Introduction
Burkitt's lymphoma is a highly invasive cancer of
the lymphatic system, particularly B lymphocytes found in the
germinal center (1). In recent
years, with the improvement of the chemotherapy regimen and the
application of rituximab, the prognosis of children with Burkitt's
lymphoma has significantly improved. The chance of successful
treatment has been significantly increased by a short-course and
high-dose combination chemotherapy (2,3).
However, in adults (especially in patients aged 40 years) and
patients with advanced disease, invasion to the central nervous
system is often present. Chemotherapy presents with a number of
side effects, and the prognosis for patients undergoing this
treatment is poor. Therefore, the identification of a low-toxic and
effective treatment method for Burkitt's lymphoma is urgently
required (3,4). Targeted therapy is currently
considered to be the best available treatment option. A previous
study observed the abnormal activation of the phosphoinositide
3-kinase/protein kinase B (PI3K/AKT) pathway in Burkitt's lymphoma
cells, and demonstrated that inhibition of PI3K/AKT pathway
activation can inhibit the growth of Burkitt's lymphoma cells
(5). Additionally, PTEN has been
indicated to negatively regulate the PI3K/AKT signaling pathway. As
a negative regulatory factor, PTEN exhibits mutations, deletions,
methylation and abnormal activity in solid tumors (6–8). PTEN
is also abnormally expressed in B-cell lymphomas, including diffuse
large B-cell lymphoma (9),
resulting in the aberrant activation of the PI3K/AKT pathway. In
NK/T lymphoma cells, the expression level of PTEN is also decreased
to activate the PI3K/AKT signaling pathway (10). Therefore, it is hypothesized that
abnormal expression of PTEN may serve an essential role in the
pathogenesis of Burkitt's lymphoma. In order to rule out the effect
of lymphoma infection of Epstein-Barr virus (EBV) on the
experimental results, EBV-positive and EBV-negative cells were
used. In the present study, the effect of PTEN on the
proliferation, apoptosis, cell cycle distribution and migration
ability of Burkitt lymphoma cells (including CA46 and RAJI; CA46 is
EBV negative; RAJI is EBV positive) was assessed. The mechanism of
PTEN in Burkitt's lymphoma was investigated by determining PTEN
downstream-related proteins.
Materials and methods
Cell lines and plasmids
RAJI cells were purchased from Jiangsu Kaiji
Bio-Technology Co., Ltd. and the CA46 cell strain was purchased
from Guangzhou Jiniou Co., Ltd. The lentiviral packaging plasmids
PLV-CMV-FLAG, pcDNA-VSVG, pCMV-Δ8/9 and the silencing vector
plasmid PLKO.1-puro were purchased from Sigma-Aldrich; Merck KGaA.
The overexpression vector plasmid GV358-PTEN and the vector plasmid
GV358 were purchased from Shanghai Genechem Co., Ltd.
Lentiviral packaging and stable strain
construction
Packaging of the overexpression
lentiviral
Recombinant overexpression lentivirus (Lv-PTEN) and
the negative control (Lv-NC) were constructed by a three-plasmid
coinfection method. In regards to the system: 293FT cells were
cotransfected with GV358/GV358-PTEN (15 µg), pCMV-Δ8/9 (15 µg) and
pcDNA-VSVG (7.5 µg), and the virus suspension was collected after
48 h.
Packaging of the silencing
lentiviral
First, the PTEN shRNA target sequence was designed.
The PTEN-shRNA double-stranded hairpin structure was designed
according to the target sequence (the primer was synthesized by
Shanghai Biosynthesis) to synthesize two shPTEN oligonucleotide
sequences (shPTEN#1 and shPTEN#2). The recombinant nucleotide
sequence was ligated to the PLKO.1-puro vector according to the
manufacturer's protocol, and the PTEN silencing vector plasmid was
successfully constructed. The silencing lentivirus (shPTEN#1 and
shPTEN#2) was also packaged using the three-plasmid cotransfection
method. In regards to the system: 293FT cells were cotransfected
with PLKO.1/PLKO.1-shPTEN#1/PLKO.1-shPTEN#2 (15 µg), pCMV-Δ8/9 (15
µg) and pcDNA-VSVG (7.5 µg), and then the virus suspension was
collected after 48 h.
Finally, CA46 and RAJI cells were infected with the
successfully packaged virus, and a stable cell line was selected by
adding a final concentration of 1 µl/ml puromycin.
Cell proliferation
Cell proliferation was analyzed by the CCK-8 assay.
The cell density was adjusted to 8×104/ml and inoculated
into 96-well plates; 100 µl of cell suspension was added to each
well, and 5 replicate wells were set for each cell group, which
included a blank group. The control group was tested at 0, 1, 2, 3
and 4 days after inoculation. CCK-8 (10 µl) was added to each well
of a 96-well plate, and incubation was carried out for 2.5 h in a
37°C incubator. The absorbance was determined using a microplate
reader. The absorbance of each well was determined and a growth
curve for each group was plotted.
Apoptosis
Hoechst 33342 and propidium iodide (PI) double
staining were used to verify apoptosis. A total of ~106
cells were collected from each group. The cell pellet was
resuspended in 0.8 ml cell staining buffer; and 5 µl Hoechst
staining solution and 5 µl PI staining solution were added. Red
fluorescence and blue fluorescence were detected using fluorescence
microscopy. Multiple fields of view were randomly selected for each
group of cells, and red and blue fluorescence images of the same
field of view were synthesized and analyzed using PS software
(Adobe Photoshop CS6; Adobe Systems, Inc).
Cell cycle distribution
The cell density was adjusted to ~106
cells/ml. The cells were mixed with 1 ml PBS and 3 ml absolute
ethanol to avoid cell clumping and fixed at −20°C overnight. The
fixed cells were collected and suspended in 1 ml PBS buffer three
times; and the supernatant was retained subsequently. The cells
were incubated for 30 min in 1 ml PBS with 4 µl RNase (10 µg/µl)
and 30 µl PI stain (1 mg/ml) at room temperature with protection
from light. Cells were strained in 200-µm mesh sieves into a
special flow cytometry centrifuge tube. The DNA content of each
group of cells was determined using flow cytometry. FlowJo™
software (FlowJo 7.6.1; BD Biosciences) was used to calculate and
analyze cell cycle distribution.
Cell migration ability
Cells were resuspended in RPMI-1640 medium at a cell
density of 106 cells/ml. RPMI-1640 medium with 10% FBS
(600 µl) was added to a 24-well plate and placed in a Transwell
chamber with 200 µl of the cell suspension. For each group of
cells, a total of three duplicate wells were incubated in 5%
CO2 at 37°C for 18 h. Once the Transwell chamber was
removed, each well was centrifuged at 100 × g and the supernatant
was discarded. The remaining 100 µl of the liquid was pipetted,
mixed, and inoculated into a 96-well plate. CCK-8 solution (10 µl)
was added to each well, and the plate was subsequently incubated
for 2 h. The absorbance of each well was measured at a wavelength
of 450 nm using a microplate reader.
Cell invasion
The cell density was adjusted to 106
cells/ml in the upper chamber of a Transwell plate that was coated
with Matrigel. The culture method was the same as that
aforementioned in the migration experiment. The lower chamber was
incubated with 4% paraformaldehyde and stained with
4′,6-diamidino-2-phenylindole (DAPI). The cells were observed under
fluorescence microscopy (magnification, ×200). Three fields of view
were randomly selected for imaging, and the number of cells was
calculated for each group to perform statistical analysis.
Western blotting
Protein lysates were separated by SDS-PAGE,
transferred to PVDF membranes and then incubated with primary
antibodies (GAPDH, PTEN, AKT, pAKT, Bad, Bax, P53, P21, CDK4, CDK6,
cyclin D3, cyclin H, E-cadherin, N-cadherin, β-catenin, TCF-8,
vimentin, Slug and Snail). The membranes were then incubated with
HRP-labeled secondary antibodies. Finally, the hybridization signal
was detected using ECL, exposed and photographed with a gel imager.
The protein extraction buffer was RIPA Lysis Buffer, which was
purchased from Shanghai Biyuntian Institute of Biotechnology. The
BCA kit was used for protein determination method, and the mass of
protein loaded per lane was 15 µg. The percentage of separated gel
was 15%, and the percentage of concentrated gel was 5%. Blocking
reagent was 5% skim milk powder PBST solution at room temperature
shock closure 2 h. The primary antibodies used were rabbit
anti-human antibodies. The secondary antibody was goat anti-rabbit
IgG(H+L) HRP. All antibodies were diluted in PBST solution. The
primary antibody was incubated for 12 h at a temperature of 4°C,
and the secondary antibody was incubated at room temperature for 2
h. All antibodies and kits were purchased from Cell Signaling
Technology (CST). The catalog numbers of anti-GAPDH, anti-PTEN,
anti-AKT, anti-pAKT, anti-Bad, anti-Bax, anti-P53, anti-P21,
anti-CDK4 and anti-CDK6 were #5157, #9188, #4685, #4060, #9268,
#5023, #2527, #2947, #12790, #13331, respectively. Anti-cyclin D3,
and anti- H were included in the Cyclin Antibody Sampler Kit
(#9869). Anti-E-cadherin, anti-N-cadherin, anti-β-catenin,
anti-TCF-8, anti-vimentin, anti-Slug and anti-Snail were from the
Epithelial-Mesenchymal Transition (EMT) Antibody Sampler Kit
(#9782).
Statistical analysis
The experimental results in the present study are
represented as images and graphs. The data are expressed as the
mean ± standard deviation (trials for each experiment were three).
Data analyses were performed using SPSS 22.0 (IBM Corp.). The
statistical analysis methods used included independent sample's
t-test, rank sum test and one-way analysis of variance (ANOVA).
P<0.01 was considered to indicate a statistically significant
result.
Results
Overexpression of PTEN inhibits the
growth and proliferation of CA46 and RAJI cells and induces
apoptosis
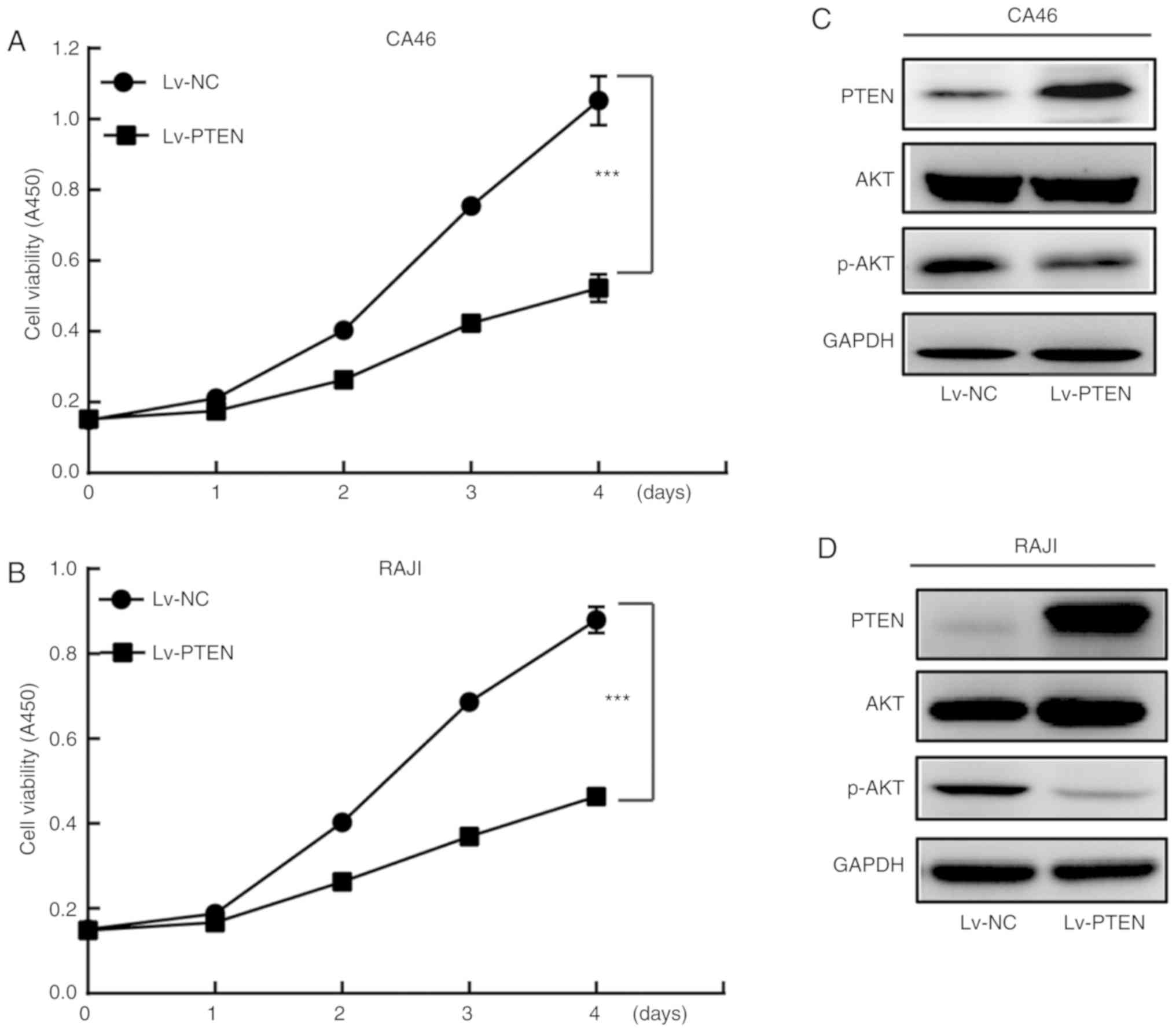
The growth rate of the Lv-PTEN cell group was
significantly lower compared with that of the Lv-NC cell group
(P<0.01) (Fig. 1A and B). In
addition, the expression level of p-AKT (Ser473) in the Lv-PTEN
cell group was lower when compared with the Lv-NC cell group in
both the CA46 and RAJI cell lines (Fig.
1C and D), suggesting that PTEN inhibited the proliferation of
Burkitt's lymphoma cells by inhibiting AKT phosphorylation.
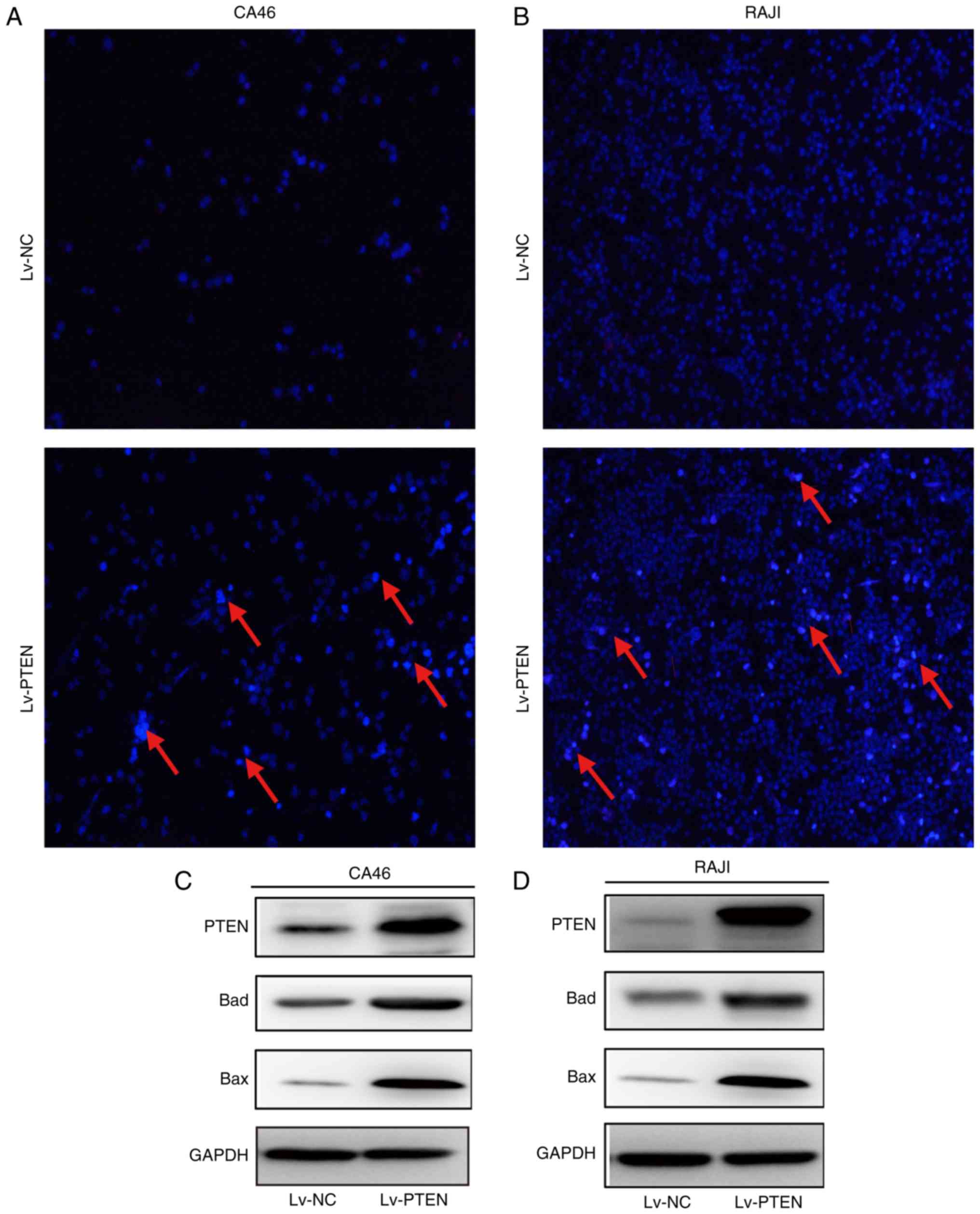
Fig. 2A and B demonstrated that the
Lv-PTEN cell group exhibited weak red and strong blue fluorescence,
which indicates the presence of apoptotic cells. No apoptotic cells
were observed in the Lv-NC cell group. The expression levels of Bad
and Bax in the PTEN cell group were higher than levels in the Lv-NC
cell group in the CA46 and RAJI cell lines (Fig. 2C and D), suggesting that PTEN may
promote the apoptosis of Burkitt's lymphoma cells by upregulating
the expression of proapoptotic proteins Bad and Bax.
Overexpression of PTEN blocks the cell
cycle progression of the CA46 and RAJI cells
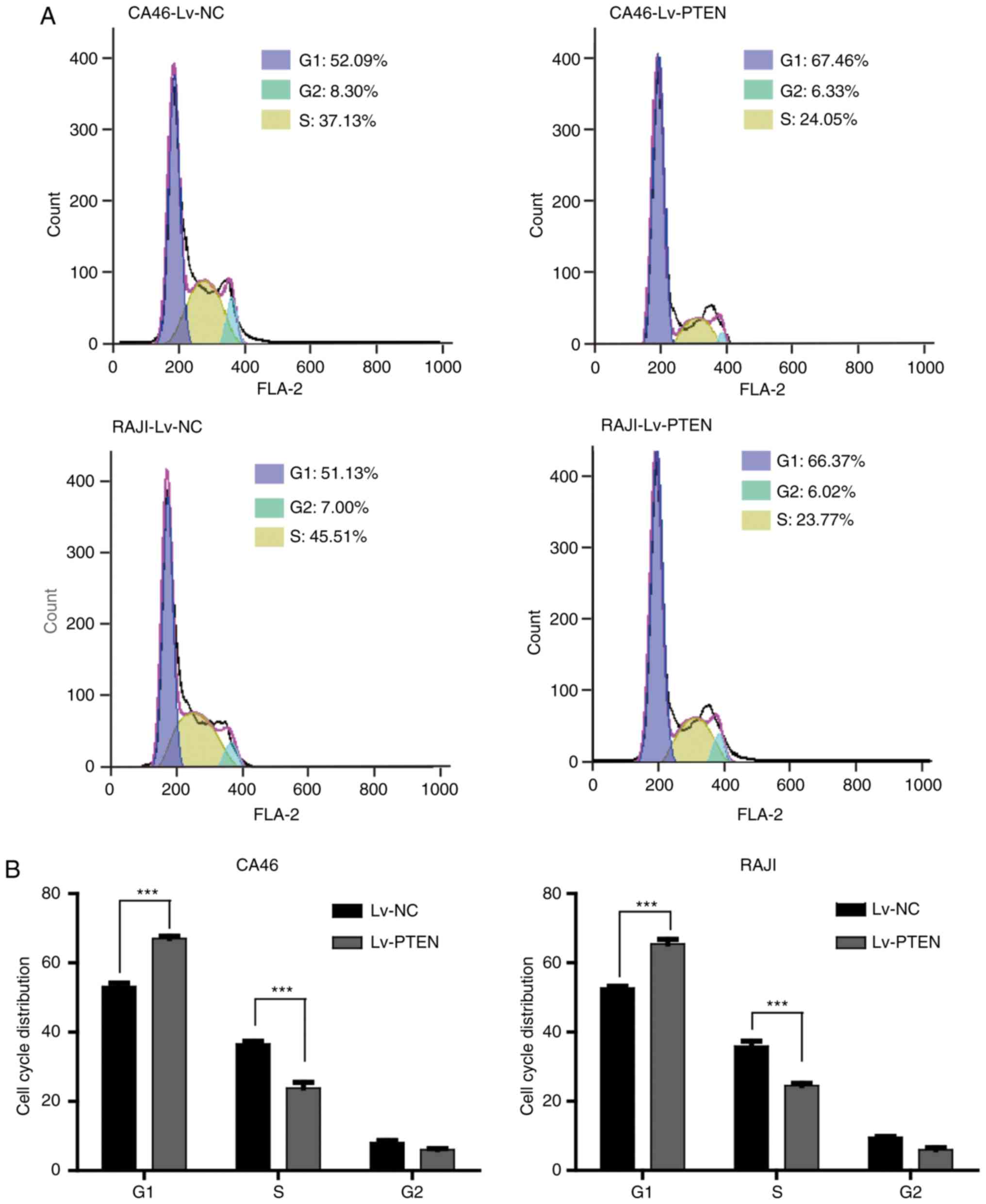
As shown in Fig. 3,
the percentage (%) of cells in the cell cycle distribution of the
CA46-Lv-NC cell group are as follows: 53.19±0.99 in G1, 36.56±0.84
in S phase, and 8.09±0.61 in G2 phase. The percentage (%) of cells
in the cell cycle distribution of the CA46-Lv-PTEN cell group are
as follows: 67.06±0.63 in G1, 23.84±1.65 in S phase, and 6.05±0.32
in G2 phase. The percentage (%) of cells in the cell cycle
distribution of the RAJI-Lv-NC cell group was as follows:
52.69±0.55 in G1, 35.89±1.47 in S phase, and 9.53±0.32 in G2 phase.
The percentage (%) of cells in the cell cycle distribution of the
RAJI-Lv-PTEN cell group was as follows: 65.45±1.31 in G1,
25.43±0.66 in S phase, and 5.99±0.58 in G2 phase. From the results
of the data analysis, the proportion of G1 phase cells in the
Lv-PTEN cell group was significantly higher compared with the Lv-NC
cell group (P<0.01), while the proportion of S phase cells in
the Lv-PTEN cell group was significantly lower compared with that
in the Lv-NC cell group (P<0.01). As presented in Fig. 3C and D, the expression levels of P53
and P21 in the Lv-PTEN cell groups were higher compared with the
Lv-NC cell groups. The expression levels of CDK4, CDK6, cyclin D3
and cyclin H in the Lv-PTEN cell groups were lower when compared
with these levels in the Lv-NC cells, suggesting that PTEN
overexpression inhibited the Burkitt's lymphoma cell cycle by
promoting the expression of P53 and P21, and inhibiting the
expression of CDK4, CDK6, cyclin D3 and cyclin H. In conclusion,
the cell cycle was arrested at the G1 phase.
Overexpression of PTEN inhibits the
migration and invasion of CA46 and RAJI cells
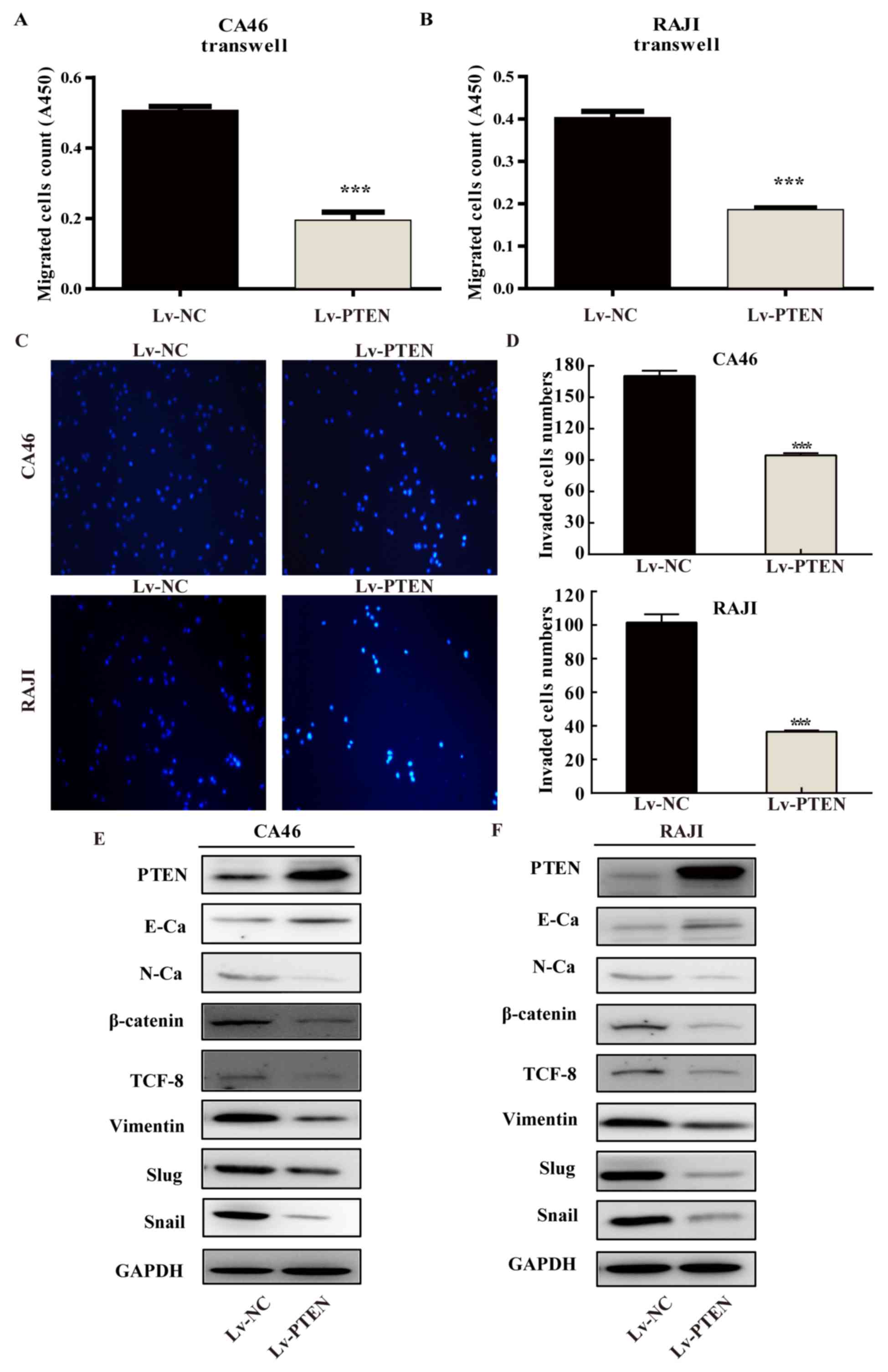
As shown in Fig. 4A and
B, the optical density (OD) values of the cells in each group
were 0.506±0.012 in the CA46-Lv-NC cell group, 0.194±0.024 in the
CA46-Lv-PTEN cell group, and 0.403±0.015 in the RAJI-Lv-NC cell
group. The OD value in the RAJI-Lv-PTEN cell group was 0.186±0.004.
The numbers of migrated cells in the Lv-PTEN cell groups were
significantly lower compared with these numbers in the Lv-NC cell
groups (P<0.01). The results of the cell invasion into Matrigel
for each group are presented in Fig. 4C
and D. The number of cells per field in the CA46-Lv-NC,
CA46-Lv-PTEN, RAJI-Lv-NC and RAJI-Lv-PTEN was 165±10.15, 93±3.51,
103.67±5.13 and 34.67±3.22, respectively. The number of invasive
cells in the Lv-PTEN cell group was significantly lower compared
with that in the Lv-NC group in both cell lines (P<0.01). As
presented in Fig. 4E and F, the
expression level of E-cadherin in the Lv-PTEN cell group was higher
when compared with the Lv-NC cell group. The expression levels of
N-cadherin, β-catenin, TCF-8, vimentin, Slug and Snail in the
Lv-PTEN cell group were lower compared with the Lv-NC cell group in
both cell lines, suggesting that PTEN upregulates E-cadherin and
downregulates N-cadherin, β-catenin, TCF-8, vimentin, Slug, and
Snail to inhibit the migration and invasion of Burkitt's lymphoma
cells.
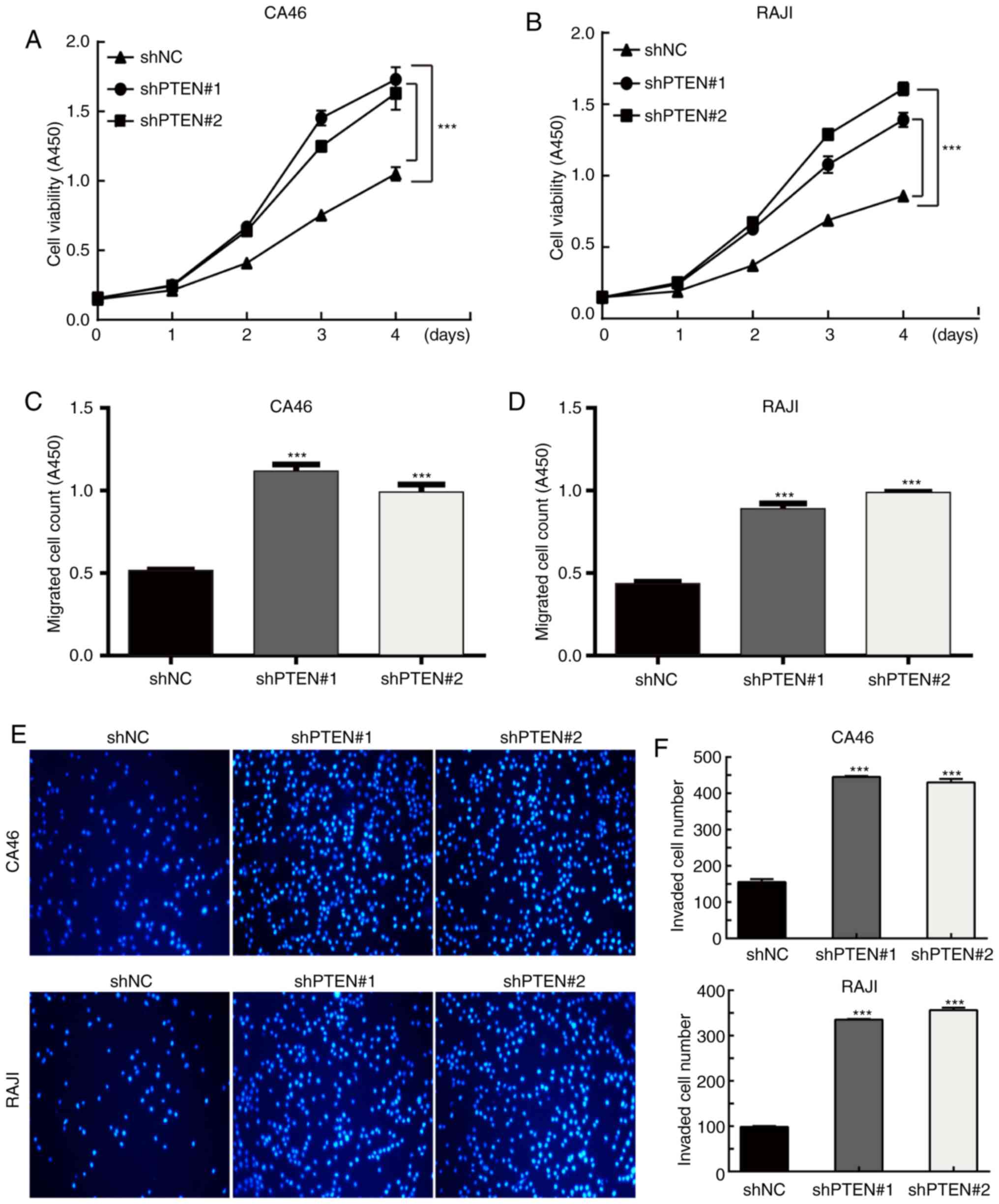
Silencing of PTEN promotes the growth,
migration and invasion of CA46 and RAJI cells
To further verify the biological function of the
silencing of PTEN on CA46 and RAJI cells, PTEN was silenced and
cell function assays were performed. As expected, silencing of PTEN
promoted the growth, migration and invasion of the two cell lines.
As presented in Fig. 5A and B, the
growth rate of the shPTEN cell groups was significantly higher
compared with the shNC cell group in both cell lines (P<0.01).
As indicated in Fig. 5C and D, the
OD values were 0.515±0.006 for the CA46-shNC cell group,
1.116±0.042 for the CA46-shPTEN#1 cell group, 0.989±0.048 for the
CA46-shPTEN#2 cell group, 0.434±0.013 for the RAJI-shNC cell group,
0.888±0.034 for the RAJI-shPTEN#1 cell group, and 0.986±0.005 for
the RAJI-shPTEN#2 cell group. The numbers of migrated cells in the
shPTEN cell groups were significantly higher compared with the shNC
cell groups for both cell lines (P<0.01). As presented in
Fig. 5E and F, the invasive number
of cells per field for each group was 154.33±6.11 for CA46-shNC,
440.67±7.77 for CA46-shPTEN#1, 426.33±9.45 for CA46-shPTEN#2;
95.00±6.25 for RAJI-shNC, 331.67±5.86 for RAJI-shPTEN#1, and
346.00±17.18 for RAJI-shPTEN#2. The numbers of invasive cells in
the shPTEN cell groups were significantly higher compared with the
shNC groups in both cell lines (P<0.01). These results
demonstrated that the silencing of PTEN produced opposite results
on cell behavior when compared with the overexpression of PTEN.
Discussion
Burkitt's lymphoma is a highly invasive cancer of
the lymphatic system, particularly B lymphocytes found in the
germinal center (11). A number of
studies have investigated Burkitt's lymphoma; however, the specific
pathogenesis of the disease is not fully understood. Our previous
studies found that inhibition of the PI3K pathway can inhibit the
proliferation of Burkitt's lymphoma, and abnormal expression of
phosphatase and tensin homolog (PTEN) is common in solid tumors,
including breast cancer, kidney cancer, esophageal cancer,
endometrial cancer (6–8) and diffuse large B cell lymphoma
(12). Therefore, a relationship
between PTEN overexpression and the incidence of Burkitt's lymphoma
has been indicated.
Research has demonstrated that increased expression
levels of PTEN inhibits the growth of breast cancer cells (13). Zhao et al (14) used PTEN as a silencing target and
performed PTEN knockdown in neuronal cells, which activated the
PI3K/AKT signaling pathway and promoted cell growth and
proliferation. The results of the present study indicated that PTEN
overexpression downregulated the expression of p-AKT, inhibited the
proliferation of Burkitt's lymphoma cells and induced apoptosis.
Silencing of PTEN promoted the growth and proliferation of
Burkitt's lymphoma cells. The results of the present study
demonstrated that PTEN inhibited the PI3K/AKT signaling pathway,
decreased the activity of AKT, upregulated the expression of
proapoptotic proteins Bad and Bax, inhibited the proliferation of
Burkitt's lymphoma cells and induced apoptosis.
The present study also indicated that overexpression
of PTEN promoted the expression of P53 and P21 and inhibited the
expression of CDK4, CDK6, cyclin D3 and cyclin H, which can arrest
the Burkitt's lymphoma cell cycle at the G1 phase. G1/S phase
checkpoints are critical for the regulation of the cell cycle
(15). The overexpression of PTEN
upregulated the expression of the tumor-suppressor gene P53 in
Burkitt's lymphoma cells, thereby activating P21 transcription. P21
is an inhibitor of CDK, which can bind to CDK4/6, and inhibit its
activity. The activity of CDK4/6 was decreased, which further
affected the expression of cyclin D3 and cyclin H, and caused G1
phase arrest of Burkitt's lymphoma cells. Zheng et al
(16) used PTEN as a therapeutic
target and applied FTY720 immunosuppressive agents to gastric
cancer cells. The inhibitors were found to suppress the PI3K/AKT
signaling pathway and increase the expression of P53 and P21 by
upregulating PTEN expression. The inhibition of CDK4/6 expression
and cell cycle arrest at the G1 phase indicated that PTEN
upregulation significantly inhibited the proliferation of gastric
cancer cells. Moon et al (17) also demonstrated that PTEN is
associated with inhibition of the growth and regulation of vascular
smooth muscle cells and regulation of the cell cycle, and this
inhibition is associated with the downregulation of cyclins and
CDKs and the upregulation of CDK inhibitors P21 and P27. Luo et
al (18) transfected airway
smooth muscle cells with a plasmid vector carrying PTEN and
revealed that PTEN overexpression blocked the G1 phase of the
airway smooth muscle cell cycle by inhibiting the expression of P21
and cyclin D. All of the aforementioned studies demonstrated that
PTEN overexpression can inhibit cell growth by blocking the cell
cycle progression of Burkitt's lymphoma cells.
The invasion and metastasis of malignant tumor cells
are attributed to poor patient prognosis and mortality (19,20).
Therefore, the prevention of tumor metastasis is essential. The
present study demonstrated that PTEN overexpression significantly
inhibited the migration and invasion of Burkitt's lymphoma cells.
In contrast, silencing of PTEN was revealed to promote the
migration and invasion of Burkitt's lymphoma cells. These results
indicated that PTEN may be a potential target for preventing the
invasion and metastasis of Burkitt's lymphoma cells. It still
remains unclear as to which molecular mechanisms are involved in
the inhibition of Burkitt's lymphoma progression by PTEN. The
epithelial-mesenchymal transition (EMT) is an important process
that is associated with tumor metastasis and invasion (21). Xu et al (22) demonstrated that treatment with
doxorubicin can upregulate the expression of PTEN and inhibit the
phosphorylation of AKT in gastric cancer cells, thereby inhibiting
the metastasis and invasion of gastric cancer. Wang et al
(23) demonstrated that knockdown
of PTEN can promote the migration of nasopharyngeal carcinoma
cells. Wang et al (24)
demonstrated that when SYNJ2BP induced PTEN degradation in breast
cancer tissues, PTEN expression levels were decreased, and the
PI3K/AKT signaling pathway was activated to induce the invasion of
breast cancer cells. In recent years, a number of studies have
revealed that activation of AKT can promote the metastasis and
invasion of tumor cells. Activated AKT binds to vimentin, which
induces the metastasis of gastric cancer cells through the
regulation of the E-cadherin/β-catenin complex (25,26).
Additionally, activated AKT regulates EMT via the
AKT/GSK-3β/β-catenin signaling pathway (27,28).
Nuclear β-catenin not only regulates proteins that are associated
with the EMT signaling pathway, but also induces transcriptional
upregulation of TCF-8, which in turn regulates DNA damage. The
aforementioned results demonstrate that activated AKT can promote
EMT and induce tumor formation and metastasis. Jin et al
(29) also indicated that
inhibition of the PTEN/PI3K/AKT signaling pathway can enhance the
sensitivity of esophageal cancer to radiotherapy and reverse the
process of EMT. This studied showed that the upregulation of PTEN
expression and inhibition of AKT phosphorylation can promote
esophageal cancer cell apoptosis, enhance the expression of
E-cadherin and downregulate the expression of N-cadherin and
vimentin to inhibit cancer cell invasion and metastasis. The
present study demonstrated that PTEN upregulates E-cadherin and
downregulates the expression of N-cadherin, β-catenin, TCF-8,
vimentin, Slug and Snail. β-catenin is an important molecule in the
Wnt pathway, and abnormal expression of β-catenin can promote tumor
metastasis and infiltration. PTEN overexpression downregulates the
expression of zinc finger transcription factors TCF-8, Slug and
Snail by reducing the accumulation of β-catenin in Burkitt's
lymphoma cells, and consequently inhibits the transition from
epithelial cadherin E-cadherin to neurokarin N-cadherin and the
expression of vimentin, thereby inhibiting the migration and
invasion of Burkitt's lymphoma cells. This process is consistent
with previous reports (22,30). These findings indicate that PTEN
overexpression can inhibit the migration and invasion of Burkitt's
lymphoma cells. However, there is also a shortcoming in our study.
We did not use normal cells as a control to detect the specificity
of the effect of PTEN on Burkitt's lymphoma. This is a limitation
to the present study. In conclusion, the present study demonstrated
that the tumor-suppressor gene PTEN inhibited the growth and
proliferation of Burkitt's lymphoma cells by inhibiting the
PI3K/AKT signaling pathway during in vitro experiments; PTEN
was indicated to induce apoptosis by upregulating proapoptotic
proteins. PTEN was also demonstrated to arrest the cell cycle of
Burkitt's lymphoma cells by regulating cell cycle-associated
proteins. Additionally, PTEN was revealed to inhibit cell invasion
by mediating EMT-like cell markers. PTEN serves an important role
in the development of Burkitt's lymphoma and the present study
provides insight into this potential clinical target for the
treatment of Burkitt's lymphoma. We studied the mechanism of PTEN
only in lymphoma, which confirms that it may become a target for
the treatment of lymphoma. In the future, tumor cells should be
induced to specifically express high PTEN using other methods,
without affecting the expression level of PTEN in normal cells;
thereby achieving the aim of treating lymphoma.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural
Science Foundation of Fujian Province (grant no. 2017J01349) and
the Natural Science Foundation of Quanzhou (grant no.
2018C063R).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
CL participated in the design of this study. YX
collected the data and performed the statistical analysis. PX
carried out the study, together with YZ, and collected important
background information. XZ drafted the manuscript. All authors read
and approved the final manuscript and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
Not applicable as only cell lines were used in the
study and no human or animal subjects were utilized in the
study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
PTEN
|
phosphatase and tensin homolog
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
AKT/PKB
|
protein kinase B
|
|
EBV
|
Epstein-Barr virus
|
|
SDS
|
sodium dodecyl sulfate
|
|
PAGE
|
polyacrylamide gel electrophoresis
|
|
PBS
|
phosphate-buffered saline
|
|
PI
|
propidium iodide
|
|
OD
|
optical density
|
|
CCK-8
|
Cell Counting Kit-8
|
|
DAPI
|
4′,6-diamidino-2-phenylindole
|
|
CDK
|
cyclin-dependent kinase
|
|
Bad
|
Bcl-2/Bcl-XL-associated death
promoter
|
|
Bax
|
Bcl-2 associated X protein
|
|
TCF8/ZEB1
|
zinc finger E-box-binding protein
1
|
|
EMT
|
epithelial-mesenchymal transition
|
References
|
1
|
Coakley D: Denis Burkitt and his
contribution to haematology/oncology. Br J Haematol. 135:17–25.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Miron I, Miron L, Lupu VV and Ignat A:
Silent presentation of multiple metastasis Burkitt lymphoma in a
child: A case report and review of the literature. Medicine
(Baltimore). 96:e75182017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bouska A, Bi C, Lone W, Zhang W, Kedwaii
A, Heavican T, Lachel CM, Yu J, Ferro R, Eldorghamy N, et al: Adult
high-grade B-cell lymphoma with Burkitt lymphoma signature: Genomic
features and potential therapeutic targets. Blood. 130:1819–1831.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oosten LEM, Chamuleau MED, Thielen FW, de
Wreede LC, Siemes C, Doorduijn JK, Smeekes OS, Kersten MJ, Hardi L,
Baars JW, et al: Treatment of sporadic Burkitt lymphoma in adults,
a retrospective comparison of four treatment regimens. Ann Hematol.
97:255–266. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li C, Xin P, Xiao H, Zheng Y, Huang Y and
Zhu X: The dual PI3K/mTOR inhibitor NVP-BEZ235 inhibits
proliferation and induces apoptosis of Burkitt lymphoma cells.
Cancer Cell Int. 15:652015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu F, Zhang C, Cui J, Liu J, Li J and
Jiang H: The prognostic value and potential drug target of
phosphatase and tensin homolog in breast cancer patients: A
meta-analysis. Medicine (Baltimore). 96:e80002017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gao T, Mei Y, Sun H, Nie Z, Liu X and Wang
S: The association of Phosphatase and tensin homolog (PTEN)
deletion and prostate cancer risk: A meta-analysis. Biomed
Pharmacother. 83:114–121. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Que WC, Qiu HQ, Cheng Y, Liu MB and Wu CY:
PTEN in kidney cancer: A review and meta-analysis. Clin Chim Acta.
480:92–98. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pfeifer M, Grau M, Lenze D, Wenzel SS,
Wolf A, Wollert-Wulf B, Dietze K, Nogai H, Storek B, Madle H, et
al: PTEN loss defines a PI3K/AKT pathway-dependent germinal center
subtype of diffuse large B-cell lymphoma. Proc Natl Acad Sci USA.
110:12420–12425. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fu X, Zhang X, Gao J, Li X, Zhang L, Li L,
Wang X, Sun Z, Li Z, Chang Y, et al: Phosphatase and tensin homolog
(PTEN) is down-regulated in human NK/T-cell lymphoma and corrects
with clinical outcomes. Medicine (Baltimore). 96:e71112017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Casulo C and Friedberg J: Treating burkitt
lymphoma in adults. Curr Hematol Malig Rep. 10:266–271. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu L, Wu H, Wu X, LI W and He D: The
expression pattern of Bcl11a, Mdm2 and Pten genes in B-cell acute
lymphoblastic leukemia. Asia Pac J Clin Oncol. 14:e124–e128. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu W and Wang W: MicroRNA-142-5p modulates
breast cancer cell proliferation and apoptosis by targeting
phosphatase and tensin homolog. Mol Med Rep. 17:7529–7536.
2018.PubMed/NCBI
|
|
14
|
Zhao T, Adams MH, Zou SP, El-Hage N,
Hauser KF and Knapp PE: Silencing the PTEN gene is protective
against neuronal death induced by human immunodeficiency virus type
1 Tat. J Neurovirol. 13:97–106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Furnari FB, Huang HJ and Cavenee WK: The
phosphoinositol phosphatase activity of PTEN mediates a
serum-sensitive G1 growth arrest in glioma cells. Cancer Res.
58:5002–5008. 1998.PubMed/NCBI
|
|
16
|
Zheng T, Meng X, Wang J, Chen X, Yin D,
Liang Y, Song X, Pan S, Jiang H and Liu L: PTEN- and p53-mediated
apoptosis and cell cycle arrest by FTY720 in gastric cancer cells
and nude mice. J Cell Biochem. 111:218–228. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moon SK, Kim HM and Kim CH: PTEN induces
G1 cell cycle arrest and inhibits MMP-9 expression via the
regulation of NF-kappaB and AP-1 in vascular smooth muscle cells.
Arch Biochem Biophys. 421:267–276. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Luo L, Gong YQ, Qi X, Lai WY, Lan H and
Luo Y: Effect of tumor suppressor PTEN gene on apoptosis and cell
cycle of human airway smooth muscle cells. Mol Cell Biochem.
375:1–9. 2013.PubMed/NCBI
|
|
19
|
Streutker CJ: Extramural venous invasion
in patients with locally advanced esophageal cancer: A reminder to
pathologists to look harder. Ann Surg Oncol. 25:1465–1466. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fang JH, Zhang ZJ, Shang LR, Luo YW, Lin
YF, Yuan Y and Zhuang SM: Hepatoma cell-secreted exosomal
microRNA-103 increases vascular permeability and promotes
metastasis by targeting junction proteins. Hepatology.
68:1459–1475. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aruga N, Kijima H, Masuda R, Onozawa H,
Yoshizawa T, Tanaka M, Inokuchi S and Iwazaki M:
Epithelial-mesenchymal transition (EMT) is correlated with
patient's prognosis of lung squamous cell carcinoma. Tokai J Exp
Clin Med. 43:5–13. 2018.PubMed/NCBI
|
|
22
|
Xu J, Liu D, Niu H, Zhu G, Xu Y, Ye D, Li
J and Zhang Q: Resveratrol reverses doxorubicin resistance by
inhibiting epithelial-mesenchymal transition (EMT) through
modulating PTEN/Akt signaling pathway in gastric cancer. J Exp Clin
Cancer Res. 36:192017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang MH, Sun R, Zhou XM, Zhang MY, Lu JB,
Yang Y, Zeng LS, Yang XZ, Shi L, Xiao RW, et al: Epithelial cell
adhesion molecule overexpression regulates epithelial-mesenchymal
transition, stemness and metastasis of nasopharyngeal carcinoma
cells via the PTEN/AKT/mTOR pathway. Cell Death Dis. 9:22018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang M, Wu H, Li S, Xu Z, Li X, Yang Y, Li
B, Li Y, Guo J and Chen H: SYNJ2BP promotes the degradation of PTEN
through the lysosome-pathway and enhances breast tumor metastasis
via PI3K/AKT/SNAI1 signaling. Oncotarget. 8:89692–89706.
2017.PubMed/NCBI
|
|
25
|
Otsuki S, Inokuchi M, Enjoji M, Ishikawa
T, Takagi Y, Kato K, Yamada H, Kojima K and Sugihara K: Vimentin
expression is associated with decreased survival in gastric cancer.
Oncol Rep. 25:1235–1242. 2011.PubMed/NCBI
|
|
26
|
Balasundaram P, Singh MK, Dinda AK, Thakar
A and Yadav R: Study of β-catenin, E-cadherin and vimentin in oral
squamous cell carcinoma with and without lymph node metastases.
Diagn Pathol. 9:1452014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang YG, Xu L, Jia RR, Wu Q, Wang T, Wei
J, Ma JL, Shi M and Li ZS: DDR2 induces gastric cancer cell
activities via activating mTORC2 signaling and is associated with
clinicopathological characteristics of gastric cancer. Dig Dis Sci.
61:2272–2283. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang C, Jin H, Wang N, Fan S, Wang Y,
Zhang Y, Wei L, Tao X, Gu D, Zhao F, et al: Gas6/Axl axis
contributes to chemoresistance and metastasis in breast cancer
through Akt/GSK-3β/β catenin signaling. Theranostics. 6:1205–1219.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jin Y, Xu K, Chen Q, Wang B, Pan J, Huang
S, Wei Y and Ma H: Simvastatin inhibits the development of
radioresistant esophageal cancer cells by increasing the
radiosensitivity and reversing EMT process via the PTEN-PI3K/AKT
pathway. Exp Cell Res. 362:362–369. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zheng Q, Lin Z, Xu J, Lu Y, Meng Q, Wang
C, Yang Y, Xin X, Li X, Pu H, et al: Long noncoding RNA MEG3
suppresses liver cancer cells growth through inhibiting β-catenin
by activating PKM2 and inactivating PTEN. Cell Death Dis.
9:2532018. View Article : Google Scholar : PubMed/NCBI
|