Introduction
Recently, breast cancer has become the primary cause
of mortality among women worldwide (1,2).
Although gene mutations have been regarded as the main cause of
breast cancer and a number of these mutations have been identified
(3–5), numerous setbacks have occurred during
the investigation into cancer and effective therapy remains to be
determined. With the development of epigenetics, abnormal
modification of genes and proteins can also affect a number of
biological processes in breast cancer cells without any gene
mutations and have been regarded as another important pathogenesis
of breast cancer (6–9).
Protein acetylation and succinylation can cause
significant changes in the alteration, structure and function of
proteins, and are closely associated with the formation and
development of a number of different types of tumors (10–12).
At present, acetylation has been one of the most focused-on
modifications, and many studies have demonstrated that enzymes
regulating acetylation and deacetylation are involved in apoptosis,
proliferation, aging and certain other biological processes of
breast cancer (13–15), indicating the close association
between protein acetylation and breast cancer. However, these
enzymes can simultaneously modulate the acetylation or
deacetylation of many proteins (16,17),
thus these studies cannot determine the key player among them.
Therefore, it is essential to determine and fully understand the
level of protein acetylation in breast cancer and to identify the
key targets among them. Lysine succinylation has been discovered in
recent years (18) and has been
reported to be involved in the initiation and development of a
number of different types of tumors, such as lung and gastric
cancer (19,20). In addition to the aforementioned
types of cancer, Liu et al revealed that succinylation may
play a significant role in regulating pentose phosphate and
endoplasmic reticulum protein processing pathway (21). However, this result remains to be
confirmed. Recently, an increasing number of studies have
demonstrated that numerous acetylated lysine sites can also be
succinylated (22,23). Meanwhile, various enzymes that
regulate acetylation can also regulate succinylation (19,24–26),
indicating the similarity in function between protein acetylation
and succinylation. Thus, it is essential to investigate protein
acetylation and succinylation collaboratively.
The present study used proteomic techniques to
detect the overall protein acetylation and succinylation levels in
breast cancer tissues and adjacent normal tissues (hereinafter
referred to as normal tissues). The results revealed that the
acetylation and succinylation levels of the majority of proteins in
breast cancer tissue were significantly higher than those in normal
tissue. Further bioinformatic analyses indicated that the modified
proteins were significantly enriched in three H2A.X complexes, and
that nucleophosmin (NPM1) may be a key player in these complexes.
The function of the H2A.X complexes in the DNA damage response
(DDR) revealed via biological process analyses, and the abnormal
DDR statues determined by protein quantification linked the
modification of H2A.X complexes and DDR in breast cancer. Finally,
the western blotting results demonstrated high acetylation and
succinylation levels in the majority of proteins, including the
modification of NPM1 and its correlation between cell viability. In
addition, the high expression level of H2A.X in breast cancer
tissues further suggested a certain association between the
modification of H2A.X complexes and DDR in breast cancer. In
conclusion, the results of the present study highlighted the
importance of protein acetylation and succinylation in the
initiation and development of breast cancer, and revealed the
potential target: H2A.X complexes and its member NPM1. These
results provide a foundation for the investigation of protein
acetylation and succinylation, and provide new research targets for
breast cancer.
Materials and methods
Sample acquisition
Ten pairs of tumor tissue and normal tissue samples
were obtained from patients at the Second Affiliated Hospital of
Qiqihar Medical University (Qiqihar, Heilongjiang), who were
diagnosed with invasive ductal breast carcinoma from January 2018
to December of 2018 but who received no previous chemotherapy or
radiotherapy. The age range of the patients was from 38 to 64 years
old and their average age was 51.9 years old. The tissue samples
were immediately stored in liquid nitrogen. All methods were
performed in accordance with relevant guidelines. All experimental
protocols were approved by the Qiqihar Medical Ethics Committee,
approval no. [2018]27. Patients provided informed consent for the
use of their samples.
Proteomic detection
Protein extraction and digestion
Tissue samples were ground quickly and dissolved in
lysis buffer [8 M urea, 1% Triton X-100, 65 mM DTT, 1% Protease
Inhibitor Cocktail, 3 µM TSA (Trichostatin A), 50 mM NAM
(nicotinamide), 50 mM Tris-HCl] for sonication three times using a
high intensity ultrasonic processor (Scientz). After centrifugation
for 10 min at 12,000 × g and 4°C, the proteins in the supernatant
were precipitated by 15% trichloroacetic acid (TCA) for 2 h at
−20°C and centrifuged for 10 min at 4°C. The precipitate was then
washed with cold acetone and dissolved in 8 M urea, 100 mM
triethylamine borane (TEAB), pH 8.0. Prior to digestion, the
protein solution was reduced in 10 mM DTT for 1 h at 37°C and then
alkylated with 20 mM iodoacetamide (IAA) for 45 min in the dark at
room temperature. After adding 100 mM TEAB to reduce the urea
concentration to <2 M, trypsin at a ratio of 1:50
trypsin-to-protein (w/w) was added into the solution for the first
digestion overnight at 37°C, and a second digestion at a ratio of
1:100 trypsin-to-protein (w/w) for 4 h at 37°C followed.
Tandem mass tag (TMT) labeling
Tryptic peptide samples were desalted using a Strata
X C18 SPE column (Phenomenex) and reconstituted in 0.5 M TEAB. The
TMT labeling was performed according to the manufacturer's protocol
for the 6-plex TMT kit. For the acetylation and succinylation
assessment, 126- and 127-TMT labeling reagents were used for the
cancer and normal tissue labeling, respectively. For the
quantitative study, three cancer tissues were labeled using 126-,
127-, 128-TMT labeling reagents, and three normal tissues were
labeled using 129-, 130-, 131-TMT labeling reagents. After
labeling, equal amounts of sample from each group were mixed. The
peptide mixtures were then desalted and dried by vacuum
centrifugation.
Affinity enrichment for acetylated and
succinylated proteins
Affinity enrichment was applied for collecting the
acetylated and succinylated peptides. First, tryptic peptides were
dissolved in NP-40 lysis buffer (100 mM NaCl, 1 mM EDTA, 50 mM
Tris-HCl and 0.5% NP-40, pH 8.0) and incubated with pre-washed
corresponding antibody beads (PTM Biolabs) at 4°C overnight with
gentle shaking. The beads were then washed four times with NP-40
lysis buffer and twice with ddH2O. The eluting buffer
was selected as 0.1% TFA and the eluted fractions were combined and
vacuum-dried. After desalting using C18 ZipTips (EMD Millipore),
the samples were ready to undergo high performance liquid
chromatography (HPLC) separation.
Separation of peptides
Peptides for the acetylation and succinylation
analysis were separated using an EASY-nLC 1000 UPLC system (Thermo
Fisher Scientific, Inc.). Samples were first dissolved in 0.1%
formic acid (FA) and loaded onto a reverse-phase pre-column
(Acclaim PepMap 100; Thermo Fisher Scientific, Inc.), and then
separated in solvent B by loading onto a reverse-phase analytical
column (Acclaim PepMap RSLC; Thermo Fisher Scientific, Inc.). The
solvent B gradient was set as follows: 6 to 22% over 24 min; 22 to
36% for 8 min; 80% for 4 min; and finally 80% for 4 min; all
performed at a constant flow rate of 280 nl/min.
Peptides for quantification were separated via high
pH reverse-phase HPLC with an Agilent 300Extend C18 column
(Agilent). Briefly, the peptides were first separated in a gradient
from 2 to 60% acetonitrile in 10 mM ammonium bicarbonate, pH 10.0
over 80 min. Meanwhile, the solution was collected every 1 min. A
total of 80 fractions were obtained and then combined into 18
fractions and dried by vacuum centrifuging.
MS/MS analysis
MS/MS analysis for separated peptides was performed
by Q Exactive™ Plus Hybrid Quadrupole-Orbitrap Mass Spectrometer
(Thermo Fisher Scientific, Inc.) equipped with an NSI (Nanospray
Ionization) source. The parameters were set as follows: Resolution
for intact and ion fragment peptides was 70,000 and 17,500,
respectively; normalized collision energy was set at 30%; electro
spray voltage was set to 2.0 kV; threshold of ion count in MS
survey scan was set to 2E4 under a 30 sec dynamic exclusion;
automatic gain control was 5E4; the m/z scan range was from
350–1,800; and the first fixed mass was 100.
Database search
For the acetylation and succinylation experiments,
the resulting MS/MS data was processed using MaxQuant (27) integrated with the Andromeda search
engine (version 1.4.1.2) (28).
Tandem mass spectra were searched for against the SwissProt_Human
database (29) (20,203 sequences)
concatenated with reverse decoy database. Trypsin/P was specified
as a cleavage enzyme allowing up to 4 missing cleavages, 5
modifications per peptide and 5 charges. The mass error was set to
20 ppm for the first search, 5 ppm for the main search and 0.02 Da
for the fragment ions. Carbamidomethylation on Cys was specified as
fixed modification and oxidation on Met, acetylation and
succinylation on Lys, and the protein N-terminal was specified as
variable modifications. The false discovery rate (FDR) threshold
for the protein, peptide and modification sites were specified at
1%. Minimum peptide length was set at 7. All the other parameters
in MaxQuant were set to default values. The site localization
probability was set as >0.75.
The Mascot search engine (version 2.3.0) was applied
for the quantification analysis. The resulting MS/MS data were
processed using the Mascot search engine (version 2.3.0). Tandem
mass spectra were searched against the Uniprot_Human database
(29) (8,274 sequences). Trypsin/P
was specified as a cleavage enzyme allowing up to 2 missing
cleavages. The mass error was set to 10 ppm for precursor ions and
0.02 Da for fragment ions. Carbamidomethyl on Cys, TMT-6plex
(N-term) and TMT-6plex (K) were specified as fixed modification,
and oxidation on Met was specified as variable modifications. FDR
was adjusted to <1% and peptide ion score was set as >20.
QC validation of MS data
The MS data validation are presented in Fig. S1. First, the mass error of all the
identified peptides were checked. The distribution of mass error
was near zero and the majority were <0.02 Da, which indicated
that the mass accuracy of the MS data fit the requirement (Fig. S1A-C). Secondly, the lengths of the
majority of the peptides were distributed between 8 and 20, which
was consistent with the lengths of tryptic peptides (Fig. S1D-F), indicating that the sample
preparation had met the standard.
Bioinformatic analysis
Complex enrichment analysis
Manually the curated CORUM protein complexes
database (30) for humans was used
for the protein complexes analysis. Enrichment complexes were
identified using a hypergeometric test for each category of
proteins. A two-tailed Fisher's exact test was employed to test the
enrichment of the proteins containing SwissProt entries against all
SwissPort human proteins. Correction for multiple hypothesis
testing was performed using standard FDR control methods and
complexes. Corrected P-value <0.05 was considered to indicate a
statistically significant difference.
Analysis of H2A.X complexes
The interaction, Venn and biological process
analyses of all members in H2A.X complexes were performed using
FunRich software (version 3.1.3) (31). Briefly, all members in the H2A.X
complexes were identified in the CORUM database (http://mips.helmholtz-muenchen.de/corum/) and the
corresponding protein ID was loaded onto the Fun_Rich software. The
Venn and biological process analyses were then performed in the
background of the UniProt database. Biological process entries with
a corrected P-value <0.05 were considered to indicate a
statistically significant difference.
The sequence conservatism analysis of NPM1 was
performed using MEGA software (version 7.0.26) (32). Briefly, the protein sequences of
NPM1 from different species were identified in the NCBI database
and saved in the Fasta file. The file was then loaded onto MEGA
software and the sequences were aligned with ClustalW to analyze
their conservatism.
Analysis of proteins associated with
DDR
The present study used Functional Annotation Tool of
DAVID Bioinformatics Resources 6.7 (33) to analyze the enriched Gene Ontology
(GO) proteins that were upregulated (fold change≥1.5) or
downregulated (fold change ≤1/1.5) in breast cancer tissues. The GO
with a corrected P-value<0.05 was considered to be significantly
different. The GO terms associated with DDR were listed and a heat
map of the proteins in these terms was created using RGui software
(version 3.4.2) (34).
Validation of proteomic results
Cell culture
MCF-7 and BT-549 cells were purchased from the Cell
Bank of the Chinese Academy of Science. MCF-7 cells were cultured
in MEM (HyClone) supplemented with 10% fetal bovine serum (HyClone;
GE Healthcare), 1 mM sodium pyruvate (Sigma-Aldrich; Merck KGaA)
and 1.74 µM bovine insulin (Sigma-Aldrich; Merck KGaA). BT-549
cells were cultured in RPMI-1640 (HyClone; GE Healthcare)
supplement with 10% fetal bovine serum (FBS), 14 mM glucose
(Sigma-Aldrich; Merck KGaA), 1 mM sodium pyruvate and 0.023 IU/ml
insulin (Sigma-Aldrich; Merck KGaA). Both cells were cultured in a
humidified atmosphere of 5% CO2 at 37°C.
Antibodies
Human polyclonal antibodies against H2A.X and NPM1
were purchased from Abcam (cat. nos. ab10530 and ab20669,
respectively), rabbit polyclonal antibody against pan-acetylation
proteins of all species, and rabbit monoclonal antibodies against
human GAPDH were purchased from Cell Signaling Technology (cat.
nos. 9441 and 8884, respectively). Rabbit polyclonal antibodies
against pan-succinylation proteins of all species were obtained
from PTM Biolab (cat. nos. PTM-401), and the secondary antibodies
against mouse IgG and rabbit IgG were purchased from Cell Signaling
Technology (cat. nos. 7076 and 7074, respectively).
Plasmid constructs and
transfection
Gene expression sequence of NPM1 was amplified using
the designed primers (Table I) via
Ex Taq HS enzyme (Takara). The PCR conditions were set as 94°C for
30 sec, 55°C for 30 sec and 72°C for 1 min and the above step was
repeated for 30 cycles finally followed by 72°C for 10 min. After
that, the sequence was cloned into the PC-DNA3.0 and PCI2 vector
respectively via T4 DNA ligase (TransGen Biotech) according to the
manufacturer's instructions. The NPM1-K27R variant was constructed
using PCI2-NPM1 as the template with Quick Mutation Site-Directed
Mutagenesis Kit (Beyotime Institute of Biotechnology) according to
the manufacturer's instructions; the primers used are presented in
Table I. All above plasmids were
transfected via Liposomal Transfection Reagent (HAN Biotechnology)
according to the manufacturer's instructions.
 | Table I.Sequence of the primers for NPM1 and
its variant NPM1-K27R. |
Table I.
Sequence of the primers for NPM1 and
its variant NPM1-K27R.
| Gene | Primer
sequences |
|---|
| NPM1 | Forward:
CCAAGCTTTGCCGCCACCCGATGGAAGATTC |
|
| Reverse:
GGGGTACCTTAAAGAGACTTCCTCCACTGCC |
|
NPM1-K27R | Forward:
GGTTGTGAACTAAAGGCCGACAGAGATTATCACTTTAAGGTGG |
|
| Reverse:
CCACCTTAAAGTGATAATCTCTGTCGGCCTTTAGTTCACAACC |
Protein extraction and western
blotting
Cells and tissues were collected and dissolved in
RIPA lysis buffer (Beyotime Biotechnology) and the proteins were
extracted according to the manufacturer's protocol. Proteins were
separated via 12% SDS-PAGE and transferred to nitrocellulose
filters (EMD Millipore). The membranes were cut into small pieces
according to the protein ladder (Thermo Scientific, Inc.) and the
parts with the target proteins were reserved. After that, the
membranes were blocked with 5% non-fat milk in TBST buffer (20 mM
Tris-HCl, pH 7.6, 150 mM NaCl and 0.05% Tween-20) for 1 h at room
temperature and then incubated overnight at 4°C with the following
primary antibodies: H2A.X (dilution 1:500), NPM1 (dilution 1:500),
pan-acetylation (dilution 1:1,000), pan-succinylation (dilution
1:1,000), GAPDH (dilution 1:1,000). The membranes were then washed
with TBST buffer and incubated with horseradish
peroxidase-conjugated secondary antibody (dilution 1:2,500) for 30
min at room temperature. After extensive washing, the proteins were
detected using an Electrochemiluminescence-Plus kit (CoWin
Biotechnology) and imaged using a Gel-imaging system (Bio-Rad) with
Image Lab Software (version 5.2; Bio-Rad Laboratories).
Cell viability detection
BT-549 cells were seeded into a 96-well at a density
of 0.8×104 cells/well. After being cultured for 24 h,
the cells were transfected into control vector PCI2, NPM1 and its
variant NPM1-K27R with Liposomal Transfection Reagent (HAN
Biotechnology) respectively and the culture was continued for 36 h.
After that, 20 µl/well MTT (5 mg/ml; Sigma-Aldrich, Merck KGaA) was
added into the cells and incubated for 4 h. Finally, the
supernatant was removed and 150 µl/well DMSO was added
(Sigma-Aldrich, Merck KGaA) and the absorbance was detected at a
wavelength of 570 nm using a microscope reader (Olympus Corp.).
Statistical analysis
Enrichment analysis was performed using a Fisher'
exact test. Average modification level analysis between breast
cancer and normal tissue was performed by Wilcoxon Signed Rank
Test. Cell viability analysis was performed by Student's t-test.
The significance level was set at P<0.05 and P<0.01. Error
bars denote the standard deviation.
Results
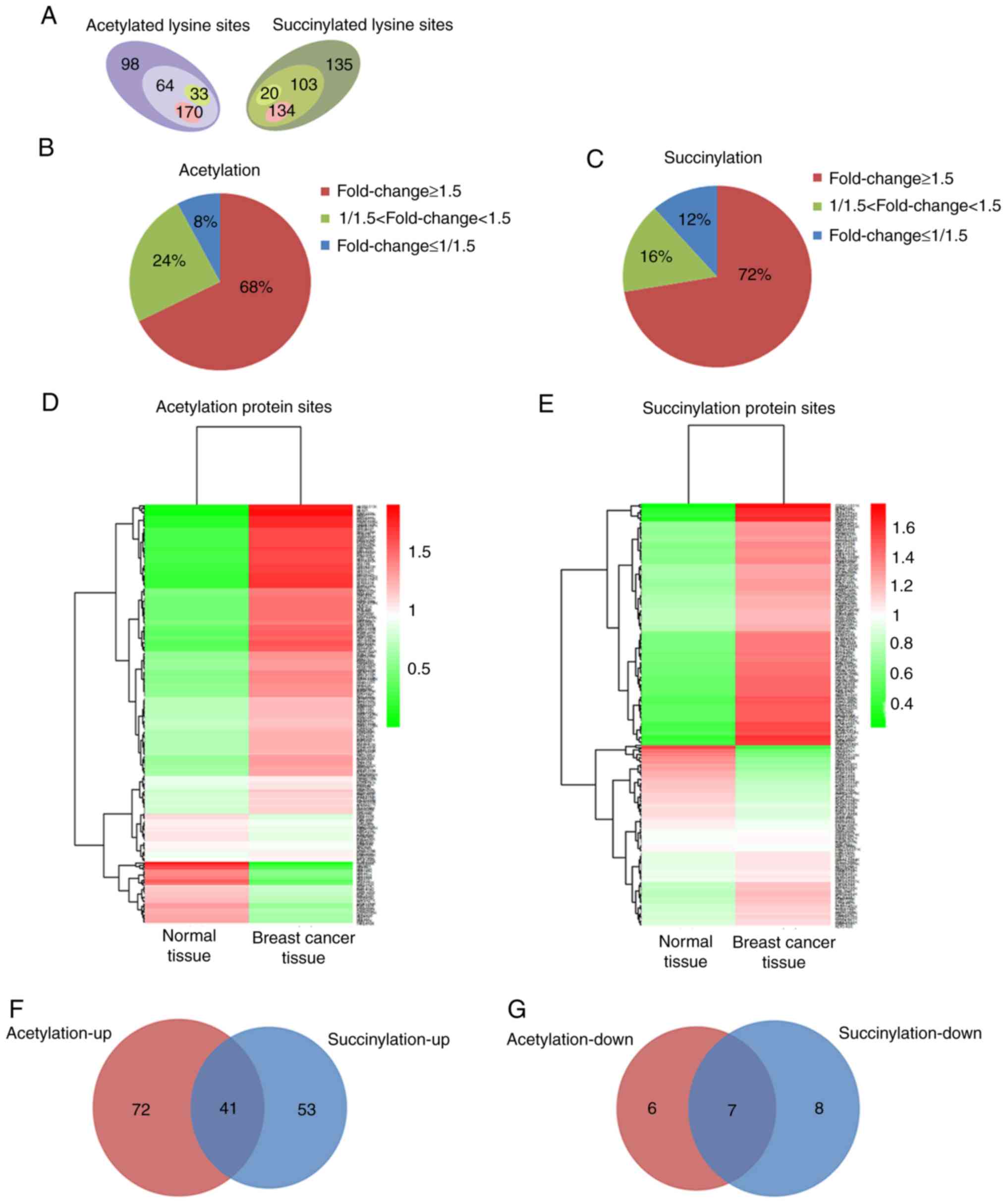
High protein acetylation and
succinylation levels in breast cancer tissue
Using TMT labeling and affinity enrichment followed
by high-resolution LC-MS/MS analysis, quantitative lysine
acetylation and succinylation analyses were performed in the
present study. Altogether, 364 lysine acetylation sites from 229
proteins and 392 succinylation sites from 226 proteins were
identified, among which 267 acetylation sites in 167 proteins and
257 succinylation sites in 130 proteins were quantified; both the
acetylation and succinylation levels showed a statistically
significant difference (Fig. 1A and
Fig. S2). Among the quantified
proteins, when setting 1.5 as the fold threshold change in
modification levels (cancer vs. normal tissue), 170 acetylation
sites in 113 proteins and 134 succinylation sites in 94 proteins
with higher acetylation and succinylation levels (Fig. 1A), respectively, reached 68 and 72%
of all identified proteins (fold change≥1.5, Fig. 1B and C). However, only 33
acetylation sites in 13 proteins and 20 succinylation sites in 15
proteins exhibited lower acetylation and succinylation levels
(Fig. 1A), accounting for 8 and 12%
of all identified proteins (fold change ≤1/1.5; Fig. 1B and C). All the quantified protein
sites and their modification levels in breast cancer and normal
tissues are presented in Fig. 1D and
E. Among the proteins whose acetylation or succinylation levels
were altered more than 1.5 fold, 41 of them were increased in both
of these two modifications (Fig.
1F), 16 of the 41 proteins changed in the same lysine sites
(Table SI); 7 of the proteins were
decreased in both of these two modifications (Fig. 1G), 4 of the 7 proteins changed in
the same lysine sites (Table SII).
These results suggest that protein acetylation and succinylation
may work together and play an important role in the initiation and
development of breast cancer.
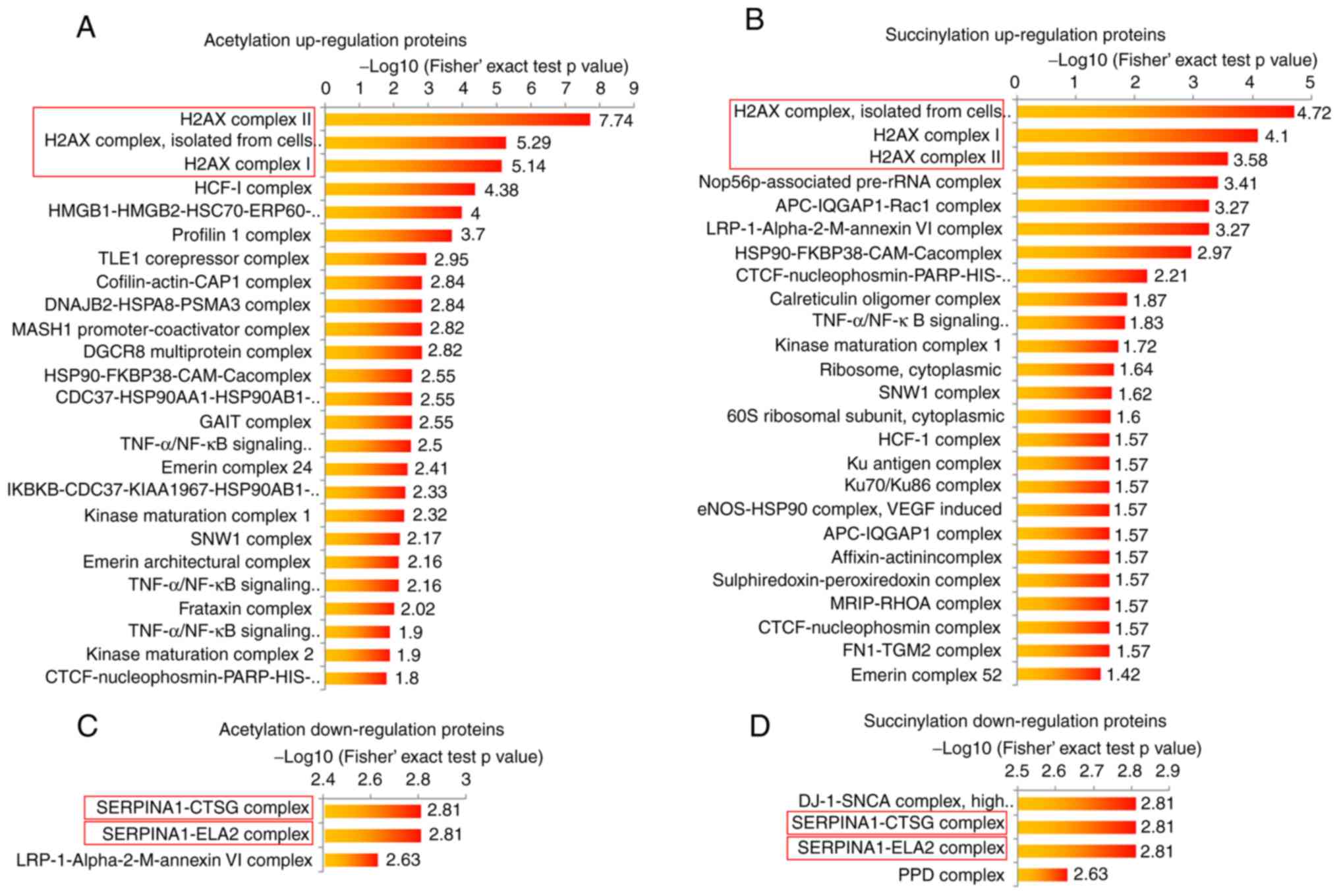
Highly acetylated or succinylated
proteins are enriched in H2A.X complexes
The present study performed bioinformatic analyses
in order to determine the potential common functions of the
acetylation and succinylation proteins in the breast cancer tissue.
The protein complex enrichment analysis revealed that both high
acetylation and succinylation proteins in breast cancer tissue were
enriched in numerous complexes, such as H2A.X, HCF-1,
Nop56p-associated pre-rRNA complexes and others (Fig. 2A and B). Due to the small protein
number, the low modification proteins in breast cancer tissue were
only observed to be enriched in 5 complexes, including
SERPINA1-CTSG, SERPINA1-ELA2, LRP-1-α-2-M-annexin VI, DJ-1-SNCA and
PPD complexes (Fig. 2C and D). It
is worth noting that either highly acetylated or succinylated
proteins in breast cancer were most commonly enriched in three
H2A.X complexes (Fig. 2A and B),
and proteins with lower modification levels were both more commonly
enriched in the SERPINA1-CTSG and SERPINA1-ELA2 complexes (Fig. 2C and D), indicating that the protein
acetylation and succinylation may exert similar functions in these
complexes. In order to investigate the function of highly modified
proteins, H2A.X complexes were selected for further analysis in the
present study.
Acetylation and succinylation of H2A.X
complexes may affect DDR in breast cancer
The present study further analyzed the function of
three H2A.X complexes. It has been reported that the three H2A.X
complexes are formed in different ionizing radiation conditions,
which can cause DDR (35).
Meanwhile, the further analysis demonstrated that all members in
the H2A.X complexes were enriched in certain biology processes,
such as nucleosome assembly, DNA repair, cellular senescence and
cellular response to γ radiation (Fig.
3A), all of which are closely associated with DDR (36–38).
This result indicated that acetylation and succinylation of
proteins in H2A.X complexes may affect DDR in breast cancer. For a
comprehensive understanding of the DDR condition in breast cancer,
the present study quantified all protein expression levels in
breast cancer tissues and compared them with those in normal
tissues. Proteins whose expression level changed significantly
(fold change≥1.5 or fold change≤1/1.5) in breast cancer tissues are
presented in Tables SIII and
SIV. The GO analysis of these
proteins revealed those biological process entries that were
associated with DDR (P<0.05; Fig. 3B
and C), corresponding proteins and their expression levels in
breast cancer and normal tissues, and these are presented in
Fig. 3D and E. Among them, the
proteins involved in DNA repair were significantly upregulated, and
those proteins that were associated with the response to certain
stimuli that can induce DNA damage were significantly
downregulated. These results indicated an abnormal form of breast
cancer that may be associated with the abnormal modification of
H2A.X complexes.
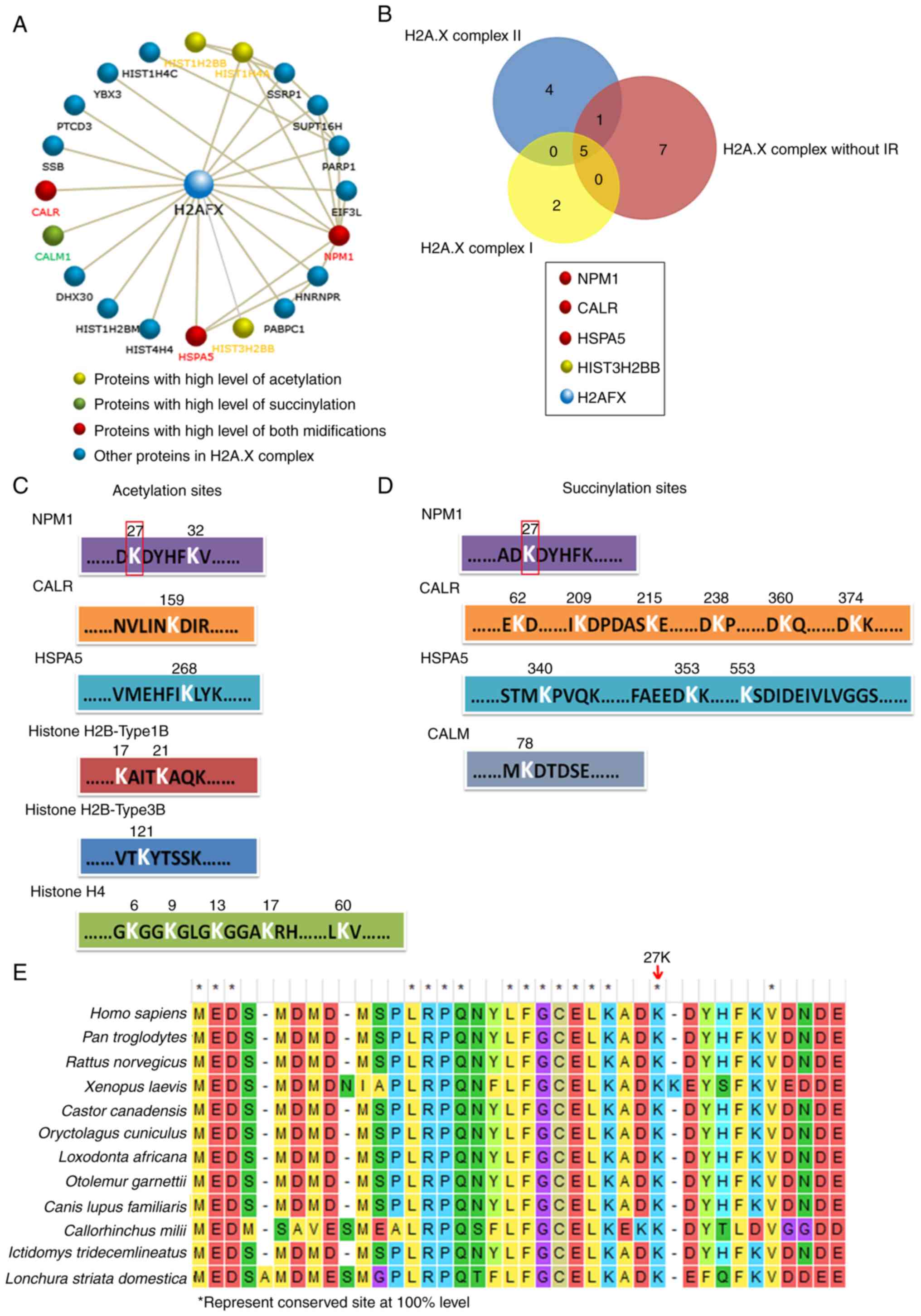
NPM1 may play an important role in
H2A.X complexes
The present study then performed further analyses of
H2A.X complexes in order to determine the key factors among them.
The CORUM database revealed the 19 proteins in the H2A.X complexes,
the majority of which can interact with H2A.X (encoded by
H2A.FX) directly, and a few of them interact with H2A.X
indirectly by combining with other proteins in these complexes
(Fig. 4A, among the 19 proteins,
Histone H4 can be encoded by three kinds of genes: HIST1H4C,
HIST1H4A and HIS4H4, so there are 21 gene members in the
figure). Histone H4 (encoded by HIST1H4A), Histone H2Btype1B
(HIST1H2BB) and Histone H2Btype3B (HIST3H2BB) were
highly acetylated, and CALM was highly succinylated. It is worth
noting that NPM1, HSPA5 and CALR were upregulated in both their
acetylation and succinylation (Fig.
4A). In addition, the Venn analysis revealed that NPM1, CALR
and HSPA5 were also the common members of the three H2A.X complexes
(Fig. 4B). The results indicated
that the modification of these three proteins may play an important
role in H2A.X complexes. The acetylation and succinylation at the
lysine sites of proteins in the H2A.X complexes are presented in
Fig. 4C and D; notably, the 27th
lysine site of NPM1 was highly acetylated and succinylated
simultaneously and it is a highly conserved amino acid site in a
number of different species (Fig.
4E). All the aforementioned results indicated that NPM1 may
play a more important role in H2A.X complexes.
Verification of proteomic results
Finally, the present study validated the proteomics
results via western blotting. The results revealed that the
acetylation and succinylation levels of the majority of proteins in
breast cancer tissues were significantly higher than that of normal
tissues (Fig. 5A and B). The
acetylation and succinylation of 40 kDa protein weights indicated
the obvious modification of NPM1 in breast cancer tissues (Fig. 5C). Meanwhile, the acetylation and
succinylation levels of the 40 kDa proteins increased following the
overexpression of NPM1 in MCF-7 and BT-549 cells (Fig. 5D and E). Moreover, when the 27th
lysine was mutated into arginine, which cannot be acetylated or
succinylated, the modification of the protein weight in 40 kDa was
restored to the normal level (Fig.
5E). This provides further evidence to support the acetylation
and succinylation of NPM1 in breast cancer cells. In addition, the
overexpression of NPM1 increased the viability of BT-549 cells,
while that of its variant NPM1-K27R lost this function (Fig. 5F). This indicated that the
acetylation and succinylation can affect some function related to
cell proliferation. Finally, it was revealed that H2A.X, the core
member of the H2A.X complexes and the marker of DDR (39,40),
was significantly upregulated in breast cancer tissues (Fig. 5G). This further proved that breast
cancer is in an abnormal DDR condition, which may be associated
with the modification of H2A.X complexes.
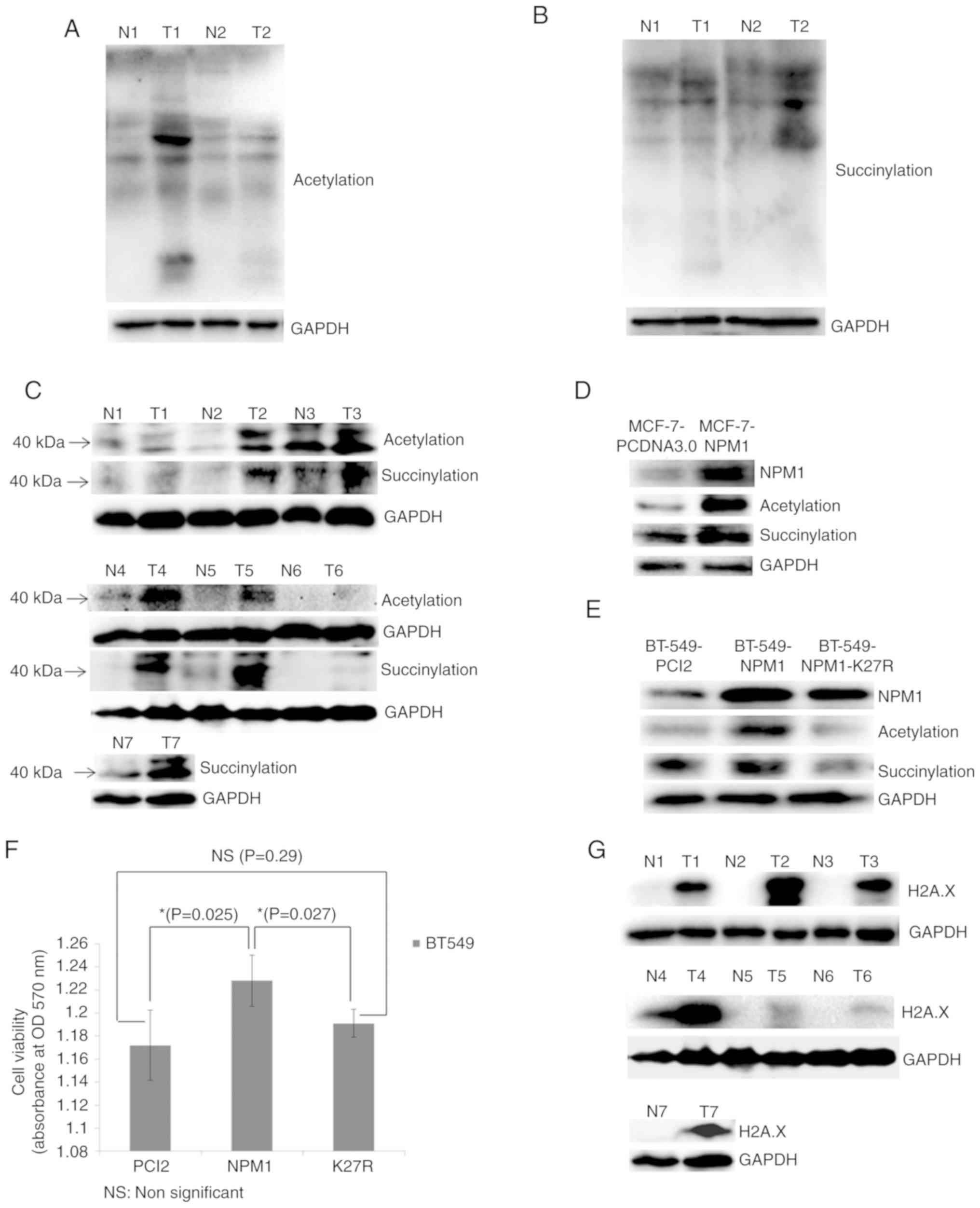 | Figure 5.Validation of proteomic results. (A)
Acetylation and (B) succinylation of whole proteins in breast
cancer tissues (T) and adjacent normal tissues (N) examined via
western blotting. GAPDH was used as a loading control. (C)
Acetylation and succinylation of proteins ~40 kDa (used for
indicating NPM1) in breast cancer tissues (T) and adjacent normal
tissues (N) examined via western blotting. GAPDH was used as a
loading control. (D) MCF-7 cells overexpressing NPM1 (MCF-7-NPM1)
and PCDNA3.0 (MCF-7-PCDNA3.0, used as control) were collected for
the protein extraction. Expression level of NPM1 and its
acetylation and succinylation were detected via western blotting.
GAPDH was used as a loading control. (E) BT549 cells overexpressing
NPM1 (BT549-NPM1), PCI2 (BT549-PCI2, used as control) and the
variants of NPM1 (NPM1-K27R, lysine mutated into arginine) were
collected for protein extraction. Expression level of NPM1 and its
acetylation and succinylation were detected via western blotting.
GAPDH was used as a loading control. (F) Viability of BT549 cells
overexpressing NPM1 (NPM1), PCI2 (PCI2, used as control) and the
variants of NPM1 (K27R, lysine mutated into arginine) was detected
via MTT methods. *P<0.05. (G) Expression of H2A.X in breast
cancer tissues (T) and adjacent normal tissues (N) were detected
via western blotting. GAPDH was used as a loading control. The
H2A.X and the succinylation bands from N4 to T6 shows non-adjacent
bands from the same gel, therefore they used the same GAPDH result.
NPM1, nucleophosmin; H2A.X, histone H2A.X. |
Discussion
Lysine acetylation and succinylation can change the
function of proteins as well as their target genes and proteins
without any gene mutation, thus, taking on another role with a
genetic code that can play an important role in the initiation and
development of a number of different types of tumors (9,41–43).
However, the information regarding the majority of proteins,
particularly for non-histone proteins, remains unknown. In
addition, extensive overlap of protein acetylation and
succinylation (22,23), as well as their common regulators,
such as SIRT5 (19,25), SIRT7 (26) and KAT2A (24) have demonstrated similarity in their
function. Thus, it is necessary to investigate protein acetylation
and succinylation both simultaneously and systematically. Through
proteomic investigation, the present study demonstrated a
significantly higher acetylation and succinylation level in the
majority of proteins in breast cancer, and observed a high
repetition rate and a similar alteration tendency of the two kinds
of modified proteins. These results proved that it is feasible to
identify potential targets for breast cancer among the modified
proteins. At present, some of the common functions of protein
acetylation and succinylation have been reported, such as abnormal
spermatogenesis, metabolism and DNA damage process. However, there
is an urgent need for precise targets for cancer investigation. Our
systematic analysis results can provide useful clues for the
double-modified target proteins in breast cancer research.
Moreover, compared with only one modification, double-modification
can also narrow the scope of the regulators, which can help us to
find or design potential anticancer drugs.
The enrichment analysis of the complexes provide
evidence to support the common functions of protein acetylation and
succinylation. The results revealed that both acetylation and
succinylation proteins were most significantly enriched in three
H2A.X complexes and they are involved in a number of processes
associated with DNA damage response (DDR). This builds a connection
between protein modification and DDR. It has previously been
reported that H2A.X complexes are the dynamic foci formed at DNA
break sites under different DNA damage conditions (35). When DNA is damaged, H2A.X is
activated and then many other proteins are recruited to form
protein foci to mediate DNA damage repair (40). What's more, this process can be
regulated by protein modification (37,44,45).
Thus, the over-acetylation and succinylation of proteins in H2A.X
complexes may affect DNA damage repair by regulating the formation
of protein foci in breast cancer. The results of the present study
demonstrated that those proteins associated with DNA damage repair
were upregulated, but those sensitive to DNA damage were
downregulated, suggesting a resistance characteristic to DNA damage
of breast cancer. This may be caused by the over-modification of
H2A.X complexes and may help to explain the resistance to
chemotherapy and radiotherapy for breast cancer.
The present study revealed that nucleophosmin (NPM1)
may be a key player of the complexes, for the following reasons: i)
NPM1 is one of the core members that were both acetylated and
succinylated in the three H2A.X complexes; ii) NPM1 can also
interact with numerous proteins in the three complexes, forming a
second regulation center in these complexes; iii) NPM1 was the only
protein whose acetylation and succinylation occurred on the same
lysine site, which is highly conserved in a number of other
different species; iv) upregulation of NPM1 expression and its
modification in breast cancer further demonstrated the importance
of NPM1 in H2A.X complexes. It has previously been reported that
NPM1 is a type of histone chaperonin that can transfer histones to
the chromosome and then stabilize the structure of the chromosome.
However, the stable structure of the chromosome can form a barrier
for the recognition of DNA damage and this can be regulated by the
modification of NPM1. Thus, the acetylation and succinylation of
NPM1 may affect DDR through the regulation of chromosome structure.
In addition, NPM1 is a type of protein that undergoes a variety of
post-translational modifications, such as phosphorylation,
acetylation, sumoylation and so on, all of these can direct its
various functions (46,47). Some of the modifications, such as
its phosphorylation and acetylation, have been reported to play an
important role in a number of different types of tumor (46,48–50).
To the best of our knowledge, there have not yet been any reports
regarding the succinylation of NPM1, and thus, the way in which its
acetylation and succinylation work in breast cancer remains
unknown. Thus, the results of the present study may provide a new
potential target for breast cancer investigation and therapy.
Supplementary Material
Supporting Data
Acknowledgements
We thank all the patients for their participation in
the study and the support funding.
Funding
The present study was supported by the National
Natural Science Foundation of China (nos. 81802834 and 81972491),
the Education Department of Heilongjiang Province (no.
2017-KYYWF-0696), the Scientific Research Startup Foundation for
Postdoctoral of Heilongjiang Province (no. LBH-Q17178), Qiqihar
Science and Technology Bureau (no. SFGG-201912) and the Doctoral
Research Startup Fund of Qiqihar Medical University
(QY2016B-04).
Availability of data and materials
Data and materials are available upon request to the
corresponding author.
Authors' contributions
LY conceived and designed the experiments. HB
conducted the participant recruitment and the tissue collection. LZ
conducted the data analysis. The experiments were performed by XG,
WZ and LL. XG and HB wrote the manuscript. All authors read and
approved the manuscript and agree to be accountable for all aspects
of the research in ensuring that the accuracy or integrity of any
part of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
All experimental protocols were approved by Qiqihar
Medical Ethics Committee [NO. (2018)27]. The patients provided
informed consent for the use of tissue samples.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
H2A.X
|
histone H2A.X
|
|
NPM1
|
nucleophosmin
|
|
DDR
|
DNA damage response
|
|
TMT
|
tandem mass tag
|
|
TEAB
|
triethylamine borane
|
|
TFA
|
trifluoroacetic acid
|
|
FA
|
formic acid
|
|
HPLC
|
high performance liquid
chromatography
|
|
FDR
|
false discovery rate
|
References
|
1
|
DeSantis CE, Fedewa SA, Goding Sauer A,
Kramer JL, Smith RA and Jemal A: Breast cancer statistics, 2015:
Convergence of incidence rates between black and white women. CA
Cancer J Clin. 66:31–42. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
DeSantis CE, Ma J, Goding Sauer A, Newman
LA and Jemal A: Breast cancer statistics, 2017, racial disparity in
mortality by state. CA Cancer J Clin. 67:439–448. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li L, Tian T and Zhang X: Mutation
mechanisms of human breast cancer. J Comput Biol. 25:396–404. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Takahashi M, Chiba N, Shimodaira H,
Yoshino Y, Mori T, Sumii M, Nomizu T and Ishioka C: OLA1 gene
sequencing in patients with BRCA1/2 mutation-negative suspected
hereditary breast and ovarian cancer. Breast Cancer. 24:336–340.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sheikh A, Hussain SA, Ghori Q, Naeem N,
Fazil A, Giri S, Sathian B, Mainali P and Al Tamimi DM: The
spectrum of genetic mutations in breast cancer. Asian Pac J Cancer
Prev. 16:2177–2185. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kulis M and Esteller M: DNA methylation
and cancer. Adv Genet. 70:27–56. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ahmadzada T, Reid G and McKenzie DR:
Fundamentals of siRNA and miRNA therapeutics and a review of
targeted nanoparticle delivery systems in breast cancer. Biophys
Rev. 10:69–86. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao QY, Lei PJ, Zhang X, Zheng JY, Wang
HY, Zhao J, Li YM, Ye M, Li L, Wei G and Wu M: Global histone
modification profiling reveals the epigenomic dynamics during
malignant transformation in a four-stage breast cancer model. Clin
Epigenetics. 8:342016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xi Y, Shi J, Li W, Tanaka K, Allton KL,
Richardson D, Li J, Franco HL, Nagari A, Malladi VS, et al: Histone
modification profiling in breast cancer cell lines highlights
commonalities and differences among subtypes. BMC Genomics.
19:1502018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wan Y, Liu J and Guo S: Effects of
succinylation on the structure and thermal aggregation of soy
protein isolate. Food Chem. 245:542–550. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dehzangi A, Lopez Y, Lal SP, Taherzadeh G,
Sattar A, Tsunoda T and Sharma A: Improving succinylation
prediction accuracy by incorporating the secondary structure via
helix, strand and coil, and evolutionary information from profile
bigrams. PLoS One. 13:e01919002018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dawson MA and Kouzarides T: Cancer
epigenetics: From mechanism to therapy. Cell. 150:12–27. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hsieh TH, Hsu CY, Tsai CF, Long CY, Wu CH,
Wu DC, Lee JN, Chang WC and Tsai EM: HDAC inhibitors target HDAC5,
upregulate microRNA-125a-5p, and induce apoptosis in breast cancer
cells. Mol Ther. 23:656–666. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma L, Yuan L, An J, Barton MC, Zhang Q and
Liu Z: Histone H3 lysine 23 acetylation is associated with oncogene
TRIM24 expression and a poor prognosis in breast cancer. Tumour
Biol. 37:14803–14812. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fermento ME, Gandini NA, Salomon DG,
Ferronato MJ, Vitale CA, Arévalo J, López Romero A, Nuñez M, Jung
M, Facchinetti MM and Curino AC: Inhibition of p300 suppresses
growth of breast cancer. Role of p300 subcellular localization. Exp
Mol Pathol. 97:411–424. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
McClure JJ, Li X and Chou CJ: Advances and
challenges of HDAC inhibitors in cancer therapeutics. Adv Cancer
Res. 138:183–211. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fiorentino F, Mai A and Rotili D: Lysine
acetyltransferase inhibitors: Structure-activity relationships and
potential therapeutic implications. Future Med Chem. 10:1067–1091.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Z, Tan M, Xie Z, Dai L, Chen Y and
Zhao Y: Identification of lysine succinylation as a new
post-translational modification. Nat Chem Biol. 7:58–63. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xiangyun Y, Xiaomin N, Linping G, Yunhua
X, Ziming L, Yongfeng Y, Zhiwei C and Shun L: Desuccinylation of
pyruvate kinase M2 by SIRT5 contributes to antioxidant response and
tumor growth. Oncotarget. 8:6984–6993. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Song Y, Wang J, Cheng Z, Gao P, Sun J,
Chen X, Chen C, Wang Y and Wang Z: Quantitative global proteome and
lysine succinylome analyses provide insights into metabolic
regulation and lymph node metastasis in gastric cancer. Sci Rep.
7:420532017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu C, Liu Y, Chen L, Zhang M, Li W, Cheng
H and Zhang B: Quantitative proteome and lysine succinylome
analyses provide insights into metabolic regulation in breast
cancer. Breast Cancer. 26:93–105. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pan J, Chen R, Li C, Li W and Ye Z: Global
analysis of protein lysine succinylation profiles and their overlap
with lysine acetylation in the marine bacterium vibrio
parahemolyticus. J Proteome Res. 14:4309–4318. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Weinert BT, Scholz C, Wagner SA,
Iesmantavicius V, Su D, Daniel JA and Choudhary C: Lysine
succinylation is a frequently occurring modification in prokaryotes
and eukaryotes and extensively overlaps with acetylation. Cell Rep.
4:842–851. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Y, Guo YR, Liu K, Yin Z, Liu R, Xia
Y, Tan L, Yang P, Lee JH, Li XJ, et al: KAT2A coupled with the
α-KGDH complex acts as a histone H3 succinyltransferase. Nature.
552:273–277. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang F, Wang K, Xu W, Zhao S, Ye D, Wang
Y, Xu Y, Zhou L, Chu Y, Zhang C, et al: SIRT5 desuccinylates and
activates pyruvate kinase M2 to block macrophage IL-1β production
and to prevent DSS-induced colitis in mice. Cell Rep. 19:2331–2344.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li L, Shi L, Yang S, Yan R, Zhang D, Yang
J, He L, Li W, Yi X, Sun L, et al: SIRT7 is a histone desuccinylase
that functionally links to chromatin compaction and genome
stability. Nat Commun. 7:122352016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tyanova S, Temu T and Cox J: The MaxQuant
computational platform for mass spectrometry-based shotgun
proteomics. Nat Protoc. 11:2301–2319. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cox J, Neuhauser N, Michalski A, Scheltema
RA, Olsen JV and Mann M: Andromeda: A peptide search engine
integrated into the MaxQuant environment. J Proteome Res.
10:1794–1805. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
UniProt Consortium: UniProt: A worldwide
hub of protein knowledge. Nucleic Acids Res. 47:D506–D515. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Giurgiu M, Reinhard J, Brauner B,
Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C and Ruepp A:
CORUM: The comprehensive resource of mammalian protein
complexes-2019. Nucleic Acids Res. 47:D559–D563. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pathan M, Keerthikumar S, Ang CS, Gangoda
L, Quek CY, Williamson NA, Mouradov D, Sieber OM, Simpson RJ, Salim
A, et al: FunRich: An open access standalone functional enrichment
and interaction network analysis tool. Proteomics. 15:2597–2601.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kumar S, Stecher G and Tamura K: MEGA7:
Molecular evolutionary genetics analysis version 7.0 for bigger
datasets. Mol Biol Evol. 33:1870–1874. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
R CoreTeam, . R: A language and
environment for statistical computing. R Foundation for Statistical
Computing; Vienna: 2017
|
|
35
|
Du YC, Gu S, Zhou J, Wang T, Cai H,
Macinnes MA, Bradbury EM and Chen X: The dynamic alterations of
H2AX complex during DNA repair detected by a proteomic approach
reveal the critical roles of Ca(2+)/calmodulin in the ionizing
radiation-induced cell cycle arrest. Mol Cell Proteomics.
5:1033–1044. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hauer MH and Gasser SM: Chromatin and
nucleosome dynamics in DNA damage and repair. Genes Dev.
31:2204–2221. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Polo SE and Jackson SP: Dynamics of DNA
damage response proteins at DNA breaks: A focus on protein
modifications. Genes Dev. 25:409–433. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bielak-Zmijewska A, Mosieniak G and Sikora
E: Is DNA damage indispensable for stress-induced senescence? Mech
Ageing Dev. 170:13–21. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Paull TT, Rogakou EP, Yamazaki V,
Kirchgessner CU, Gellert M and Bonner WM: A critical role for
histone H2AX in recruitment of repair factors to nuclear foci after
DNA damage. Curr Biol. 10:886–895. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Palla VV, Karaolanis G, Katafigiotis I,
Anastasiou I, Patapis P, Dimitroulis D and Perrea D: Gamma-H2AX:
Can it be established as a classical cancer prognostic factor?
Tumour Biol. 39:10104283176959312017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wagner VP, Martins MD and Castilho RM:
Histones acetylation and cancer stem cells (CSCs). Methods Mol
Biol. 1692:179–193. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu B, Wang T, Wang H, Zhang L, Xu F, Fang
R, Li L, Cai X, Wu Y, Zhang W and Ye L: Oncoprotein HBXIP enhances
HOXB13 acetylation and co-activates HOXB13 to confer tamoxifen
resistance in breast cancer. J Hematol Oncol. 11:262018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fang X, Lu G, Ha K, Lin H, Du Y, Zuo Q, Fu
Y, Zou C and Zhang P: Acetylation of TIP60 at K104 is essential for
metabolic stress-induced apoptosis in cells of hepatocellular
cancer. Exp Cell Res. 362:279–286. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sharma GG, So S, Gupta A, Kumar R, Cayrou
C, Avvakumov N, Bhadra U, Pandita RK, Porteus MH, Chen DJ, et al:
MOF and histone H4 acetylation at lysine 16 are critical for DNA
damage response and double-strand break repair. Mol Cell Biol.
30:3582–3595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Murr R, Loizou JI, Yang YG, Cuenin C, Li
H, Wang ZQ and Herceg Z: Histone acetylation by Trrap-Tip60
modulates loading of repair proteins and repair of DNA
double-strand breaks. Nat Cell Biol. 8:91–99. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shandilya J, Swaminathan V, Gadad SS,
Choudhari R, Kodaganur GS and Kundu TK: Acetylated NPM1 localizes
in the nucleoplasm and regulates transcriptional activation of
genes implicated in oral cancer manifestation. Mol Cell Biol.
29:5115–5127. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Okuwaki M: The structure and functions of
NPM1/Nucleophsmin/B23, a multifunctional nucleolar acidic protein.
J Biochem. 143:441–448. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Swaminathan V, Kishore AH, Febitha KK and
Kundu TK: Human histone chaperone nucleophosmin enhances
acetylation-dependent chromatin transcription. Mol Cell Biol.
25:7534–7545. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Destouches D, Sader M, Terry S, Marchand
C, Maillé P, Soyeux P, Carpentier G, Semprez F, Céraline J, Allory
Y, et al: Implication of NPM1 phosphorylation and preclinical
evaluation of the nucleoprotein antagonist N6L in prostate cancer.
Oncotarget. 7:69397–69411. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Box JK, Paquet N, Adams MN, Boucher D,
Bolderson E, O'Byrne KJ and Richard DJ: Nucleophosmin: From
structure and function to disease development. BMC Mol Biol.
17:192016. View Article : Google Scholar : PubMed/NCBI
|