Introduction
Lung cancer is a highly malignant disease, and lung
cancer-associated deaths and morbidity are increasing yearly. The
World Health Organization divides lung cancer into small-cell lung
cancer (SCLC) and non-small cell lung cancer (NSCLC), with
small-cell lung cancer accounting for ~13 percent of lung cancer
patients (1). Small-cell lung
cancer is characterized by rapid growth and high invasiveness. If
the patient is not treated in a timely and effective manner, they
will likely die within a few months. For decades, clinical trials
for a variety of treatment regimens have failed to find a cure for
small-cell lung cancer, and most patients relapse or succumb to
metastasis after treatment (2,3).
Effective therapeutic targets and solutions remain an unmet need in
the treatment of small-cell lung cancer, largely because we still
know little concerning the molecular mechanisms driving the
development of this disease.
The TP53 tumor suppressor gene is mutated in
the majority of human tumors, and mutant p53 protein is highly
expressed in >50% of these tumors, inhibiting the activity of
wild-type p53 and acquiring oncogenic functions. The incidence of
TP53 mutation in small-cell lung cancer is as high as 75–90%
(4). Mutation inhibits the normal
transcriptional activity of p53, thereby preventing the ability of
p53 to protect cells from malignant transformation (5). Regulation of p53 protein translation
is considered to be the main mechanism by which the transcriptional
activity and tumor suppressive functions of the protein are
regulated. Sirtuin 3 (SIRT3) is a member of the Sirtuin family of
nicotinamide adenine dinucleotide (NAD+)-dependent
deacetylases (6). SIRT3 is a major
mitochondrial stress-reactive protein, and participates in the
binding and deacetylation of enzymes that regulate important
mitochondrial functions. In recent years, an increasing number of
studies have revealed that SIRT3 plays an important role in tumor
biology (7). The function of SIRT3
may vary depending on cell and tumor type, and SIRT3 may therefore
exhibit the characteristics of either an oncogene or tumor
suppressor gene. In recent years, the association between SIRT3 and
p53 signaling has received increasing attention. There are several
studies that have reported the regulation of p53 post-translational
modification by SIRT3. Zhang et al proposed that
overexpression of SIRT3 upregulated the protein level of p53 by
attenuating MDM2-mediated p53 degradation in liver cancer (8). The balance between acetylated and
deacetylated p53 within the cell determines the overall
transcriptional activity of the protein, and this has consequences
for the downstream transcription of p53-responsive genes associated
with cancer. Therefore, it is important to explore the role of
SIRT3-dependent deacetylation in the regulation of p53. Indeed,
some studies suggest that acetylation also affects the activity of
mutant p53 (9–11).
In a previous study using the human non-small cell
lung cancer cell line A549, we revealed that SIRT3 overexpression
could both enhance the expression and reduce the acetylation of p53
within the cell, indicating that SIRT3 may affect the balance of
acetylated-to-deacetylated p53. In addition, our clinical data
revealed a negative correlation between mutant p53 and SIRT3
expression (unpublished data). We therefore speculate that SIRT3
may be involved in the post-translational modification of mutant
p53, thereby affecting the stability of the protein and,
consequently, the growth of lung cancer cells.
In the present study, human small-cell lung cancer
cell line NCI-H446, which is positive for mutant p53, was used to
examine the effects of SIRT3 expression on cell behavior and mutant
p53 expression. These findings provide additional insights into the
regulation of mutant p53 in small-cell lung cancer, highlighting
new possibilities for the development of treatment strategies for
this disease.
Materials and methods
Cell culture
NCI-H446 cells were obtained from ATCC and stored
under liquid nitrogen at the Prostate Diseases Prevention and
Treatment Research Center, Jilin University, Changchun, China. The
cells were cultured in Dulbecco's modified Eagle's medium (DMEM;
Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal
bovine serum (Biological Industries). Cells were grown in a
humidified incubator maintained at 37°C and supplied with 5%
CO2. Cells were passaged by trypsinization (0.25%
EDTA-free trypsin; Sangon Biotech Co., Ltd.) and reseeded in new
culture dishes at one half of their original density. The day after
cultures had reached a confluency of 70%, cells were transiently
transfected with NC or SIRT3 expression constructs. RNA and protein
were then extracted from cells 48 h post-transfection, for use in
downstream experiments.
MTT assay
NCI-H446 cells were seeded in 96-well plates at a
density of 6×103 cells/well. After 24 h, the cells were
transfected with either empty control plasmid (NC group) or plasmid
encoding SIRT3 (SIRT3 group). Plasmids were constructed by Shanghai
GeneChem Co., Ltd. Cells were then maintained under normal culture
conditions until analysis of viability by MTT assay. To assess
viability, 20 µl of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sangon Biotech Co., Ltd.) was added per well of the 96-well plate.
After a 4-hour incubation, 150 µl dimethyl sulfoxide (Sangon
Biotech Co., Ltd.) was then added to each well. The absorbance of
each well at 490 nm was then measured using a FLUOstar Omega
microplate reader (BMG LABTECH). Cell viability was assessed at 24,
48 and 72 h post-transfection.
Western blotting
To prepare protein lysates for western blotting
assays, cultured cells were washed with PBS, detached with a cell
scraper and then lysed in 100–200 µl RIPA buffer (Beyotime
Institute of Biotechnology) containing the protease inhibitor
phenylmethylsulfonyl fluoride (Roche Diagnostics). Cell lysates
were then sonicated on ice to disrupt protein aggregates by using
ultrasonic disruptor UD-201 (TOMY SEIKO Co., Ltd.). Samples were
then clarified by centrifugation at 15,000 × g for 20 min at 4°C,
and the supernatants were retained. Clarified protein lysates (20
µg per lane) were then resolved by electrophoresis on 12%
polyacrylamide gels and subsequently transferred onto
polyvinylidene fluoride membranes (EMD Millipore). The membranes
were blocked in Tris-buffered saline/0.5% Tween-20 (TBST)
containing 5% non-fat milk for 1 h at room temperature, washed
three times in TBST and then incubated with primary antibodies
overnight at 4°C. The membranes were then washed three times in
TBST before incubation with secondary antibodies for 1 h. After
washing three times with TBST, the membranes were incubated with
Hypersensitive ECL chemiluminescence reagent (Beyotime Institute of
Biotechnology), and immunoreactivity was subsequently detected
using a Syngene HR chemiluminescence imaging system (Synoptics).
Primary antibodies, their respective dilutions and manufacturers,
were as follows: anti-SirT3 (1:1,000; clone D22A3; product #5490),
cleaved caspase-3 (1:1,000; product #9664), cleaved caspase-9
(1:1,000; product #9509), and BID (1:1,000; product #2002S) were
purchased from Cell Signaling Technology, Inc.; anti-β-actin
(1:1,000, cat. no. 66009-1-Ig) was obtained from Proteintech Group;
anti-Bcl-2 (1:1,000, ab32124), anti-RIP (1:1,000; product code
ab125072), RIP3 (1:1,000; product code ab152130), MLKL (1:1,000;
product code ab183770), p-MLKL (1:1,000; product code ab196436),
LC3 II (1:1,000; product code ab48394), and mutant p53 antibodies
[Y5] (1:1,000; product code ab32049) were obtained from Abcam;
anti-Mcl-1 (1:200; cat. no. sc-819), Bax (1:200; cat. no. sc-7480),
and Ubiquitin (1:200; sc-8017) antibodies were purchased from Santa
Cruz Biotechnology, Inc.; anti-Beclin-1 (1:1,000; cat. no. 612112)
was obtained from BD Biosciences. Horseradish peroxidase goat
anti-mouse (1:2,000; cat. no. SA00001-1) and goat anti-rabbit
(1:2,000; cat. no. SA00001-2) antibodies were purchased from
Proteintech Group.
Caspase activity
Caspase-Glo 3/7 Assay (Promega Corporation) were
used to determine cellular caspase-3/7 activities according to the
manufacturer's instructions.
Reverse transcription PCR
(RT-PCR)
Total RNA was extracted from tissue samples and cell
lines using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.)
based on standard protocols. NCI-H446 cells with SIRT3
overexpression treatment were cultured in a 6-well plate, washed
twice with cold PBS and the culture medium was discarded. Then 500
µl of TRIzol per well was added and incubation followed for 5 min
at room temperature. Chloroform, isopropanol and 75% ethanol were
added successively, and mRNA was obtained by drying at room
temperature after 7,500 × g centrifugation repeatedly. Subsequent
generation of cDNA was performed using the EasyScript First-Strand
cDNA Synthesis SuperMix kit according to the manufacturer's
instructions (Beyotime Institute of Biotechnology). Briefly, in all
reactions, 50–500 ng RNA was used as the starting template. The
RT-PCR reaction was performed at 42°C for 30 min followed by an
enzyme inactivation step at 85°C for 5 sec. Primer information and
reaction conditions are presented in Tables I and II.
 | Table I.Primer sequences for reverse
transcription polymerase chain reaction. |
Table I.
Primer sequences for reverse
transcription polymerase chain reaction.
| Gene | Forward primer
sequence | Reverse primer
sequence |
|---|
| GAPDH |
5′-AGAAGGCTGGGGCTCATTTG-3′ |
5′-AGGGGCCATCCACAGTCTTC-3′ |
| TP53 |
5′-CCATCTACAAGCAGTCACAG-3′ |
5′-CAAATCTACAAGCAGTCACAG-3′ |
| BCL2 |
5′-GACTTCGCCGAGATGTCCAGC-3′ |
5′-GCGTTACGATCGCCTCCATCA-3′ |
| BAX |
5′-CGGCGAATTGGAGATGAACTG-3′ |
5′-AGCAAAGTAGAAGAGGGCAACC-3′ |
| CASP3 |
5′-ATGGACAACAACGAAACCTCCGTG-3′ |
5′-CCACTCCCAGTCATTCCTTTTAGTG-3′ |
| Table II.Reaction conditions for reverse
transcription polymerase chain reaction. |
Table II.
Reaction conditions for reverse
transcription polymerase chain reaction.
| Gene | Denaturation | Annealing | Extension | No. of cycles |
|---|
| GAPDH | 94°C, 30 sec | 55°C, 30 sec | 55°C, 30 sec | 26 |
| SIRT3 | 94°C, 30 sec | 55°C, 30 sec | 55°C, 30 sec | 28 |
| TP53 | 94°C, 30 sec | 56°C, 30 sec | 72°C, 30 sec | 30 |
| BCL2 | 94°C, 30 sec | 55°C, 30 sec | 72°C, 30 sec | 30 |
| BAX | 94°C, 30 sec | 54°C, 30 sec | 72°C, 30 sec | 30 |
| CASP3 | 94°C, 30 sec | 55°C, 30 sec | 72°C, 30 sec | 28 |
Flow cytometric analysis of
apoptosis
NCI-H446 cells were seeded in 6-well plates at a
density of 3×105 cells/well. After 24 h, the cells were
transfected with either empty plasmid (NC group) or plasmid
encoding SIRT3 (SIRT3 group). After 48 h, the cells were collected,
washed twice with PBS, and apoptosis was then evaluated using the
FITC Annexin V Apoptosis Detection kit (BD Biosciences). Briefly,
cells were resuspended in 100 µl of 1X Binding Buffer before the
addition of 5 µl Annexin V-FITC and 5 µl propidium iodide. The
cells were then incubated in the dark for 15 min. An additional 400
µl of 1X Binding Buffer was added before analysis of apoptosis
using a BD FACSCalibur flow cytometer (BD Biosciences).
Detection of mitochondrial membrane
potential
Mitochondrial membrane potential was assessed using
the Mitochondrial membrane potential assay kit with JC-1 (Beyotime
Institute of Biotechnology), which accumulates in healthy
mitochondria. NCI-H446 cells were seeded into 6-well plates at a
density of 3×105 cells/well. After experimental
treatments, cells were harvested by digestion with EDTA-free
trypsin. The cells were then resuspended in 1 ml serum-free DMEM
containing 1 µl JC-1 (5 µg/ml) and incubated at 37°C for 20 min. At
3–5 min intervals, the cells were removed from the incubator and
mixed by inversion to ensure uniform uptake of the dye. The cells
were then pelleted by centrifugation at 1,500–2,000 × g for 5 min,
washed with PBS and then resuspended in 400 µl DMEM. The
fluorescence intensity of dye-labelled cells was analyzed using a
BD FACSCalibur flow cytometer.
Proteasome inhibition assay
NCI-H446 cells were divided into five groups as
follows; untransfected cells (CON group), cells transfected with
empty plasmid (NC group), cells transfected with SIRT3 plasmid
(SIRT3 group), cells treated with MG132 only (MG132 group), and
cells transfected with SIRT3 plasmid and subsequently treated with
MG132 (MG132+SIRT3 group). NCI-H446 cells were seeded into 6-well
plates and 24 h later the appropriate groups were transfected with
their respective plasmids and treated with MG132 inhibitor as
indicated. After an additional 24 h, the cells were collected and
lysates were prepared for analysis of protein expression by western
blotting.
Analysis of protein half-life
Cycloheximide (CHX) was used to evaluate changes in
mutant p53 protein half-life. NCI-H446 cells were transiently
transfected with a plasmid encoding SIRT3 48 h prior to CHX
experiments. Cells were then treated with 50 µg/ml CHX, and cell
lysates were subsequently obtained at 0, 0.5, 1, and 2-h
time-points for analysis of protein expression by western
blotting.
Immunoprecipitation assays
After NCI-H446 cells had been subjected to the
indicated treatments, they were washed twice with PBS and lysed in
RIPA buffer. Cell lysates were incubated for 30 min on ice with
rotation. Lysates were then clarified by centrifugation at 14,000 ×
g for 15 min at 4°C, and the supernatants were subsequently
retained and transferred to a fresh tube. Approximately 30–50 µl of
clarified lysate was retained for analysis of input. Samples
containing 1 mg total protein were incubated with 2 µg antibody
overnight on ice. The following day, protein A/G-conjugated beads
were added and the samples were then incubated for an additional
night on ice. On the third day, the beads were pelleted by
centrifugation for 5 min at 1,000 × g at 4°C and the supernatants
were discarded. The beads were then washed 3–5 times with PBS.
Finally, 60 µl RIPA buffer was added to 20 µl of bead slurry and 20
µl of sample was used/well for subsequent polyacrylamide gel
electrophoresis.
Animal experiments
Salmonella typhimurium A1-R is auxotrophic
for arg and leu, which enables improved targeting and toxicity
towards tumors, and this strain has previously been used in cancer
treatment (12). Mouse-attenuated
salmonella typhimurium phoP/phoQ was used in this study to
deliver expression constructs to tumor cells in vivo.
Attenuated salmonella typhimurium was induced into
competence. Competent cells (100 µl) were mixed with 1 µl SIRT3
plasmid, and then transferred to a cold MicroPulser electroporation
cuvette (0.1-cm gap). The cuvette was placed in a Gene Pulser
Transfection Apparatus (product no. 1652100; Bio-Rad Laboratories,
Inc.), and pulsed once. After hearing the beep, 1 ml LB liquid
medium was immediately added into the cuvette. After gently
resuspending the cells, they were transferred to a new centrifuge
tube, cultured at 37°C for 1 h, and shaked at 220 RPM. Then the
cells were plated on LB plates with ampicillin at 37°C for 12 h.
The following parameters were used for electroporation: 2.5 kV, 25
µF, 200 Ω, 5 msec.
The animal study was approved by the Animal Ethics
Review Committee of Basic Medical Sciences, Jilin University, in
accordance with the regulations of the Institutional Committee for
the Care and Use of Laboratory Animals of the Experimental Animal
Center of Jilin University.
A total of 12 male 4 to 6-week-old BALB/C nu/nu nude
mice weighing between 18–20 g were used for in vivo
experiments. Animals were purchased from Huafukang Bioscience Co.,
Inc. Each mouse was injected subcutaneously with 2×106
cells in a final suspension volume of 100 µl. Tumor size was then
monitored daily by Vernier caliper until they had reached
approximately 5 mm in diameter. Ten tumor-bearing mice were then
randomly divided into two groups; the PQ group (animals injected
with attenuated Salmonella transformed with empty plasmid)
and the PQ-SIRT3 group (animals injected with attenuated
Salmonella transformed with SIRT3 plasmid). Mice were
intraperitoneally injected every 14 days with 5×107 cfu
of PQ or PQ-SIRT3-harboring bacteria. After treatment, the behavior
and changes in water intake and weight of the animals were
monitored. The maximum length (L) and short diameter (W) of the
tumor was measured by Vernier caliper to calculate the tumor volume
using the formula V=0.52 × L × W2. The tumor growth
index was used to record and plot tumor growth and was calculated
using the formula: Tumor growth
index=(Vn-V1)/V1 (where
V1 is the tumor volume of the first day, and
Vn is the tumor volume of the nth day).
After 25 days of treatment, the nude mice were
euthanized by cervical dislocation after intraperitoneal injection
of 20% urethane (0.1 ml per mouse) and the tumors were removed,
weighed and photographed. A small portion of each tumor was fixed
in 10% formaldehyde solution for subsequent hematoxylin and eosin
(H&E) staining. The remaining tumors were stored in liquid
nitrogen for subsequent RNA and protein extraction.
Histology and
immunohistochemistry
The tumors were fixed in 10% formaldehyde solution
for 48 h, and then embedded in paraffin. Histological sections were
prepared by standard conventional processing and stained with
H&E. The sections (0.2 mm) were incubated with 2% bovine serum
albumin (BSA) for 30 min at RT to block nonspecifc binding, and
then incubated with rabbit polyclonal anti-SIRT3 (1:200; product
no. 2627; Cell Signaling Technology, Inc.) and anti-mutant p53
antibodies [Y5] (1:1,000; product no. ab32049), followed by
incubation with goat anti-rat immunoglobulin G (IgG; 1:400; cat.
no. SA00001-15; ProteinTech Group, Inc.) for 1 h at RT. Between
each incubation step, the sections were washed 3 times with
Tris-buffered saline with Tween-20 (12.5 mM Tris/HCl, pH 7.6, 137
mM NaCl and 0.1% Tween-20).
Statistical analysis
All the data were obtained after at least three
independent experiments. Data are presented as the mean ± standard
deviation. A Student's t-test was used to compare two groups and
ANOVA with Tukey post hoc test was used to enable multiple
comparisons between groups. P<0.05 was considered to indicate a
statistically significant difference in data.
Results
SIRT3 overexpression inhibits the
growth of NCI-H446 small-cell lung cancer cells
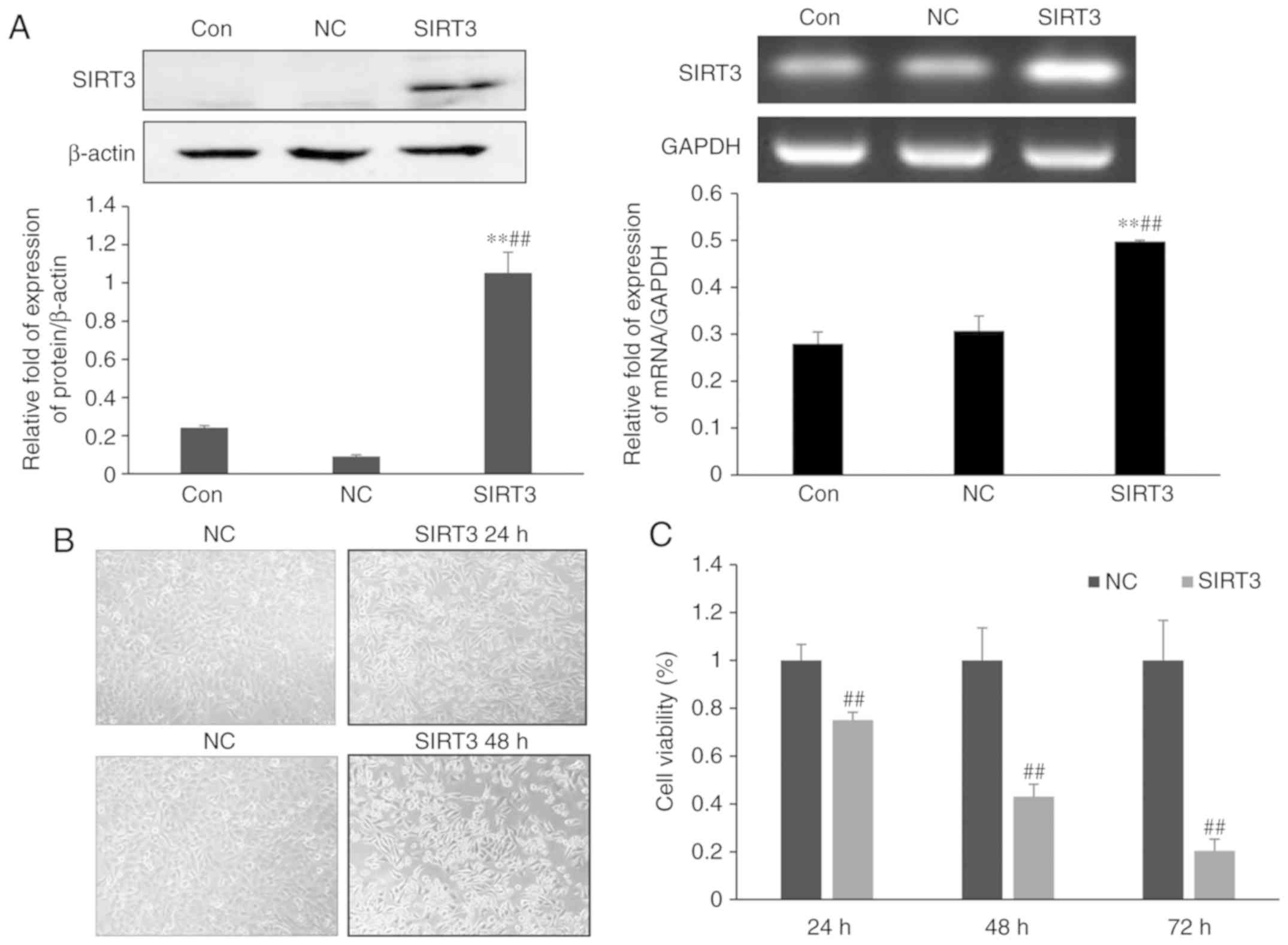
To study the effect of SIRT3 expression on the
growth of small-cell lung cancer cells, NCI-H446 cells were
transfected with either a control plasmid (NC group) or a plasmid
encoding SIRT3 (SIRT3 group). Western blotting and RT-PCR analysis
confirmed an increase in expression of SIRT3 in cells of the SIRT3
group, when compared with those of the NC group (P<0.01),
indicating that the transfection was successful. A microscopy-based
analysis of cell morphology revealed an increase in cell death
among cells of the SIRT3-group at 24 h post-transfection (Fig. 1B). By 48 h post-transfection the
difference in cell death between the control and SIRT3 groups was
even more pronounced. SIRT3 overexpression promoted cell rounding
and detachment, resulting in increased numbers of cells floating in
the culture media. MTT assays revealed that cell viability in the
SIRT3 group was reduced to 75% compared to the control group 24 h
after transfection (Fig. 1C). By 48
and 72 h post-transfection, viability of the SIRT3 group was
reduced to 43 and 20%, respectively, indicating that overexpression
of SIRT3 in lung cancer NCI-H446 cells inhibited the growth of
NCI-H446 cells in lung cancer.
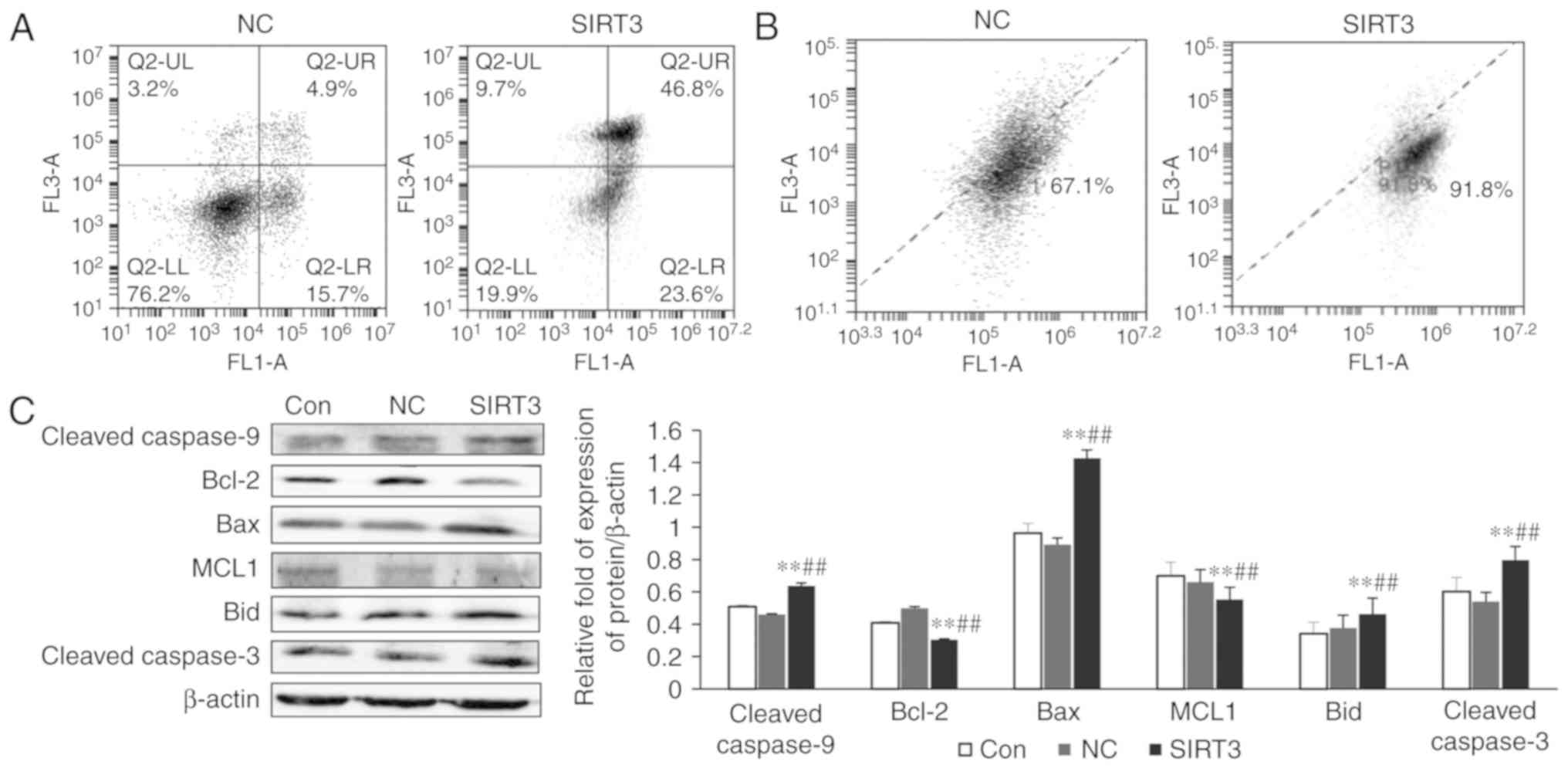
SIRT3 overexpression results in
apoptosis of small-cell lung cancer NCI-H446
Cell apoptosis was detected by flow cytometry BD
Accuri C6. The upper right quadrant represents late apoptosis and
necroptosis, and the lower right quadrant represents early
apoptosis. It can be observed from Fig.
2A that early apoptosis of the SIRT3 group was increased
compared to the NC group, and late apoptosis and necroptosis were
also increased (P<0.01). The mitochondrial membrane potential
was also examined by flow cytometry, and it was revealed to be
significantly decreased in the SIRT3-overexpressing cells when
compared with the control (Fig. 2B;
P<0.05). The results of western blotting experiments revealed
that the expression of the apoptosis-related proteins cleaved
caspase-9, Bax, Bid, and cleaved caspase-3 was significantly
increased when compared with controls (Fig. 2C; P<0.01), and that the
expression of the anti-apoptotic proteins Bcl-2 and MCL1 was
significantly decreased (Fig. 2C;
P<0.01). RT-PCR analysis also revealed a significant increase in
the expression of the pro-apoptotic genes TP53, BAX and
CASP3 (P<0.05) and a decrease in the expression of the
pro-survival gene BCL2 (P<0.01) in SIRT3-overexpressing
cells (Fig. 2D). Furthermore, after
24 h of treatment with SIRT3 overexpression, a significant increase
in caspase-3/7 activity was revealed in NCI-H446 cells (Fig. 2E).
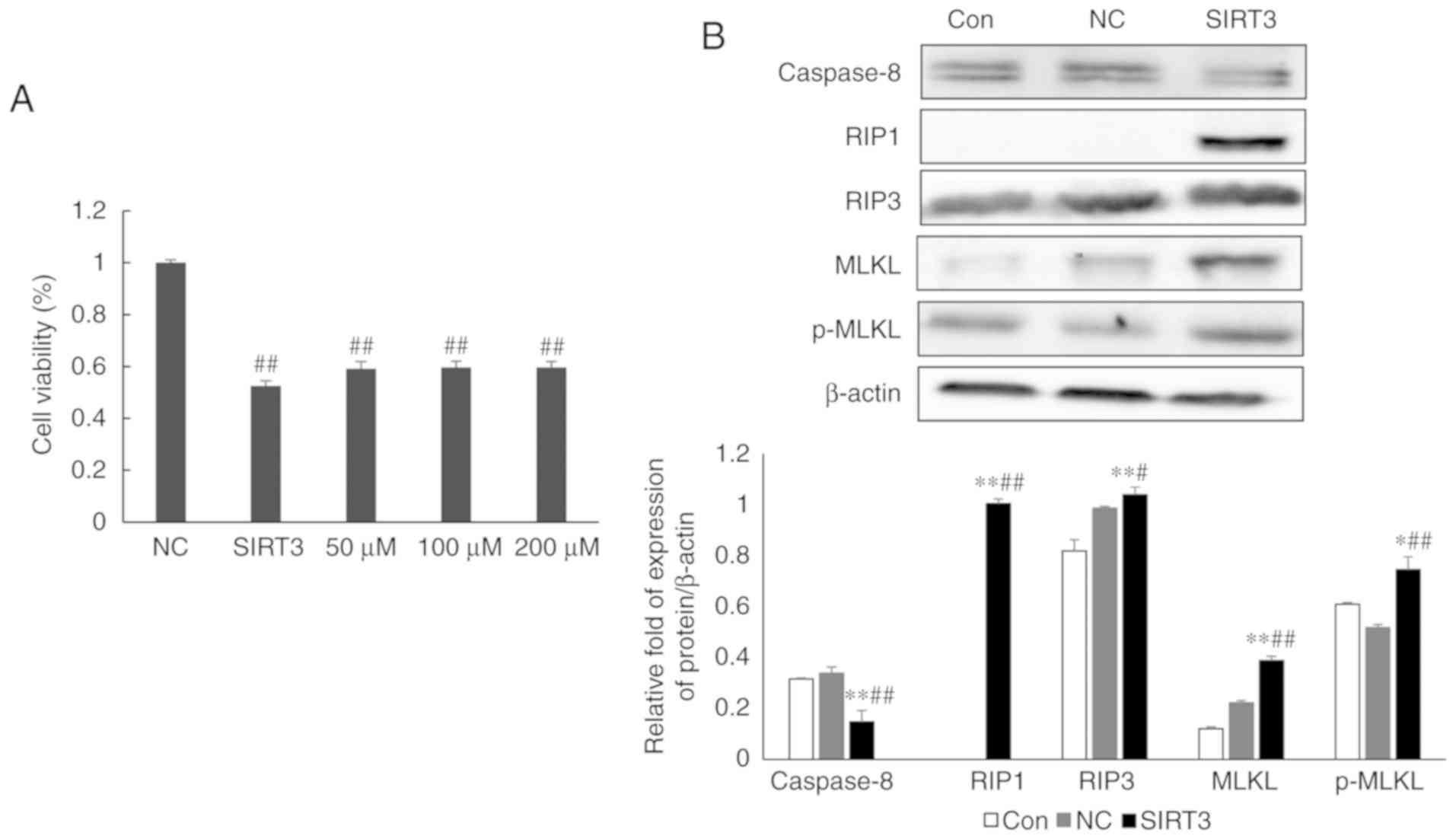
SIRT3 overexpression promotes necrotic
apoptosis in NCI-H446 small-cell lung cancer cells
Nec-1, an inhibitor of necrotic apoptosis, was used
to determine whether SIRT3-dependent cell death involved the
necrotic pathway. Twenty-four hours post-transfection with control
or SIRT3 expression plasmids, NCI-H446 cells were treated with
different concentrations of Nec-1, and cell survival was
subsequently assessed by MTT assay. This experiment revealed that
Nec-1 treatment could partially reverse the effects of
SIRT3-overexpression on cell death (Fig. 3A; P<0.01). However, the effects
of Nec-1 treatment did not increase with increasing drug
concentration. Western blot analysis of the expression of RIP1,
RIP3, MLKL and p-MLKL, four important regulators of programmed
necroptosis, revealed that their expression was significantly
increased in SIRT3-overexpressing cells, when compared with
controls (Fig. 3B; P<0.01).
Collectively, these data indicated that SIRT3 overexpression
promoted necroptosis in NCI-H446 cells.
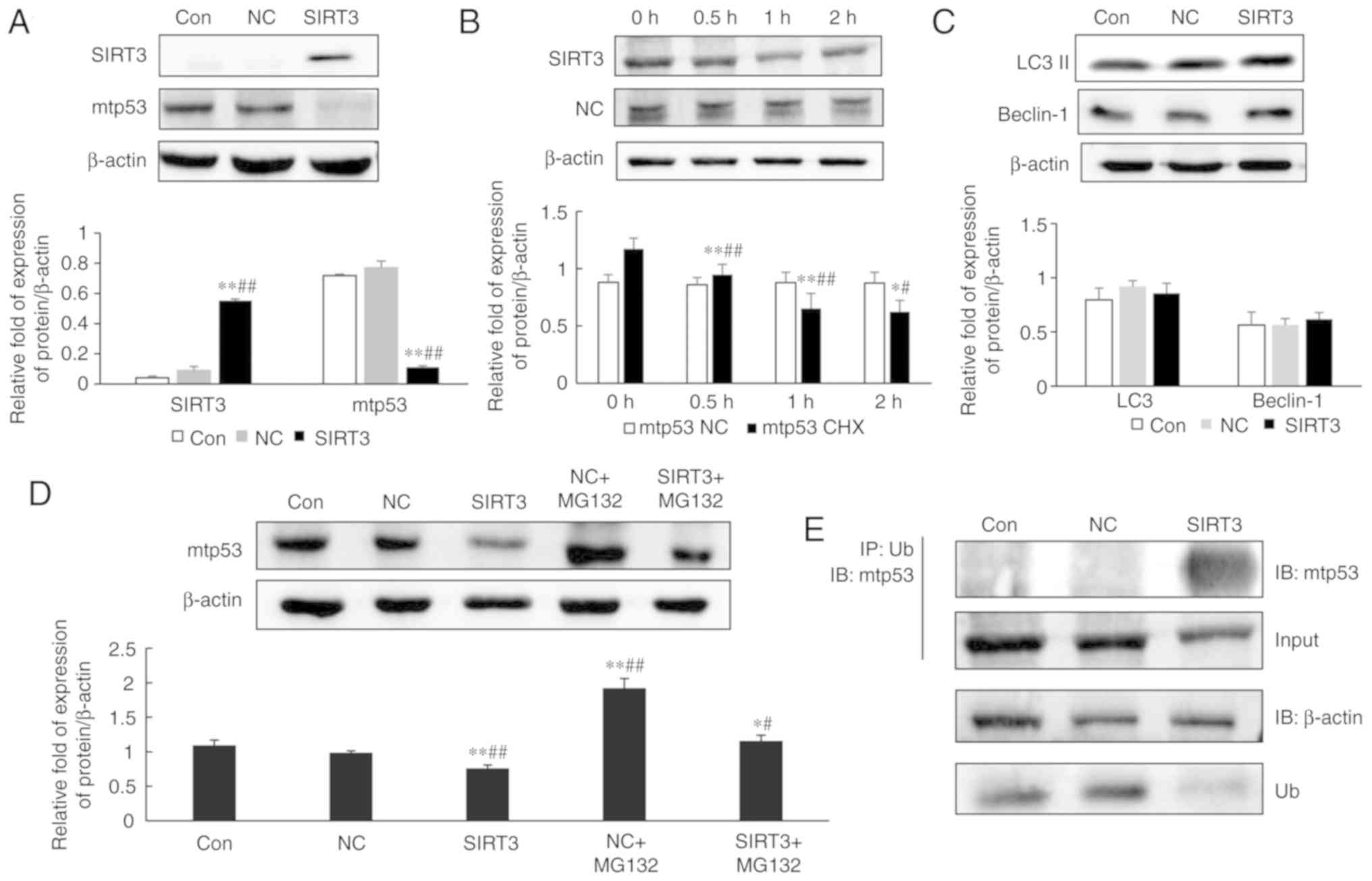
SIRT3-dependent changes in mutant p53
expression and the underlying mechanism
Next, mutant p53 expression in SIRT3-overexpressing
NCI-H446 cells was evaluated by western blotting. As revealed in
Fig. 4A, mutant p53 protein levels
were significantly decreased in cells overexpressing SIRT3, when
compared with controls (P<0.05). SIRT3 overexpression resulted
in a reduction in the half-life of mutant p53 protein (Fig. 4B). Therefore, the mechanism
underlying this SIRT3-dependent decrease in mutant p53 expression
was investigated. Protein degradation in eukaryotic cells is
primarily controlled by the lysosomal and ubiquitin proteasome
pathways. To determine whether the lysosomal pathway was implicated
in mutant p53 degradation, the expression of the
autophagy-associated proteins LC3 II and Beclin-1 were examined in
SIRT3-overexpressing cells and it was revealed that their levels
remained unchanged, when compared with the control NC group
(Fig. 4C). Therefore, it was
hypothesized that mutant p53 was degraded by the ubiquitin
proteasome pathway.
MG132 is a potent inhibitor of protein degradation
mediated by the ubiquitin proteasome pathway (13). SIRT3-overexpressing NCI-H446 cells
were treated with MG132 for 24 h, and then protein lysates were
prepared for analysis of mutant p53 expression. Western blot
analysis revealed that mutant p53 protein expression in the
NC+MG132 group was significantly increased, when compared with the
NC group (Fig. 4D; P<0.05). An
interaction between mutant p53 and SIRT3 was demonstrated by
immunoprecipitation assay (Fig.
4E). Immunoprecipitation assays also revealed that mutant p53
in SIRT3-overexpressing cells was more highly ubiquitinated than in
control cells. These data indicated that overexpression of SIRT3
promoted degradation of mutant p53 by the ubiquitin proteasome
pathway.
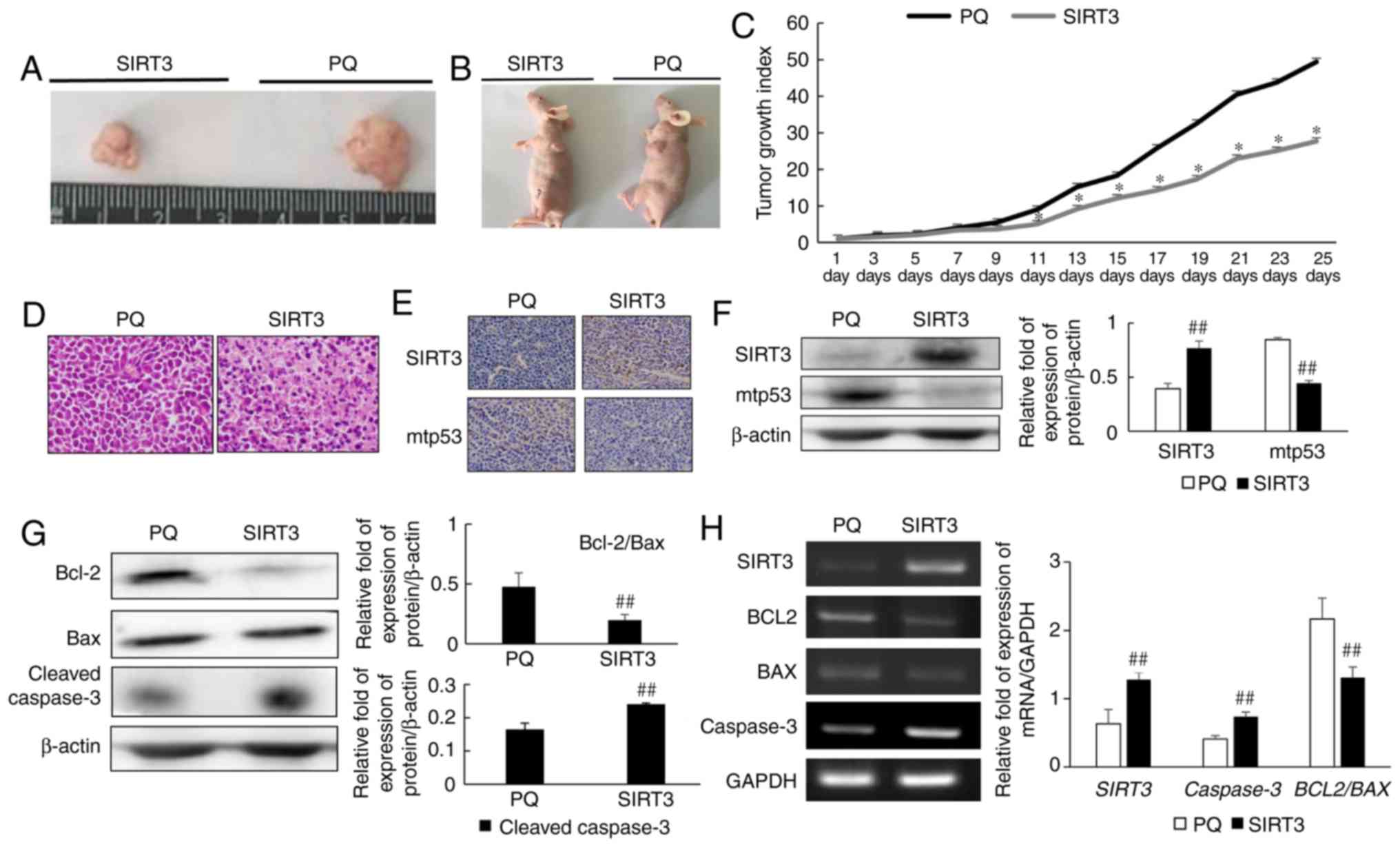
SIRT3 inhibits NCI-H446 tumor growth
in vivo
To further study the effects of SIRT3 on NCI-H446
cells, an in vivo experiment was conducted. NCI-H446 cells
were injected subcutaneously into mice, and tumors were then
allowed to establish and grow until approximately 5 mm in diameter.
After 25 days, mice and tumors were photographed and differences in
tumor size were evaluated. SIRT3 overexpression was revealed to
inhibit tumor growth in vivo (Fig. 5A and B). The tumor growth index also
confirmed that SIRT3 inhibited tumor growth (Fig. 5C). H&E staining revealed
increased apoptosis in the tumor tissues of the SIRT3 group
(Fig. 5D). Immunohistochemical
analysis demonstrated that SIRT3 protein expression was enhanced
and that mutant p53 expression was decreased in the SIRT3 group,
when compared with the PQ control group (Fig. 5E). Western blot analysis of tumor
tissue extracts confirmed that SIRT3 treatment resulted in reduced
mutant p53 expression (Fig. 5F), a
finding that was consistent with our in vitro data, and
which indicated that SIRT3 inhibited the development of small-cell
lung cancer. Analysis of the expression of apoptosis-related
proteins by western blotting revealed that the ratio of Bcl-2 to
Bax protein in SIRT3 tumors was reduced, when compared with PQ
control tumors (Fig. 5G). Cleaved
caspase-3 expression was also upregulated in SIRT3 tumors. In
addition, RT-PCR was used to evaluate changes in the expression of
apoptosis-related genes (Fig. 5H).
In agreement with the protein expression data, a decrease in
BCL2 to BAX gene expression, and increased
CASP3 gene expression was observed in SIRT3 tumors, when
compared with controls.
Discussion
Researching the role of SIRT3 in tumors is becoming
increasingly popular. However, Ashraf et al reported that
increased SIRT3 transcription was associated with node-positive
breast cancer (14). The function
of SIRT3 may therefore differ according to cell and tumor type.
Consequently, SIRT3 may function both as an oncogene and a tumor
suppressor gene.
In a study using A549 lung adenocarcinoma cells,
SIRT3 was revealed to exhibit tumor suppressive properties in the
regulation of cell proliferation. High SIRT3 expression was
revealed to induce apoptosis through a mechanism involving
increased Bax and Bad-dependent sequestration of Bc1-2 and Bcl-XL,
respectively, thereby promoting AIF translocation to the nucleus.
High expression of SIRT3 in A549 cells also resulted in an increase
in p53 and p21 protein levels, and a decrease in the intracellular
ROS level (15). We previously
revealed that SIRT3 may also function as a tumor suppressor in
small-cell lung cancer, a complex and highly malignant disease that
lacks effective treatment options. It is therefore particularly
important to explore the mechanism of the tumor suppressive
function of SIRT3 in small-cell lung cancer, since this may
facilitate the development of new therapeutic strategies for the
treatment of this disease.
In the present study, it was demonstrated that SIRT3
can inhibit growth and promote apoptosis and necroptosis in
NCI-H446 small-cell lung cancer cells using MTT and flow cytometric
assays. In apoptosis mediated by the mitochondrial pathway, a
change in the permeability of the outer mitochondrial membrane
causes the release of cytochrome c. Cytoplasmic cytochrome
c activates Apaf-1, which further combines with procaspase-9
to form an apoptosis complex that activates procaspase-9. Once
activated, caspase-9 can activate other caspases, such as
caspase-3. Mitochondrial membrane permeabilization is controlled by
the Bcl-2 protein family. One such member is Bax, an
apoptosis-stimulating factor which is activated at the outer
mitochondrial membrane to promote membrane channel formation and
release of cytochrome c. Bcl-2 can inhibit the activation of
caspase-9 by inhibiting the release of cytochrome c, and
therefore functions as an inhibitor of apoptosis. In addition, MCL1
can inhibit Bax and thus antagonize apoptosis signaling, while Bid
can promote mitochondrial membrane permeability, and thus is an
activator of apoptosis. The present flow cytometric data revealed
that SIRT3 overexpression induced cell apoptosis and necroptosis.
Necroptosis is now considered as a regulated cellular process, an
additional method of programmed cell death. The inhibition of
caspase-8 can activate receptor interaction protein kinase 1 (RIP1)
and 3 (RIP3), and mixed lineage kinase domain-like (MLKL) which is
regulated by RIP3 to be phosphorylated (p-MLKL). The activated MLKL
is transferred from the cytoplasm to the cell membrane and the
integrity of the cell membrane is damaged, resulting in cell
necroptosis. (16,17). In order to determine whether
necroptosis also contributed to cellular death after SIRT3
overexpression, cells were treated with the necroptosis inhibitor,
Nec-1, and cell survival was evaluated by MTT assay. MTT assays
revealed that cell survival was increased in the presence of Nec-1,
while western blotting assays revealed downregulation of caspase-8
and upregulation of RIP1, RIP3, MLKL and p-MLKL. This inhibition of
caspase-8 and induction of RIP1, RIP3, MLKL and p-MLKL protein
expression indicated that necrotic apoptosis was triggered after
SIRT3 expression. In an in vivo model of small-cell lung
cancer it was also revealed that SIRT3 expression could inhibit
tumor growth through induction of tumor cell apoptosis. The present
in vitro data indicated that a reduction in the ratio of
Bcl-2 to Bax and an associated activation of cleaved caspase-3
provided the mechanism driving apoptosis in SIRT3-overexpressing
cells.
Subsequently, the expression of mutant p53 after
overexpression of SIRT3 in NCI-H446 cells was detected.
The results revealed that overexpression of SIRT3
could reduce the expression of mutant p53. In addition, it has been
reported that the decrease of the level of mutant p53 can lead to
an increase of apoptosis (18–20).
The present study revealed for the first time that SIRT3 may
promote cell apoptosis by leading to decreased levels of mutant
p53. Studies indicated that SIRT3 can modify a large number of
cellular proteins through it deacetylase activity. There are
studies that have reported that SIRT3 can use its function of
acetylation to modify many cellular proteins. Xiong et al
revealed that, following SIRT3 overexpression in A549 cells, SIRT3
interacted with p53 and that these proteins co-localized in cells.
Furthermore, SIRT3 was revealed to regulate the p53 degradation
pathway by deacetylation (21).
However, the mechanism by which SIRT3 regulates mutant p53 remains
unknown.
TRRAP is a large multidomain protein that is a
component of many histone acetyltransferase (HAT) multiprotein
complexes. Silencing of TRRAP expression has been revealed to
reduce p53 accumulation in lymphoma and colon cancer models.
Conversely, overexpression of TRRAP has been revealed to increase
mutant p53 levels, while the inhibition of histone deacetylase
(HDAC) has been revealed to decrease p53 levels (22). The present study therefore provides
a link between deacetylation and mutant p53 stability.
SIRT3-mediated FOXO3 deacetylation has also been
revealed to reduce FOXO3 phosphorylation, ubiquitination and
degradation, thereby increasing protein stability (23). This suggests that SIRT3 activity may
also influence the ubiquitination status of proteins.
Given these previous studies, it was hypothesized
that the downregulation of mutant p53 observed in
SIRT3-overexpressing NCI-H446 cells was a consequence of the
SIRT3-dependent deacetylation of mutant p53, which may impact the
mutant p53 ubiquitination pathway. In NCI-H446 cells, both the
expression and half-life of the mutant p53 protein were reduced
following overexpression of SIRT3, a finding that indicated
increased degradation of the protein. Protein degradation in
eukaryotes is primarily mediated by the lysosomal and ubiquitin
proteasome pathways. LC3 was the first protein to be identified as
a marker of autophagy. Autophagosome formation requires the
conversion of LC3 I into LC3 II (24) and, consequently, LC3 II levels
increase during autophagy. Another autophagy-related protein,
Beclin-1, which is the mammalian homolog of yeast Apg6/Vps30, forms
a complex with the Class III PI3K Vps34, and together these
proteins play an important role in activating autophagy (25). In the present study, it was revealed
that LC3 II and Beclin-1 expression levels remained unchanged in
SIRT3-overexpressing cells, indicating that the autophagy pathway
was unlikely to be involved in the degradation of mutant p53.
However, treatment of cells with the proteasome inhibitor MG132
resulted in a recovery of mutant p53 levels, while
immunoprecipitation assays revealed that mutant p53 was
ubiquitinated. Collectively, these results demonstrated that mutant
p53 degradation was mediated by the ubiquitin proteasome
pathway.
The present study revealed an inhibitory role for
SIRT3 in small-cell lung cancer, whereby SIRT3 promoted cell
apoptosis and necroptosis via a mechanism that may require
degradation of mutant p53 by the ubiquitin proteasome degradation
pathway. It is therefore possible that SIRT3-dependent loss of
mutant p53 oncogene function provides a mechanism for inhibiting
the development of small-cell lung cancer.
Acknowledgements
We would like to thank professor Xu Deqi of the Food
and Drug Administration (FDA) for the donation of attenuated
Salmonella typhimurium phoP/phoQ. We would also like to
thank Dr James Monypenny, for editing the English text of a draft
of this manuscript.
Funding
The present study was supported by the Research Fund
of the National Natural Science Foundation of China (grant nos.
81501982, 81572927, 81772794, 81472419 and 81672948), the Jilin
Province Research Foundation for the Development of Science and
Technology Project (grant nos. 20150414025GH, 20170623021TC and
20160414005GH) and the Jilin University Bethune Plan B Projects
(grant no. 2015222), the Education Department of Jilin Province
(no. JJKH20170834KJ), the Department of Science and Technology of
Jilin Province, China (grant no. 20160520151JH) and the Department
of Education of Jilin Province, China (grant no. 2016456).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
XT, YLi, LS and YLiu participated in the research
design. XT, LL, RG, PZ and YiZ conducted the experiments and
contributed to the data acquisition and analysis. YoZ, JZ and JS
performed the data analysis. XT and YLi contributed to the writing
of the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The animal study was approved by the Animal Ethics
Review Committee of Basic Medical Sciences, Jilin University, in
accordance with the regulations of the Institutional Committee for
the Care and Use of Laboratory Animals of the Experimental Animal
Center of Jilin University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Aridgides PD, Movsas B and Bogart JA:
Thoracic radiotherapy for limited stage small cell lung carcinoma.
Curr Probl Cancer. 36:88–105. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
William WN Jr and Glisson BS: Novel
strategies for the treatment of small-cell lung carcinoma. Nat Rev
Clin Oncol. 8:611–619. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nickolich M, Babakoohi S, Fu P and Dowlati
A: Clinical trial design in small cell lung cancer: Surrogate end
points and statistical evolution. Clin Lung Cancer. 15:207–212.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Byers LA and Rudin CM: Small cell lung
cancer: Where do we go from here? Cancer. 121:664–672. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xiong S, Tu H, Kollareddy M, Pant V, Li Q,
Zhang Y, Jackson JG, Suh YA, Elizondo-Fraire AC, Yang P, et al:
Pla2g16 phospholipase mediates gain-of-function activities of
mutant p53. Proc Natl Acad Sci USA. 111:11145–11150. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nguyen GT, Schaefer S, Gertz M, Weyand M
and Steegborn C: Structures of human sirtuin 3 complexes with
ADP-ribose and with carba-NAD+ and SRT1720: Binding
details and inhibition mechanism. Acta Crystallogr D Biol
Crystallogr. 69:1423–1432. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guarente L: Introduction: Sirtuins in
aging and diseases. Methods Mol Biol. 1077:3–10. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang YY and Zhou LM: Sirt3 inhibits
hepatocellular carcinoma cell growth through reducing Mdm2-mediated
p53 degradation. Biochem Biophys Res Commun. 423:26–31. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Minamoto T, Buschmann T, Habelhah H,
Matusevich E, Tahara H, Boerresen-Dale AL, Harris C, Sidransky D
and Ronai Z: Distinct pattern of p53 phosphorylation in human
tumors. Oncogene. 20:3341–3347. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Warnock LJ, Raines SA and Milner J: Aurora
A mediates cross-talk between N- and C-terminal post-translational
modifications of p53. Cancer Biol Ther. 12:1059–1068. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rodriguez OC, Choudhury S, Kolukula V,
Vietsch EE, Catania J, Preet A, Reynoso K, Bargonetti J, Wellstein
A, Albanese C and Avantaggiati M: Dietary downregulation of mutant
p53 levels via glucose restriction: Mechanisms and implications for
tumor therapy. Cell Cycle. 11:4436–4446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pawelek JM, Low KB and Bermudes D:
Bacteria as tumour- targeting vectors. Lancet Oncol. 4:548–556.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
O'Leary EM and Igdoura SA: The therapeutic
potential of pharmacological chaperones and proteosomal inhibitors,
Celastrol and MG132 in the treatment of sialidosis. Mol Genet
Metab. 107:173–185. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ashraf N, Zino S, Macintyre A, Kingsmore
D, Payne AP, George WD and Shiels PG: Altered sirtuin expression is
associated with node-positive breast cancer. Br J Cancer.
95:1056–1061. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xiao K, Jiang J, Wang W, Cao S, Zhu L,
Zeng H, Ouyang R, Zhou R and Chen P: Sirt3 is a tumor suppressor in
lung adenocarcinoma cells. Oncol Rep. 30:1323–1328. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oberst A: Death in the fast lane: What's
next for necroptosis? FEBS J. 283:2616–2625. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pasparakis M and Vandenabeele P:
Necroptosis and its role in inflammation. Nature. 517:311–320.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Braicu C, Pileczki V, Irimie A and
Berindan-Neagoe I: p53siRNA therapy reduces cell proliferation,
migration and induces apoptosis in triple negative breast cancer
cells. Mol Cell Biochem. 381:61–68. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lim LY, Vidnovic N, Ellisen LW and Leong
CO: Mutant p53 mediates survival of breast cancer cells. Br J
Cancer. 101:1606–1612. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ali A, Wang Z, Fu J, Ji L, Liu J, Li L,
Wang H, Chen J, Caulin C, Myers JN, et al: Differential regulation
of the REGү-proteasome pathway by p53/TGF-β signalling and mutant
p53 in cancer cells. Nat Commun. 4:26672013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xiong Y, Wang L, Wang S, Wang M, Zhao J,
Zhang Z, Li X, Jia L and Han Y: SIRT3 deacetylates and promotes
degradation of P53 in PTEN-defective non-small cell lung cancer. J
Cancer Res Clin Oncol. 144:189–198. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jethwa A, Słabicki M, Hüllein J, Jentzsch
M, Dalal V, Rabe S, Wagner L, Walther T, Klapper W; MMML Network
Project, ; et al: TRRAP is essential for regulating the
accumulation of mutant and wild-type p53 in lymphoma. Blood.
131:2789–2802. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tseng AH, Wu LH, Shieh SS and Wang DL:
SIRT3 interactions with FOXO3 acetylation, phosphorylation and
ubiquitinylation mediate endothelial cell responses to hypoxia.
Biochem J. 464:157–168. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hemelaar J, Lelyveld VS, Kessler BM and
Ploegh HL: A single protease, Apg4B, is specific for the
autophagy-related ubiquitin-like proteins GATE-16, MAP1-LC3,
GABARAP, and Apg8L. J Biol Chem. 278:51841–51850. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Boutouja F, Brinkmeier R, Mastalski T, El
Magraoui F and Platta HW: Regulation of the tumor-suppressor BECLIN
1 by distinct ubiquitination cascades. Int J Mol Sci. 18:E25412017.
View Article : Google Scholar : PubMed/NCBI
|