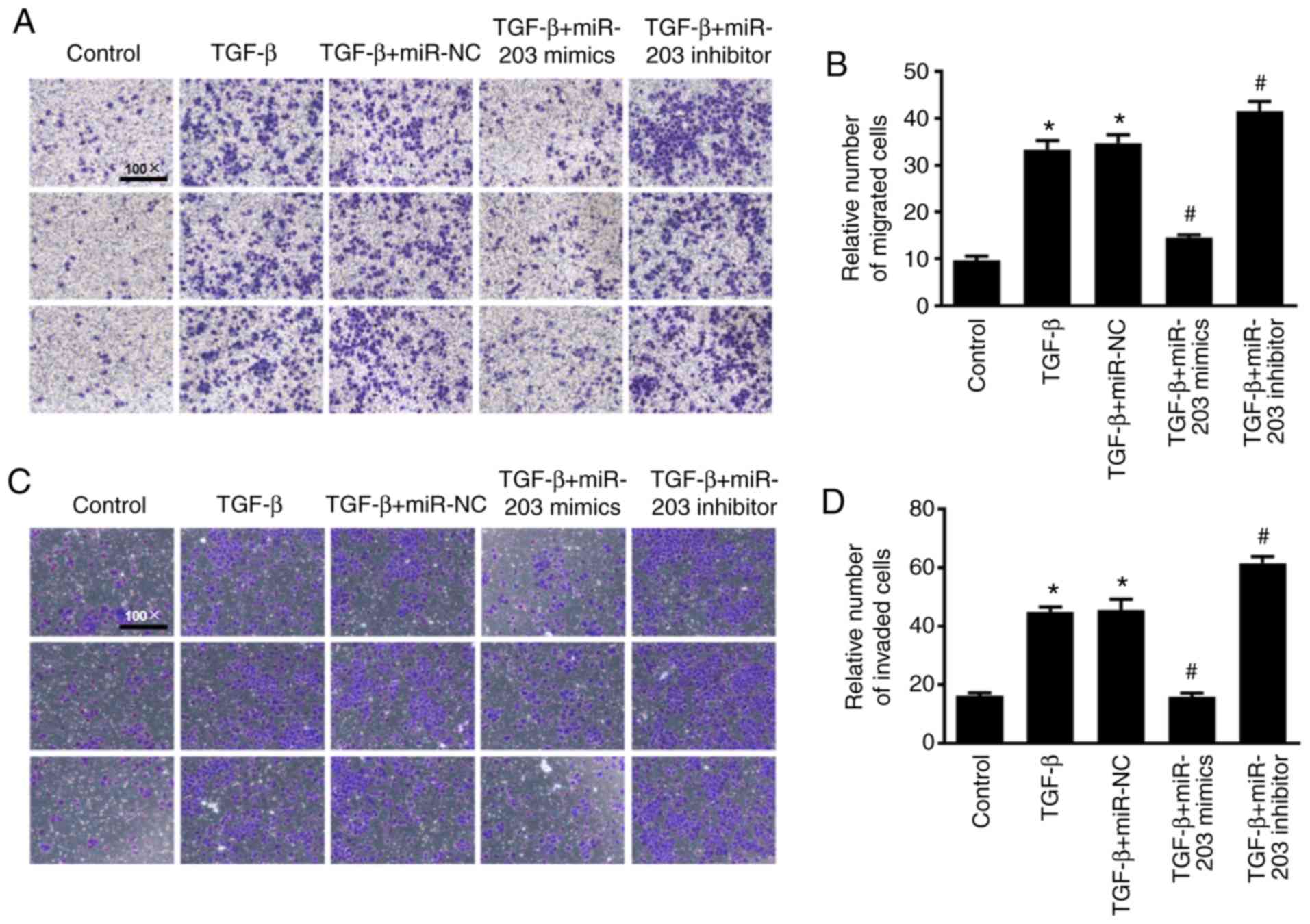
Effect of miR-203 on the migration and
invasion of NSCLC cells
To investigate the effect of miR-203 on the
migration and invasion of NSCLC cells, Transwell cell migration and
invasion assays were performed and the results were quantified
using ImageJ software to count the relative number of cells. As
revealed in Fig. 4, under the
induction of TGF-β, the migration and invasion abilities of the
cells were significantly enhanced, while the migration and invasion
abilities of H226 cells were significantly decreased after
overexpression of miR-203 (P<0.05). In contrast, the migration
and invasion abilities in the miR-203 inhibitor group were
significantly increased (P<0.05).
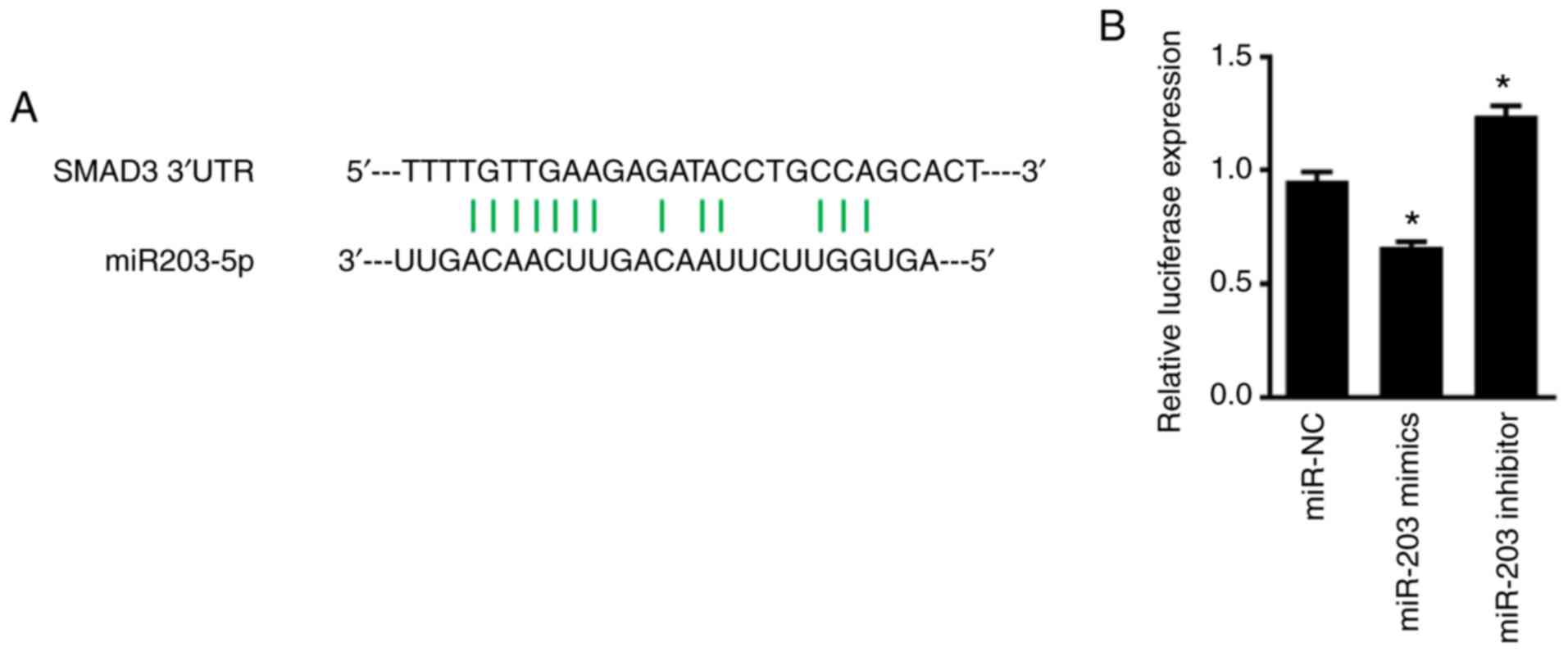
miR-203 inhibits SMAD3 expression by
targeting specific sites of SMAD 3′-UTR
To determine whether miR-203 directly binds to SMAD,
the luciferase plasmid of SMAD 3′-UTR was synthesized, and
co-transfected NSCLC cells with miR-203. As revealed in Fig. 5, miR-203 significantly suppressed
the fluorescence activity of the luciferase plasmid of SMAD 3′-UTR
in H226 cells (P<0.05). When the expression of miR-203 was
inhibited, however, the fluorescence activity of the luciferase
plasmid of SMAD 3′-UTR was significantly increased (P<0.05).
Overall, these findings indicated that the inhibitory effect of
miR-203 on SMAD3 expression was exerted by interaction with a
specific region of its 3′-UTR.
Discussion
In the present study, the regulatory mechanism of
miR-203 on SMAD3 in TGF-β-induced EMT in NSCLC and cell migration
and invasion abilities were investigated. The results revealed that
miR-203 inhibited SMAD3 by interacting with specific regions of the
3′-UTR of SMAD3, thereby inhibiting TGF-β-induced EMT progression
and migration and invasion of NSCLC cells.
EMT occurs early in the process of
epithelial-derived tumor metastasis (9), which involves changes in cell
phenotype, the cytoskeleton, and some protein expression, and
ultimately leads to the ability of tumor cells to migrate and
invade. Previous studies have revealed that EMT is not only
associated with tumor migration and invasion, but is also related
with resistance of NSCLC (33,34).
For advanced lung cancer cases, ionizing radiation is one of the
main treatment methods. However, EMT promotes radioactive fibrosis
and related metastasis after radiotherapy (35–37),
and plays an important role in tumor development and follow-up
treatment. Downregulated expression of epithelial marker E-cadherin
and upregulated expression of mesenchymal markers N-cadherin,
vimentin and Snail are the main molecular biological markers of EMT
(38,39). As an essential factor in the
induction of EMT, TGF-β plays an important role in promoting the
migration and invasion of NSCLC cells. Many studies have revealed
that TGF-β is highly expressed in many types of tumors (40,41),
and its signaling pathway can induce the progression of EMT
(21,42–44).
As a significant mediator of the TGF-β pathway in tumor cells,
SMAD3 plays different roles in the development and progression of
cancer by regulating different transcriptional reactions, depending
on the type of tumor cells and the clinical stage of cancer
(15). Studies have suggested that
SMAD3 plays a key role in promoting EMT (45,46),
and downregulation of SMAD3 or downregulation of its activated form
p-SMAD3 significantly inhibited the TGF-β-mediated EMT and slowed
the progression of pulmonary fibrosis (47). Yang et al (14) also revealed that TGF-β/SMAD3 can
directly transcribe and activate the expression of N-cadherin,
thereby promoting the EMT process of NSCLC cells. In the present
study, after TGF-β induced H226 cells, p-SMAD3 protein expression
was significantly increased, the mRNA levels of E-cadherin were
decreased, Snail, N-cadherin and vimentin mRNA expression was
upregulated, and these changes were statistically significant. In
addition, the migration and invasion abilities of the cells were
significantly enhanced. The aforementioned results indicated that
TGF-β promoted SMAD3 activation, thereby stimulating the occurrence
of EMT and enhancing the migration and invasion abilities of tumor
cells, which was consistent with previous studies.
More than 500 miRNAs have been identified through
current research, and miRNAs can participate in the regulation of
various biological processes, including proliferation,
differentiation, and apoptosis (48,49).
Evidence has demonstrated that miRNAs regulate cancer metastasis by
targeting different key proteins (50). During regulation, the target gene is
silenced or degraded mainly by binding to the 3-UTR region of the
target gene mRNA (51,52). The miR-203 gene sequence is located
on chromosome 14q32.33 and encodes ~12% of the miRNA known to
humans, and has been revealed to express abnormalities in many
types of tumors (53). Zhou et
al revealed that miR-203 could directly target the LIN28B gene
to enhance the biosynthesis of the tumor suppressor let-7 in lung
cancer and exert its anticancer effect (54). Wang et al revealed that
miR-203 inhibited the expression of SRC as well as the
proliferation and migration of lung cancer cells and promoted
apoptosis of lung cancer cells (30). The signaling pathway between TGF-β
and the transcription factor SNAI2 was revealed to inhibit the
expression of miR-203 and promote EMT and tumor metastasis
(9). The aforementioned studies
have revealed that the miR-203 regulatory mechanism and target
genes are diverse, suggesting that it may play a role in multiple
signaling pathways and target genes. Notably, further
identification and clarification of the target genes of miR-203 are
particularly important for understanding the entire regulatory
network of miR-203 and then carrying out targeted intervention. The
3′-UTR region of SMAD3 has a complementary pairing region with
miR-203, indicating that SMAD3 may be a target gene for miR-203.
Therefore, the luciferase plasmid of SMAD 3′-UTR was synthesized
and NSCLC cells were co-transfected with miR-203. The results
revealed that miR-203 significantly inhibited its fluorescence
activity, and the fluorescence activity of the luciferase plasmid
was significantly increased after inhibition of the expression of
miR-203. In addition, studies have revealed that miR-203 can
inhibit the migration of lung cancer cells by directly targeting
PKCα (26). Chen et al also
demonstrated that miR-203 inhibits the migration and invasion of
NSCLC cells by targeting Bmi1 (29). In the present study, it was revealed
that miR-203 significantly inhibited TGF-β-induced EMT and protein
expression of SMAD3 and p-SMAD3, and the migration and invasion
abilities of NSCLC H226 cells were significantly attenuated
(P<0.05). Conversely, after inhibition of miR-203, the EMT
process was further aggravated, and the expression of p-SMAD3 was
significantly upregulated, and the migration and invasion abilities
of the H226 cell line was significantly enhanced (P<0.05). These
results indicated that miR-203 targeted the 3′-UTR region of SMAD3
and blocked SMAD3 phosphorylation to inhibit TGF-β-induced EMT
progression as well as migration and invasion of NSCLC cells.
In NSCLC tissues, it was revealed that miR-203 was
significantly downregulated, while SMAD3 mRNA and protein
expression were significantly upregulated, which confirmed that
miR-203 played a tumor suppressor role in the development of NSCLC,
clinically. Additionally, the possibility that other miRNAs may
affect the expression of SMAD3 cannot be excluded. Yang et
al revealed that miR-136 can target SMAD3, thereby inhibiting
the migration and invasion of lung adenocarcinoma cells,
accompanied by increased epithelial marker expression and decreased
mesenchymal marker expression (55). In a recent study on pancreatic
ductal adenocarcinoma, it was reported that miR-323-3p could also
target the inhibition of SMAD3 expression, thereby inhibiting the
invasion and metastasis of cancer cells (56). These findings demonstrated that
SMAD3 plays an important role in tumor EMT and migration and
invasion (19). In addition to
exploring the effects of miR-203 and SMAD3, the present study also
investigated the mechanism of action between miR-203 and EMT marker
molecules induced by TGF-β, which has not been reported
previously.
Considering the important role of EMT in
tumorigenesis and subsequent treatment, the present study
demonstrated that miR-203 can directly target SMAD3 and inhibit
TGF-β-induced migration and invasion of EMT and NSCLC cells,
providing a theoretical basis for the development of new drugs for
tumor invasion and metastasis. Moreover, the present study also
offered insights into targeted treatment strategies to solve
problems related to lung cancer resistance.
Acknowledgements
Not applicable.
Funding
The present study was supported by Zhejiang Natural
Science Fund Youth Project (LQ18H010004) and the Wenzhou Science
and Technology Bureau (Y20160047).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WH was responsible for the conception and design of
the study, the data analysis and interpretation and the manuscript
revision. YW was responsible for the collection and assembly of
data, data analysis and interpretation, and manuscript writing. DC
was responsible for the collection and assembly of data, and
revised the manuscript. ZH carried out the collection and assembly
of data, wrote and reviewed the manuscript. All authors read and
approved the final manuscript and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
All patients provided their written informed consent
and ethics approval was obtained from the Ethics Committees of the
First Affiliated Hospital of Wenzhou Medical University (2017063).
The study adhered to the ethical standards of the Helsinki
Declaration.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD, Fedewa SA, Ahnen DJ,
Meester RGS, Barzi A and Jemal A: Colorectal cancer statistics,
2017. CA Cancer J Clin. 67:177–193. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen W, Zhang S and Zou X: Estimation and
projection of lung cancer incidence and mortality in China.
Zhongguo Fei Ai Za Zhi. 13:488–493. 2010.(In Chinese). PubMed/NCBI
|
|
4
|
Robinson KW and Sandler AB: The role of
MET receptor tyrosine kinase in non-small cell lung cancer and
clinical development of targeted anti-MET agents. Oncologist.
18:115–122. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rosell R and Karachaliou N: Lung cancer:
Maintenance therapy and precision medicine in NSCLC. Nat Rev Clin
Oncol. 10:549–550. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Verdecchia A, Francisci S, Brenner H,
Gatta G, Micheli A, Mangone L and Kunkler I; EUROCARE-4 Working
Group, : Recent cancer survival in Europe: A 2000-02 period
analysis of EUROCARE-4 data. Lancet Oncol. 8:784–796. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Molina JR, Yang P, Cassivi SD, Schild SE
and Adjei AA: Non-small cell lung cancer: Epidemiology, risk
factors, treatment, and survivorship. Mayo Clin Proc. 83:584–594.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gal A, Sjöblom T, Fedorova L, Imreh S,
Beug H and Moustakas A: Sustained TGF beta exposure suppresses Smad
and non-Smad signalling in mammary epithelial cells, leading to EMT
and inhibition of growth arrest and apoptosis. Oncogene.
27:1218–1230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ding X, Park SI, McCauley LK and Wang CY:
Signaling between transforming growth factor β (TGF-β) and
transcription factor SNAI2 represses expression of microRNA miR-203
to promote epithelial-mesenchymal transition and tumor metastasis.
J Biol Chem. 288:10241–10253. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: At the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Moustakas A and Heldin CH: Signaling
networks guiding epithelial-mesenchymal transitions during
embryogenesis and cancer progression. Cancer Sci. 98:1512–1520.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang HJ, Wang HY, Zhang HT, Su JM, Zhu J,
Wang HB, Zhou WY, Zhang H, Zhao MC, Zhang L and Chen XF:
Transforming growth factor-β1 promotes lung adenocarcinoma invasion
and metastasis by epithelial-to-mesenchymal transition. Mol Cell
Biochem. 355:309–314. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang H, Wang L, Zhao J, Chen Y, Lei Z, Liu
X, Xia W, Guo L and Zhang HT: TGF-β-activated SMAD3/4 complex
transcriptionally upregulates N-cadherin expression in non-small
cell lung cancer. Lung Cancer. 87:249–257. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Millet C and Zhang YE: Roles of Smad3 in
TGF-beta signaling during carcinogenesis. Crit Rev Eukaryot Gene
Expr. 17:281–293. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Attisano L and Wrana JL: Signal
transduction by the TGF-beta superfamily. Science. 296:1646–1647.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang G and Yang X: Smad4-mediated TGF-beta
signaling in tumorigenesis. Int J Biol Sci. 6:1–8. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Levy L and Hill CS: Alterations in
components of the TGF-beta superfamily signaling pathways in human
cancer. Cytokine Growth Factor Rev. 17:41–58. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roberts AB, Tian F, Byfield SD, Stuelten
C, Ooshima A, Saika S and Flanders KC: Smad3 is key to
TGF-beta-mediated epithelial-to-mesenchymal transition, fibrosis,
tumor suppression and metastasis. Cytokine Growth Factor Rev.
17:19–27. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xue J, Lin X, Chiu WT, Chen YH, Yu G, Liu
M, Feng XH, Sawaya R, Medema RH, Hung MC and Huang S: Sustained
activation of SMAD3/SMAD4 by FOXM1 promotes TGF-β-dependent cancer
metastasis. J Clin Invest. 124:564–579. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vincent T, Neve EP, Johnson JR, Kukalev A,
Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL,
et al: A SNAIL1-SMAD3/4 transcriptional repressor complex promotes
TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol.
11:943–950. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang H, Sun P, Lei Z, Li M, Wang Y, Zhang
HT and Liu J: miR-145 inhibits invasion and metastasis by directly
targeting Smad3 in nasopharyngeal cancer. Tumour Biol.
36:4123–4131. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhao W, Zou J, Wang B, Fan P, Mao J, Li J,
Liu H, Xiao J, Ma W, Wang M, et al: microRNA-140 suppresses the
migration and invasion of colorectal cancer cells through targeting
Smad3. Zhonghua Zhong Liu Za Zhi. 36:739–745. 2014.(In Chinese).
PubMed/NCBI
|
|
24
|
Huang S, Wu S, Ding J, Lin J, Wei L, Gu J
and He X: MicroRNA-181a modulates gene expression of zinc finger
family members by directly targeting their coding regions. Nucleic
Acids Res. 38:7211–7218. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen K and Rajewsky N: Natural selection
on human microRNA binding sites inferred from SNP data. Nat Genet.
38:1452–1456. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang C, Wang X, Liang H, Wang T, Yan X,
Cao M, Wang N, Zhang S, Zen K, Zhang C and Chen X: miR-203 inhibits
cell proliferation and migration of lung cancer cells by targeting
PKCα. PLoS One. 8:e739852013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schneider MR: MicroRNAs as novel players
in skin development, homeostasis and disease. Br J Dermatol.
166:22–28. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Butz H, Rácz K, Hunyady L and Patócs A:
Crosstalk between TGF-β signaling and the microRNA machinery.
Trends Pharmacol Sci. 33:382–393. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen T, Xu C, Chen J, Ding C, Xu Z, Li C
and Zhao J: MicroRNA-203 inhibits cellular proliferation and
invasion by targeting Bmi1 in non-small cell lung cancer. Oncol
Lett. 9:2639–2646. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang N, Liang H, Zhou Y, Wang C, Zhang S,
Pan Y, Wang Y, Yan X, Zhang J, Zhang CY, et al: miR-203 suppresses
the proliferation and migration and promotes the apoptosis of lung
cancer cells by targeting SRC. PLoS One. 9:e1055702014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dickie LJ, Aziz AM, Savic S, Lucherini OM,
Cantarini L, Geiler J, Wong CH, Coughlan R, Lane T, Lachmann HJ, et
al: Involvement of X-box binding protein 1 and reactive oxygen
species pathways in the pathogenesis of tumour necrosis factor
receptor-associated periodic syndrome. Ann Rheum Dis. 71:2035–2043.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yauch RL, Januario T, Eberhard DA, Cavet
G, Zhu W, Fu L, Pham TQ, Soriano R, Stinson J, Seshagiri S, et al:
Epithelial versus mesenchymal phenotype determines in vitro
sensitivity and predicts clinical activity of erlotinib in lung
cancer patients. Clin Cancer Res. 11:8686–8698. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shintani Y, Okimura A, Sato K, Nakagiri T,
Kadota Y, Inoue M, Sawabata N, Minami M, Ikeda N, Kawahara K, et
al: Epithelial to mesenchymal transition is a determinant of
sensitivity to chemoradiotherapy in non-small cell lung cancer. Ann
Thorac Surg. 92:1794–1804. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jung JW, Hwang SY, Hwang JS, Oh ES, Park S
and Han IO: Ionising radiation induces changes associated with
epithelial-mesenchymal transdifferentiation and increased cell
motility of A549 lung epithelial cells. Eur J Cancer. 43:1214–1224.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhou YC, Liu JY, Li J, Zhang J, Xu YQ,
Zhang HW, Qiu LB, Ding GR, Su XM, Mei-Shi and Guo GZ: Ionizing
radiation promotes migration and invasion of cancer cells through
transforming growth factor-beta-mediated epithelial-mesenchymal
transition. Int J Radiat Oncol Biol Phys. 81:1530–1537. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Theys J, Jutten B, Habets R, Paesmans K,
Groot AJ, Lambin P, Wouters BG, Lammering G and Vooijs M:
E-Cadherin loss associated with EMT promotes radioresistance in
human tumor cells. Radiother Oncol. 99:392–397. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zeisberg M and Neilson EG: Biomarkers for
epithelial-mesenchymal transitions. J Clin Invest. 119:1429–1437.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Derynck R and Akhurst RJ: Differentiation
plasticity regulated by TGF-beta family proteins in development and
disease. Nat Cell Biol. 9:1000–1004. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Thiery JP: Epithelial-mesenchymal
transitions in development and pathologies. Curr Opin Cell Biol.
15:740–746. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang L, Yang H, Lei Z, Zhao J, Chen Y,
Chen P, Li C, Zeng Y, Liu Z, Liu X and Zhang HT: Repression of
TIF1γ by SOX2 promotes TGF-β-induced epithelial-mesenchymal
transition in non-small-cell lung cancer. Oncogene. 35:867–877.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cho HJ, Baek KE, Saika S, Jeong MJ and Yoo
J: Snail is required for transforming growth factor-beta-induced
epithelial-mesenchymal transition by activating PI3 kinase/Akt
signal pathway. Biochem Biophys Res Commun. 353:337–343. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shintani Y, Maeda M, Chaika N, Johnson KR
and Wheelock MJ: Collagen I promotes epithelial-to-mesenchymal
transition in lung cancer cells via transforming growth factor-beta
signaling. Am J Respir Cell Mol Biol. 38:95–104. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bruna A, Darken RS, Rojo F, Ocaña A,
Peñuelas S, Arias A, Paris R, Tortosa A, Mora J, Baselga J and
Seoane J: High TGFbeta-Smad activity confers poor prognosis in
glioma patients and promotes cell proliferation depending on the
methylation of the PDGF-B gene. Cancer Cell. 11:147–160. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu RY, Zeng Y, Lei Z, Wang L, Yang H, Liu
Z, Zhao J and Zhang HT: JAK/STAT3 signaling is required for
TGF-β-induced epithelial-mesenchymal transition in lung cancer
cells. Int J Oncol. 44:1643–1651. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang C, Song X, Li Y, Han F, Gao S, Wang
X, Xie S and Lv C: Low-dose paclitaxel ameliorates pulmonary
fibrosis by suppressing TGF-β1/Smad3 pathway via miR-140
upregulation. PLoS One. 8:e707252013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cao H, Yang CS and Rana TM: Evolutionary
emergence of microRNAs in human embryonic stem cells. PLoS One.
3:e28202008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Krichevsky AM, King KS, Donahue CP,
Khrapko K and Kosik KS: A microRNA array reveals extensive
regulation of microRNAs during brain development. RNA. 9:1274–1281.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nakayama K, Nakayama N, Katagiri H and
Miyazaki K: Mechanisms of ovarian cancer metastasis: Biochemical
pathways. Int J Mol Sci. 13:11705–11717. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cuesta R, Martínez-Sánchez A and Gebauer
F: miR-181a regulates cap-dependent translation of p27(kip1) mRNA
in myeloid cells. Mol Cell Biol. 29:2841–2851. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Dai Y, Huang YS, Tang M, Lv TY, Hu CX, Tan
YH, Xu ZM and Yin YB: Microarray analysis of microRNA expression in
peripheral blood cells of systemic lupus erythematosus patients.
Lupus. 16:939–946. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Abella V, Valladares M, Rodriguez T, Haz
M, Blanco M, Tarrío N, Iglesias P, Aparicio LA and Figueroa A:
miR-203 regulates cell proliferation through its influence on Hakai
expression. PLoS One. 7:e525682012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhou Y, Liang H, Liao Z, Wang Y, Hu X,
Chen X, Xu L and Hu Z: miR-203 enhances let-7 biogenesis by
targeting LIN28B to suppress tumor growth in lung cancer. Sci Rep.
7:426802017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yang Y, Liu L, Cai J, Wu J, Guan H, Zhu X,
Yuan J, Chen S and Li M: Targeting Smad2 and Smad3 by miR-136
suppresses metastasis-associated traits of lung adenocarcinoma
cells. Oncol Res. 21:345–352. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang C, Liu P, Wu H, Cui P, Li Y, Liu Y,
Liu Z and Gou S: MicroRNA-323-3p inhibits cell invasion and
metastasis in pancreatic ductal adenocarcinoma via direct
suppression of SMAD2 and SMAD3. Oncotarget. 7:14912–14924.
2016.PubMed/NCBI
|