Introduction
Drug resistance is a common obstacle in successfully
treating cancer. Searching for new molecular targets which will
increase treatment efficiency leading to key discoveries which
could drive the field of oncology forward, and thus improving
patient outcomes is crucial. Patients who suffer from malignant
melanoma, the deadliest skin-related cancer, are still waiting for
improved treatment regimen. The 5-year survival of metastatic
melanoma patients is still relatively low, leading to the urgent
need for research in this area (1).
Cyclin F is a non-canonical cyclin involved in the
degradation of various molecular targets through ubiquitin-mediated
proteolysis. The first identified target recognized by cyclin F was
ribonucleotide reductase subunit M2 (RRM2). The cyclin F-RRM2 axis
provides a pool of DNA which can then be utilized for DNA synthesis
and repair. In the G2/M phase, when DNA synthesis is complete, RRM2
is targeted for degradation. Presence of genotoxic stress induces
ATR-dependent degradation of cyclin F and RRM2 can translocate to
the nucleus, facilitating the accumulation of a nucleotide pool for
use in DNA repair. Overexpression of RRM2 has been observed in
various cancer types including lung cancer, head and neck cancer,
and melanoma (2–4). Targeting RRM2 sensitizes cancer cells
for drug treatment, reducing aggressiveness. The role of cyclin F
in cancer development and treatment response, however, is still
elusive. Some studies have revealed that cyclin F acts as a tumor
suppressor, whereas other studies have revealed that cyclin F
promotes cancer progression. To study changes in cyclin F following
drug exposure primary and metastatic melanoma cells lines were
treated with cisplatin, a compound with a well-known mechanism of
action. The present study revealed that cisplatin differentially
impacted cyclin F expression in the primary A375 melanoma cell line
and the metastatic RPMI-7951 cell line. This initial study presents
cyclin F as a new factor which may determine cellular response
during drug intervention.
Materials and methods
Antibodies
The following primary antibodies were used: Cyclin F
(cat. no. sc-515207; 1:100, Santa Cruz Biotechnology Inc.). RRM2
(cat. no. ab57653; 1:200, Abcam), p53 (Pab 240; cat. no. 13-4100;
1:100), p-ATR (cat. no. 720107; 1:200), p-H2.AX (cat. no. MA1-2022;
1:100), GAPDH (cat. no. MA5-15738; 1:500; all from Life
Technologies; Thermo Fisher Scientific, Inc.). The following
secondary antibodies were used: Alexa Fluor 594 goat anti-mouse
(cat. no. A11005; 1:200), Alexa Fluor 594 donkey anti-rabbit (cat.
no. A21207; 1:200), Alexa Fluor 647 anti-rabbit (cat. no. A31573;
1:500), Alexa Fluor 488 anti-mouse (cat. no. A11029; 1:500, Life
Technologies; Thermo Fisher Scientific, Inc.).
Cell culture
Two melanoma cell lines, A375 and RPMI-7951, were
purchased from ATCC. The cells were cultured in DMEM (A375) or EMEM
(RPMI-7951) supplemented with 10% FBS and 50 µg/ml gentamycin and
were incubated in a humidified atmosphere of 95% air/5%
CO2 at 37°C. The cell culture was tested for Mycoplasma,
based on the rapid uptake of DAPI by cellular DNA. All tests were
negative. All in vitro studies were performed on low passage
number cells (P<5). The RPMI-7951 cell line bears a TP53
homozygous mutation (c.497C>A) (5).
Apoptosis analysis
The presence of apoptotic cells was determined using
Alexa Fluor™ 488 Annexin V/Dead Cell Apoptosis Kit (Life
Technologies; Thermo Fisher Scientific, Inc.) following the
manufacturer's instructions. Cells were analyzed using Guava
EasyCyte 6HT-2L Cytometer (Merck KGaA). FCS files were analyzed
using FlowJo software (version 10.07; FlowJo LLC).
Cell cycle analysis
Cells were fixed in ethanol for 24 h in −20°C. The
cells were then washed with PBS and incubated for 30 min in
FxCycle™ PI/RNase Staining Solution (Life Technologies; Thermo
Fisher Scientific, Inc). After 24 and 48 h, cells were analyzed
using Guava EasyCyte 6HT-2L Cytometer. FCS files were analyzed
using InCyte software (version 3.3; Merck KGaA).
Immunofluorescence
Cells were stained using the standard protocol
described in a previous study (6).
Briefly, the cells were fixed with 4% paraformaldehyde blocked with
4% BSA and stained with appropriate primary and secondary
antibodies. F-actin was stained using Alexa Fluor 488 phalloidin
(cat. no. A12379; 1:40; Life Technologies; Thermo Fisher
Scientific, Inc) (6).
Western blot assay
Whole cell lysates were prepared using RIPA buffer
(Merck KGaA). Following normalization of the protein concentration,
using the BCA protein assay kit (Thermo Fisher Scientific, Inc.),
equal amounts of protein (25 µg of total protein per lane) were
separated using 4–12% NuPAGE Bis-Tris Gel (Novex/Life Technologies;
Thermo Fisher Scientific, Inc.) and transferred onto nitrocellulose
membranes using the iBlot dry transfer system (Invitrogen; Thermo
Fisher Scientific, Inc.). The membrane was processed in room
temperature using iBind Flex Western Blot system (Thermo Fisher
Scientific, Inc.) as described by the manufacturer. Bands were
stained using 1-Step™ Ultra TMB-Blotting solution (Thermo Fisher
Scientific, Inc.). Densitometry analysis was performed using ImageJ
software (version 1.52q; National Institiutes of Health).
Statistical analysis
Analyses was performed using statistical software
(GraphPad Prism 6; GraphPad Software, Inc.). The data were compared
with the non-parametric Mann-Whitney U test or nonparametric
Kruskal-Wallis test with Dunn's multiple comparisons test, and the
changes were considered to indicate a statistically significant
difference at a level of P<0.05.
Results
RPMI-7951 cell line is more
susceptible to cisplatin treatment
Tumor protein p53 (TP53) is a potent tumor
suppressor. In the presence of DNA damage, p53 plays a dual role in
the regulation of cell fate. Through the p21 pathway, p53 drives
cell cycle arrest and permits the cell to repair any DNA damage
(7). When the DNA damage is severe
and cannot be repaired, p53 then triggers apoptosis (8). To elucidate the impact of p53 on
cisplatin treatment, two cell lines which differ in p53 status were
selected, A375 with functional p53 and p53-mutated, RPMI-7951.
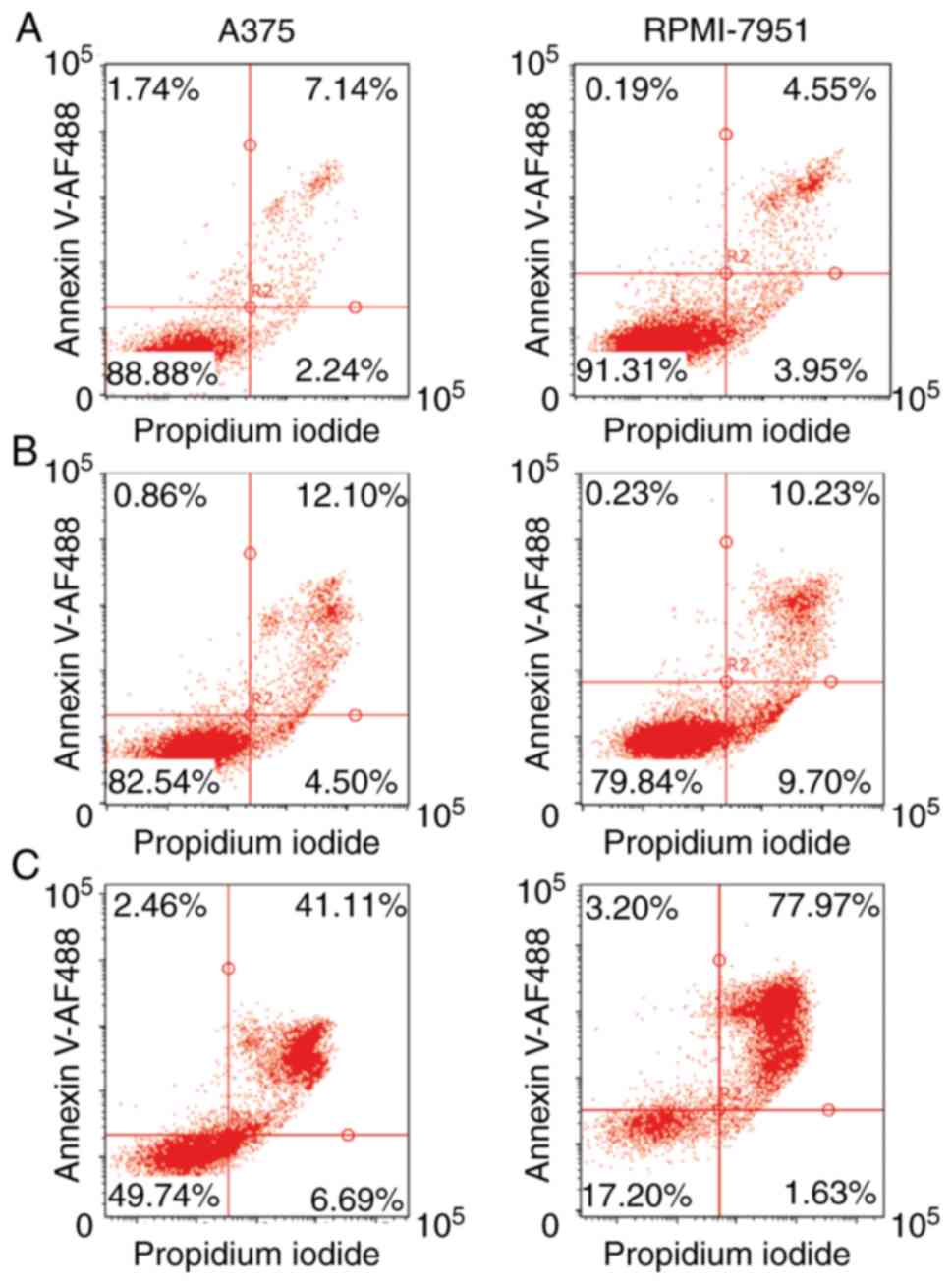
After 24 h of cisplatin treatment, both A375 and RPMI-7951 cell
lines exhibited similar, high viability with a low extent of
Annexin V-positive cells. However, with prolonged, 48 h of
treatment, the RPMI-7951 line contained a significantly higher
percentage of Annexin V-positive cells compared to the A375 cell
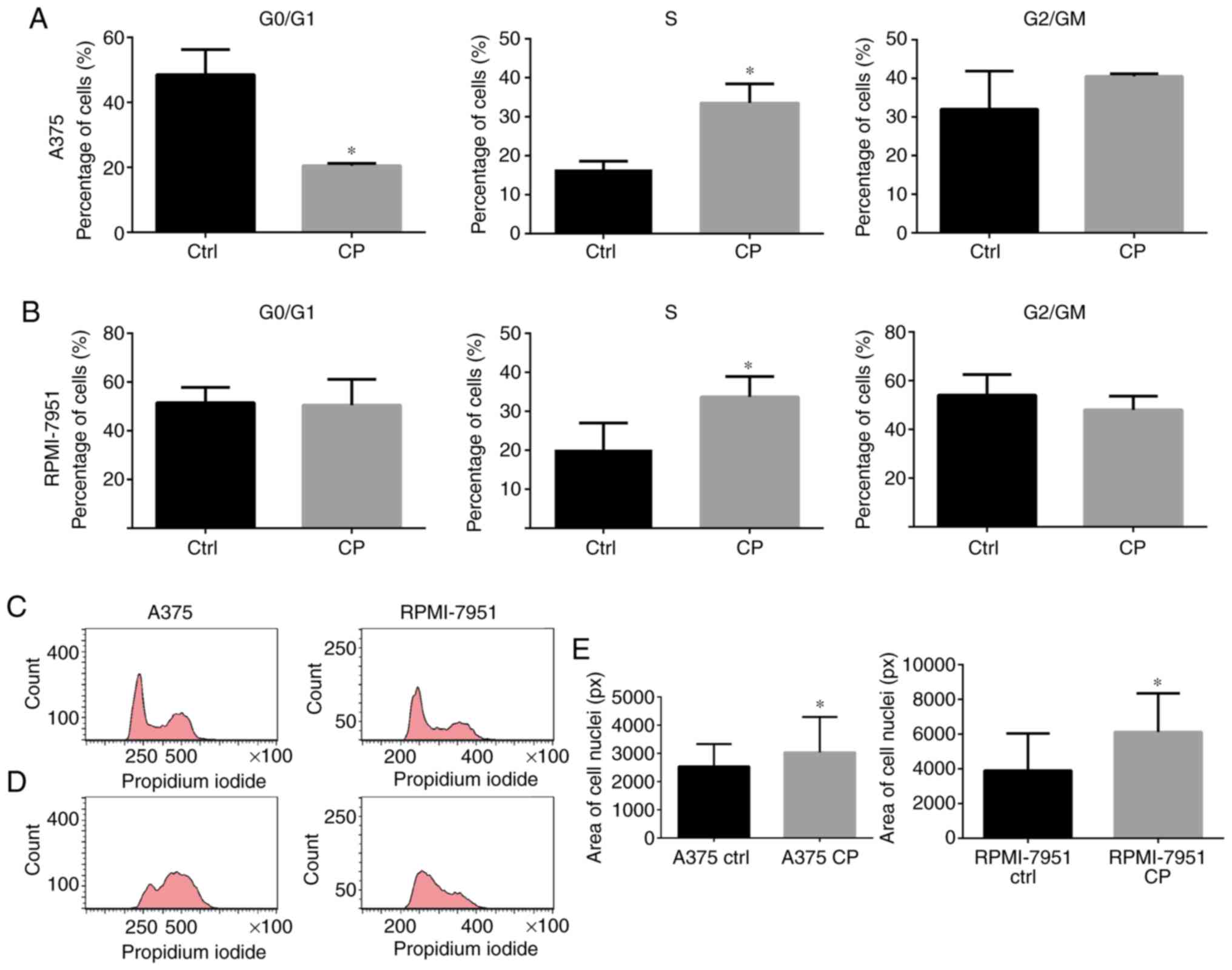
line (Fig. 1A-C). The DNA content
analysis revealed that cell cycle arrest in the S and G2/M phase
was more marked in the A375 cell line in comparison to RPMI-7951
cell line (Fig. 2A-D). An increased
nuclei size corresponded with cell cycle arrest in both cell lines
(Fig. 2E).
Cisplatin activates the p53 pathway in
the A375 cell line
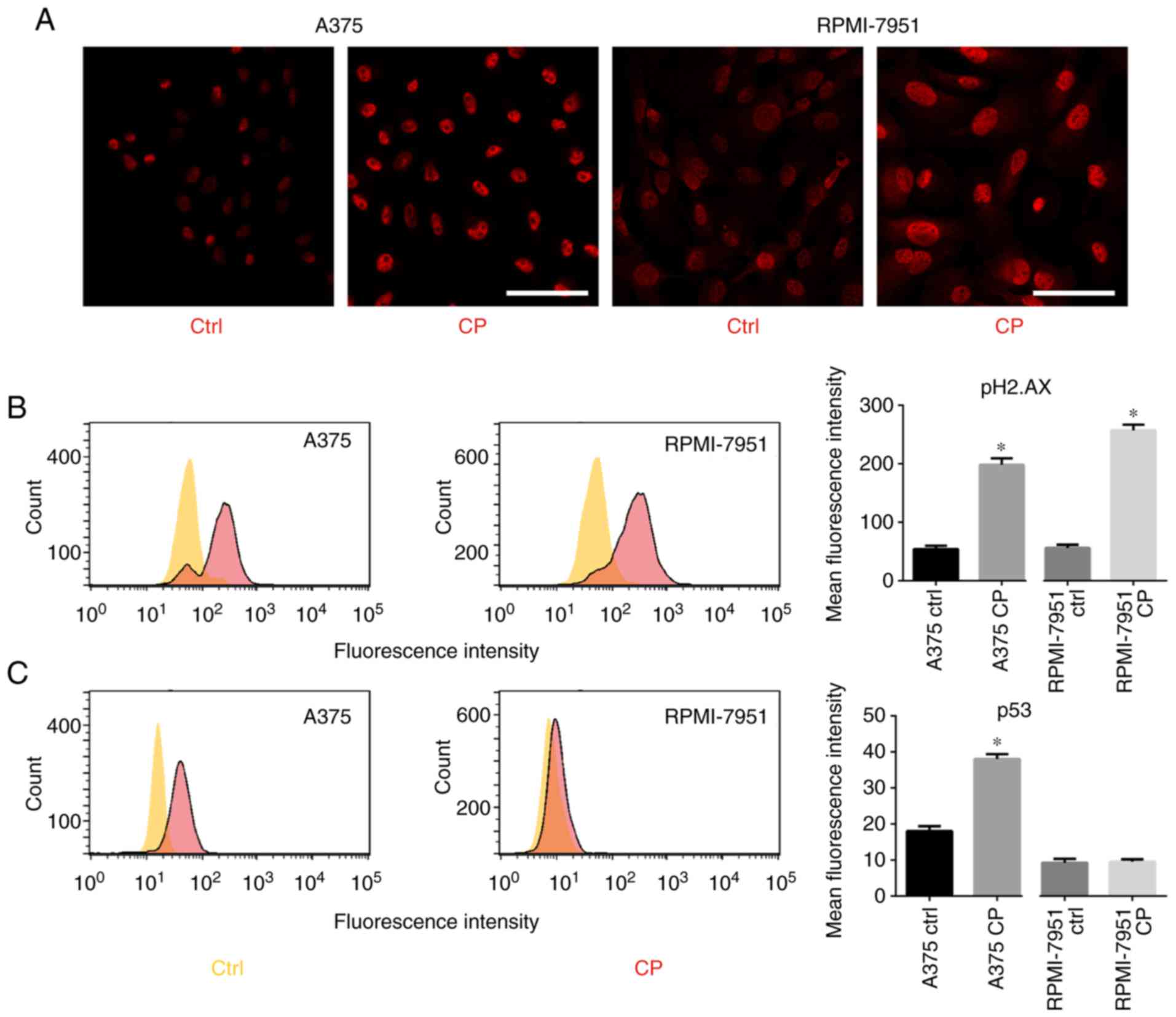
D'Angiolella et al revealed that when DNA
damage occurs, cyclin F is downregulated, likely in an
ATR-dependent manner (9). Since
cisplatin induces DNA damage, p53, pATR, and pH2.AX expression was
analyzed. After cisplatin treatment, an increase in p-ATR and
p-H2.AX levels (Fig. 3A and B) was
observed. RPMI-7951 cells bear a nonsense mutation in the p53
locus, thus an increased expression of p53 was only observed in the
A375 cell line (Fig. 3C).
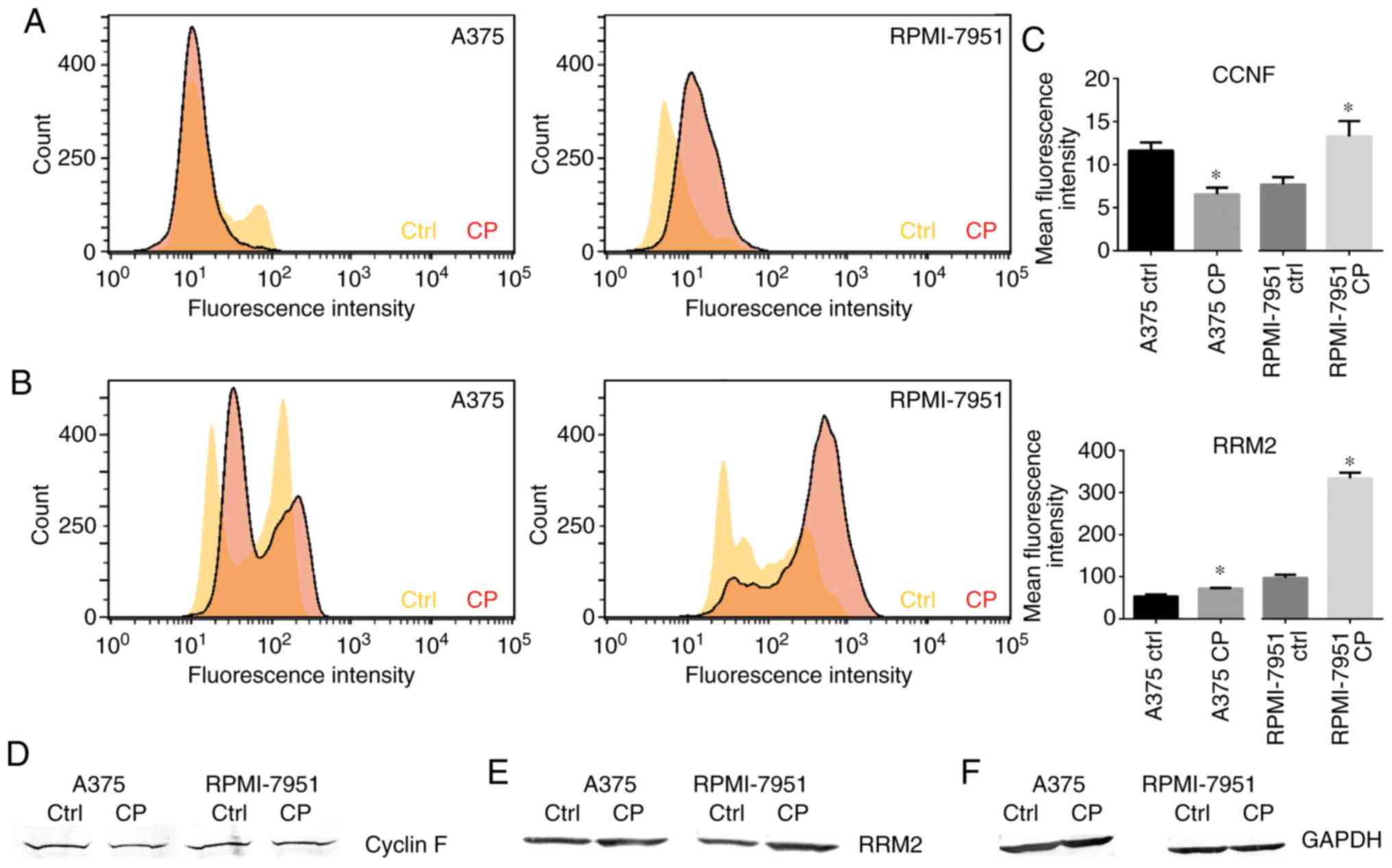
Cisplatin downregulates cyclin F in
A375 cell line but not in RPMI-7951 cells
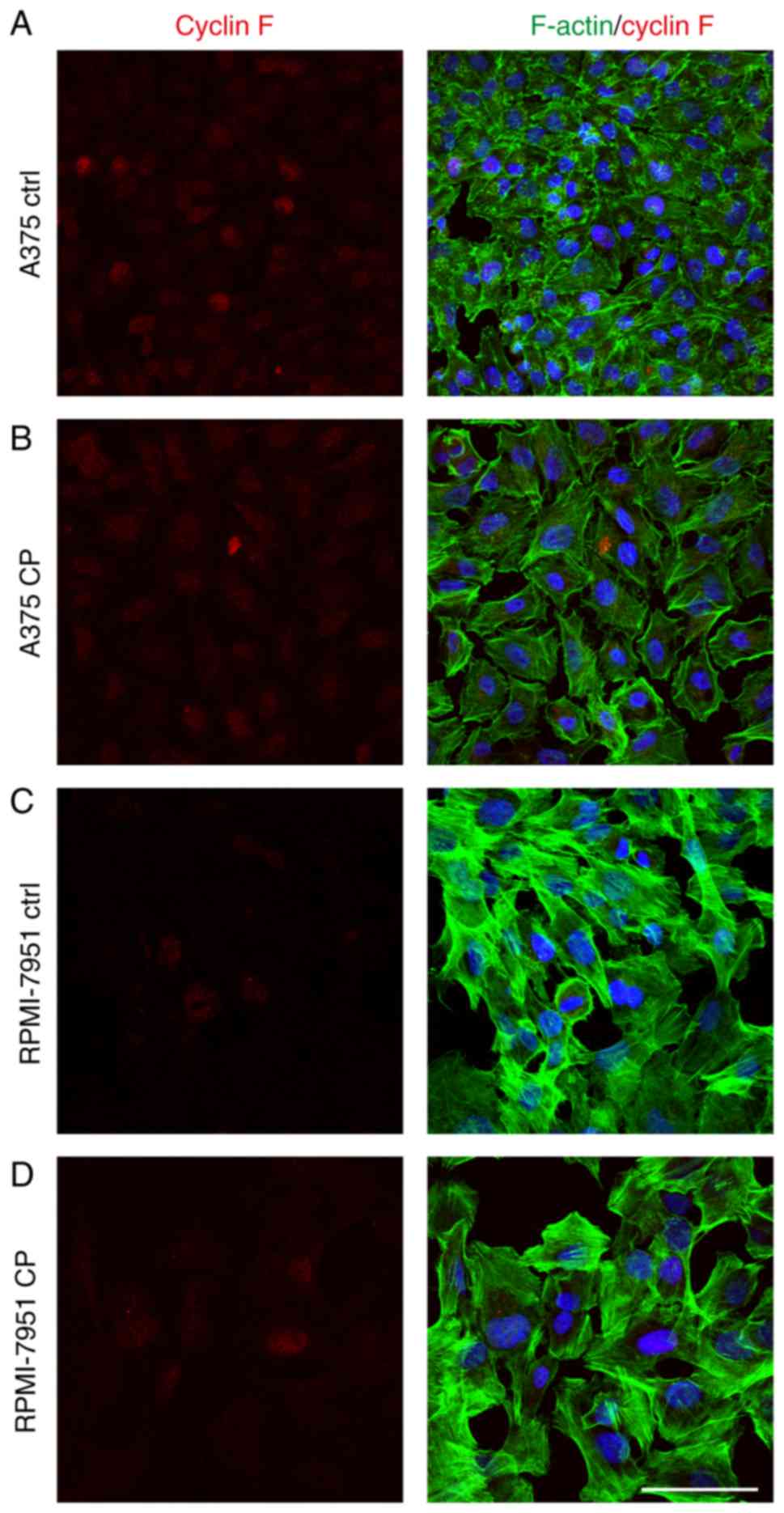
Following cisplatin treatment, a significant
decrease in cyclin F expression in the A375 cell line was observed.
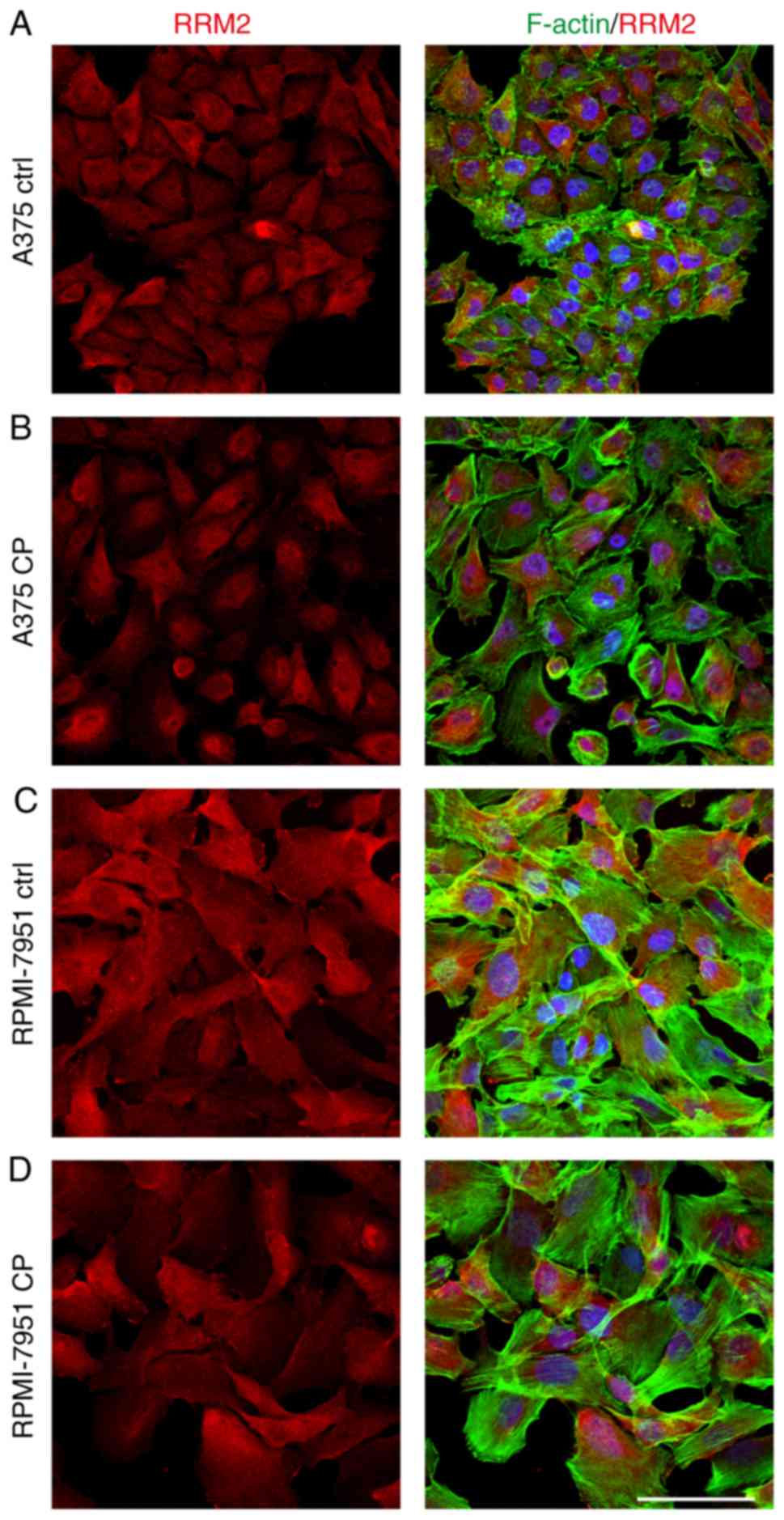
Immunofluorescence staining revealed that the percentage of treated
cells with a strong nuclear cyclin F signal was significantly lower
than observed in the control cells (Fig. 4). Notably, this effect was not
observed in the RPMI-7951 cell line. Flow cytometric analysis
revealed a significant increase in mean fluorescence intensity in
the RPMI-7951 cell line after the cisplatin treatment compared to
the control group (Fig. 5A and C).
While both cell lines exhibited nuclear localization of cyclin F,
with weak but positive cytoplasmic staining, this was more marked
in the RPMI-7951 cell line (Fig.
6). Western blot analysis confirmed the aforementioned
observations (Fig. 5D).
Cisplatin upregulates the RRM2
expression
Cisplatin is a well-characterized DNA damage-inducer
and it is well-known that alterations in the DNA repair pathway can
drive resistance to DNA-focused agents. RRM2 plays a central role
in the synthesis of deoxyribonucleotides from ribonucleotides
(10). Overexpression of RRM2 has
been associated with drug resistance and worse prognosis for cancer
patients (11). The degradation of
RRM2 is regulated by cyclin F, a functional axis which controls
genome integrity. To investigate how the p53 status impacts RRM2
flow cytometry was performed and western blot assays to assess the
level of RRM2 in the A375 and RPMI-7951 cell lines. After the
treatment with cisplatin, western blot analysis revealed an
increased expression of RRM2 in both melanoma cell lines compared
to the controls (Fig. 5E).
Moreover, flow cytometry was also conducted to assess the mean
fluorescence intensity of the RRM2 protein. The flow cytometric
data revealed a markedly higher increase of RRM2 in the RPMI-7951
cell line relative to the A375 cell line (Fig. 5B and C). The immunofluorescence
staining revealed a shift of RRM2 from cytoplasmic to cellular
localization (Fig. 6).
Discussion
Understanding the molecular mechanisms responsible
for cancer development, metastasis and migration of tumor cells, as
well as cancer aggressiveness are crucial in designing improved
treatment strategies to benefit cancer patients. Drug resistance is
a significant obstacle in achieving satisfactory effects of
therapy. D'Angiolella et al described the functional axis
which comprises genome stability and undisturbed cell proliferation
as associated with cyclin F (9).
Cyclin F regulates the pool of nucleotides available for DNA
synthesis and repair, through proteasome-mediated degradation of
RRM2. The overexpression of RRM2 is common in multiple cancer types
including lung, head and neck, and gastric cancer. Furthermore,
patients with high RRM2 expression are characterized with a worse
prognosis (12). While the role of
RRM2 in cancer development and progression is well-established,
little is known about how changes in cyclin F expression may affect
cancer development and drug response. Fu et al revealed that
low cyclin F expression is associated with a worse prognosis for
hepatocarcinoma patients. Downregulation of cyclin F was correlated
with tumor size, differentiation, clinical stage, and tumor
multiplicity (13). Conversely, in
glioma cells, cyclin F was reported as a tumor-suppressive factor.
A study by Deshmukh et al revealed that gliomas are
characterized by lower cyclin F expression in comparison with
normal brain tissue. Moreover, depletion of cyclin F using shRNA
resulted in increased tumor size and formation of numerous
metastatic nodules in the lungs and liver. Additionally, a decrease
in cyclin F expression coincided with increased circulating tumor
cells, affected epithelial markers including E-cadherin, and
increased expression of mesenchymal markers such as vimentin and
fibronectin (14). On the other
hand, cyclin F has been reported as an oncogene in ovarian cancer.
Through the OCT4-Nipp1/Ccnf-PP1-pRb axis, cyclin F was involved
with the phosphorylation of retinoblastoma protein. Activation of
the pathway resulted in enhanced tumor proliferation and increased
expression of the chromosomal passenger complex (CPC) elements such
as Aurora B, survivin and borealin. The present study indicated
that treatment of melanoma cells with cisplatin resulted in a
greater decrease in cyclin F expression in A375 compared to the
p53-mutant RPMI-7951 cell line. These results were consistent with
the work of D'Angiolella et al (9) revealing downregulation of cyclin F
after cisplatin treatment. The abundance of functional p53 results
in ineffective cyclin F elimination and increased apoptosis due to
the inability of cells to undergo cell cycle arrest. These findings
support the notion that cyclin F acts as a tumor suppressor.
However, the exact mechanism of the oncogenic properties of this
protein remain unclear. Cyclin F is reported to provide genome
stability through ubiquitin-mediated proteolysis of CP110, NUSAP,
and RRM2. Enrichment of the CP110 protein leads to overduplication
of the centrosome and mitotic aberrations. CP110 is stabilized by
the USP33 protein, which de-ubiquitinates and prevents degradation
via cyclin F-mediated proteolysis. Centrosome amplification is a
common event in melanoma cells (15). It has been proposed that most of the
amplified centrosomes are a result of centriole overduplication
(16). The downregulation of cyclin
F can contribute to genome instability and development of cells
with malignant properties.
The faithful replication of DNA cannot be conducted
without an appropriate pool of nucleotides delivered in proper
time. RRM2 is a functional part of the ribonucleotidase reductase
enzyme. During cell cycle progression, the activity of RRM2
increases and peaks at the S phase, when demand for the nucleotides
used in DNA synthesis is the highest. During the G2/M phase, RRM2
is phosphorylated and directed to proteasome-mediated degradation.
This degradation of RRM2 is regulated by cyclin F (9). While cisplatin is not a cell
cycle-specific drug, cells are the most susceptible for treatment
during the G1 phase. Cisplatin alternates the expression of
cell-cycle related genes and creates subsequent cell cycle arrest
in the G2 phase (17). Cisplatin
induces the p53 protein and functions as a p21CIP/WAF1
inhibitor, which stops cell cycle progression. It has been
demonstrated that mutation in the p53 gene may significantly
increase sensitivity, while the accumulation of p53 is partially
responsible for cisplatin resistance (18,19).
The efficient repair of DNA damage after the genotoxic stress via
RRM2 requires downregulation of cyclin F. In the present study,
higher RRM2 fluorescence intensity after cisplatin treatment was
observed in the RPMI-7951, mutant p53 cell line. The basal level of
cyclin F in the RPMI-7951 cell line was slightly higher compared to
control cells, indicating that the degradation rate of RRM2 should
also be greater. However, this phenomenon did not occur. Since the
degradation of RRM2 is p53-dependent, p53-mutated RPMI-7951 cells
accumulate RRM2, even in the presence of cyclin F. The lack of
functional p53 prevents cell cycle arrest, leading to the high
pools of nucleotides which cannot be utilized for DNA repair.
Increased susceptibility for cisplatin treatment in the RPMI-7951
p53-mutated metastatic melanoma cell line compared to the p53
wild-type A375 primary melanoma cell line was observed. The
inhibition of RRM2 by p53 follows suppression of mammalian target
of rapamycin complex 1 (mTORC1) (20). Moreover, mutations in p53 have been
revealed to cause increased levels of RRM1 and RRM2 in various
cancer cell lines (20,21). This data indicated the paramount
role of p53 in the regulation of RRM2 expression. Effective DNA
damage repair depends on the ability of the cell to pause cell
cycle progression through the activation of the cell cycle
checkpoints. If the cells enter the next cycle phase with
unrepaired DNA, this may trigger unfaithful DNA replication or
improper cell division, leading to cancer. When cells are exposed
to ionizing radiation (IR), cyclin F promotes entry into a cell
cycle checkpoint, suppressing the oncogenic B-Myb protein. The
interaction between cyclin F and B-Myb prevents cyclin A-mediated
phosphorylation of B-Myb and suppresses its activity, which is
necessary for recovery from cell cycle arrest after genotoxic
stress (22). Moreover, the
degradation of cyclin F via β-TrCP is required for G2/M transition
and activates the transcription of the mitosis-related enzymatic
machinery (23).
The present observations support the role of cyclin
F as a tumor suppressor in melanoma. However, there are several
studies which indicate the potential oncogenic effects of cyclin F.
Cancer stem-like cells are identified in many tumor types. The
presence of stem-like cells confers to increased tumor-initiating
potential, chemoresistance, apoptosis resistance, and enhanced
EMT-associated events (24–26). Oct-4 and Nanog are important
transcription factors, essential for the self-renewal of embryonic
stem cells (27). It has been
reported that the expression of Oct-4 and Nanog in several cancers
increases malignancy and is associated with poor prognosis. It has
also been revealed that Oct-4 expression is a strong prognostic
marker which can be utilized to predict poor clinicopathological
and prognostic characteristics in non-small cell lung cancer
(28). Oct-4 has been revealed to
drive Nanog and cyclin F expression, both inhibitors of protein
phosphatase 1 (PP1), preventing the dephosphorylation of Rb and
increasing the cell proliferation rate (29). It is possible that cyclin F, under
specific circumstances, can act as an oncogene rather than a tumor
suppressor gene. Activation of Oct4/Nanog signaling was revealed to
enhance stem-like properties such as spindle shape, foci formation,
and increased levels of CD133. A549 cells overexpressing OCT4
achieved the ability to form spheres in suspension and were
characterized by higher resistance to cisplatin (30). Oct4 overexpression was also revealed
to contribute to gefitinib resistance and increased self-renewal
capacity in PC9 and HCC827 cell lines (28). However, strong OCT4 expression in
the A375 or RPMI-7951 cell lines was not observed (data not shown).
Other investigators have indicated a crucial role of Oct-4 in
carcinogenesis and metastasis events in malignant melanoma.
Expression of Nanog and Oct-4 markers were revealed to be
associated with higher tumor aggressiveness and invasiveness. Oct-4
overexpression induced ameboid migration markers and increased
extravasation and transmigration capacities in melanoma cell lines
(31).
It has been revealed that A549 non-small cell lung
cancer cells grown on 3D scaffolds were characterized by higher
cyclin F expression and were more radio-resistance compared to 2D
cultured cells (32).
Hepatocellular carcinoma HepG2 cells treated with polysaccharides
from abalone had stimulatory potential and exhibited increased mRNA
levels of cyclin B, CDK1, and cyclin F and reduced levels of cyclin
E and CDK6 (33). It is possible
that the expression profile of cyclin F and its oncogenic or tumor
suppressor effect depends on the cellular state and cannot be
described with simple relationships. Choudhury et al
revealed that cyclin F is a substrate for oncogenic kinase AKT1.
The phosphorylation of cyclin F resulted in increased stability and
promoted the G1/S transition. Stabilization of cyclin F promoted
Cdh1 degradation, allowing S-phase entry. Cyclin F may act as an
oncogene promoting the degradation of Cdh1 and AKT1 activation
(34,35). Conversely, at the end of the S
phase, cyclin F takes part in Cdc6 degradation, preventing DNA
re-replication. The absence of cyclin F provoked genome instability
and allowed more than one replication event per cell cycle
(36). Additionally, cyclin F was
revealed to target SLBP for proteasome-mediated degradation in the
G2 phase. In the presence of SLBP, G2 phase translation of H2A.X
histone mRNA increased. Elevated H2A.X levels promoted apoptosis
upon the genotoxic stress. The degradation of SLBP via cyclin F led
to increased cell proliferation and decreased cytotoxicity of
agents. These data revealed the potential role of cyclin F in drug
response (37). All the presented
data indicate a dual role of cyclin F in integrating cell cycle
control and maintaining genome stability. Few studies on cancer
cells have revealed that low cyclin F expression is associated with
worse prognosis and increased proliferation of the cells. Thus, is
important to define the circumstances when cyclin F bears oncogenic
properties. Our previous research analyzed The Cancer Genome Atlas
data and revealed that high expression of cyclin F mRNA was
associated with poor prognosis and increased activity of pathways
related to the cell cycle and DNA damage repair (38).
In conclusion, it was demonstrated that cyclin F was
involved in the response of melanoma cell lines to the cisplatin
treatment. The change in cyclin F expression in response to
cisplatin treatment was significantly different in A375, a primary
melanoma cell line, and RPMI-7951, a metastatic melanoma cell line.
The observed difference may be related to the p53 mutation in the
RPMI-7951 cell line, which results in increased levels of cyclin F
and a simultaneous increase in RRM2 (5). Further investigations must be
conducted to elucidate the role of cyclin F in drug response and
regulation of the tumor invasiveness.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the
National Science Centre, Poland (grant no. 2016/21/B/NZ7/01121 to
AG).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
AK and AG conceived and designed the study. AK, MG,
AŻ and MHW performed the experiments and AK and MG wrote the
manuscript. DG performed the statistical analysis. AK and MHW
reviewed the manuscript and AG supervised the project. All authors
read and approved the manuscript and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Maverakis E, Cornelius LA, Bowen GM, Phan
T, Patel FB, Fitzmaurice S, He Y, Burrall B, Duong C, Kloxin AM, et
al: Metastatic melanoma-a review of current and future treatment
options. Acta Dermato Venereol. 95:516–524. 2015. View Article : Google Scholar
|
|
2
|
Fatkhutdinov N, Sproesser K, Krepler C,
Liu Q, Brafford PA, Herlyn M, Aird KM and Zhang R: Targeting RRM2
and mutant BRAF is a novel combinatorial strategy for melanoma. Mol
Cancer Res. 14:767–775. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rahman MA, Amin AR, Wang D, Koenig L,
Nannapaneni S, Chen Z, Wang Z, Sica G, Deng X, Chen ZG and Shin DM:
RRM2 regulates Bcl-2 in head and neck and lung cancers: A potential
target for cancer therapy. Clin Cancer Res. 19:3416–3428. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang L, Meng L, Wang XW, Ma GY and Chen
JH: Expression of RRM1 and RRM2 as a novel prognostic marker in
advanced non-small cell lung cancer receiving chemotherapy. Tumor
Biol. 35:1899–1906. 2014. View Article : Google Scholar
|
|
5
|
Bamford S, Dawson E, Forbes S, Clements J,
Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR
and Wooster R: The COSMIC (catalogue of somatic mutations in
cancer) database and website. Br J Cancer. 91:355–358. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Żuryń A, Krajewski A,
Klimaszewska-Wiśniewska A, Grzanka A and Grzanka D: Expression of
cyclin B1, D1 and K in non-small cell lung cancer H1299 cells
following treatment with sulforaphane. Oncol Rep. 41:1313–1323.
2019.PubMed/NCBI
|
|
7
|
Levine AJ: p53, the cellular gatekeeper
for growth and division. Cell. 88:323–331. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lowe SW, Ruley HE, Jacks T and Housman DE:
p53-dependent apoptosis modulates the cytotoxicity of anticancer
agents. Cell. 74:957–967. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
D'Angiolella V, Donato V, Forrester FM,
Jeong YT, Pellacani C, Kudo Y, Saraf A, Florens L, Washburn MP and
Pagano M: Cyclin F-mediated degradation of ribonucleotide reductase
M2 controls genome integrity and DNA repair. Cell. 149:1023–1034.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shao J, Zhou B, Chu B and Yen Y:
Ribonucleotide reductase inhibitors and future drug design. Curr
Cancer Drug Targets. 6:409–431. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Su YF, Wu TF, Ko JL, Tsai HT, Tee YT,
Chien MH, Chou CH, Lin WL, Low HY, Chou MY, et al: The expression
of ribonucleotide reductase M2 in the carcinogenesis of uterine
cervix and its relationship with clinicopathological
characteristics and prognosis of cancer patients. PLoS One.
9:e916442014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhong Z, Cao Y, Yang S and Zhang S:
Overexpression of RRM2 in gastric cancer cell promotes their
invasiveness via AKT/NF-κB signaling pathway. Pharmazie.
71:280–284. 2016.PubMed/NCBI
|
|
13
|
Fu J, Qiu H, Cai M, Pan Y, Cao Y, Liu L,
Yun J and Zhang CZ: Low cyclin F expression in hepatocellular
carcinoma associates with poor differentiation and unfavorable
prognosis. Cancer Sci. 104:508–515. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Deshmukh RS, Sharma S and Das S: Cyclin
F-dependent degradation of RBPJ inhibits
IDH1R132H-mediated tumorigenesis. Cancer Res.
78:6386–6398. 2018.PubMed/NCBI
|
|
15
|
Li J, D'Angiolella V, Seeley ES, Kim S,
Kobayashi T, Fu W, Campos EI, Pagano M and Dynlacht BD: USP33
regulates centrosome biogenesis via deubiquitination of the
centriolar protein CP110. Nature. 495:255–259. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Denu RA, Shabbir M, Nihal M, Singh CK,
Longley BJ, Burkard ME and Ahmad N: Centriole overduplication is
the predominant mechanism leading to centrosome amplification in
melanoma. Mol Cancer Res. 16:517–527. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Donaldson KL, Goolsby GL and Wahl AF:
Cytotoxicity of the anticancer agents cisplatin and taxol during
cell proliferation and the cell cycle. Int J Cancer. 57:847–855.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Perego P, Giarola M, Righetti SC, Supino
R, Caserini C, Delia D, Pierotti MA, Miyashita T, Reed JC and
Zunino F: Association between cisplatin resistance and mutation of
p53 gene and reduced bax expression in ovarian carcinoma cell
systems. Cancer Res. 56:556–562. 1996.PubMed/NCBI
|
|
19
|
Hawkins DS, Demers GW and Galloway DA:
Inactivation of p53 enhances sensitivity to multiple
chemotherapeutic agents. Cancer Res. 56:892–898. 1996.PubMed/NCBI
|
|
20
|
He Z, Hu X, Liu W, Dorrance A, Garzon R,
Houghton PJ and Shen C: P53 suppresses ribonucleotide reductase via
inhibiting mTORC1. Oncotarget. 8:41422–41431. 2017.PubMed/NCBI
|
|
21
|
Kollareddy M, Dimitrova E, Vallabhaneni
KC, Chan A, Le T, Chauhan KM, Carrero ZI, Ramakrishnan G, Watabe K,
Haupt Y, et al: Regulation of nucleotide metabolism by mutant p53
contributes to its gain-of-function activities. Nat Commun.
6:73892015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Klein DK, Hoffmann S, Ahlskog JK, O'Hanlon
K, Quaas M, Larsen BD, Rolland B, Rösner HI, Walter D, Kousholt AN,
et al: Cyclin F suppresses B-Myb activity to promote cell cycle
checkpoint control. Nat Commun. 6:58002015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mavrommati I, Faedda R, Galasso G, Li J,
Burdova K, Fischer R, Kessler BM, Carrero ZI, Guardavaccaro D,
Pagano M and D'Angiolella V: β-TrCP- and casein kinase II-mediated
degradation of cyclin F controls timely mitotic progression. Cell
Rep. 24:3404–3412. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen T, You Y, Jiang H and Wang ZZ:
Epithelial-mesenchymal transition (EMT): A biological process in
the development, stem cell differentiation, and tumorigenesis. J
Cell Physiol. 232:3261–3272. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Phi LTH, Sari IN, Yang YG, Lee SH, Jun N,
Kim KS, Lee YK and Kwon HY: Cancer stem cells (CSCs) in drug
resistance and their therapeutic implications in cancer treatment.
Stem Cells Int. 2018:54169232018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu Y, So C, Lam HM, Fung MC and Tsang SY:
Apoptosis reversal promotes cancer stem cell-like cell formation.
Neoplasia. 20:295–303. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Loh YH, Wu Q, Chew JL, Vega VB, Zhang W,
Chen X, Bourque G, George J, Leong B, Liu J, et al: The Oct4 and
Nanog transcription network regulates pluripotency in mouse
embryonic stem cells. Nat Genet. 38:431–440. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li SJ, Huang J, Zhou XD, Zhang WB, Lai YT
and Che GW: Clinicopathological and prognostic significance of
Oct-4 expression in patients with non-small cell lung cancer: A
systematic review and meta-analysis. J Thorac Dis. 8:1587–1600.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Comisso E, Scarola M, Rosso M, Piazza S,
Marzinotto S, Ciani Y, Orsaria M, Mariuzzi L, Schneider C,
Schoeftner S and Benetti R: OCT4 controls mitotic stability and
inactivates the RB tumor suppressor pathway to enhance ovarian
cancer aggressiveness. Oncogene. 36:4253–4266. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chiou SH, Wang ML, Chou YT, Chen CJ, Hong
CF, Hsieh WJ, Chang HT, Chen YS, Lin TW, Hsu HS and Wu CW:
Coexpression of Oct4 and nanog enhances malignancy in lung
adenocarcinoma by inducing cancer stem cell-like properties and
epithelial-mesenchymal transdifferentiation. Cancer Res.
70:10433–10444. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Borrull A, Ghislin S, Deshayes F, Lauriol
J, Alcaide-Loridan C and Middendorp S: Nanog and Oct4
overexpression increases motility and transmigration of melanoma
cells. J Cancer Res Clin Oncol. 138:1145–1154. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pan D, Chen Y, Du Y, Ren Z, Li X and Hu B:
Methylation of promoter of RBL1 enhances the radioresistance of
three dimensional cultured carcinoma cells. Oncotarget.
8:4422–4435. 2017.PubMed/NCBI
|
|
33
|
Wang YM, Wu FJ, Du L, Li GY, Takahashi K,
Xue Y and Xue CH: Effects of polysaccharides from abalone (Haliotis
discus hannai Ino) on HepG2 cell proliferation. Int J Biol
Macromol. 66:354–361. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Choudhury R, Bonacci T, Wang X, Truong A,
Arceci A, Zhang Y, Mills CA, Kernan JL, Liu P and Emanuele MJ: The
E3 ubiquitin ligase SCF(Cyclin F) transmits AKT signaling to the
cell-cycle machinery. Cell Rep. 20:3212–3222. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Choudhury R, Bonacci T, Arceci A, Lahiri
D, Mills CA, Kernan JL, Branigan TB, DeCaprio JA, Burke DJ and
Emanuele MJ: APC/C and SCF (cyclin F) constitute a reciprocal
feedback circuit controlling s-phase entry. Cell Rep. 16:3359–3372.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Walter D, Hoffmann S, Komseli ES,
Rappsilber J, Gorgoulis V and Sørensen CS: SCF(Cyclin F)-dependent
degradation of CDC6 suppresses DNA re-replication. Nat Commun.
7:105302016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dankert JF, Rona G, Clijsters L, Geter P,
Skaar JR, Bermudez-Hernandez K, Sassani E, Fenyö D, Ueberheide B,
Schneider R and Pagano M: Cyclin F-mediated degradation of SLBP
limits H2A.X accumulation and apoptosis upon genotoxic stress in
G2. Mol Cell. 64:507–519. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gagat M, Krajewski A, Grzanka D and
Grzanka A: Potential role of cyclin F mRNA expression in the
survival of skin melanoma patients: Comprehensive analysis of the
pathways altered due to cyclin F upregulation. Oncol Rep.
40:123–144. 2018.PubMed/NCBI
|