Introduction
Currently, there are 163 reported
post-transcriptional modifications of RNAs, including mRNA, tRNA,
rRNA, snRNA and others (1). These
processes can directly influence the structure of RNAs, which
ensures a diversity of functions. The modifications of mRNAs, such
as N1-methyladenosine, N6-methyladenosine
(m6A), N7-methylguanosine, 5-methylcytosine and
2′-O-methylation, serve fundamental roles in the regulation of gene
expression (2–4). Among these, the m6A RNA modification
is the most prevalent post-transcriptional modification of internal
mRNA in eukaryotes, accounting for 0.1–0.4% of total adenosine
residues (5).
Although m6A modification has been known for over
four decades (6), its distribution
and function were largely unexplored until the development of m6A
RNA immunoprecipitation sequencing (7,8).
Preferentially enriched around stop codons and the long internal
exons in the 3′-untranslated region, m6A RNA methylation is
evolutionarily conserved between human and mouse, indicating an
essential role for this modification (7,8).
The status of m6A RNA methylation is mainly
regulated by m6A RNA methylation regulators; ≥20 m6A RNA
methylation regulators have been identified and termed ‘writers’,
‘erasers’ and ‘readers’ (5,9–11).
‘Writers’ refer to m6A methyltransferases including Wilms tumor
1-associated protein (WTAP), zinc finger CCCH domain-containing
protein 13 (ZC3H13), KIAA1429, methyltransferase like 3 (METTL3),
METTL14 and RNA-binding motif protein 15 (RBM15). ‘Erasers’ are
demethylases including Fat mass and obesity-associated protein
(FTO), alkB homolog 3 (ALKBH3) and ALKBH5. ‘Readers’ function as
binding proteins and include YTH domain-containing 1 (YTHDC1),
YTHDC2, YTH N6-methyladenosine RNA binding protein 1
(YTHDF1), YTHDF2, YTHDF3, insulin-like growth factor 2 mRNA-binding
protein 1 (IGF2BP1), IGF2BP2, IGF2BP3, heterogeneous nuclear
ribonucleoprotein C (HNRNPC), heterogeneous nuclear
ribonucleoprotein A2/B1 (HNRNPA2B1) and RNA binding motif protein
X-linked (RBMX). The interactions among m6A RNA methylation
regulators contribute to the dynamic role of m6A methylation in
multiple physiological processes, including stem cell renewal,
differentiation, carcinogenesis, neurogenesis, circadian clock
functions and DNA damage response (12–15).
Increasing evidence has suggested that dysregulated
expression of m6A RNA methylation regulators affects the initiation
and progression of cancers, such as glioblastoma (16,17),
acute myeloid leukemia (AML) (18),
hepatocellular carcinoma (19) and
breast cancer (20). Recently, m6A
RNA methylation regulator expression levels were demonstrated to be
effective biomarkers for discrimination among RCC subtypes
(21), and genetic alterations of
m6A RNA methylation regulators contributed to malignant progression
and poor clinical characteristics in patients with clear cell renal
cell carcinoma (ccRCC) (22). In
addition, studies revealed that the levels of m6A RNA methylation
were decreased in ccRCC tissues compared with adjacent non-tumor
tissues, which promoted the invasion and migration of renal cancer
cells and led to poor prognoses in patients with ccRCC (10,22,23).
Despite recent advances, a limited number of studies have
comprehensively explored the expression of m6A methylation
regulators in patients with ccRCC (21,24)
exhibiting different clinicopathological features, their
involvement in malignant progression and prognostic predictions. In
addition, which m6A RNA methylation regulators may be more suitable
for prognostic stratification in ccRCC has not been
investigated.
The present study aimed to examine the expression of
19 RNA methylation regulators in a comprehensive manner with the
clinicopathological features and RNA sequencing data of a ccRCC
cohort from The Cancer Genome Atlas (TCGA) to explore the
association between the gene expression and clinicopathological
features. In addition, the present study aimed to construct a
m6A-related risk signature and nomograms for prognostic prediction.
Finally, this study aimed to detect the expression of seven hub m6A
RNA methylation regulators and m6A RNA modification levels in RCC
cell lines and a normal renal tubular epithelial cell line.
Materials and methods
Data processing
The mRNA (RNA sequencing) Fragments Per Kilobase of
transcript per Million Fragments standardized expression data and
corresponding clinicopathological features were retrieved for 489
ccRCC tissues and 72 adjacent non-tumor tissues from TCGA
(http://cancergenome.nih.gov/) and 91
ccRCC tissues from the International Cancer Genome Consortium
(ICGC; http://icgc.org/). Patients without prognostic
information were excluded from analysis. Overall survival (OS) and
disease-free survival (DFS) were the primary end points of this
study. OS was defined as the time between diagnosis and death or
was censored at the last follow-up. DFS was defined as the time
between diagnosis or surgery and the recurrence of disease or death
(for any reason) at the last follow-up.
m6A RNA methylation regulators
A total of 19 m6A RNA methylation regulators that
were available in TCGA and ICGC cohorts were selected. The
expression of 19 m6A RNA methylation regulators was systematically
compared in 539 ccRCC tissues with different pathological features
and with 72 adjacent non-tumor tissues.
Bioinformatics analyses
To identify the optimal molecular subgroups, the
present study divided patients with ccRCC into different clusters
by applying the ‘ConsensusClusterPlus’ package in R v.1.50.0
(http://www.bioconductor.org/packages/release/bioc/html/ConsensusClusterPlus.html)
based on the expression of the 19 m6A RNA methylation regulators.
The resampling program was used to sample 80% of the samples 50
times; the similarity distance between samples was estimated by the
Euclidean distance (25), and ‘km
dist’ was used as the clustering algorithm to select the reliable
and stable subgroup classification. The criteria to determine the
optimal number of clusters were a relatively high consistency
between clusters and no appreciable rise in the area under the
cumulative distribution function curve. The ‘proportion of
ambiguous clustering’ was used to select an optimal value of
clusters (k value) (26). Venn
online software (http://bioinformatics.psb.ugent.be/webtools/Venn/) was
used to identify the overlapping differentially expressed genes
(DEGs) between clusters. Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathway enrichment and Gene Ontology (GO) analyses were
performed and visualized using the Database for Annotation,
Visualization and Integrated Discovery software (DAVID; version
6.7; http://david.abcc.ncifcrf.gov/home.jsp) to provide
comprehensive pathway interpretations and functional annotation of
DEGs (|log2FC|>2 and adjusted P value <0.05)
between different clusters. Search Tool for the Retrieval of
Interacting Genes software (STRING; http://string-db.org/) was used to analyze the
interactions and evaluate the level of interactions (including
interactions determined by experiments or obtained from curated
databases, and interactions determined by text mining or
co-expression analyses) among the m6A RNA methylation
regulators.
Univariate Cox regression analysis was used to
identify the prognostic value of the expression of the m6A RNA
methylation regulators in the training cohort (TCGA). Regulators
associated with OS in univariate analyses were subsequently
selected for least absolute shrinkage and selection operator
(LASSO) Cox regression to construct a m6A-related risk signature
for clinical prognosis (27). As a
result, seven m6A RNA methylation regulators with their
corresponding coefficients were determined by the minimum mean
cross-validated error, choosing the optimal penalty parameter λ
related to the minimum 10-fold cross validation within the training
set. The risk score of each patient with ccRCC in the training and
validation (ICGC) cohorts was calculated using the following
formula:
Risk score=∑i=1nCoefi×xi
Where xi is the standardized expression
value of each selected m6A RNA methylation regulator, and
Coefi is the corresponding coefficient of the gene. All
patients were divided into low- and high-risk groups based on the
median value of the risk scores in the training and validation
cohorts.
Cell culture
The human RCC cell lines SW839, SN12C, 786-O and
OSRC-2 and human normal renal tubular epithelial cell line HK-2
were obtained from Cell Bank of the Chinese Academy of Sciences.
The human RCC cell lines were cultured in RPMI-1640 (Gibco; Thermo
Fisher Scientific, Inc.) with 10% FBS (HyClone; GE Healthcare Life
Sciences) and 1% penicillin/streptomycin (P/S; Gibco; Thermo Fisher
Scientific, Inc.). HK-2 cells were cultured in Keratinocyte Medium
(ScienCell Research Laboratories, Inc.) with 1% Keratinocyte Growth
Supplement (ScienCell Research Laboratories, Inc.) and 1% P/S
(ScienCell Research Laboratories, Inc.). All cells were cultured at
37°C in 5% CO2.
RNA isolation and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was isolated from cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. Reverse
transcription was performed using SuperScript III Reverse
Transcriptase (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions, and qPCR was
performed using a SYBR® Green PCR Master Mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.) and a 7900HT Fast
Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The thermocycling conditions were as follows: 95°C for 10
min, followed by 40 cycles of 95°C for 10 sec and at 60°C for 1
min. Relative mRNA levels were normalized against β-actin. Data
were analyzed using the 2−ΔΔCq method (28). Primer sequences are presented in
Table SI.
Total m6A RNA modification
detection
Total m6A RNA modification was detected in 200 ng
aliquots of total RNA extracted from cells using the EpiQuik m6A
RNA Methylation Quantification Kit (EpiGentek Group, Inc.)
according to the manufacturer's instructions. Briefly, total RNA
was bound to wells using the RNA Binding Solution. m6A RNA
modification was detected using capture and detection antibodies.
The detected signal was enhanced and quantified by reading
absorbance at 450 nm using a microplate reader (BioTek Instruments,
Inc.). The amount of m6A RNA modification was proportional to the
OD intensity. The experiments were performed in triplicate.
Statistical analysis
The differences in the expression levels of m6A RNA
methylation regulators between ccRCC tissues and adjacent non-tumor
tissues were compared using Wilcoxon test. Differences in the
expression levels of m6A RNA methylation regulators and m6A RNA
modification levels between HK-2 and human RCC cell lines were
analyzed using one-way ANOVA followed by Dunnett's post-hoc test.
The distributions of age, sex, histological grade and TNM stage
between clusters and between risk subgroups were analyzed using the
Kruskal-Wallis test and the Chi-square test, respectively. One-way
ANOVA was performed to compare the risk scores in ccRCC with
different T stages. Student's t-test was employed to compare the
risk scores in patients grouped by binary clinical variables.
Univariate and multivariate Cox regression analyses were performed
to identify the prognostic value of the risk score and other
clinicopathological features. Comparisons of survival between the
clusters or risk groups were performed using Kaplan-Meier curves
with the log-rank test, followed by pairwise comparisons between
clusters using the ‘survminer’ R package v.0.4.6 with Bonferroni
correction. Receiver operating characteristic curves (ROCs) were
used to test the prediction accuracy of the risk score and
clinicopathological features for 5-year survival and recurrence.
Variables significant in the multivariate Cox regression analyses
were used to construct prognostic nomograms validated by the
concordance index (c-index) and calibration plots using the ‘Rms’
package (https://cran.r-project.org/web/packages/rms/index.html)
in R. SPSS 22.0 (IBM Corp.), GraphPad Prism 6 (GraphPad Software,
Inc.) and R v3.6.1 (https://www.r-project.org/) were used for statistical
analysis. P<0.05 was considered to indicate a statistically
significant difference, with a Bonferroni correction (p<0.05/3)
applied for pairwise comparisons.
Results
Associations among the expression of
m6A RNA methylation regulators and clinicopathological
features
Among 19 m6A RNA methylation regulators, 15 DEGs
were identified, including 9 upregulated and 6 downregulated genes
in 539 ccRCC tissues compared with 72 adjacent non-tumor tissues in
TCGA cohort (Fig. 1A), which
indicated that m6A RNA methylation regulators exerted important
biological functions in the tumorigenesis of ccRCC. The main
clinicopathological characteristics of patients with ccRCC are
presented in Table I. In the
present study, G1-G2 was defined as low histological grade, G3-G4
was defined as high histological grade, stage I–II was defined as
low pathological stage, and stage III–IV was defined as high
pathological stage. The associations between the expression levels
of m6A regulatory regulators and the clinicopathological features
of ccRCC, including histological grade and pathological stage, were
analyzed in TCGA cohort. The results demonstrated that the
expression of most m6A RNA methylation regulators was significantly
associated with histological grade (Fig. 1B) and pathological stage (Fig. 1C). In addition, different expression
levels of FTO, IGF2BP2, IGF2BP3, KIAA1429, YTHDC1 and ZC3H13 were
identified by quantitative analyses according to histological grade
or pathological stage (Fig. 1D and
E). These results suggested that the m6A RNA methylation
regulators contributed to the malignant progression of ccRCC.
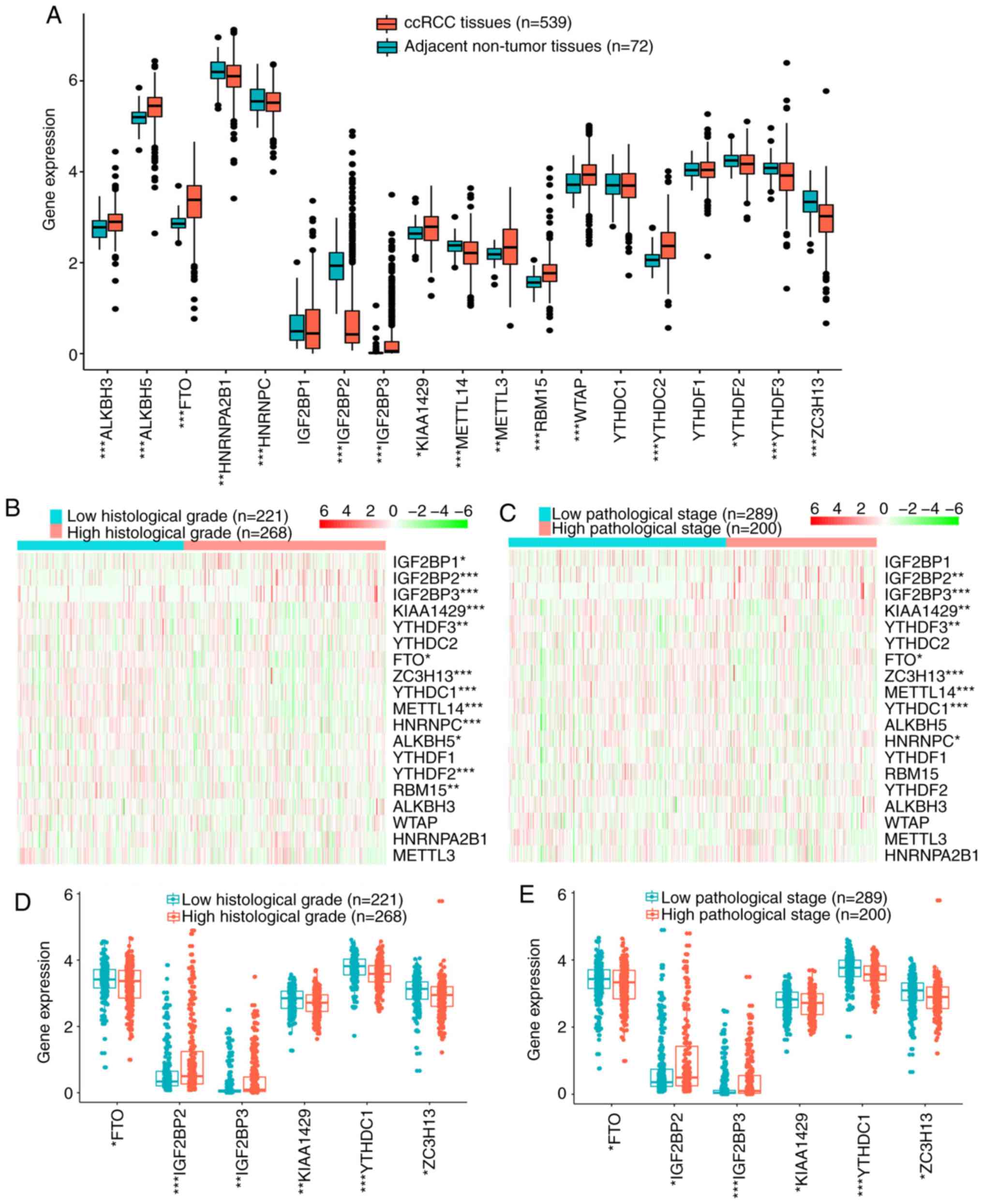 | Figure 1.Expression of m6A RNA methylation
regulators. (A) Expression levels of 19 m6A RNA methylation
regulators in ccRCC and adjacent non-tumor tissues. (B and C) Heat
maps of expression levels of 19 m6A RNA methylation regulators in
ccRCC with different (B) histological grades or (C) pathological
stages. (D and E) Expression levels of FTO, IGF2BP2, IGF2BP3,
KIAA1429, YTHDC1 and ZC3H13 in ccRCC with different (D)
histological grades or (E) pathological stages. *P<0.05,
**P<0.01, ***P<0.001. m6A, N6-methyladenosine;
ccRCC, clear cell renal cell carcinoma; FTO, fat mass and
obesity-associated protein; YTHDC1, YTH domain-containing 1;
IGF2BP, insulin-like growth factor 2 mRNA-binding protein; ZC3H13,
zinc finger CCCH-type-containing 13. |
 | Table I.Clinicopathological characteristics
of patients in TCGA and ICGC cohorts. |
Table I.
Clinicopathological characteristics
of patients in TCGA and ICGC cohorts.
|
| TCGA cohort | ICGC cohort |
|---|
|
|
|
|
|---|
| Characteristic | Number | % | Number | % |
|---|
| Age, years |
|
>65 | 169 | 34.6 | 28 | 30.8 |
|
≤65 | 320 | 65.4 | 63 | 69.2 |
| Sex |
|
Male | 323 | 66.1 | 52 | 57.1 |
|
Female | 166 | 33.9 | 39 | 42.9 |
| Pathological
stage |
| I | 238 | 48.7 | 52 | 57.1 |
| II | 51 | 10.4 | 13 | 14.3 |
|
III | 120 | 24.5 | 15 | 16.5 |
| IV | 80 | 16.4 | 9 | 9.9 |
| NA | 0 | 0 | 2 | 2.2 |
| Histological
grade |
| G1 | 10 | 2.0 | NA | NA |
| G2 | 211 | 43.1 |
|
|
| G3 | 195 | 39.9 |
|
|
| G4 | 73 | 14.9 |
|
|
| T stage |
| T1 | 244 | 49.9 | 54 | 59.3 |
| T2 | 62 | 12.7 | 13 | 14.3 |
| T3 | 172 | 35.2 | 22 | 24.2 |
| T4 | 11 | 2.2 | 2 | 2.2 |
| N stage |
| N0 | 232 | 47.4 | 85 | 93.4 |
| N1 | 14 | 2.9 | 2 | 2.2 |
| Nx | 243 | 49.7 | 4 | 4.4 |
| M stage |
| M0 | 412 | 84.3 | 81 | 89 |
| M1 | 77 | 15.7 | 9 | 9.9 |
| Mx | 0 | 0 | 1 | 1.1 |
Associations among the m6A RNA
methylation regulators
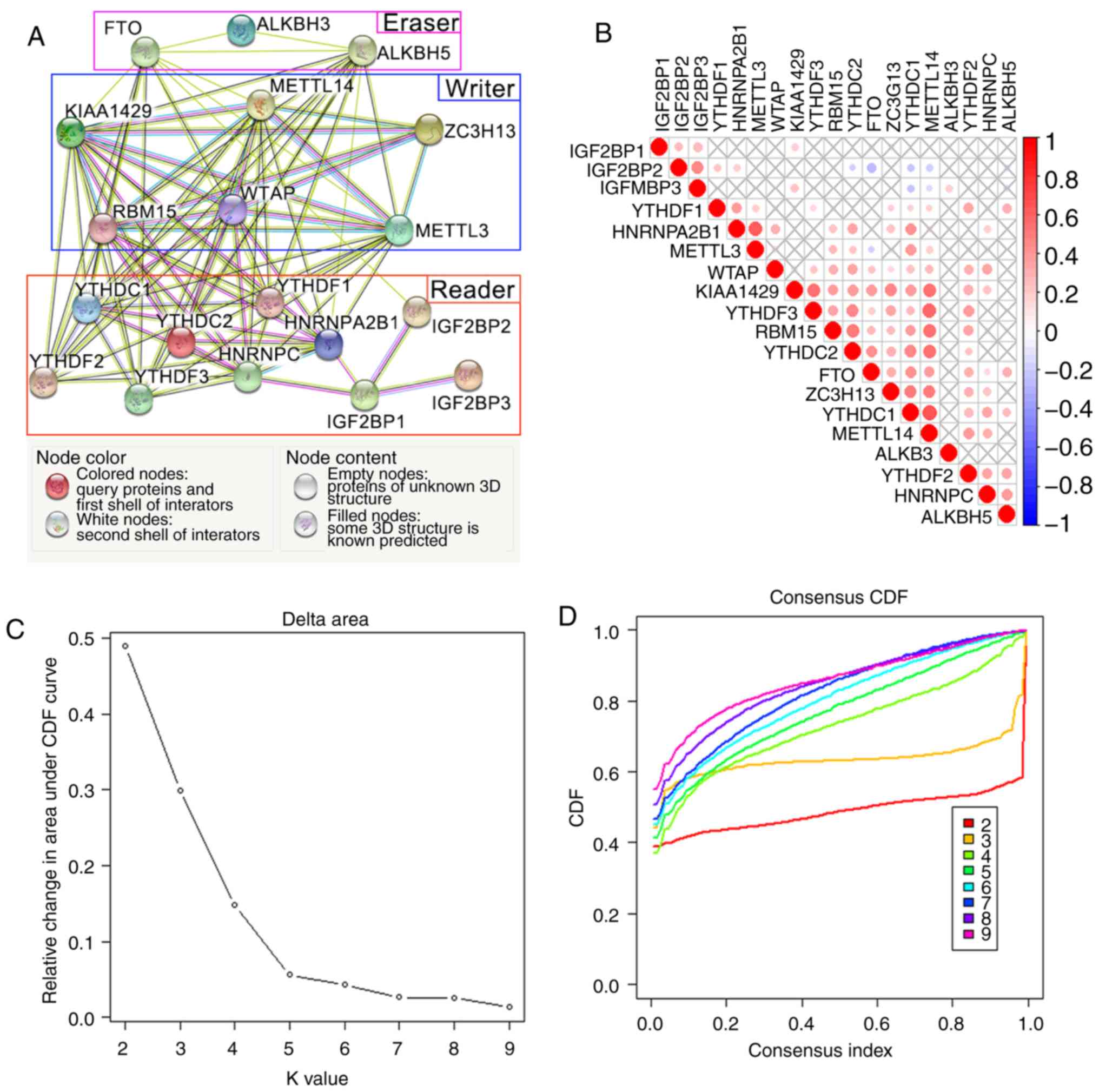
The network and correlation analyses among 19 m6A
RNA methylation regulators were conducted to determine their
interactions (Fig. 2A and B).
‘Writers’ had a wide range of interactions, including interactions
determined by experiments or obtained from curated databases, with
the m6A RNA methylation regulators. In addition, the ‘writers’
METTL14, KIAA1429, WTAP, RBM15 and ZC3H13 were co-expressed with
more than half of the 19 m6A RNA methylation regulators. Based on
these results, ‘writers’ were considered to be the hub gene set in
the network.
WTAP was the only ‘writer’ with various known
interactions with the other five ‘writers’, revealing that it may
be a hub gene of the ‘writers’. WTAP expression was also positively
associated with the ‘writers’ KIAA1429, ZC3H13 and RBM15 in ccRCC
(Fig. 2B). In addition, the
expressions levels of KIAA1429, YTHDF3, RBM15, YTHDC2, FTO, ZC3H13,
YTHDC1, METTL14 and YTHDF2 were positively associated with each
other.
By contrast, the interactions between ‘erasers’ and
other regulators were identified by text mining or co-expression
analyses, but lacked known interactions. With the exception of FTO,
the ‘erasers’ ALKBH3 and ALHBH5 lacked co-expression with other m6A
RNA methylation regulators. Independent interaction groups were
identified within the ‘readers’, indicating the diverse functions
of the ‘readers’; YTHDC1, YTHDC2, YTHDF1, IGF2BP2 and HNRNPA2B1
exhibited co-expression with each other in ccRCC.
Clinicopathological features and
biological processes of three clusters of patients with ccRCC
According to the ‘proportion of ambiguous
clustering’ method and the criteria for selecting the number of
clusters, k=3 was selected as the optimal value in TCGA cohorts
(Fig. 2C-E). Thus, the patients
from TCGA cohort were divided into three clusters, namely cluster
1, 2 and 3. Differences in clinicopathological features and
survival distributions between different clusters were identified,
with the expression values of the m6A RNA methylation regulators
screened out as heatmaps (Fig. 2F).
Cluster 1/2/3 subgroups were significantly associated with sex
(P<0.001), M stage (P=0.048), N stage (P=0.039), pathological
stage (P=0.021) and survival outcome (P<0.001) (Table II). Additionally, patients in
cluster 1 exhibited the shortest OS (P=0.02), followed by cluster 3
and cluster 2 (Fig. 2G). These
results revealed that patients with ccRCC in different clusters
exhibited significantly diverse clinicopathological features, and
the clustering results were associated with survival.
| Table II.Clinicopathological features between
clusters in The Cancer Genome Atlas cohort. |
Table II.
Clinicopathological features between
clusters in The Cancer Genome Atlas cohort.
| Characteristic | Cluster 1, n
(%) | Cluster 2, n
(%) | Cluster 3, n
(%) | P-value |
|---|
| Age, years |
|
>65 | 46 (39.0) | 80 (32.7) | 43 (34.1) | 0.453 |
|
≤65 | 72 (61.0) | 165 (67.3) | 83 (65.9) |
|
| Sex |
|
Male | 70 (59.3) | 151 (61.6) | 102 (19.0) |
<0.001c |
|
Female | 48 (40.7) | 94 (38.4) | 24 (81.0) |
|
| Pathological
stage |
| I | 47 (39.8) | 127 (51.8) | 64 (50.8) | 0.021a |
| II | 17 (14.4) | 20 (8.2) | 14 (11.1) |
|
|
III | 27 (22.9) | 60 (24.5) | 33 (26.2) |
|
| IV | 27 (22.9) | 38 (15.5) | 15 (11.9) |
|
| Histological
grade |
| G1 | 3 (2.5) | 6 (2.4) | 1 (0.8) | 0.058 |
| G2 | 44 (37.3) | 117 (47.8) | 50 (39.7) |
|
| G3 | 49 (41.5) | 92 (37.6) | 54 (42.9) |
|
| G4 | 22 (18.6) | 30 (12.2) | 21 (16.7) |
|
| T stage |
| T1 | 49 (41.5) | 129 (52.7) | 66 (52.4) | 0.058 |
| T2 | 20 (16.9) | 25 (10.2) | 17 (13.5) |
|
| T3 | 43 (36.4) | 87 (35.5) | 42 (33.3) |
|
| T4 | 6 (5.1) | 4 (1.6) | 1 (0.8) |
|
| N stage |
| N0 | 56 (47.5) | 120 (49.0) | 56 (44.4) | 0.039a |
| N1 | 6 (5.1) | 4 (1.6) | 4 (3.2) |
|
| Nx | 56 (47.5) | 121 (49.4) | 66 (52.4) |
|
| M stage |
| M0 | 92 (78.0) | 209 (85.3) | 111 (88.1) | 0.048a |
| M1 | 26 (22.0) | 36 (14.7) | 15 (11.9) |
|
| Outcome |
|
Alive | 64 (54.2) | 179 (73.1) | 85 (67.5) | 0.002b |
|
Dead | 54 (45.8) | 66 (26.9) | 41 (32.5) |
|
To identify different biological processes among the
three clusters, DEGs were identified among the clusters, and their
pathway interpretations and functional annotations were visualized.
As pathological features between cluster 2 and cluster 3 were
similar, and patients with ccRCC in cluster 1 exhibited the worst
survival outcomes among all clusters and largely different
clinicopathological features from cluster 2 and cluster 3, DEGs
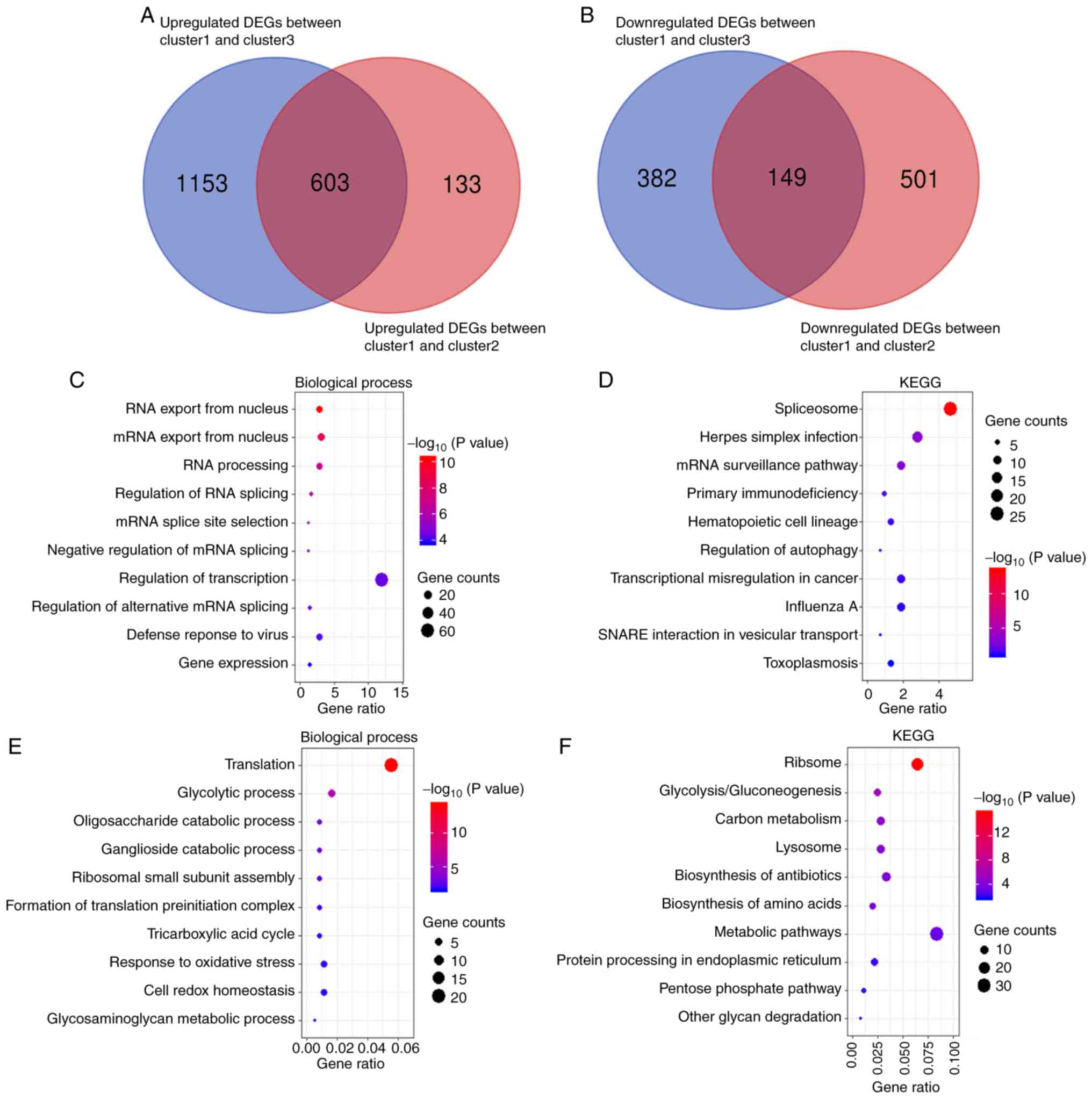
between cluster 1 and cluster 2 and between cluster 1 and cluster 3
were investigated. A total of 1,386 DEGs (736 upregulated and 650
downregulated) were identified between cluster 1 and cluster 2, and
2,287 DEGs (1,756 upregulated and 531 downregulated) were
identified between cluster 1 and cluster 3. Venn online software
was used to identify the overlapping DEGs between the two groups of
DEGs (Fig. 3A and B). As a result,
603 upregulated and 149 downregulated overlapping DEGs were
selected and analyzed by DAVID software to perform GO enrichment
and KEGG pathway analyses.
The results of GO biological processes analysis
demonstrated that upregulated and downregulated overlapping DEGs
were mainly enriched in RNA metabolism, including ‘RNA export from
the nucleus’, ‘mRNA export from the nucleus’, ‘RNA processing’,
‘regulation of RNA splicing’, ‘mRNA splice site selection’ and
‘translation’ (Fig. 3C and E). In
KEGG pathway analysis, similar changes were observed in the
corresponding signaling pathways, including ‘spliceosome’,
‘ribosome’, ‘transcriptional misregulation in cancer’, ‘mRNA
surveillance pathway’ and other malignancy-related pathways
including ‘primary immunodeficiency’, ‘regulation of autophagy’ and
‘response to oxidative stress’ (Fig. 3D
and F). These results suggested that cluster 1/2/3 subgroups
were associated not only with the clinicopathological features and
survival, but also with malignancy-related biological
processes.
Survival analysis of the m6A RNA
methylation regulators in patients with ccRCC
The prognostic significance of the m6A RNA
methylation regulators in patients with ccRCC was further analyzed.
In TCGA cohort, univariate Cox regression analysis identified 14
and 11 m6A RNA methylation regulators significantly associated with
OS and DFS, respectively (Fig. 4A and
B). Among the 14 m6A RNA methylation regulators in OS, six
regulators were associated with poor OS (IGF2BP1, IGF2BP2, IGF2BP3,
HNRNPA2B1, METTL3 and ALKBH3), and eight were associated with
favorable OS (YTHDF2, YTHDF3, FTO, YTHDC1, YTHDC2, ZC3H13, KIAA1429
and METTL14). For DFS, two regulators were associated with poor DFS
(IGF2BP2 and IGF2BP3), and nine regulators were associated with
favorable DFS (YTHDF1, YTHDF2, YTHDF3, YTHDC1, YTHDC2, ZC3H13,
KIAA1429, METTL14 and RBM15). The prognostic values of the m6A RNA
methylation regulators in patients with different histological
grades and pathological stages in TCGA cohort were subsequently
analyzed by univariate Cox regression. In both low and high
histological grade ccRCC, IGF2BP3 (P=0.030 and P<0.001,
respectively) and KIAA1429 (P=0.045 and P=0.031, respectively) were
significantly associated with OS; in both low and high pathological
stage, METTL14 was significantly associated with OS (P=0.035 and
P=0.003, respectively) (Fig.
S1).
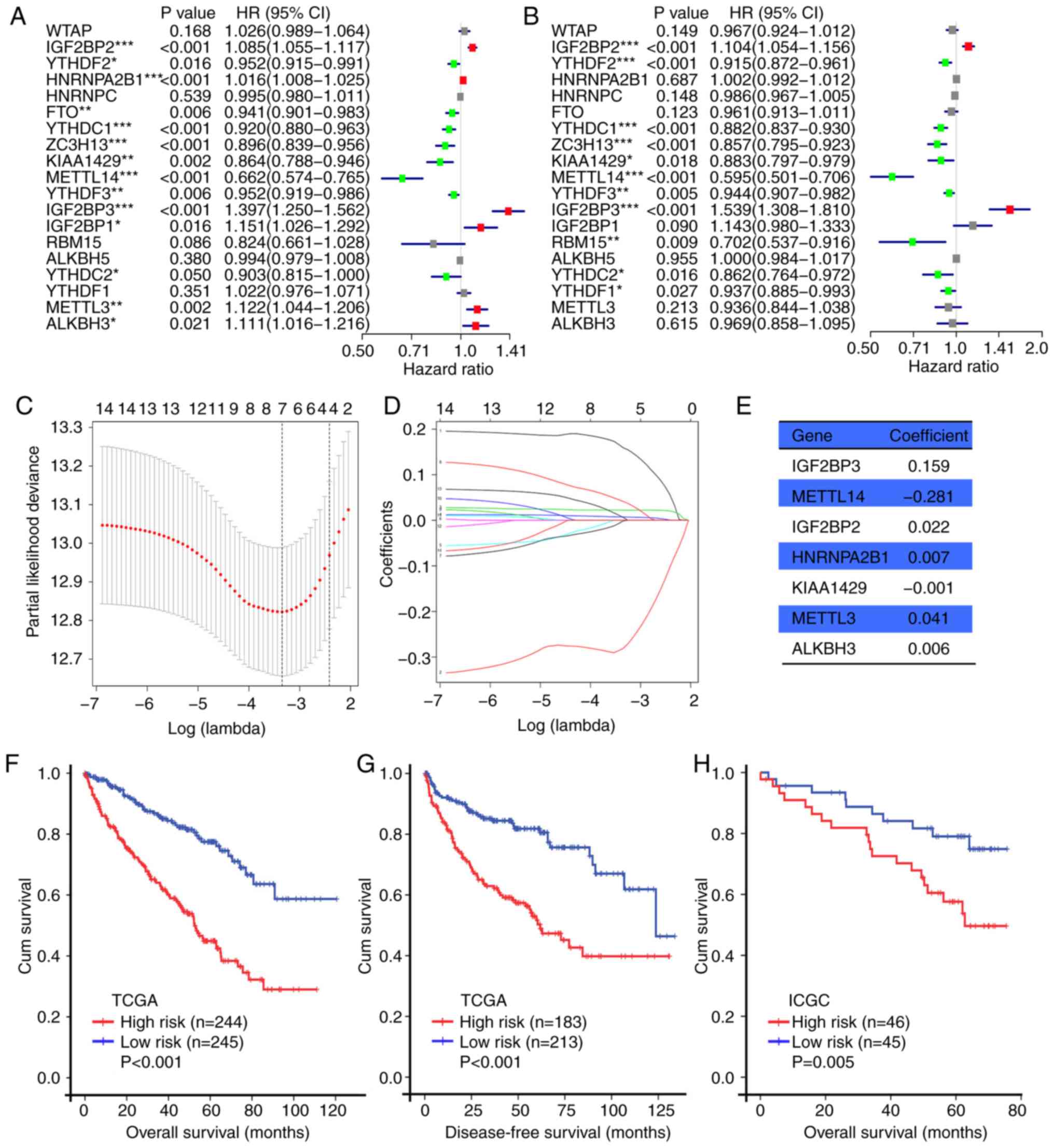 | Figure 4.Prognostic value of m6A RNA
methylation regulators in ccRCC. (A) OS-associated m6A RNA
methylation regulators in TCGA cohort. (B) DFS-associated m6A RNA
methylation regulators in TCGA cohort. (C) Plots of the
cross-validation error rates in TCGA cohort. (D) Distribution of
LASSO coefficients of 14 m6A RNA methylation regulators. (E)
Coefficient values of each of the seven selected genes. (F and G)
Kaplan-Meier OS and DFS curves for patients in TCGA cohort assigned
to the high- and low-risk groups. (H) Kaplan-Meier OS curve for
patients in the ICGC cohort assigned to the high- and low-risk
groups. *P<0.05, **P<0.01, ***P<0.001. m6A,
N6-methyladenosine; ccRCC, clear cell renal cell carcinoma; OS,
overall survival; DFS, disease-free survival; TCGA, The Cancer
Genome Atlas; ICGC, International Cancer Genome Consortium; HR,
hazard ratio; CI, confidence interval; LASSO, least absolute
shrinkage and selection operator. |
Construction and validation of the
m6A-related risk signature based on the m6A RNA methylation
regulators
Since the OS data was available in both TCGA and
ICGC cohorts, the OS-associated m6A RNA methylation regulators in
TCGA cohort were used to construct a m6A-related risk signature,
and its accuracy was validated in the ICGC cohort. As a result,
seven genes (IGF2BP2, IGF2BP3, METTL3, METTL14, HNRNPA2B1, KIAA1429
and ALKBH3) were identified by LASSO Cox regression in the training
cohort (TCGA) to build the m6A-related risk signature (Fig. 4C and D). Subsequently, the risk
score of each patient with ccRCC in the two cohorts was calculated
using these seven genes and the formula presented in Materials and
methods. Patients with ccRCC from the two cohorts were divided into
low- and high-risk subgroups based on the median risk score. In
TCGA cohort, the Kaplan-Meier method revealed that patients with
high risk scores exhibited shorter OS (P<0.001; Fig. 4F) and DFS (P<0.001, Fig. 4G) compared with those with low risk
scores. In the ICGC cohort, patients with high risk scores also
exhibited shorter OS (P=0.005; Fig.
4H) compared with those with low risk scores.
Survival analyses were performed to compare the two
risk groups of patients with different histological grades and
pathological stages. In TCGA cohort, patients in the high-risk
group exhibited poor OS and DFS compared with patients in the
low-risk group regardless of histological grade and pathological
stage (Fig. S2A-H). In the ICGC
cohort, patients with high risk scores exhibited shorter OS
compared with patients with low risk scores with a low pathological
stage, but not a high pathological stage (P=0.001 and P=0.549,
respectively; Fig. S2I and J).
ICGC data of histological grade and DFS were unavailable.
Based on these results, the risk signature derived
from the seven m6A regulators was identified to be a powerful
prognostic tool for patients with ccRCC. Further research is needed
to evaluated whether the prognostic value of the risk signature was
also independent of the known prognostic factors, such as
histological grade and pathological stage.
Associations between the prognostic
risk scores and clinicopathological features
The heat map of the expression levels of the seven
selected m6A RNA methylation regulators in the high- and low-risk
subgroup patients in TCGA cohort is presented in Fig. 5A and B. Patients in the high-risk
group had a higher proportion of males (P=0.015), higher
pathological stage (P<0.001), higher histological grade
(P<0.001), higher T stage (P<0.001), higher M stage
(P<0.001), higher N stage (P=0.012) and poorer OS compared with
patients in the low-risk group (Table
III). The levels of risk score according to different
clinicopathological features were compared; significantly different
risk scores were determined between patients stratified by
pathological stage, histological grade, T stage, N stage and M
stage in TCGA cohort (all P<0.001; Fig. 5C-H).
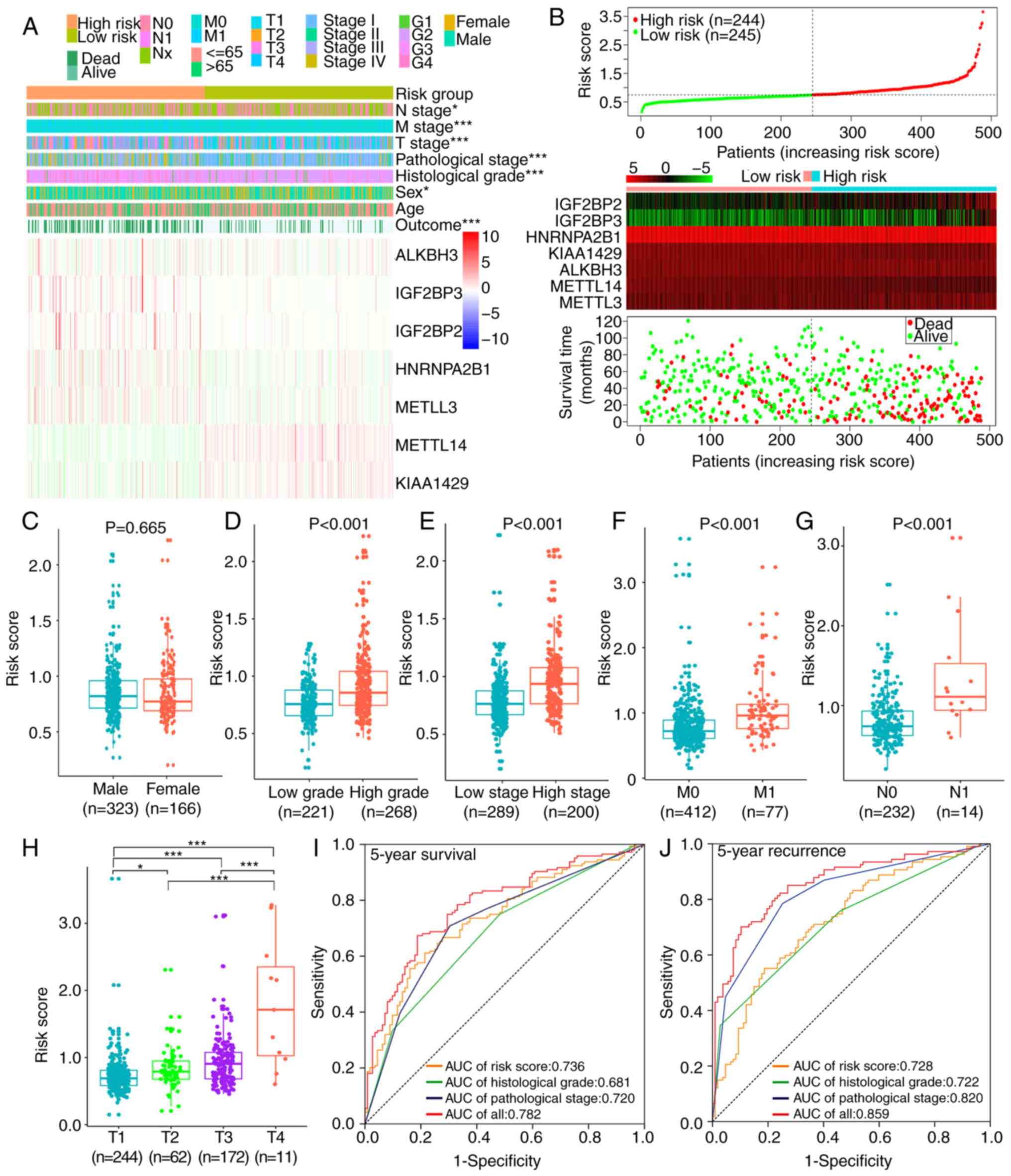 | Figure 5.Associations among risk scores,
clinicopathological features and prognoses in TCGA cohort. (A)
Clinicopathological features and expression levels of seven m6A RNA
methylation regulators were compared between the low- and high-risk
groups. (B) Risk score, expression heat maps of seven m6A RNA
methylation regulators and distribution of patient survival status
between the low- and high-risk groups. (C-H) Distribution of risk
scores stratified by (C) sex, (D) histological grade, (E)
pathological stage, (F) M stage, (G) N stage and (H) T stage. (I
and J) ROCs for risk scores, histological grade, pathological stage
and their combination for (I) 5-year survival and (J) 5-year
recurrence. *P<0.05, ***P<0.001. TCGA, The Cancer Genome
Atlas; m6A, N6-methyladenosine; ROC, receiver operating
characteristic; AUC, area under the curve. |
| Table III.Clinicopathological characteristics
in the low- and high-risk groups in The Cancer Genome Atlas
cohort. |
Table III.
Clinicopathological characteristics
in the low- and high-risk groups in The Cancer Genome Atlas
cohort.
| Characteristic | Low-risk group, n
(%) | High-risk group, n
(%) | P-value |
|---|
| Age, years |
|
>65 | 86 (35.1) | 83 (34.0) | 0.801 |
|
≤65 | 159 (64.9) | 161 (66.0) |
|
| Sex |
|
Male | 151 (61.6) | 172 (70.5) | 0.039a |
|
Female | 94 (38.4) | 72 (70.5) |
|
| Pathological
stage |
| I | 154 (62.9) | 84 (34.4) |
<0.001b |
| II | 25 (10.2) | 26 (10.7) |
|
|
III | 47 (19.2) | 73 (29.9) |
|
| IV | 19 (7.8) | 61 (25.0) |
|
| Historical
grade |
| G1 | 8 (3.3) | 2 (0.8) |
<0.001c |
| G2 | 132 (53.9) | 79 (32.4) |
|
| G3 | 90 (36.7) | 105 (43.0) |
|
| G4 | 15 (6.1) | 58 (23.8) |
|
| T stage |
| T1 | 157 (64.1) | 87 (35.7) |
<0.001b |
| T2 | 27 (11.0) | 35 (14.3) |
|
| T3 | 60 (24.5) | 112 (45.9) |
|
| T4 | 1 (0.4) | 10 (4.1) |
|
| N stage |
| N0 | 120 (49.0) | 112 (45.9) | 0.024a |
| N1 | 2 (0.8) | 12 (4.9) |
|
| Nx | 123 (50.2) | 120 (49.2) |
|
| M stage |
| M0 | 227 (92.7) | 185 (75.8) |
<0.001b |
| M1 | 18 (7.3) | 59 (24.2) |
|
| Outcome |
|
Alive | 199 (81.2) | 129 (52.9) |
<0.001b |
|
Dead | 46 (18.8) | 155 (47.1) |
|
ROCs were used to determine the prognostic accuracy
of the risk score, histological grade and pathological stage in
patients with ccRCC. The results revealed that in TCGA cohort, the
accuracy of the risk score [area under the curve (AUC), 0.736] was
superior compared with those of histological grade (AUC, 0.681) and
pathological stage (AUC, 0.720) in predicting 5-year survival
(Fig. 5I). In predicting 5-year
recurrence, the risk score (AUC, 0.728) was superior compared with
histological grade (AUC, 0.722) but inferior to pathological stage
(AUC, 0.820) (Fig. 5J). The
combination of these three characteristics improved the accuracy of
predicting the 5-year survival and recurrence of the same cohort
(AUC, 0.782 and 0.859, respectively). Therefore, the risk score was
able to correctly predict the prognosis for patients with
ccRCC.
Construction of nomograms based on the
m6A-related signature
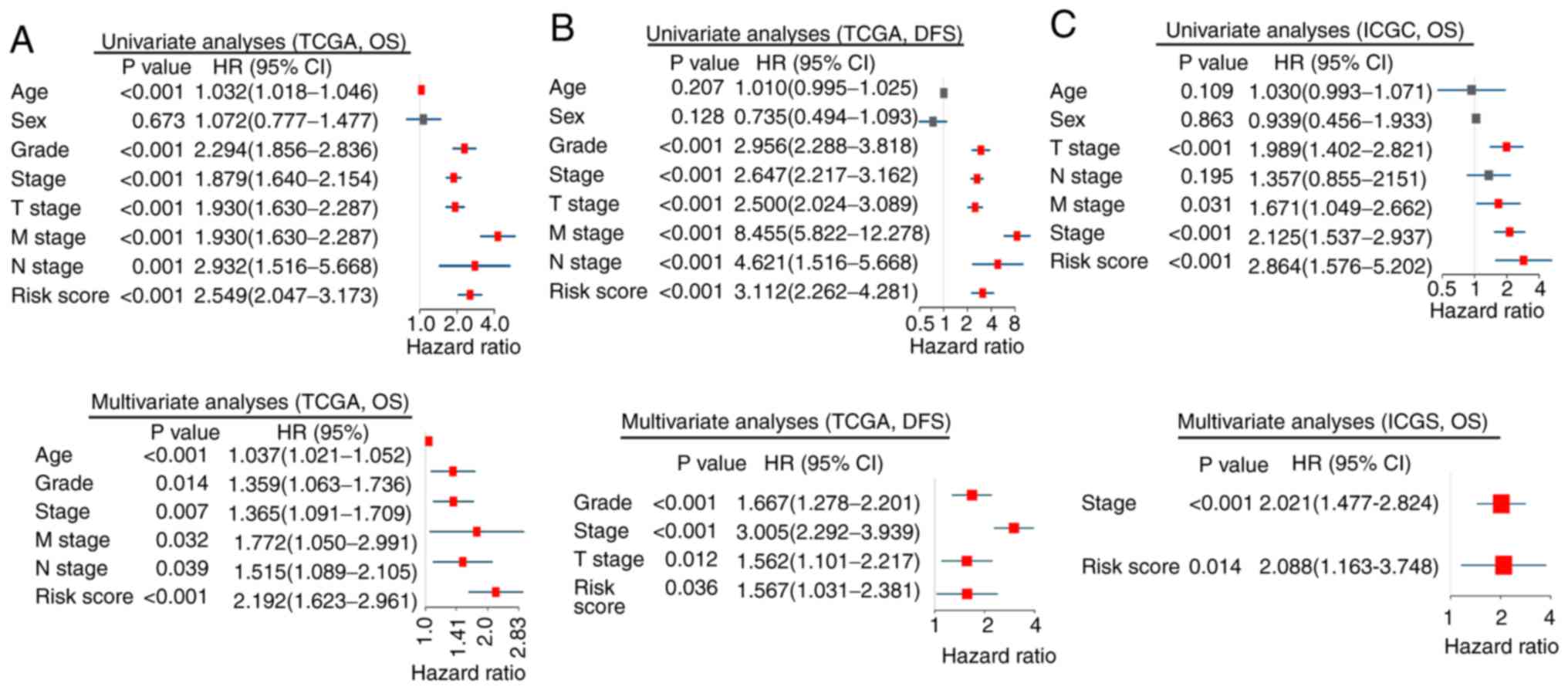
In TCGA cohort, univariate Cox regression analyses
demonstrated that the risk score, pathological stage, histological
grade, T, M and N stage had prognostic value in OS and DFS, whereas
age was associated exclusively with OS (Fig. 6A and B). These factors were used for
multivariate Cox regression analysis. The risk score, histological
grade and pathological stage remained significantly associated with
OS and DFS. Additionally, age, M and N stage were independent
prognostic factors of OS, whereas T stage was an independent
prognostic factor of DFS (Fig. 6A and
B). Similar results were observed in the validation cohort,
where the risk score (P=0.014) and pathological stage (P<0.001)
were associated with OS in multivariate analyses (Fig. 6C).
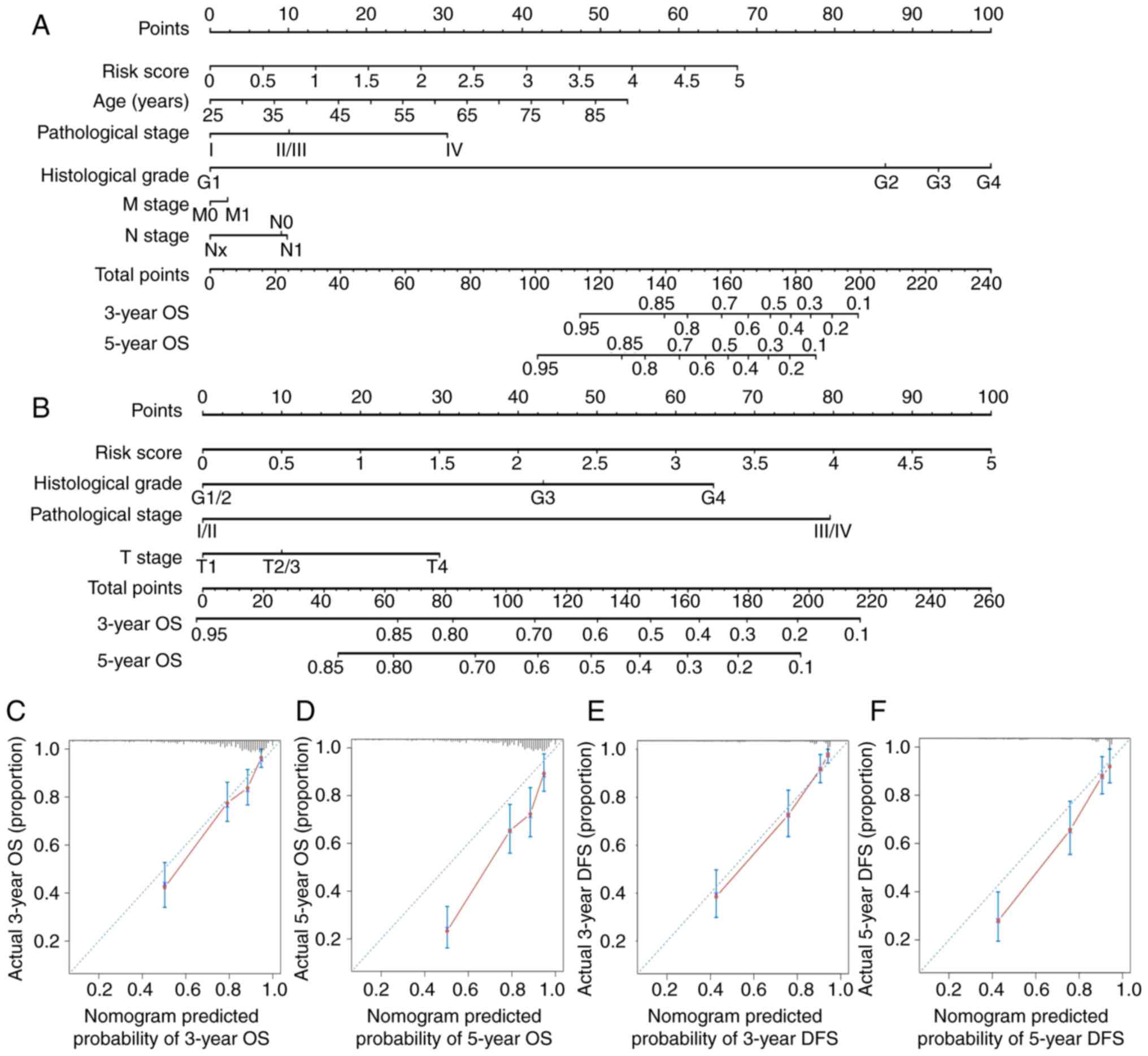
Nomograms for 3- and 5-year OS and DFS were
constructed based on the m6A-related signature and other
independent prognostic factors in multivariate Cox regression
analyses (Fig. 7A and B). The
c-indices of nomograms were 0.783±0.018 (mean ± SEM) and
0.819±0.018 for OS and DFS, respectively, indicating that the
prognostic prediction of nomograms was largely consistent with the
actual OS and DFS in patients with ccRCC. Additionally, calibration
plots demonstrated that nomogram prediction exhibited an agreement
with actual 3-year OS and DFS (Fig. 7C
and E) and a relative agreement with actual 5-year OS and DFS
(Fig. 7D and F).
Validation of m6A RNA methylation by
in vitro experiments
RT-qPCR was used to validate the expression levels
of the seven selected m6A RNA methylation regulators in four human
RCC cell lines (SW839, SN12C, 786-O and OSRC-2) and one human
normal renal tubular epithelial cell line (HK-2). The results
demonstrated significant differences in the expression levels of
five m6A RNA methylation regulators (KIAA1429, ALKBH3, HNRNPA2B1,
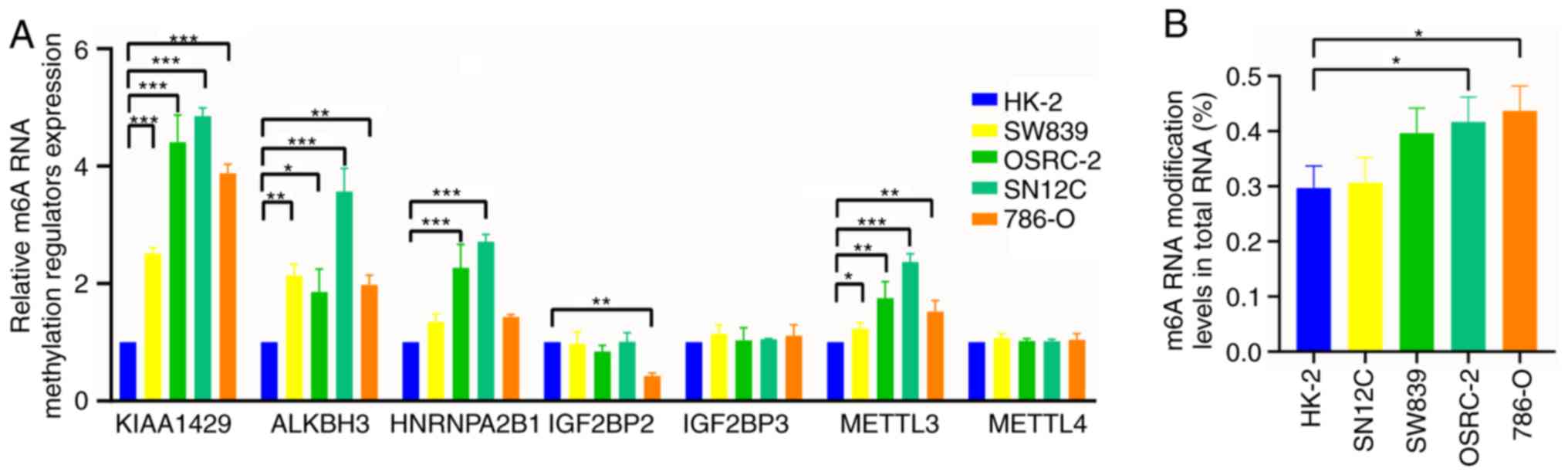
IGF2BP2 and METTL3) between RCC and normal renal cells (Fig. 8A). In addition, m6A RNA modification
levels in the cell lines were detected. The m6A RNA modification
levels in RCC cell lines SW839 and 786-O were significantly higher
compared with that in HK-2 cells (Fig.
8B).
Discussion
As the most common type of adult kidney cancer,
ccRCC is characterized by poor prognosis and a high risk of
metastasis and recurrence (29).
Epigenetic modifications, including RNA modification (21,22,24),
DNA methylation (30), histone
modification (31) and microRNA
changes (32,33), serve diverse functions in the
malignant progression and prognosis of ccRCC. As the most prevalent
type of internal RNA modification, m6A RNA methylation has gained
increasing attention over the past decade (5,7,8,12).
At present, the functions of m6A RNA methylation regulators in
ccRCC are unclear. The present study investigated the associations
between the expression of these genes and clinicopathological
features including prognosis, explored the potential biological
processes and constructed a m6A-related signature and nomograms for
prognostic prediction of ccRCC.
A total of 15 of the 19 analyzed m6A RNA methylation
regulators were differentially expressed between ccRCC and adjacent
non-tumor tissues, indicating that the m6A RNA methylation
regulators served important roles in the tumorigenesis of ccRCC.
Among the m6A RNA methylation regulators, ‘writers’ are considered
to be the main regulators of m6A in ccRCC (22), and Lobo et al (21) reported that the mRNA deregulation of
the ‘writers’ was associated with clinicopathological features and
survival of patients with RCC. The present study also confirmed the
major role of ‘writers’ by analyzing the network and associations
among 19 m6A RNA methylation regulators. In the present study, WTAP
served an important role among the ‘writers’ and was significantly
upregulated in ccRCC tissues. In a study by Tang et al
(24), the expression of WTAP was
also significantly upregulated in RCC cell lines and tissues, and
high expression of WTAP was associated with poor OS in patients
with ccRCC. These results further suggested that WTAP was a hub
gene among the ‘writers’ and served an oncogenic role in ccRCC.
A previous study has reported that WTAP can interact
and colocalize with the METTL3-METTL14 heterodimer to affect m6A
RNA methylation on nuclear RNA (34). The results of the present study
demonstrated a wide range of interactions, especially known
interactions within the ‘writers’, and the expression of WTAP was
also associated with the expression levels of the ‘writers’ METTL3
and METTL14.
As members of the ‘writers’, METTL3 and METTL14
exhibit completely opposite functions in different types of tumors.
In acute myeloid leukemia, METTL3 and METTL14 serve oncogenic
roles; compared with normal hematopoietic cells, the expression of
METTL3 and METTL14 is significantly upregulated in acute myeloid
leukemia cells (35). By contrast,
METTL3 and METTL14 are regarded as suppressor genes in
glioblastoma, and knockdown of METTL3 and METTL14 promotes the
growth, self-renewal and tumorigenesis of human glioblastoma stem
cells (16). The oncogenic role of
METTL3 has been validated in hepatocellular carcinoma (19), and the present study also revealed
that METTL3 was more abundant in ccRCC compared with adjacent
non-tumor tissues, and that patients with ccRCC with upregulated
METTL3 exhibited a shorter OS. METTL14 acts as a suppressor gene in
7 of the 37 types of cancer in TCGA (10). In hepatocellular carcinoma,
downregulation of METTL14 can significantly promote tumor
metastasis in vivo and in vitro (36). In ccRCC, the expression of METTL14
has been reported to be significantly decreased in ccRCC tissues
compared with non-tumor tissues, and METTL14 may inhibit renal
cancer cell migration and invasion by abrogating the expression of
purinergic receptor P2RX6 (23).
The results of the present study revealed that the expression of
METTL14 was significantly decreased in ccRCC compared with adjacent
non-tumor tissues, especially in high histological grade and high
pathological stage ccRCC tissues, and patients with ccRCC with low
expression of METTL14 exhibited longer OS and DFS. These results
suggested that the m6A RNA methylation regulators may serve diverse
roles in different tumors, and that even the same regulator may
exert different effects depending on the tissue specificity. In the
present study, METTL3 and METTL14 exhibited opposite functions in
ccRCC, which was also reflected in the opposite values of
coefficients. Although KIAA1429 was upregulated in ccRCC compared
to adjacent non-tumor tissues, it was significantly prone to be
upregulated in low pathological stage and low histological grade
ccRCC tissues, which contributed to the negative coefficient and
its role as a suppressor gene in ccRCC.
The ‘reader’ genes IGF2BP1, IGF2BP2, IGFBP3 and
HNRNPA2B1 were upregulated in high pathological stage and high
histological grade ccRCC tissues. In addition, upregulation of each
gene was significantly associate with poor OS or DFS. The oncogenic
features of these four genes also have been identified in at least
seven types of cancer based on the 33 types of cancer in TCGA
(10). Among them, IGF2BP3
functions as an oncogene in 13 of the 33 types of cancer in TCGA
and 33 cohorts across seven types of tissues in the Gene Expression
Omnibus. HNRNPA2B1 is associated with a high risk in nine types of
cancer and a proactive role in four types of cancer in TCGA
(10). However, only IGF2BP2,
IGF2BP3 and HNRNPA2B1 were selected by LASSO Cox regression
analyses to build the m6A-related signature. Of note, IGF2BP1,
IGF2BP2 and IGF2BP3 were positively associated with each other
(Fig. 5A), which may attribute to
the removal of IGF2BP1 as a factor.
The expression levels of the m6A methylation
‘erasers’ FTO, ALKBH3 and ALKBH5 in ccRCC tissues were higher
compared with those in adjacent non-tumor tissues, although their
expression levels were not associated with the histological grade
and pathological stage of ccRCC, and only patients with high
expression levels of ALKBH3 exhibited a poor prognosis. Thus,
ALKBH3 was the only ‘eraser’ selected for further analysis.
According to the expression similarities of the m6A
RNA methylation regulators in the present study, the patients from
TCGA cohort were divided into three clusters by consensus
clustering. GO and KEGG analyses demonstrated that the clustering
results were closely associated with the malignancy of ccRCC via
RNA metabolism and malignancy-related pathways, including primary
immunodeficiency, regulation of autophagy and response to oxidative
stress. The m6A RNA methylation regulators can influence almost
every step of RNA metabolism (11),
and their alteration is associated with the alterations of p53 and
VHL (7,22). Due to the vital roles of RNA
biology, p53 and VHL in the development of ccRCC (32,37),
the dysregulated expression of the m6A RNA methylation regulators
may result in abnormal RNA metabolism and alteration of p53 and
VHL, which may affect the initiation and progression of ccRCC. The
results of the bioinformatics analysis in the present study were in
accordance with this assumption.
Building a risk signature based on the expression
levels of m6A RNA methylation regulators may help predict the
clinical survival outcomes of patients with ccRCC, which is
regarded as a vital topic of research (9,10,17).
The present study constructed a m6A-related risk signature, which
achieved good performance in prognostic stratification in the
training (TCGA) and validation (ICGC) cohorts. In addition, the
risk signature correctly predicted the prognosis and stratified the
OS and DFS for patients with ccRCC with different histological
grades and pathological stages. This m6A-related risk signature and
other independent prognostic factors were used to build nomograms
for 3- and 5-year OS and DFS. Calibration plots and the c-indices
revealed that the prognostic prediction of the nomograms was
largely consistent with the actual OS and DFS, especially the
actual 3-year OS and DFS. Nomograms based on the m6A-related risk
signature may serve as a useful evaluation tool to perform
personalized recurrence and mortality risk identification in
patients with ccRCC.
The results of RT-qPCR in the present study
indicated that five of the seven m6A RNA methylation regulators
exhibited dysregulated expression between RCC cell lines and a
normal renal tubular epithelial cell line, suggesting that
dysregulated expression levels of m6A RNA methylation regulators
served an important role in the carcinogenesis of ccRCC. The
‘writer’ genes KIAA1429 and METTL3 were upregulated in RCC cell
lines compared with the normal renal tubular epithelial cell line.
It has been reported that the upregulation of ‘writers’, including
KIAA1429 and METTL3, is associated with invasiveness of cancer
cells, aggressive clinicopathological features and poor prognosis
in lung, liver, gastric and prostate cancer (7,19,21,38–40).
Mechanistically, this could partially explain the result of the
present study that m6A RNA modification levels were higher in RCC
cell lines compared with the normal renal tubular epithelial cell
line. A similar increase in the levels of m6A RNA modification was
also reported in gastric, prostate and pancreatic cancer (39–41).
Thus, higher levels of m6A RNA modification in total RNA may result
in the development of cancer-related features.
In conclusion, the present study systematically
demonstrated the prevalent dysregulated expression, biological
function and prognostic value of m6A RNA methylation regulators in
ccRCC. The dysregulated expression of the m6A RNA methylation
regulators was associated with differential expression of genes
enriched in RNA metabolism- and malignancy-related pathways. In
future studies, methylated m6A RNA immunoprecipitation sequencing
and m6A-sequencing may help identify the definite target mRNAs of
the m6A RNA modifications during ccRCC initiation and progression.
For clinical practices, the present study constructed m6A-related
nomograms that effectively predicted the outcome of patients with
ccRCC. Dysregulated expression of the m6A RNA methylation
regulators and dysregulated m6A RNA methylation levels were also
validated in multiple RCC cells by in vitro experiments.
Functional studies and mechanistic analyses of m6A regulators may
be helpful to the development of m6A-targeted treatments for
ccRCC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was funded by the Shanghai Science
Committee Foundation (grant no. 19411967700) and the Natural
Science Foundation of China (grant no. 81472389).
Availability of data and materials
Data of the mRNA expression of m6A RNA methylation
regulators and corresponding clinicopathological features were
retrieved from TCGA (http://cancergenome.nih.gov/) and ICGC (https://icgc.org/), which are openly available.
Authors' contributions
ZTZ and XDY conceived and designed the study. XDY
acquired the funding. ZTZ, SYM and YDG collected and collated the
data. All authors were involved in the analysis and interpretation
of data. ZTZ and SYM performed all the experiments. ZTZ wrote the
manuscript. XDY, SYM and WTZ critically reviewed and revised the
manuscript. JL and CL designed the tables and figures. All authors
read and approved the manuscript and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work were appropriately investigated
and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
m6A
|
N6-methyladenosine
|
|
WTAP
|
Wilms tumor 1-associated protein
|
|
ZC3H13
|
zinc finger CCCH domain-containing
protein 13
|
|
METTL3
|
methyltransferase like 3
|
|
RBM15
|
binding motif protein 15
|
|
FTO
|
fat mass and obesity-associated
protein
|
|
ALKBH3
|
alkB homolog 3
|
|
YTHDC1
|
YTH domain-containing 1
|
|
YTHDF1
|
YTH N6-methyladenosine
RNA-binding protein 1
|
|
IGF2BP1
|
insulin-like growth factor 2
mRNA-binding protein 1
|
|
HNRNPC
|
heterogeneous nuclear
ribonucleoprotein C
|
|
HNRNPA2B1
|
heterogeneous nuclear
ribonucleoprotein A2/B1
|
|
RBMX
|
RNA-binding motif protein X-linked
|
|
ccRCC
|
clear cell renal cell carcinoma
|
|
TCGA
|
The Cancer Genome Atlas
|
|
ICGC
|
International Cancer Genome
Consortium
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
GO
|
gene ontology
|
|
DAVID
|
Database for Annotation, Visualization
and Integrated Discovery
|
|
DEG
|
differentially expressed gene
|
|
OS
|
overall survival
|
|
LASSO
|
least absolute shrinkage and selection
operator
|
|
DFS
|
disease-free survival
|
|
ROC
|
Receiver operating characteristic
|
|
AUC
|
area under the curve
|
References
|
1
|
Boccaletto P, Machnicka MA, Purta E,
Piatkowski P, Baginski B, Wirecki TK, de Crecy-Lagard V, Ross R,
Limbach PA, Kotter A, et al: MODOMICS: A database of RNA
modification pathways. 2017 update. Nucleic Acids Res.
46:D303–D307. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lewis CJ, Pan T and Kalsotra A: RNA
modifications and structures cooperate to guide RNA-protein
interactions. Nat Rev Mol Cell Biol. 18:202–210. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gilbert WV, Bell TA and Schaening C:
Messenger RNA modifications: Form, distribution, and function.
Science. 352:1408–1412. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Karijolich J, Kantartzis A and Yu YT: RNA
Modifications: A Mechanism that Modulates Gene Expression. Methods
Mol Biol. 629:1–19. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang S, Sun C, Li J, Zhang E, Ma Z, Xu W,
Li H, Qiu M, Xu Y, Xia W, et al: Roles of RNA methylation by means
of N6-methyladenosine (m6A) in human cancers.
Cancer Lett. 408:112–120. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Desrosiers R, Friderici K and Rottman F:
Identification of methylated nucleosides in messenger RNA from
Novikoff hepatoma cells. Proc Natl Acad Sci USA. 71:3971–3975.
1974. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dominissini D, Moshitch-Moshkovitz S,
Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K,
Jacob-Hirsch J, Amariglio N, Kupiec M, et al: Topology of the human
and mouse m6A RNA methylomes revealed by m6A-seq. Nature.
485:201–206. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Meyer KD, Saletore Y, Zumbo P, Elemento O,
Mason CE and Jaffrey SR: Comprehensive analysis of mRNA methylation
reveals enrichment in 3′ UTRs and near stop codons. Cell.
149:1635–1646. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun T, Wu R and Ming L: The role of m6A
RNA methylation in cancer. Biomed Pharmacother. 112:1086132019.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Y, Xiao J, Bai J, Tian Y, Qu Y, Chen X,
Wang Q, Li X, Zhang Y and Xu J: Molecular characterization and
clinical relevance of m6A regulators across 33 cancer
types. Mol Cancer. 18:1372019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dai D, Wang H, Zhu L, Jin H and Wang X:
N6-methyladenosine links RNA metabolism to cancer progression. Cell
Death Dis. 9:1242018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Niu Y, Zhao X, Wu YS, Li MM, Wang XJ and
Yang YG: N6-methyl-adenosine (m6A) in RNA: An old modification with
a novel epigenetic function. Genomics Proteomics Bioinformatics.
11:8–17. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fustin JM, Doi M, Yamaguchi Y, Hida H,
Nishimura S, Yoshida M, Isagawa T, Morioka MS, Kakeya H, Manabe I,
et al: RNA-methylation-dependent RNA processing controls the speed
of the circadian clock. Cell. 155:793–806. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Geula S, Moshitch-Moshkovitz S,
Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V,
Peer E, Mor N, Manor YS, et al: Stem cells. m6A mRNA methylation
facilitates resolution of naive pluripotency toward
differentiation. Science. 347:1002–1006. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xiang Y, Laurent B, Hsu CH, Nachtergaele
S, Lu Z, Sheng W, Xu C, Chen H, Ouyang J, Wang S, et al: RNA
m6A methylation regulates the ultraviolet-induced DNA
damage response. Nature. 543:573–576. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, Sun
G, Lu Z, Huang Y, Yang CG, et al: m6A RNA methylation
regulates the self-renewal and tumorigenesis of glioblastoma stem
cells. Cell Rep. 18:2622–2634. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chai RC, Wu F, Wang QX, Zhang S, Zhang KN,
Liu YQ, Zhao Z, Jiang T, Wang YZ and Kang CS: m6A RNA
methylation regulators contribute to malignant progression and have
clinical prognostic impact in gliomas. Aging. 11:1204–1225. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li Z, Weng H, Su R, Weng X, Zuo Z, Li C,
Huang H, Nachtergaele S, Dong L, Hu C, et al: FTO plays an
oncogenic role in acute myeloid leukemia as a
N6-methyladenosine RNA demethylase. Cancer Cell.
31:127–141. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen M, Wei L, Law CT, Tsang FH, Shen J,
Cheng CL, Tsang LH, Ho DW, Chiu DK, Lee JM, et al: RNA
N6-methyladenosine methyltransferase-like 3 promotes liver cancer
progression through YTHDF2-dependent posttranscriptional silencing
of SOCS2. Hepatology. 67:2254–2270. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang C, Samanta D, Lu H, Bullen JW, Zhang
H, Chen I, He X and Semenza GL: Hypoxia induces the breast cancer
stem cell phenotype by HIF-dependent and ALKBH5-mediated
m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA.
113:E2047–E2056. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lobo J, Barros-Silva D, Henrique R and
Jeronimo C: The emerging role of epitranscriptomics in cancer:
Focus on urological tumors. Genes (Basel). 9(pii): E5522018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou J, Wang J, Hong B, Ma K, Xie H, Li L,
Zhang K, Zhou B, Cai L and Gong K: Gene signatures and prognostic
values of m6A regulators in clear cell renal cell carcinoma-a
retrospective study using TCGA database. Aging (Albany NY).
11:1633–1647. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gong D, Zhang J, Chen Y, Xu Y, Ma J, Hu G,
Huang Y, Zheng J, Zhai W and Xue W: The m6A-suppressed
P2RX6 activation promotes renal cancer cells migration and invasion
through ATP-induced Ca2+ influx modulating ERK1/2
phosphorylation and MMP9 signaling pathway. J Exp Clin Cancer Res.
38:2332019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tang J, Wang F, Cheng G, Si S, Sun X, Han
J, Yu H, Zhang W, Lv Q, Wei JF and Yang H: Wilms' tumor
1-associating protein promotes renal cell carcinoma proliferation
by regulating CDK2 mRNA stability. J Exp Clin Cancer Res.
37:402018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ghosh A and Barman S: Application of
Euclidean distance measurement and principal component analysis for
gene identification. Gene. 583:112–120. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Senbabaoglu Y, Michailidis G and Li JZ:
Critical limitations of consensus clustering in class discovery.
Sci Rep. 4:62072014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Simon N, Friedman J, Hastie T and
Tibshirani R: Regularization paths for cox's proportional hazards
model via coordinate descent. J Stat Softw. 39:1–13. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Amin MB, Amin MB, Tamboli P, Javidan J,
Stricker H, de-Peralta Venturina M, Deshpande A and Menon M:
Prognostic impact of histologic subtyping of adult renal epithelial
neoplasms: An experience of 405 cases. Am J Surg Pathol.
26:281–291. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ricketts CJ, Morris MR, Gentle D, Brown M,
Wake N, Woodward ER, Clarke N, Latif F and Maher ER: Genome-wide
CpG island methylation analysis implicates novel genes in the
pathogenesis of renal cell carcinoma. Epigenetics. 7:278–290. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mosashvilli D, Kahl P, Mertens C,
Holzapfel S, Rogenhofer S, Hauser S, Buttner R, Von Ruecker A,
Muller SC and Ellinger J: Global histone acetylation levels:
Prognostic relevance in patients with renal cell carcinoma. Cancer
Sci. 101:2664–2669. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xing T and He H: Epigenomics of clear cell
renal cell carcinoma: Mechanisms and potential use in molecular
pathology. Chin J Cancer Res. 28:80–91. 2016.PubMed/NCBI
|
|
33
|
Yoshino H, Yonezawa T, Yonemori M,
Miyamoto K, Sakaguchi T, Sugita S, Osako Y, Tatarano S, Nakagawa M
and Enokida H: Downregulation of microRNA-1274a induces cell
apoptosis through regulation of BMPR1B in clear cell renal cell
carcinoma. Oncol Rep. 39:173–181. 2018.PubMed/NCBI
|
|
34
|
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang
L, Jia G, Yu M, Lu Z, Deng X, et al: A METTL3-METTL14 complex
mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem
Biol. 10:93–95. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Weng H, Huang H, Wu H, Qin X, Zhao BS,
Dong L, Shi H, Skibbe J, Shen C, Hu C, et al: METTL14 inhibits
hematopoietic stem/progenitor differentiation and promotes
leukemogenesis via mRNA m6A Modification. Cell Stem
Cell. 22:191–205.e9. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH,
Wang F, Wang TT, Xu QG, Zhou WP and Sun SH: METTL14 suppresses the
metastatic potential of hepatocellular carcinoma by modulating
N6-methyladenosine-dependent primary MicroRNA
processing. Hepatology. 65:529–543. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Harlander S, Schonenberger D, Toussaint
NC, Prummer M, Catalano A, Brandt L, Moch H, Wild PJ and Frew IJ:
Combined mutation in Vhl, Trp53 and Rb1 causes clear cell renal
cell carcinoma in mice. Nat Med. 23:869–877. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lan T, Li H, Zhang D, Xu L, Liu H, Hao X,
Yan X, Liao H, Chen X, Xie K, et al: KIAA1429 contributes to liver
cancer progression through N6-methyladenosine-dependent
post-transcriptional modification of GATA3. Mol Cancer. 18:1862019.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cai J, Yang F, Zhan H, Situ J, Li W, Mao Y
and Luo Y: RNA m6A Methyltransferase METTL3 promotes the
growth of prostate cancer by regulating hedgehog pathway. Onco
Targets Ther. 12:9143–9152. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu T, Yang S, Sui J, Xu SY, Cheng YP,
Shen B, Zhang Y, Zhang XM, Yin LH, Pu YP and Liang GY: Dysregulated
N6-methyladenosine methylation writer METTL3 contributes to the
proliferation and migration of gastric cancer. J Cell Physiol.
235:548–562. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xia T, Wu X, Cao M, Zhang P, Shi G, Zhang
J, Lu Z, Wu P, Cai B, Miao Y and Jiang K: The RNA m6A
methyltransferase METTL3 promotes pancreatic cancer cell
proliferation and invasion. Pathol Res Pract. 215:1526662019.
View Article : Google Scholar : PubMed/NCBI
|