Introduction
With the current advances in determining the
mechanisms underlying estrogen-dependent cell proliferation in
breast cancer, it has been possible to treat patients with in
situ (or even invasive) ductal carcinoma, which are estrogen
receptor (ER)α positive, through hormone therapy with antagonist
molecules, such as tamoxifen or fulvestrant (1). Although these treatments are partially
responsible for an ~40% decrease in the mortality rate over the
last three decades (2), one-third
of patients with breast cancer are not positive for hormone nuclear
receptors and, therefore, do not respond favorably to such
treatments. These treatments may be detrimental for certain
intrinsic molecular subtypes (3).
In the last 20 years, signal transduction through G protein-coupled
receptor 30 (GPR30) has been considered a mechanism involved in the
resistance to endocrine therapy, owing to its ability to trigger
signaling pathways induced by antiestrogens (4). Additionally, it has been reported that
GPR30 serves a regulatory role in several cellular processes, such
as migration (5), proliferation
(6) and cell survival (7) in breast cancer.
Although GPR30 was initially classified as an orphan
receptor, subsequent biochemical studies demonstrated that this
membrane receptor is strongly stimulated by estrogens, as well as
by other compounds, including insecticides (i.e., DDT),
phytoestrogens (i.e., genistein), xenoestrogens (i.e., bisphenol A)
and antagonistic modulators of ERα and ERβ (i.e., fulvestrant and
tamoxifen) (8,9). In addition, selective binding
compounds for GPR30, such as the agonists G1
[1-(4-[6-bromobenzo-(1,3)-dioxol-5-yl]-3a,4,5,9b-tetrahydro-3H-cyclopenta-[c]-quinolin-8-yl)-ethanone]
and G36
[4-(6-bromo-benzo-[1,3]-dioxol-5-yl)-8isopropyl-3a,4,5,9b-tetrahydro-3H-cyclopenta-(c)-quinoline],
and the antagonist G15
[4-(6-bromo-benzo-[1,3]-dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta-(c)-quinoline],
have been synthesized to monitor the specific biological activity
of GPR30 (10–12).
Previous studies on GPR30 function have reported the
involvement of this receptor in the induction of the epidermal
growth factor receptor/ERK signaling pathway that promotes
tamoxifen resistance (13), as well
as in the transactivation of genes linked to proliferation and
migration in breast cancer, such as connective tissue growth factor
and N-acylsphingosine amidohydrolase 1 (14,15).
There is also evidence that GPR30 expression in breast cancer can
be regulated by a positive feedback loop with hypoxia-inducible
factor 1 (HIF-1) (16). Recently,
it was also reported that the activity of both proteins, GPR30 and
HIF-1, is linked to a complex crosstalk process with the Notch1
signaling pathway, which in turn induces epithelial-mesenchymal
transition (EMT) (17). However,
the data reported thus far from GPR30 expression and function
studies during the course of breast cancer is contradictory; for
example, GPR30 expression has been reported to be downregulated
during breast cancer development, and variously reported to
function as a suppressor or inducer of proliferation or migration
(14,18–26).
Considering the importance of the expression profile of GPR30 for
the integral understanding of its role in the progress of breast
cancer, the present study aimed to determine the transcriptional
mechanisms that regulate GPR30 expression in cellular models of
different breast cancer subtypes (metastatic and
non-metastatic).
Materials and methods
Cell cultures
The non-metastatic MCF-7 [HTB-22; American Type
Culture Collection (ATCC)] and metastatic MDA-MB-231 (HTB-26; ATCC)
breast cancer cell lines, as well as the normal breast cell line
MCF-10A (CRL-10317; ATCC), were cultured in DMEM (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS (Gibco; Thermo
Fisher Scientific, Inc.), Leibovitz's L-15 medium (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS and Mammary
Epithelial Cells Growth Medium (Lonza Walkersville, Inc.)
supplemented with cholerae toxin (Sigma-Aldrich; Merck KGaA),
respectively. All cultures were incubated at 37°C in a 5%
CO2 atmosphere. All cell lines were tested for
mycoplasma and cellular authentication by commercial PCR Mycoplasma
Detection Set (Takara Bio, Inc.), according with to manufacturer's
instructions, and capillary electrophoresis in a 3500 Genetic
Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.),
respectively.
Western blotting
For GPR30 immunoblotting, total protein was isolated
from 1.5×106 cells of MCF-10A, MCF-7, MDA-MB-231 by
lysis with RIPA buffer (1 M Tris/HCl pH 7.4; 6.5 M EDTA pH 8.0; 5 M
NaCl; 0.4% deoxycholate; 0.8% IGEPAL-CA-630), PMSF and Complete
Mini protease inhibitor cocktail tablets (Roche Diagnostics). For
the ETS translocation variant (ETV)1, ETV4 and ETV5 immunoblots,
nuclear extracts were obtained from MCF-10A, MCF-7 and MDA-MB-231
cell lines, following the protocol described by Schreiber et
al (27). The total and nuclear
protein extracts (80 µg) were separated by 10% SDS-PAGE and
transferred to PVDF membranes (EMD Millipore). Subsequently, the
membranes were incubated overnight with the following primary
antibodies in TBS-Tween 20 (TBST; 50 mM Tris; 140 mM NaCl; 0.1%
Tween-20) with 4% BSA: Anti-GPR30 (1:625; cat. no. ab39742; Abcam),
anti-E26 transformation-specific (ETS) translocation variant (ETV4;
1:100; cat. no. sc-113; Santa Cruz Biotechnology, Inc.), anti-ETV5
(1:250; cat. no. sc-22807; Santa Cruz Biotechnology, Inc.) and
anti-ETV1 (1:250; cat. no. ab81086; Abcam). As loading control,
GAPDH expression was detected with anti-GAPDH (1:10,000; cat. no.
MAB374; Merck KGaA) for total extracts. Commercial nuclear extracts
of MCF-7 (cat. no. sc-2149; Santa Cruz Biotechnology, Inc.) and
K-562 (cat. no. sc-2130; Santa Cruz Biotechnology, Inc.) were used
as positive controls (40 µg) for ETV5 and ETV4, respectively. In
the case of ETV1 and GPR30, 80 µg of total mouse brain and OVCAR3
nuclear extracts (courtesy of Ms. Isis Santos Paniagua) were used
as positive controls, respectively. The membranes were washed 3
times (15 min each) with TBST buffer and subsequently incubated
with the following horseradish peroxidase-conjugated secondary
antibodies: Goat anti-rabbit immunoglobulin G (IgG; 1:12,000; cat.
no. 111-035-003, Jackson ImmunoResearch Laboratories, Inc.) and
goat anti-mouse IgG (1:10,000; cat. no. 115-035-003; Jackson
ImmunoResearch Laboratories, Inc.). Immune complexes were
visualized by enhanced chemiluminescence with SuperSignal West Pico
Chemiluminescent Substrate (Pierce; Thermo Fisher Scientific,
Inc.), according to the manufacturer's instructions in a C-DiGit
Blot Scanner (LI-COR Biosciences) during an exposure of 12 min and
analyzed by Image Studio v2.1 software (LI-COR Biosciences).
Cell viability assay
MCF-7 and MDA-MB-231 cells were seeded into 96-well
culture plates at a density of 7.5×103 cells/well. The
adhered cells were subjected to a starvation period of 16 h and
stimulated afterwards with either 10 µM or 20 µM G15 (Azano
Biotech) or 10 µM G1 (Azano Biotech) (Fig. S1) or 10−6 M retinoic
acid (RA; Sigma-Aldrich; Merck KGaA) or vehicle (DMSO) incorporated
into the corresponding growth medium. Cells were subsequently fixed
at 0, 24, 48 and 72 h with 1.1% formaldehyde for 15 min, washed
with water three times and left to dry at room temperature. The
cells were stained with crystal violet for 20 min, washed three
times with water and allowed to dry at room temperature. Finally,
10% acetic acid was added for 20 min. The absorbance of each well
was determined at 590 nm using a Synergy HT microplate reader
(BioTek Instruments, Inc.).
Migration assay
A migration assay was performed on the MCF-7
(Fig. S2) and MDA-MB-231
metastatic cell lines. Cells were seeded in 6-well culture plates
with a 7.5×105 cell density and grown in Leibovitz's
L-15 medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% FBS, until they reached confluence. The cell monolayer was
carefully wounded using a 100 µl pipette tip and the detached cells
were removed by washing with PBS. The wounded monolayer cultures
were subjected to a 24 h starvation period and were incubated
afterwards for 48 h in serum-free medium containing 5 µM G1 (Azano
Biotech) or 5 µM G1 + 10 µM G15 (Azano Biotech). Images of the
wounds were capture at 0, 24 and 48 h using an Axiovert 25
microscope (Carl Zeiss AG) with a CP-Achromat 10×/0.25 Ph1
objective (Carl Zeiss AG) and a PowerShot A580 digital photographic
camera (Canon, Inc.) equipped with a DC150 camera adapter (Leica
Microsystems GmbH). The wound areas were analyzed with the Icy
bioimage analysis software v2.0.3.0 (http://icy.bioimageanalysis.org).
GPR30 5′ flanking region cloning
MCF-7 and MDA-MB-231 cells seeded and grown to 75%
confluence in 100 mm plates and genomic DNA was isolated according
to the procedure described by Sambrook et al (28). A 1,987 bp DNA fragment from both
genetic materials was amplified by PCR using the following
thermocycling conditions: Initial denaturation of 94°C for 30 sec;
followed by 30 cycles of 94°C for 30 sec, 50.4°C for 1 min, and
68°C for 4 min, followed by a final extension at 68°C for 7 min.
PCR was carried out with an Expand Long Template PCR system (Roche
Diagnostics) and the pair of specific primers described in Table I. The amplification product was
separated by 1.0% agarose gel electrophoresis, visualized by Visual
Violet Gel Additive (Amresco, LLC), purified with a Gene Clean III
kit (Bio 101; MP Biomedicals, LLC) and ligated with T4 DNA Ligase
(Promega Corporation) into a pGEM-T vector (Promega Corporation),
according to the manufacturer's instructions. The recombinant
plasmid was propagated in JM109 Escherichia coli (Promega
Corporation) and purified with a Wizard Plus SV Minipreps system
(Promega Corporation). To obtain a reliable sequence of the cloned
fragment, three different recombinant plasmids of each cell line
were sequenced with a Big Dye Terminator System and an ABI PRISM 77
automatic sequencer (Applied Biosystems; Thermo Fisher Scientific,
Inc.).
 | Table I.Oligonucleotides used in the
different applications. |
Table I.
Oligonucleotides used in the
different applications.
| Gene | Oligonucleotide
sequence (5′→3′) | Length (nt) |
|---|
| Genomic DNA |
|
|
|
5FGPR30-s |
AAGCAATAGGTCAACAAATCTCTAG | 25 |
|
5FGPR30-as |
GTCTCTGCACCGTGCAGCTTTCAAGA | 26 |
| Reporter
construction |
|
|
|
pGPR2.0 |
ACGCAGATCTTGTCAACAAATCTCTAG | 27 |
|
pGPR1.5 |
ACGCAGATCTTCTTGGGCACCTGTCCTAG | 29 |
|
pGPR1.0 |
ACGCAGATCTTGACTCTCTCCCTGGAG | 27 |
|
pGPR0.5 |
ACGCAGATCTTTTCCCACAGGCGACTC | 27 |
|
pGPR0.2 |
ACGCAGATCTTAGCATCTGTTCTTCCC | 27 |
|
pGPR-as |
ACGAAGCTTGTCTCTGCACCGTGCAGC | 27 |
| Reverse
transcription-PCR |
|
|
|
GPR30 | F:
TTCAGCAGTGCCGTGTAGA | 19 |
|
| R:
GTGTGCAGCTCCCGAGTC | 18 |
|
ETV5 | F:
GTTGTGCCTGAGAGACTGGAAG | 22 |
|
| R:
CATTGGCTGGGTCATCAAGAAG | 22 |
|
ETV4 | F:
CCCAACAAATGCCCATTTCATTGC | 24 |
|
| R:
AACGCTCACCAGCCACCTTC | 20 |
|
ETV1 | F:
GTACCACGACCCAGTGTATGAAC | 23 |
|
| R:
GGATGAGCCAGGAAGCCTTC | 20 |
|
GAPDH | F:
GCTCTCTGCTCCTCCTGTTTC | 21 |
|
| R:
ACGACCAAATCGTTGACTC | 19 |
| Mutagenesis |
|
|
|
ETSµ-s |
TACCTTCATTGCCCCCTGGGCCTGCTC | 27 |
|
ETSµ-as |
GAGCAGGCCCAGGGGGCAATGAAGGTA | 27 |
|
SOXµ-s |
ATTTCCCAAAACCATGACCCCTTTC | 25 |
|
SOXµ-as |
GAAAGGGGTCATGGAAAAGGGAAAT | 25 |
|
ERR1µ-s |
TCCCAAAACAATTTCCCCTTTCACTC | 26 |
|
ERR1µ-as |
GAGTGAAAGGGGAAATTGTTTTGGGA | 26 |
|
NGREµ-s |
ACTCTCTCCCTGTTGTTTCTTCCTAG | 26 |
|
NGREµ-as |
CTAGGAAGAAACAACAGGGAGAGAGT | 26 |
|
ETSc-s |
TACCTTCATTACTTCCGGTGCCTGCTC | 27 |
|
ETSc-as |
GAGCAGGCACCGGAAGTAATGAAGGTA | 27 |
Sequence analysis
The nucleotide sequences obtained from the fragment
of the 5′ flanking region of GPR30 cloned from the MCF-7 and
MDA-MB-231 cell lines was analyzed using BLAST (National Center for
Biotechnology Information; http://blast.ncbi.nlm.nih.gov). Both sequences were
compared with each other and with the chromosome 7 sequence
reported with the AC091729 accession number in the EMBL nucleotide
sequence database (https://www.ncbi.nlm.nih.gov/nuccore/18643712). In
addition, the binding sites for transcription factors were scanned
with the prediction program in the JASPAR website (http://jaspar.genereg.net).
Reporter gene constructs
The recombinant plasmid harboring the human GPR30
promoter from MCF-7 cells was used as template to amplify five
segments of different lengths (0.2, 0.5, 1.0, 1.5 and 2.0 kbp),
using specific forward and reverse primers with BglII and
HindIII restriction sites, respectively. Each amplicon was
purified using a GeneClean III Kit (MP Biomedicals, LLC) and
subsequently digested with BglII and HindIII
restriction enzymes (Roche Diagnostics). Each digested segment was
again purified and cloned into a pGL3-Enhancer firefly luciferase
reporter vector (Promega Corporation), previously digested with the
same restriction enzymes. The resulting constructs (pGPR2.0,
pGPR1.5, pGPR1.0, pGPR0.5, and pGPR0.2) were amplified by
transformation in competent JM109 E. coli cells (Promega
Corporation). The putative cis-acting elements located at
−628, −754, −892- and −896 bp from the translation start codon
(TSC) were selected based on their high similarity with consensus
sequences for ETS, negative glucocorticoid response element (NGRE),
estrogen-related receptor α (ERR1), SRY-box transcription factor
(SOX), and by site-directed mutagenesis in their ‘core’ sequence
using the reporter construct pGPR1.0 as the template and the
specific oligonucleotides described in Table I. Reporter constructs with
loss-of-function mutations (µ) were indicated as ETSµ, NGREµ, ERRµ
and SOXµ, and the construct with the only gain-of-function mutation
was named ETSc. Three clones of each plasmid were selected to
verify the fidelity of the mutagenesis procedure by incorporation
of fluorescent dideoxynucleotides using the Big Dye Terminator
system (Applied Biosystems; Thermo Fisher Scientific, Inc.),
according to the manufacturer's instructions.
Transfections
MCF-10A, MCF-7 and MDA-MB-231 cell lines were
cultured in 96-well plates with a density of 1.5×104
cells/well. The three cell lines were transiently transfected with
100 ng of each reporter gene construct, using the FuGENE HD
Transfection reagent (Roche Diagnostics) according to the
manufacturer's protocol. A total of 100 ng of pGL3 Enhancer
(Promega Corporation) plasmid was transfected to determine the
background luminescence, and the pRL-TK Renilla luciferase
control reporter plasmid (25 ng) was co-transfected as a
normalization control. After 48 h of expression, luciferase
activity was measured using the Dual-Luciferase Reporter assay
system (Promega Corporation) in a Sirius L Single Tube Luminometer
(Titertek-Berthold).
Transient expression assays with
phosphorylation inhibitors
To assess the role of the main phosphorylation
pathways activating the ETS factors associated with breast cancer
in the regulation of the GPR30 promoter region with the functional
ETS binding site, p38 kinase inhibitor SB203580, protein kinase A
(PKA) inhibitor H89, and ERK inhibitor PD98059 (all from Merck
KGaA) were used at a 10 µM concentration each in transient
expression assays. Transient transfections were performed in the
three cell seeded (1.5×104 cells/well) in 96-well
plates. Each well was transfected with 100 ng of the reporter
constructs pGPR0.5 and pGPR1.0 with or without inhibitors. The
empty pGL3-Enhancer vector (100 ng/well) and pRL-TK (25 ng/well)
were transfected as the background and internal control,
respectively. After 22 h of starvation, each of the three
inhibitors was added to the growth medium. Luciferase activity was
measured 48 h after the application of the inhibitors using the
Dual-Luciferase Reporter assay system in a Sirius L Single Tube
Luminometer, aforementioned.
Stimulation assays with RA
MCF-7 cells were cultured in 96-well plates at a
density of 1.5×104 cells/well, and transfected with
pGPR0.5 or pGPR1.0, aforementioned, and subsequently synchronized
for 24 h with DMEM without phenol red (Sigma-Aldrich; Merck KGaA),
supplemented with inactivated FBS (HyClone; GE Healthcare Life
Sciences) and stimulated with increasing RA concentrations in DMSO
(0, 10−12, 10−11, 10−10,
10−9, 10−8, 10−7 and
10−6 M) for 48 h. Reporter gene activity was measured
using the Dual-Luciferase Reporter assay system in a Sirius L
Single Tube Luminometer, aforementioned.
Reverse transcription (RT)-PCR
assays
MCF-7 cells were seeded at a density of
7.5×105 cells in 60 mm dishes with DMEM supplemented
with 10% FBS and incubated at 37°C in a 5% CO2 humid
atmosphere for 24 h. Subsequently, the cells were washed with PBS
and synchronized for 24 h with free-serum DMEM medium without
phenol red. Later the cells were treated with RA (10−6
M) or with vehicle (DMSO) for 48 h. At the end of the stimulation,
cells were washed with cold PBS and lysed with 1 ml of
Trizol® (Invitrogen; Thermo Fisher Scientific, Inc.).
The cell lysate was mixed vigorously with 0.2 ml of chloroform (MP
Biomedicals, LLC) for 30 sec and centrifuged for 40 min at 14,000
rpm and 4°C. The aqueous phase was recovered and mixed in 1 ml of
isopropanol (Amresco, LLC) to precipitate RNA, followed by
centrifugation at 14,000 rpm and 4°C for 1 h. The RNA pellet was
washed with 75% ethanol and centrifuged at 14,000 rpm and 4°C for
12 min. Finally, the total RNA was resuspended in water treated
with diethyl pyrocarbonate (Thermo Fisher Scientific, Inc.). cDNA
was synthesized using the Phusion RT-PCR kit (Finnzymes; Thermo
Fisher Scientific, Inc.), and PCR assays were performed using the
cDNA as template and specific oligonucleotides (Table I) for amplification of the
transcripts corresponding to GPR30, ETV1, ETV4, ETV5 and
housekeeping gene GAPDH. PCR was carried out with Clone Amp HiFi
PCR mix (Clontech Laboratories, Inc.) with the following
thermocycling conditions: Initial denaturation of 94°C for 30 sec;
followed by 30 cycles of 94°C for 30 sec, 58°C for 45 sec and 73°C
for 1 min, followed by a final extension period of 5 min at 74°C.
The amplification products were resolved in a 10% polyacrylamide
gel, stained with ethidium bromide (0.5 µg/ml) and visualized in a
UV transilluminator.
Statistical analysis
The results are expressed as the mean ± standard
deviation. Comparisons between >2 groups were made using ANOVA
followed by Tukey's post hoc test, or by Dunnett's post hoc test
for comparisons between treatments and the vehicle control.
Statistical analyzes were performed using the SigmaPlot 12.0
(Systat Software, Inc.) software for Windows. Each assay was
performed in triplicate. P<0.05 was considered to indicate a
statistically significant difference.
Results
GPR30 regulates cell viability and
migration of breast cancer cells
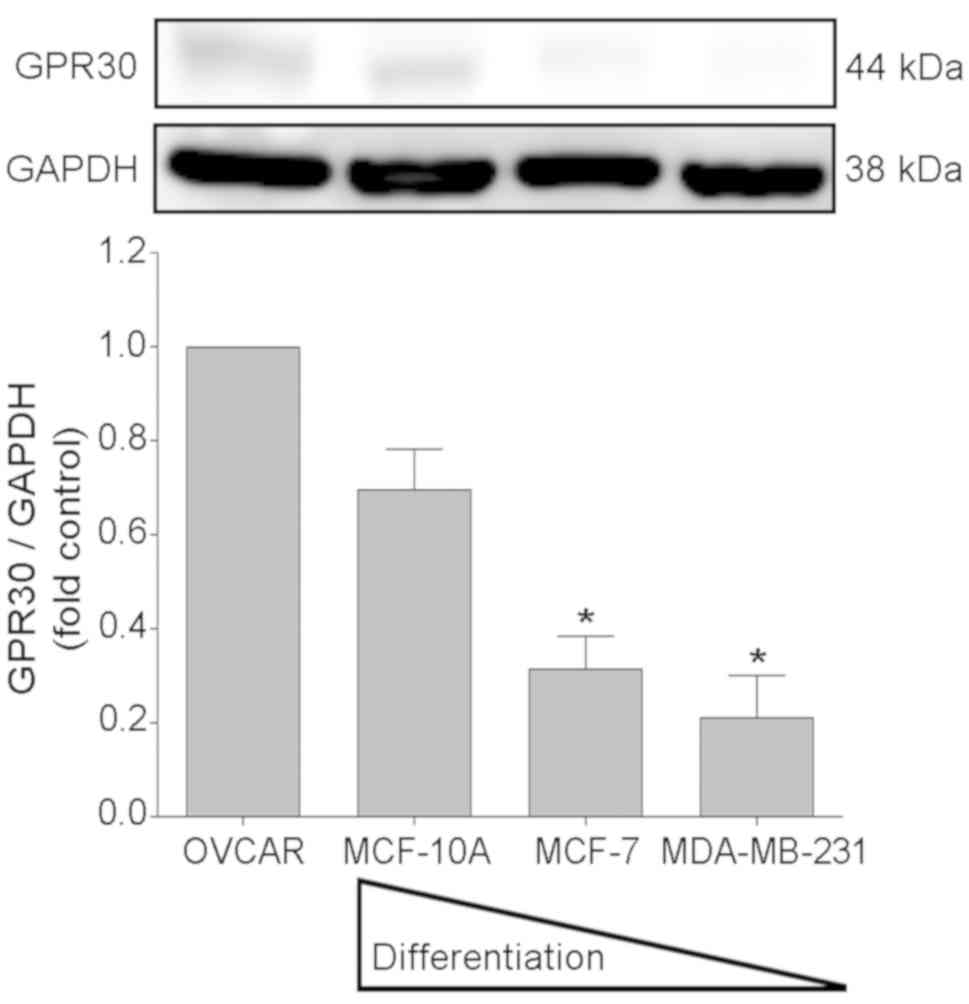
The assays carried out for the immunodetection of
GPR30 revealed a 44 kDa band corresponding to the molecular weight
of this receptor (29) in the
extracts of non-malignant epithelial cells (MCF-10A) and in
dedifferentiated malignant cells (Fig.
1). Densitometric evaluation of the bands obtained indicated
that after the control cell line (ovarian cancer cell line OVCAR3),
the non-tumorigenic mammary gland cell line MCF-10A exhibited
highest relative expression level of GPR30 (7.3%), followed by the
non-metastatic breast cancer cell line MCF-7 (2.4%) and the
metastatic MDA-MB-231 line (1.2%).
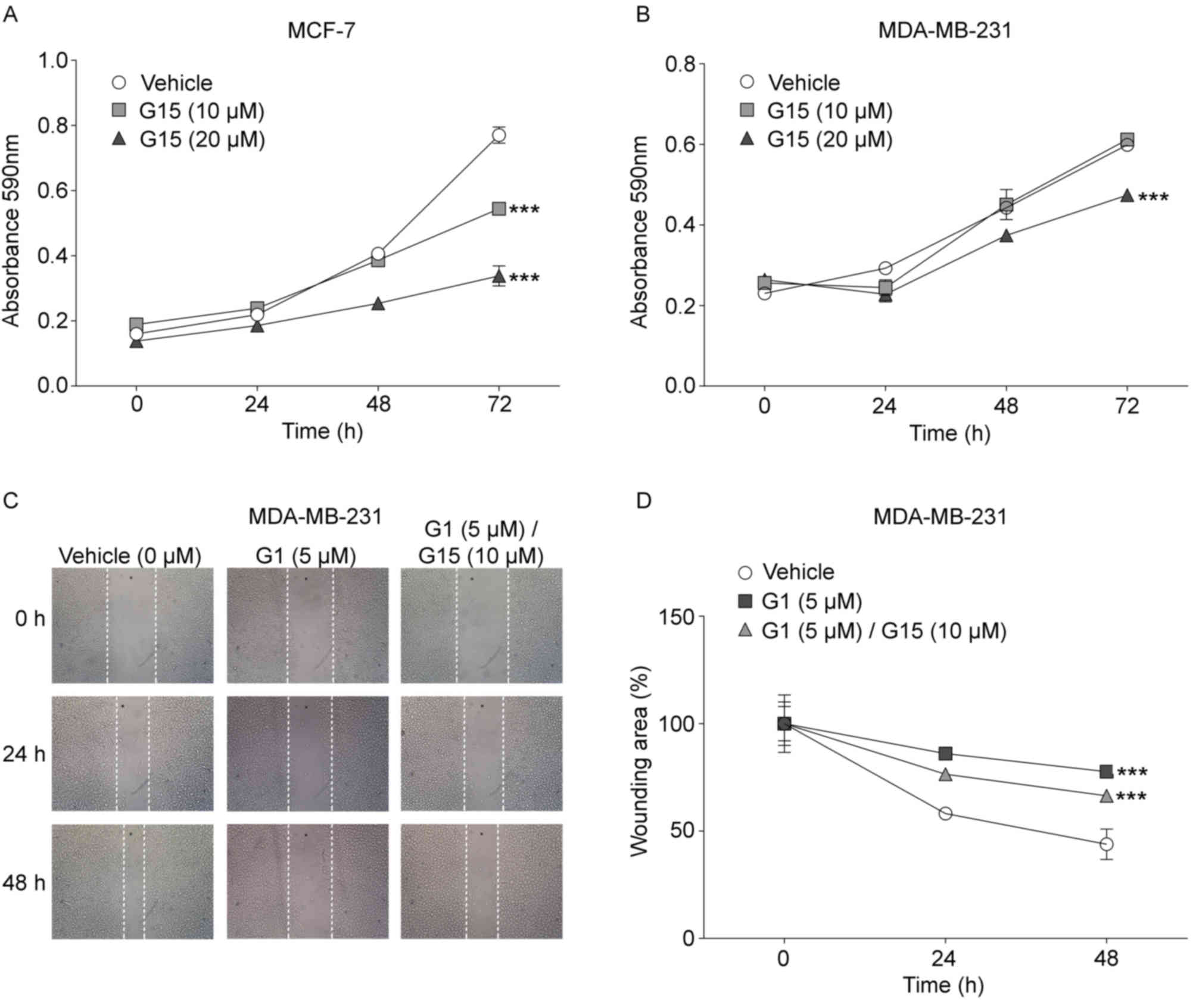
The effects of the specific GPR30 antagonist, G15,
on MCF-7 and MDA-MB-231 viability was evaluated at 0, 24, 48 and 72
h (Fig. 2). The viability of MCF-7
cells treated with G15 was significantly reduced at 72 h compared
with the vehicle-treated control group (Fig. 2A). MCF-7 cells that were cultured
for 72 h in the absence of G15 increased their absorbance 4.8-fold
compared with the 0 h time point, whereas G15-treated cells
increased their absorbance 2.8-fold with a 10 µM concentration and
2.4-fold with a 20 µM concentration. These data indicated that in
cell cultures treated for 72 h with 10 and 20 µM G15, cell
viability decreased regarding untreated cells 40 and 49%,
respectively. However, although an increase in viability in the
MDA-MB-231 cell cultures was also observed with and without G15
treatment, there was a significant decrease in viability in cells
treated with 20 µM G15 for 72 h compared with vehicle-treated cells
at the same time point (Fig. 2B).
The absorbance obtained in untreated-cells after 72 h of culture
was 2.6-fold greater compared with cells at 0 h, whereas the
increase in absorbance in cells treated with 10 and 20 µM G15 was
2.3 and 1.7-fold compared with the respective 0 h time point. These
results indicated that MDA-MB-231 cell viability decreased 31% when
cells were treated with 20 µM G15 compared with control cells at 72
h.
In the cell viability assays, untreated MCF-7 cell
viability was increased 4.8-fold at 72 h compared with the 0 h time
point, whereas the viability of cells treated with the GPR30
agonist (10 µM G1) increased only 2.6-fold over the same time
period (Fig. S1A). In the case of
MDA-MB-231 cells, viability increased 1.9-fold after 72 h without
treatment, whereas the G1-treated cells increased only 1.35-fold
after the same time elapsed (Fig.
S1B). These data demonstrated that the viability of MCF-7 and
MDA-MB-231 cells decreased significantly by 74 and 64%,
respectively, after 72 h of treatment with G1, compared with the
respective control at the same time point.
The role of GPR30 in cell migration was examined by
wound-healing assays with the metastatic MDA-MB-231 cell line
(Fig. 2C and D), since the MCF-7
cell line showed no significant changes in its migration (Fig. S2). In untreated MDA-MB-231 cells,
the wound area was reduced by 41% after 24 h and 56% after 48 h.
Conversely, in G1-treated cells the wound area reduced only by 13%
at 24 h and 22% at 48 h. To confirm that the suppressive effect of
G1 on migration was through GPR30, the effect of its antagonist,
G15, on migration impaired by G1 was also assayed. For this, a G15
concentration twice as high as the minimum G1 concentration that
was required to observe an effect on migration was used, since the
affinity of the antagonist for the receptor is three times lower
compared with that of G1 (10). The
cells treated simultaneously with both G1 and G15, diminish the
wound area 23% at 24 h and 33% at 48 h. This means that G1-treated
cells lost 39% of its migratory capacity with respect to untreated
cells, while cells treated with both agonist and antagonist
recovered 20% of its migratory capacity regarding cells treated
only G1.
Structural analysis of the 5′ flanking
region of the GPR30 encoding gene
Through structural and predictive analysis of the
1,987 bp sequence cloned from the 5′ flanking region of the gene
encoding the GPR30 receptor, it was possible to characterize the
GPR30 promoter fragment in three regions. A proximal region ranging
from −1 to −1,009 bp from TSC with a heterogeneous sequence, and
two distal regions ranging from −1,010 to −1,512 bp and −1,513 to
−1,987 bp from the TSC, which have a high GC content (70%) and a
high AT content (72%), respectively (Fig. 3). The sequence alignment of the
GPR30 promoter cloned from both MCF-7 and MDA-MB-231, with the
chromosome 7 sequence did not reveal any mismatch (data not shown).
In addition, 14 putative cis-regulatory elements with a
similarity greater than 98% in relation to consensus sequences for
forkhead box P1 (FOXP1), ERR1, NGRE, brain-specific homeobox (BSX)
and member of the ETS, SOX and hypoxia-inducible factors (HIF)
families, were found by JASPAR program. Given its high nucleotide
heterogeneity, the distribution of these possible sites of
transcriptional regulation are concentrated in the proximal region,
whereas owing to the low nucleotide heterogeneity of distal
regions, it was only possible to find three sites in one of them
(Fig. 3).
 | Figure 3.Nucleotide sequence of the cloned 5′
flanking region of the GPR30 gene. Fragment of 1987 bp upstream of
the translation start codon of GPR30 gene cloned from MCF-7 and
MDA-MB-231. The putative cis-regulatory elements predicted by
JASPAR are indicated by bold, lowercase letters. The sequence
corresponding to the oligonucleotides that were synthesized to
build the different reporter constructs are indicated in shaded
letters. The arrows indicate the beginning of the different regions
in which GPR30 was divided. BSX, brain-specific homeobox; DR1,
distal region 1; DR2, distal region 2; ERR1, estrogen-related
receptor α; ETS, E26 transformation-specific; FOXP1, forkhead box
P1; GPR30, G protein-coupled receptor 30; HIF, hypoxia-inducible
factor; NGRE, negative glucocorticoid response element; PR,
proximal region; SOX, SRY box transcription factor. |
Transcriptional regulation pattern of
the GPR30 promoter in breast cancer
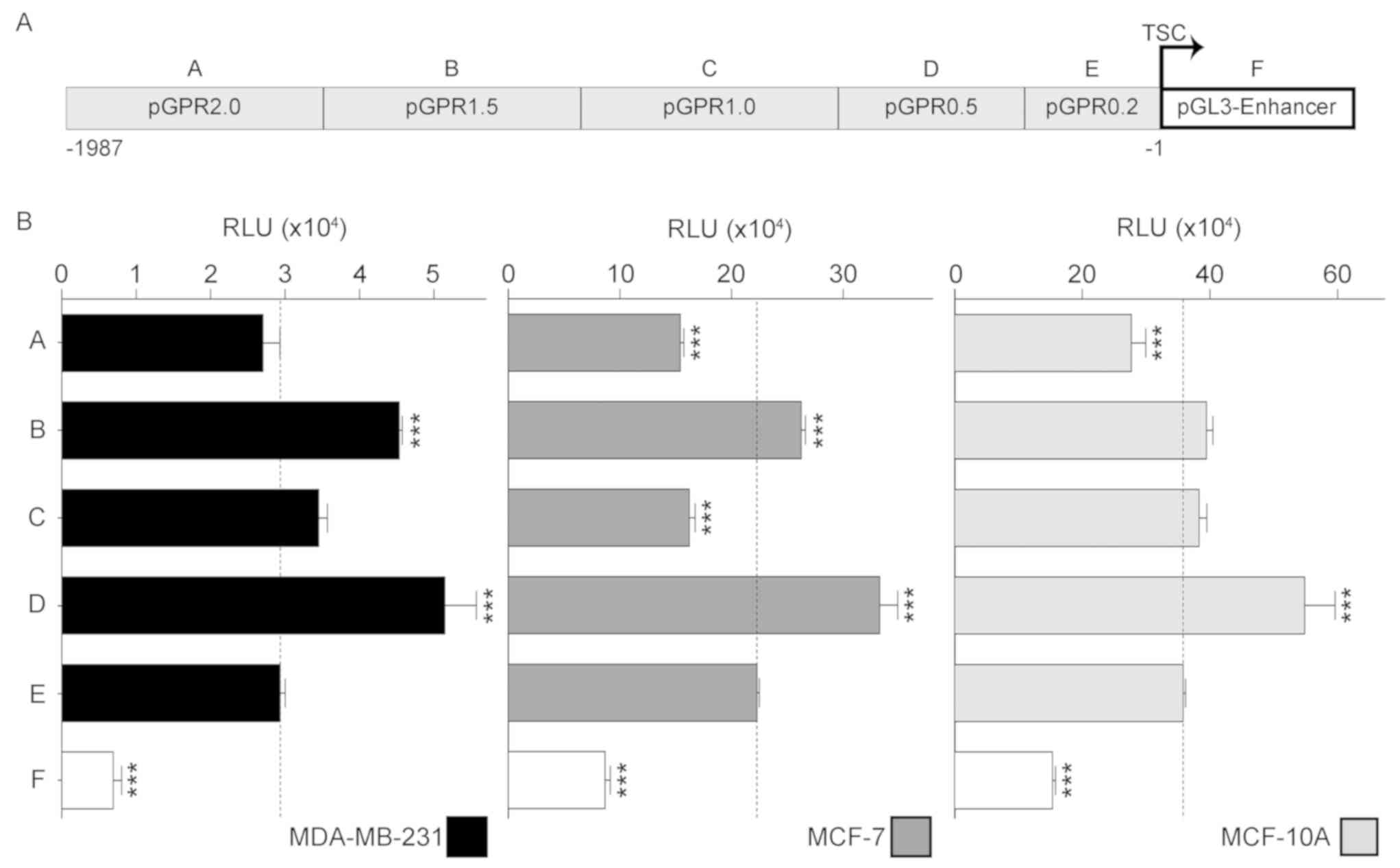
The transient expression of the five reporter
constructs governed by different length versions of the GPR30
promoter truncated at the 5′ end (Fig.
3), revealed that even without the background activity
generated by the empty vector, the transcriptional regulation
pattern of the promoter turned out to be very similar in all three
mammary gland cell lines. All reporter constructs transfected in
the three cell lines, except pGPR1.0 in MCF-10A, produced a similar
change pattern in luciferase activity. The maximum activity of
GPR30 promoter was find in pGPR0.5 being this 1.6-fold higher in
MCF-10A compared with in MCF-7, and approximately 8.8-fold higher
compared with MDA-MB-231 (Fig. 4).
Using the luciferase activity induced solely by the initial 200 bp
(pGPR0.2) of the GPR30 promoter in each cell line as the basal
promoter activity, it was demonstrated that in MCF-10A cells the
activity of pGPR0.5 significantly increased 93%, whereas activity
with pGPR2.0 significantly decreased by 22% of basal activity of
promoter. In the case of MCF-7, luciferase activity with pGPR1.5
and pGPR0.5 significantly increased 80 and 29% of basal activity,
respectively. Conversely, the reporter activity of pGPR2.0 and
pGPR1.0 in the same cell line, significantly decreased 44 and 50%
of basal activity, respectively. Similarly, luciferase activity in
MDA-MB-231 also significantly increase 98 and 71% of basal activity
with pGPR1.5 and pGPR0.5, respectively. Although the activity of
the reporter gene with pGPR2.0 and pGPR1.0 in MDA-MB-231 had a
similar pattern of change as was observed in MCF-7, such changes
were not significant (Fig. 4).
Altogether, the changes observed in the three cell lines, suggest
that the GPR30 promoter fragments ranging from 2.0 to 1.5 kbp and
1.0 to 0.5 kbp could contain transcription suppressing elements,
while fragments ranging from 1.5 to 1.0 kbp and 0.5 to 0.2 kbp
could host transcription activating elements.
A cis-regulatory element that
resembles the polyomavirus enhancer activator 3 homolog (PEA3)
subfamily consensus sequence, activates the GPR30 promoter
Bearing in mind the role of the sequence between
−1,009 to −511 bp contained in the construct pGPR1.0 in the
suppression of the transcription, functional analysis of the
putative cis-regulatory elements for negative regulation
factors located in this region was conducted. This was done by the
transient expression of the ETSµ, NGREµ, ERRµ and SOXµ reporter
constructs. None of the mutations exhibited increased luciferase
activity, as expected; however, the ETSµ mutation significantly
decreased the reporter gene activity by 24% in MCF-10A, 65.8% in
MCF-7 and 46% in MDA-MB-231 compared with the wild-type construct
pGPR1.0 (Fig. 5A). Although with
the site-directed mutations in the ERRµ, NGREµ and SOXµ constructs
also is observed a decrease in the reporter gene activity in any of
transfected cell lines, these changes were not statistically
significant. The functionality of a putative ETS
cis-regulatory element was confirmed by comparing the
activity of pGPR1.0 and the construct containing an ETS factor
consensus elements (ETSc) in the MDA-MB-231 cell line (Fig. 5B). As expected, the luciferase
activity obtained with ETSc was 1.27-fold greater compared with the
pGPR1.0 construct containing the wild-type GPR30 promoter.
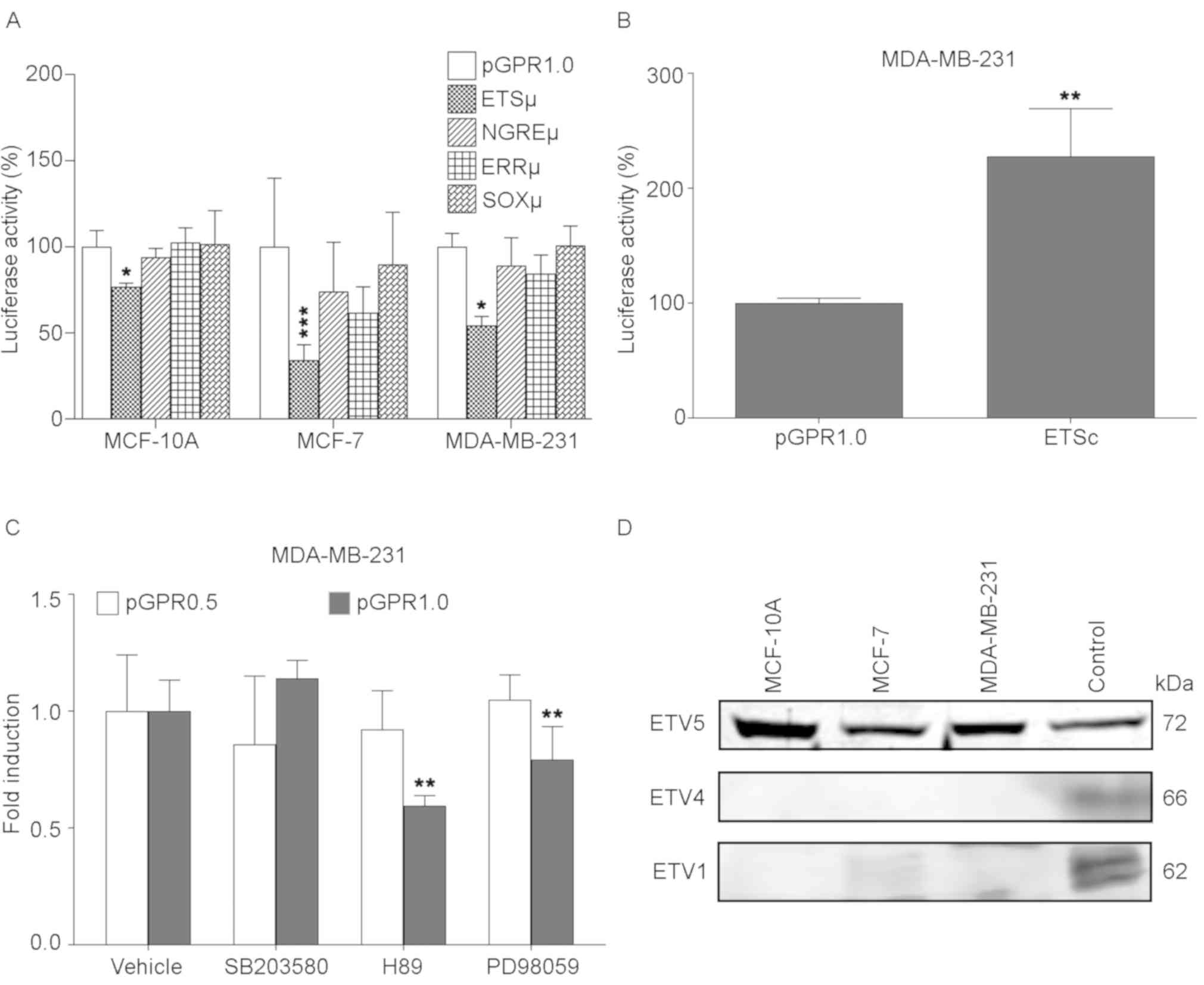 | Figure 5.PEA3 subfamily is involved in GPR30
transactivation in breast cancer cells. Reporter gene activity
(Firefly luciferase) induced by the pGPR1.0 construct and its (A)
loss-of-function mutant versions ETSµ, NGREµ, ERRµ, and SOXµ in
MCF-10A, MCF-7 and MDA-MB-231 cells and (B) gain of function
mutant, ETSc, in MDA-MB-231 cells. *P<0.05, **P<0.01 and
***P<0.001 vs. pGPR0.1. (C) Effects of the inhibitors of p38
(SB203580), PKA (H89) and ERK1/2 (PD98059) on reporter gene
activity of the GPR30 promotor constructs pGPR0.5 without the
functional ETS site (white) and pGPR1.0 containing ETS site (black)
in MDA-MB-231 cells. *P<0.05, **P<0.01 and ***P<0.001 vs.
Vehicle. (D) Immunoblots of ETV5, ETV4 and ETV1 performed with
nuclear extracts of MCF-10A, MCF-7, MDA-MB-231 and positive
controls as was described in the Materials and methods. Data are
presented as the mean ± SD of three independent experiments in
triplicate. ETS, E26 transformation-specific; NGRE, negative
glucocorticoid response element; ERR1, estrogen-related receptor α;
SOX, SRY-box transcription factor; PKA, protein kinase A; PEA3,
polyomavirus enhancer activator 3 homolog; GPR30, G protein-coupled
receptor 30. |
In addition, to gather additional evidence regarding
the participation of any of the factors of the ETS family in the
positive regulation of the GPR30 promoter observed with the ETS
element studied, the common phosphorylation pathways of such
factors were inhibited to verify if GPR30 promoter activity
decreases. For this, transient expression assays performed with
pGPR1.0 (with ETS sites) and pGPR0.5 (without an ETS site) on
MDA-MB-231 cells, treated with SB203580 (p38 inhibitor), H89 (PKA
inhibitor) and PD98059 (ERK1/2 inhibitor) demonstrated
statistically significant changes only with the 1.0 kbp GPR30
promoter in presence of H89 and PD98059, reducing its activity to
0.59 and 0.79-fold, respectively (Fig.
5C). Similar assays using the pGPR0.5 construct had no
significant effect on the activity of the reporter gene, since the
ETS factors of the PEA3 subfamily require the phosphorylation of
PKA and ERK to perform their transcriptional activity (30–32),
and given that the functional ETS element identified is located in
the region affected by these inhibitors (pGPR1.0); the results
obtained from assays with protein kinases inhibitors point to ETV1,
ETV4 and ETV5 as the factors with the greatest possibility of
interacting with this element.
The activity of the factors of the PEA3 subfamily
have been closely related to the development of breast cancer
(33–37), but the expression of each of them
may be committed to specific phenotypes. To confirm which of these
factors may be active in the cellular models in the present study,
and possibly involved in the activation of the GPR30 promoter
through the inducing ETS element, their expressions were verified
qualitatively by immunoblotting assays (38). ETV5 has been previously reported as
a factor expressed by MCF-7 (39).
Therefore, the immunoblotting with anti-ETV5 was carried out using
a commercial MCF-7 nuclear extract as positive control. This assay
confirmed the expression of a 72 kDa protein that correspond to the
molecular weight of ETV5 factor (40) in the nuclear extracts of all mammary
gland cell lines used in the previous experiments and in the
nuclear extract of positive control as expected. Conversely, a 66
kDa protein corresponding to the molecular weight of ETV4 was
detected with anti-ETV4 only in the positive control extract
(K-562) (40). In addition, a 62
kDa protein corresponding to the molecular weight of ETV1 (40) was also detected with anti-ETV1 in
the nuclear extracts of the MCF-7 cell line and the positive
control (mouse brain) (Fig. 5D).
These results suggested that ETV5 and ETV1 may be able to
transactivate GPR30 expression through the ETS element found as
functional in pGPR0.1
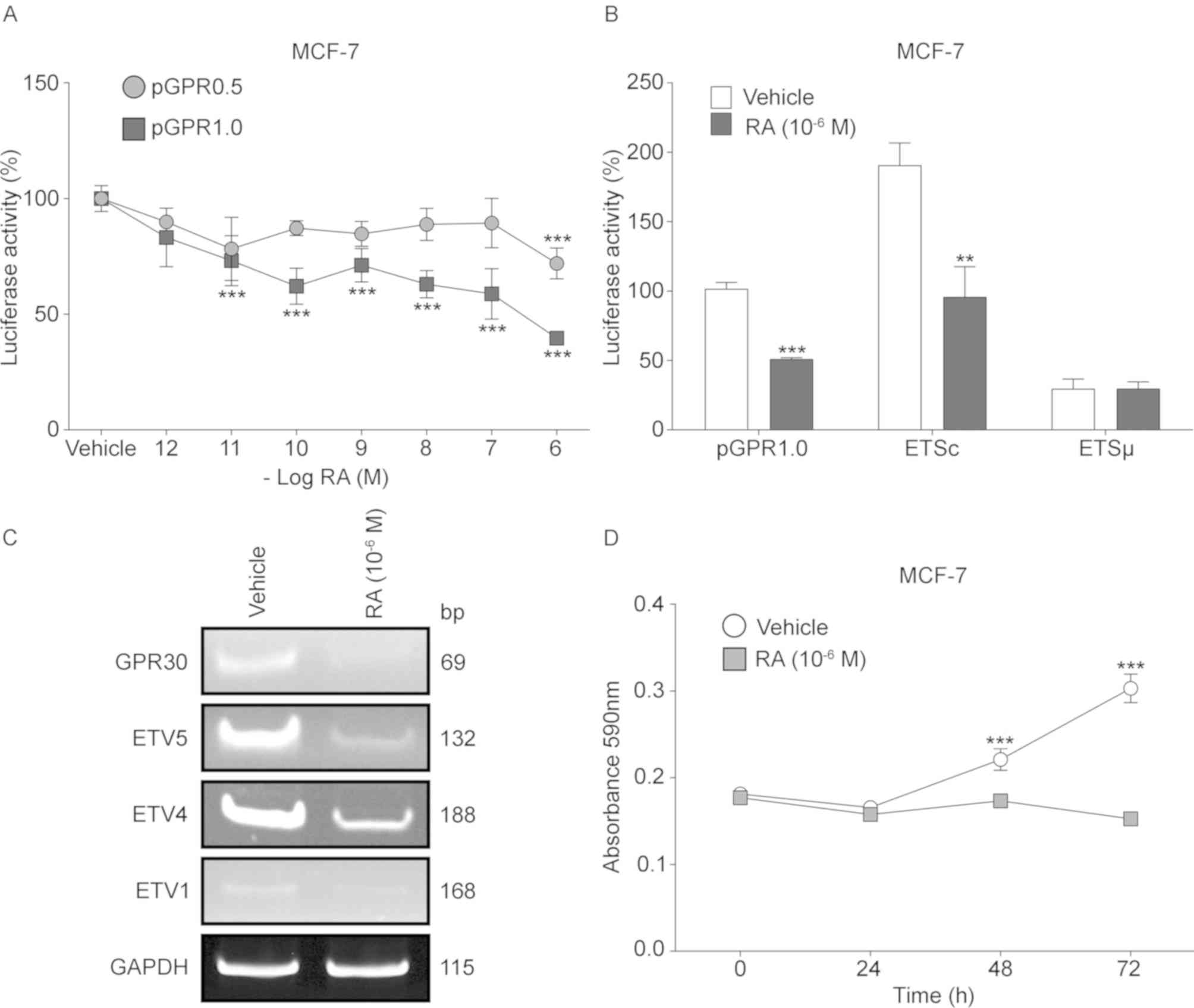
RA suppresses the activity of the
GPR30 promoter and its ETS site located at −630 bp
Since RA suppresses proliferation in ER-positive
breast cancer (41,42) and regulates the expression of
several factors of the ETS family (43), dose-response assays with this
metabolite were carried out on the MCF-7 cell line transfected with
pGPR0.5 (without ETS site) and pGPR1.0 (with ETS site). In the case
of the pGPR0.5 construct, the reporter gene activity exhibited a
statistically significant suppression of 28.1% only when it was
exposed to a final concentration of 10−6 M. By contrast,
with the pGPR1.0 construction containing the inducer element for
ETS factors, the activity of the promoter decreased significantly
from 10−11 M concentration and continued significantly
decreasing up to 60% with the highest concentration (Fig. 6A). In addition, the transient
expression assays in RA-treated and untreated MCF-7 cells with the
transfected pGPR1.0, ETSc or ETSµ constructs, exhibited a
statistically significant decrease of ~50% in luciferase activity
promoted by pGPR1.0 and ETSc, when the cells are treated with RA,
while with ETSµ no effect was observed (Fig. 6B).
The RT-PCR assays of GPR30 and its possible
transcriptional regulators, performed with total RNA isolated from
MCF-7 cells exhibited a notable decrease in amplicon production of
GPR30, ETV5, ETV4 and ETV1 transcripts in RA-treated cells compared
with the vehicle-treated cells (Fig.
6C). Similarly, viability assays using RA-treated MCF-7 cells
showed no significant changes in absorbance during different
culture times, whereas the absorbance of vehicle-treated MCF-7
cells increased 21% at 48 h and 67% at 72 h of cultivation. No
significant differences in viability were identified in RA-treated
cells at any time point compared with the respective 0 h control,
which indicated a complete depletion of viability in MCF-7 cells
associated with RA (Fig. 6D).
Discussion
A few years after the identification of GPR30 as an
orphan receptor in 1996 (44), the
expression and function of this membrane receptor in breast cancer
was intensively studied. However, although it has been reported
that the expression of GPR30 in the mammary gland is null (45) or restricted to myoepithelial cells
(46), Scaling et al
(47) have provided evidence that
suggests the involvement of GPR30 in upregulation of the in
vitro proliferation of breast tissue epithelial cells.
Conversely, data reported by other research groups regarding the
role of GPR30 in breast cancer not allowed to infer its therapeutic
importance (6,48–50).
The lack of a well-defined expression pattern of GPR30 in the
course of breast cancer hinders the correct interpretation of
evidence concerning the role that this receptor may serve in the
development of this malignancy (7,51–53).
In the present study, the GPR30 immunoblotting assays showed that
in the most differentiated mammary gland cell line (MCF-10A) there
is a greater expression of GPR30 compared with expression levels in
the cellular models of breast cancer that have a partially (MCF-7)
or completely (MDA-MB-231) lost cell polarity. In turn, the
difference observed between the GPR30 expression in MCF-7 and
MDA-MB-231, helps to explain why the GPR30-dependent induction on
proliferation deduced from viability assays with G15 in MCF-7 cells
is more evident than in MDA-MB-231 cells. It is important to
mention that the effect caused by G1 on the viability of the two
models of breast cancer was not considered for the interpretation
of the GPR30 activity, since it has been recently reported that
this agonist suppresses proliferation through an GPR30-independent
pathway (54). Overall, these
results, together with the gradual loss observed in transcriptional
regulation of the GPR30 promoter in breast cancer cell lines,
indicate that mammary gland epithelial cells such as MCF-10A have
GPR30 activity and that this can be extinguished in the course of
breast cancer as a result of gene expression reprogramming that the
malignant cells undergo during their dedifferentiation. In this
regard, several research groups have reported that this can occur
with GPR30 in breast cancer through epigenetic mechanisms that act
at the promoter level (49,51,55).
However, the viability and migration assays of the
present study show that despite the role epigenetic silencing may
be playing in the expression of GPR30, poorly differentiated breast
cancer cells, such as MDA-MB-231, still retain responsiveness to
both the agonist and the GPR30 antagonist. Contrasting the results
obtained from the viability assays with those of migration, it is
interesting to note that agonist and antagonist of GPR30 promote
contrary effect in the migration, which indicated that G1 may
reduce migratory ability through GPR30 and that G15 may be able to
at least partially block this suppressive effect. These results
also suggested that GPR30 induced proliferation through a different
pathway to the one with which it suppresses migration, which could
help to explain how the antagonism proposed by some authors between
proliferation and metastasis could happen (56). It was not possible to evaluate the
effects of G1 and G15 on the migration of MCF-7 because this cell
line showed no ability to migrate from untreated cells, which had
been previously reported by other authors (57). It was also verified that the doses
used in the different assays of viability and migration did not
have toxic effects that would alter the results obtained (data not
shown). Therefore, since the results indicated that GPR30
expression and function are subject to changes in cell
differentiation in the course of breast cancer, it is possible to
better interpret why there are some studies that report the
expression of GPR30 as a risk factor for the use of hormonal
therapy (13,58) and other studies that postulate this
receptor as a favorable prognostic factor for disease-free survival
of patients with ER-positive breast cancer (50,51,59).
Confirmation of GPR30 biological activity at
different differentiation stages in breast cancer led to the
investigation of the mechanisms that regulate GPR30 expression at
the transcriptional level. The present study aimed to determine if
there are different transcriptional mechanisms of GPR30 that match
with its performance in the different phenotypes studied, since the
studies in this regard are scarce and the majority have focused on
establishing the role of epigenetic regulation in GPR30 expression
(16,51,55,60,61).
The predictive analysis of cis-regulatory elements performed
on the 5′ flanking region of the GPR30 gene obtained of the two
malignant cell lines found only three putative sites in DR1 (GC
70%) and none in DR2 (AT 72%) probably due to its low
heterogeneity, whereas in the proximal region of greatest
heterogeneity 11 putative sites were identified for diverse
transcription factors previously related to breast cancer.
According to the data obtained from the transient expression assays
carried out with pGPR0.2, the basal activity in MDA-MB-231 cells
resulted in six-fold lower than MCF-7 and eleven-fold lower than
MCF-10A. These results support the notion that GPR30 could be
downregulated by EMT epigenetic reprogramming that malignant cells
may undergo (51,55,61).
However, this same epigenetic reprogramming could at the same time
be indirectly involved in the positive regulation of GPR30
expression, since a specific cis-regulatory element for
members of the ETS family found between −631 and −625 bp of the TSC
in the promoter of GPR30 exhibited the greatest activity in MCF-7
and MDA-MB-231 breast cancer cells compared with activity in
non-malignant mammary gland cells (MCF-10A), suggesting an
incorporation of transcriptional regulators that were absent in the
program of gene expression.
To determine possible transcription factors of the
ETS family that may be involved in the transactivation of GPR30
through cis-regulatory element located between −631 and −625
bp from TSC, those repressor factors of this family, such as Ets-2
repressor factor, ETV6, ETV7, ETS-like gene 3, and E74-like factor
1, were identified through previous reports on their predominant
function (62,63). Subsequently, taking as inclusion
criterion their relationship with the progression of breast cancer,
only the activators ETS1 (64),
ETS2 (65) and the PEA3 subfamily
(66) remained as possible
interactors with the ETS binding site of the GPR30 promoter. To
complete this analysis, transient expression assays were conducted
with the reporter constructs pGPR1.0 and pGPR0.5 in untreated
MDA-MB-231 cells and in cells treated with inhibitors of the main
phosphorylation pathways, including SB203580 (p38), H89 (PKA),
PD98059 (ERK1/2); exposure to H89 and PD98059 produced the expected
suppressor effect on the pGPR1.0 construct. Several previous
studies have reported that the PKA and ERK phosphorylation pathways
are overactivated during the development of breast cancer, which
promotes the expression, stability and activation of several
oncogenes (67–70). However, ETS1 and ETS2 are factors
that only need to be phosphorylated by the ERK pathway to be
activated (71,72), whereas PEA3 subfamily factors can be
phosphorylated by both PKA and ERK (73,74).
Western blotting data from the present study demonstrated that only
ETV5 is expressed in the three mammary gland cell lines, where the
activity of the ETS binding site was observed. Although these
results suggested that ETV5 has the highest possibility of
interacting with the GPR30 promoter in MCF-10A and MDA-MB-231, it
does not imply that other factors, such as ETV1 in MCF-7, are not
involved in the transactivation of GPR30.
Since cell growth is a process favored by the gene
expression program of malignant cells with a still epithelial
phenotype (56), as well as by
GPR30 activation, it was proposed to investigate whether RA, an
anti-proliferative agent frequently used as an adjuvant in the
treatment of non-invasive breast cancer, exerted a suppressive
effect on GPR30 expression. Transient expression assays in MCF-7
revealed that even though the GPR30 promoter does not harbor an RA
response elements, this metabolite significantly decreased the
activity not only of the promoter but also of ETS binding site
found between −631 and −625 bp from TSC. The data of the
RA-dependent reduction in the expression of GPR30 and the
transcription factors possibly involved with its transcriptional
regulation indicated that RA suppressed the activity of the
transcription factors that bind to the cis-regulatory
element in MCF-7 cells. The discrepancy between the results
obtained from immunoblot and those from RT-PCR for the ETV4
expression may be due to a poor performance in the translation of
their mRNA, since with the nuclear extracts of positive control
(K562) was detected a 66 kDa protein corresponding with the
molecular weight of ETV4 (40).
In conclusion, the results suggested a possible
mechanism by which GPR30-dependent cell viability in non-invasive
and ER-positive breast cancer cells may be downregulated by RA, but
with a risk of triggering dedifferentiation. This indicated that
specific activation of GPR30 may contribute to the treatment of
non-invasive breast cancer tumors, preventing the transition to a
more aggressive phenotype or may intervene by reversing the
invasiveness of more aggressive tumors for the use of a more
conventional treatment. Given the implications that the mechanism
proposed in the present study may have for the prognosis and
optimization of treatments for breast cancer at specific stages, it
is also important to consider that the evidence reported here comes
only from in vitro assays. Therefore, it is required that in
subsequent studies, the functional ETS site between −631 and −625
bp from the TSC in the GPR30 promoter, as well as its possible
transfactors, be included in assays that have a closer approach to
the in vivo conditions, such as RNA interference or
CRISPR.
Supplementary Material
Supporting Data
Acknowledgements
The authors wish to thank Ms. Isis Santos Paniagua
[Medical Research Unit in Reproductive Medicine, Instituto Mexicano
del Seguro Social (IMSS)] for the total mouse brain and nuclear
OVCAR3 extracts, as well as for her guidance in the standardization
of cell viability assays with crystal violet. We would also like to
thank Dr Guadalupe Maya Nuñez (Medical Research Unit in
Reproductive Medicine, IMSS) for the technical advice provided
during the reverse transcription-PCR assays, and Dr Arturo Aguilar
Rojas (Medical Research Unit in Reproductive Medicine, IMSS) for
the MDA-MB-231 cell line (American Type Culture Collection) and for
his technical advice during the standardization of wound-healing
assays.
Funding
The present study was supported by Fondo de
Investigación en Salud IMSS (grant. nos. FIS/IMSS/PROT/G12/1154,
FIS/IMSS/PROT/G12/1150 and FIS/IMSS/PROT/G17-2/1720) and IMSS and
CONACYT scholarships from the Universidad Autónoma
Metropolitana-Iztapalapa to DSB.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon request.
Authors' contributions
DSB, MAPS and AO conceived and designed the
experiments. DSB, MAPS and PCG performed the experiments. DSB, MAPS
and AO analyzed the data. MAPS, ZS and EB substantial contributed
to the conception and drafting of work and revising it critically
for important intellectual content. DSB, MAPS, PCG and AO wrote the
paper. All authors read and approved the manuscript and agreed to
be accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Vergote I, Amant F, Leunen K, Van Gorp T,
Berteloot P and Neven P: Metastatic breast cancer: Sequencing
hormonal therapy and positioning of fulvestrant. Int J Gynecol
Cancer. 16 (Suppl 2):S524–S526. 2006. View Article : Google Scholar
|
|
2
|
DeSantis CE, Ma J, Gaudet MM, Newman LA,
Miller KD, Goding Sauer A, Jemal A and Siegel RL: Breast cancer
statistics, 2019. CA Cancer J Clin. 69:438–451. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fan W, Chang J and Fu P: Endocrine therapy
resistance in breast cancer: Current status, possible mechanisms
and overcoming strategies. Future Med Chem. 7:1511–1519. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barton M, Filardo EJ, Lolait SJ, Thomas P,
Maggiolini M and Prossnitz ER: Twenty years of the G
protein-coupled estrogen receptor GPER: Historical and personal
perspectives. J Steroid Biochem Mol Biol. 176:4–15. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ruan SQ, Wang SW, Wang ZH and Zhang SZ:
Regulation of HRG-β1-induced proliferation, migration and invasion
of MCF-7 cells by upregulation of GPR30 expression. Mol Med Rep.
6:131–138. 2012.PubMed/NCBI
|
|
6
|
Girgert R, Emons G and Gründker C:
17β-estradiol-induced growth of triple-negative breast cancer cells
is prevented by the reduction of GPER expression after treatment
with gefitinib. Oncol Rep. 37:1212–1218. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Molina L, Figueroa CD, Bhoola KD and
Ehrenfeld P: GPER-1/GPR30 a novel estrogen receptor sited in the
cell membrane: Therapeutic coupling to breast cancer. Expert Opin
Ther Targets. 21:755–766. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thomas P and Dong J: Binding and
activation of the seven-transmembrane estrogen receptor GPR30 by
environmental estrogens: A potential novel mechanism of endocrine
disruption. J Steroid Biochem Mol Biol. 102:175–179. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Long N, Long B, Mana A, Le D, Nguyen L,
Chokr S and Sinchak K: Tamoxifen and ICI 182,780 activate
hypothalamic G protein-coupled estrogen receptor 1 to rapidly
facilitate lordosis in female rats. Horm Behav. 89:98–103. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bologa CG, Revankar CM, Young SM, Edwards
BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck
NP, Sklar LA, et al: Virtual and biomolecular screening converge on
a selective agonist for GPR30. Nat Chem Biol. 2:207–212. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dennis MK, Burai R, Ramesh C, Petrie WK,
Alcon SN, Nayak TK, Bologa CG, Leitao A, Brailoiu E, Deliu E, et
al: In vivo effects of a GPR30 antagonist. Nat Chem Biol.
5:4212009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dennis MK, Field AS, Burai R, Ramesh C,
Petrie WK, Bologa CG, Oprea TI, Yamaguchi Y, Hayashi S, Sklar LA,
et al: Identification of a GPER/GPR30 antagonist with improved
estrogen receptor counterselectivity. J Steroid Biochem Mol Biol.
127:358–366. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mo Z, Liu M, Yang F, Luo H, Li Z, Tu G and
Yang G: GPR30 as an initiator of tamoxifen resistance in
hormone-dependent breast cancer. Breast Cancer Res. 15:R1142013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pandey DP, Lappano R, Albanito L, Madeo A,
Maggiolini M and Picard D: Estrogenic GPR30 signalling induces
proliferation and migration of breast cancer cells through CTGF.
EMBO J. 28:523–532. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lucki NC and Sewer MB: Genistein
stimulates MCF-7 breast cancer cell growth by inducing acid
ceramidase (ASAH1) gene expression. J Biol Chem. 86:19399–19409.
2011. View Article : Google Scholar
|
|
16
|
Recchia AG, De Francesco EM, Vivacqua A,
Sisci D, Panno ML, Andò S and Maggiolini M: The G protein-coupled
receptor 30 is up-regulated by hypoxia-inducible factor-1alpha
(HIF-1alpha) in breast cancer cells and cardiomyocytes. J Biol
Chem. 286:10773–10782. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
De Francesco EM, Maggiolini M and Musti
AM: Crosstalk between Notch, HIF-1α and GPER in Breast Cancer EMT.
Int J Mol Sci. 19(pii): E20112018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ariazi EA, Brailoiu E, Yerrum S, Shupp HA,
Slifker MJ, Cunliffe HE, Black MA, Donato AL, Arterburn JB, Oprea
TI, et al: The G protein-coupled receptor GPR30 inhibits
proliferation of estrogen receptor-positive breast cancer cells.
Cancer Res. 70:1184–1194. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen ZJ, Wei W, Jiang GM, Liu H, Wei WD,
Yang X, Wu YM, Liu H, Wong CK, Du J and Wang HS: Activation of GPER
suppresses epithelial mesenchymal transition of triple negative
breast cancer cells via NF-κB signals. Mol Oncol. 10:775–788. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Filardo EJ, Quinn JA, Frackelton AR Jr and
Bland KI: Estrogen action via the G protein-coupled receptor,
GPR30: Stimulation of adenylyl cyclase and cAMP-mediated
attenuation of the epidermal growth factor receptor-to-MAPK
signaling axis. Mol Endocrinol. 16:70–84. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Filigheddu N, Sampietro S, Chianale F,
Porporato PE, Gaggianesi M, Gregnanin I, Rainero E, Ferrara M,
Perego B, Riboni F, et al: Diacylglycerol kinase α mediates
17-β-estradiol-induced proliferation, motility, and
anchorage-independent growth of Hec-1A endometrial cancer cell line
through the G protein-coupled estrogen receptor GPR30. Cell Signal.
23:1988–1996. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ge C, Yu M and Zhang C: G protein-coupled
receptor 30 mediates estrogen-induced proliferation of primordial
germ cells via EGFR/Akt/β-catenin signaling pathway. Endocrinology.
153:3504–3516. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hsu LH, Chu NM, Lin YF and Kao SH:
G-protein coupled estrogen receptor in breast cancer. Int J Mol
Sci. 20(pii): E3062019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Poola I, Abraham J, Liu A, Marshalleck JJ
and DeWitty RL: The cell surface estrogen receptor, G
protein-coupled receptor 30 (GPR30), is markedly down regulated
during breast tumorigenesis. Breast Cancer (Auckl). 1:65–78.
2008.PubMed/NCBI
|
|
25
|
Wei W, Chen ZJ, Zhang KS, Yang XL, Wu YM,
Chen XH, Huang HB, Liu HL, Cai SH, Du J and Wang HS: The activation
of G protein-coupled receptor 30 (GPR30) inhibits proliferation of
estrogen receptor-negative breast cancer cells in vitro and in
vivo. Cell Death Dis. 5:e14282014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu T, Liu M, Luo H, Wu C, Tang X, Tang S,
Hu P, Yan Y, Wang Z and Tu G: GPER mediates enhanced cell viability
and motility via non-genomic signaling induced by 17β-estradiol in
triple-negative breast cancer cells. J Steroid Biochem Mol Biol.
143:392–403. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schreiber E, Matthias P, Müller MM and
Schaffner W: Rapid detection of octamer binding proteins with
‘mini-extracts’, prepared from a small number of cells. Nucleic
Acids Res. 17:64191989. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sambrook J, Fritsch EF and Maniatis T:
Molecular cloning: A laboratory manual. Cold Spring Harbor
Laboratory Press; pp. 1–3. 1989
|
|
29
|
Filardo EJ and Thomas P: Minireview: G
protein-coupled estrogen receptor-1, GPER-1: Its mechanism of
action and role in female reproductive cancer, renal and vascular
physiology. Endocrinology. 153:2953–2962. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Janknecht R, Monté D, Baert JL and de
Launoit Y: The ETS-related transcription factor ERM is a nuclear
target of signaling cascades involving MAPK and PKA. Oncogene.
13:1745–1754. 1996.PubMed/NCBI
|
|
31
|
Wu J and Janknecht R: Regulation of the
ETS transcription factor ER81 by the 90-kDa ribosomal S6 kinase 1
and protein kinase A. J Biol Chem. 277:42669–42679. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang JW, Kim MR, Kim HG, Kim SK, Jeong HG
and Kang KW: Differential regulation of ErbB2 expression by
cAMP-dependent protein kinase in tamoxifen-resistant breast cancer
cells. Arch Pharm Res. 31:350–356. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Barrett JM, Puglia MA, Singh G and Tozer
RG: Expression of Ets-related transcription factors and matrix
metalloproteinase genes in human breast cancer cells. Breast Cancer
Res Treat. 72:227–232. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chotteau-Lelièvre A, Révillion F,
Lhotellier V, Hornez L, Desbiens X, Cabaret V, de Launoit Y and
Peyrat JP: Prognostic value of ERM gene expression in human primary
breast cancers. Clin Cancer Res. 10:7297–7303. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Goueli BS and Janknecht R: Upregulation of
the catalytic telomerase subunit by the transcription factor ER81
and oncogenic HER2/Neu, Ras, or Raf. Mol Cell Biol. 24:25–35. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jiang J, Wei Y, Liu D, Zhou J, Shen J,
Chen X, Zhang S, Kong X and Gu J: E1AF promotes breast cancer cell
cycle progression via upregulation of Cyclin D3 transcription.
Biochem Biophys Res Commun. 358:53–58. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shepherd TG, Kockeritz L, Szrajber MR,
Muller WJ and Hassell JA: The pea3 subfamily ETS genes are required
for HER2/Neu-mediated mammary oncogenesis. Curr Biol. 11:1739–1748.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wu X and Sukumar S: ETS genes in breast
cancer: A step in the right direction. Cancer Biol Ther. 6:83–84.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hong H, Yu H, Yuan J, Guo C, Cao H, Li W
and Xiao C: MicroRNA-200b impacts breast cancer cell migration and
invasion by regulating Ezrin-Radixin-Moesin. Med Sci Monit.
22:1946–1952. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Baert JL, Monté D, Musgrove EA, Albagli O,
Sutherland RL and Launoit Y: Expression of the PEA3 group of
ETS-related transcription factors in human breast-cancer cells. Int
J Cancer. 70:590–597. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Müller P, Kietz S, Gustafsson JA and Ström
A: The anti-estrogenic effect of all-trans-retinoic acid on the
breast cancer cell line MCF-7 is dependent on HES-1 expression. J
Biol Chem. 277:28376–28379. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Müller P, Crofts JD, Newman BS,
Bridgewater LC, Lin CY, Gustafsson JA and Ström A: SOX9 mediates
the retinoic acid-induced HES-1 gene expression in human breast
cancer cells. Breast Cancer Res Treat. 120:317–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Park S-W, Do H-J, Ha WT, Han MH, Song H,
Uhm SJ, Chung HJ and Kim JH: Differential expression of ETS family
transcription factors in NCCIT human embryonic carcinoma cells upon
retinoic acid-induced differentiation. Biol Pharm Bull. 37:659–665.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Owman C, Blay P, Nilsson C and Lolait SJ:
Cloning of human cDNA encoding a novel heptahelix receptor
expressed in Burkitt's lymphoma and widely distributed in brain and
peripheral tissues. Biochem Biophys Res Commun. 228:285–292. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Olde B and Leeb-Lundberg LM: GPR30/GPER1:
Searching for a role in estrogen physiology. Trends Endocrinol
Metab. 20:409–416. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhou X, Wang S, Wang Z, Feng X, Liu P, Lv
XB, Li F, Yu FX, Sun Y, Yuan H, et al: Estrogen regulates Hippo
signaling via GPER in breast cancer. J Clin Invest. 125:2123–2135.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Scaling AL, Prossnitz ER and Hathaway HJ:
GPER mediates estrogen-induced signaling and proliferation in human
breast epithelial cells and normal and malignant breast. Horm
Cancer. 5:146–160. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lappano R, Pisano A and Maggiolini M: GPER
function in breast cancer: An overview. Front Endocrinol
(Lausanne). 5:662014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Weißenborn C, Ignatov T, Poehlmann A, Wege
AK, Costa SD, Zenclussen AC and Ignatov A: GPER functions as a
tumor suppressor in MCF-7 and SK-BR-3 breast cancer cells. J Cancer
Res Clin Oncol. 140:663–671. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Martin SG, Lebot MN, Sukkarn B, Ball G,
Green AR, Rakha EA, Ellis IO and Storr SJ: Low expression of G
protein-coupled oestrogen receptor 1 (GPER) is associated with
adverse survival of breast cancer patients. Oncotarget.
9:25946–25956. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Manjegowda MC, Gupta PS and Limaye AM:
Hyper-methylation of the upstream CpG island shore is a likely
mechanism of GPER1 silencing in breast cancer cells. Gene.
614:65–73. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ignatov T, Weißenborn C, Poehlmann A,
Lemke A, Semczuk A, Roessner A, Costa SD, Kalinski T and Ignatov A:
GPER-1 expression decreases during breast cancer tumorigenesis.
Cancer Invest. 31:309–315. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Steiman J, Peralta EA, Louis S and Kamel
O: Biology of the estrogen receptor, GPR30, in triple negative
breast cancer. Am J Surg. 206:698–703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lv X, He C, Huang C, Hua G, Wang Z,
Remmenga SW, Rodabough KJ, Karpf AR, Dong J, Davis JS and Wang C:
G-1 inhibits breast cancer cell growth via targeting
colchicine-binding site of tubulin to interfere with microtubule
assembly. Mol Cancer Ther. 16:1080–1091. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Weissenborn C, Ignatov T, Nass N, Kalinski
T, Dan Costa S, Zenclussen AC and Ignatov A: GPER promoter
methylation controls GPER expression in breast cancer patients.
Cancer Invest. 35:100–107. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kohrman AQ and Matus DQ: Divide or
conquer: Cell cycle regulation of invasive behavior. Trends Cell
Biol. 27:12–25. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kamath L, Meydani A, Foss F and Kuliopulos
A: Signaling from protease-activated receptor-1 inhibits migration
and invasion of breast cancer cells. Cancer Res. 61:5933–5940.
2001.PubMed/NCBI
|
|
58
|
Ignatov A, Ignatov T, Roessner A, Costa SD
and Kalinski T: Role of GPR30 in the mechanisms of tamoxifen
resistance in breast cancer MCF-7 cells. Breast Cancer Res Treat.
123:87–96. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Broselid S, Cheng B, Sjöström M, Lövgren
K, Klug-De Santiago HL, Belting M, Jirström K, Malmström P, Olde B,
Bendahl PO, et al: G protein-coupled estrogen receptor is apoptotic
and correlates with increased distant disease-free survival of
estrogen receptor-positive breast cancer patients. Clin Cancer Res.
19:1681–1692. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Vivacqua A, Lappano R, De Marco P, Sisci
D, Aquila S, De Amicis F, Fuqua SA, Andò S and Maggiolini M: G
protein-coupled receptor 30 expression is up-regulated by EGF and
TGF alpha in estrogen receptor alpha-positive cancer cells. Mol
Endocrinol. 23:1815–1826. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Manjegowda MC and Limaye AM: DNA
methylation dependent suppression of GPER1 in colorectal cancer.
Med Res Arch. 6:2018.
|
|
62
|
Lelièvre E, Lionneton F, Soncin F and
Vandenbunder B: The Ets family contains transcriptional activators
and repressors involved in angiogenesis. Int J Biochem Cell Biol.
33:391–407. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Mavrothalassitis G and Ghysdael J:
Proteins of the ETS family with transcriptional repressor activity.
Oncogene. 19:6524–6532. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kim GC, Kwon HK, Lee CG, Verma R, Rudra D,
Kim T, Kang K, Nam JH, Kim Y and Im SH: Upregulation of Ets1
expression by NFATc2 and NFKB1/RELA promotes breast cancer cell
invasiveness. Oncogenesis. 7:912018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wallace JA, Li F, Balakrishnan S,
Cantemir-Stone CZ, Pecot T, Martin C, Kladney RD, Sharma SM,
Trimboli AJ, Fernandez SA, et al: Ets2 in tumor fibroblasts
promotes angiogenesis in breast cancer. PLoS One. 8:e715332013.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
de Launoit Y, Chotteau-Lelievre A,
Beaudoin C, Coutte L, Netzer S, Brenner C, Huvent I and Baert JL:
The PEA3 group of ETS-related transcription factors: Role in breast
cancer metastasis. Adv Exp Med Biol. 480:107–116. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
De Luca A, Maiello MR, D'Alessio A,
Pergameno M and Normanno N: The RAS/RAF/MEK/ERK and the PI3K/AKT
signalling pathways: Role in cancer pathogenesis and implications
for therapeutic approaches. Expert Opin Ther Targets. 16 (Suppl
2):S17–S27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Jiang P, Enomoto A and Takahashi M: Cell
biology of the movement of breast cancer cells: Intracellular
signalling and the actin cytoskeleton. Cancer Lett. 284:122–130.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Nguyen DH, Hussaini IM and Gonias SL:
Binding of urokinase-type plasminogen activator to its receptor in
MCF-7 cells activates extracellular signal-regulated kinase 1 and 2
which is required for increased cellular motility. J Biol Chem.
273:8502–8507. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Palorini R, Votta G, Pirola Y, De Vitto H,
De Palma S, Airoldi C, Vasso M, Ricciardiello F, Lombardi PP,
Cirulli C, et al: Protein kinase A activation promotes cancer cell
resistance to glucose starvation and anoikis. PLoS Genet.
12:e10059312016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Svensson S, Jirström K, Rydén L, Roos G,
Emdin S, Ostrowski MC and Landberg G: ERK phosphorylation is linked
to VEGFR2 expression and Ets-2 phosphorylation in breast cancer and
is associated with tamoxifen treatment resistance and small tumours
with good prognosis. Oncogene. 24:4370–4379. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Switzer CH, Cheng RY, Ridnour LA, Glynn
SA, Ambs S and Wink DA: Ets-1 is a transcriptional mediator of
oncogenic nitric oxide signaling in estrogen receptor-negative
breast cancer. Breast Cancer Res. 14:R1252012. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Baert J-L, Beaudoin C, Coutte L and De
Launoit Y: ERM transactivation is up-regulated by the repression of
DNA binding after the PKA phosphorylation of a consensus site at
the edge of the ETS domain. J Biol Chem. 277:1002–1012. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Keld R, Guo B, Downey P, Cummins R,
Gulmann C, Ang YS and Sharrocks AD: PEA3/ETV4-related transcription
factors coupled with active ERK signalling are associated with poor
prognosis in gastric adenocarcinoma. Br J Cancer. 105:124–130.
2011. View Article : Google Scholar : PubMed/NCBI
|