Introduction
Colorectal cancer (CRC), the fourth leading cause of
cancer-related deaths worldwide, resulted in 50,630 deaths in 2018
in the United States (1,2). With the improvement of screening
technologies and therapeutic strategies, the death rate from CRC
has been dropping over the past decades (3,4).
However, reduced sensitivity to conventional treatments, such as
chemotherapy and radiotherapy, is a major obstacle in the treatment
of advanced and metastatic CRC (5,6).
Therefore, it is urgent to develop novel effective therapeutic
strategies.
It has been widely demonstrated that aberrant
activation of the Ras/Raf-1/extracellular-regulated kinase (ERK)
signaling pathway plays pivotal roles in the initiation and
progression of various tumors (7–10).
Spred2 is an important member of the sprouty-related EVH1 domain
proteins, which are known to be negative regulators of growth
factor-induced Ras/Raf-1/ERK activation (11,12).
As anticipated, Spred2 was revealed to be significantly
downregulated in various tumor cells, including hematological
malignancies and solid tumors (13,14).
Moreover, the Spred2 expression level was demonstrated to be
closely associated with distal metastasis and survival in the
clinic (15,16). Notably, strategies to increase
Spred2 expression would be beneficial to enhance antitumor
responses. For example, our group reported that imatinib could
upregulate Spred2, and induce apoptosis and growth arrest in
chronic myeloid leukemia (CML) cells (17). However, the roles of Spred2 in CRC
are still unexplored.
In the present study, whether Spred2 expression is
obviously decreased in the tumor tissues of CRC patients was first
explored. Then, a fiber-modified replication-deficient adenoviral
vector expressing Spred2, Ad.Spred2, was prepared as reported
previously (17). Ad.Spred2
mediated Spred2 overexpression in CRC cells inhibited cellular
proliferation, survival and migration, and reduced
epithelial-mesenchymal transition (EMT). Finally, it was revealed
that Spred2 inhibited EMT in CRC cells by blocking the ERK
signaling pathway, with or without reduced TGFβ/SMAD signaling.
Materials and methods
Spred2 mRNA expression in tumor
tissues from CRC patients
Surgical specimens were obtained from 26 CRC
patients, including 11 male and 15 female patients from 49 to 86
years old, that underwent clinical pathological examination at
Beijing Friendship Hospital, an affiliate of Capital Medical
University, from August 2010 to December 2013. Beijing Friendship
Hospital is our cooperative partner in the field of cancer
research. All patients provided written consent and all the
procedures were approved by the Ethics Committee of Beijing
Friendship Hospital. The mRNA expression of Spred2, in tumor
tissues and distal normal tissues, was analyzed by real-time
reverse transcription polymerase chain reaction (RT-PCR).
Cell lines
The SW480, SW620, RKO and Colo205 and HCT116 human
CRC cell lines were purchased from the cell bank at the Shanghai
Institutes for Biological Sciences, Chinese Academy of Sciences.
Human embryonic kidney cell line, 293, was obtained from American
Type Culture Collection (ATCC). SW480, SW620, Colo205 and HCT116
cells were maintained in RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal calf serum (FCS),
while 293 cells were cultured in Dulbecco's minimal essential
medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.) plus 10% FCS.
The RKO cells were maintained in Eagle's Minimum Essential Medium
(EMEM; Gibco; Thermo Fisher Scientific, Inc.) containing 10%
FCS.
Adenoviruses
An adenoviral vector expressing Spred2, Ad.Spred2,
was constructed and prepared using a fiber modified Ad5/F11p
adenoviral system as described previously (18). In addition, an Ad5/F11p adenoviral
system-based adenoviral vector without any transgene, Ad.Null was
prepared and used as a control. All the adenoviruses were purified
by CsCl density gradient centrifugation, and the infectious titers
were measured by median tissue culture infective dose
(TCID50).
Analysis of biological
characteristics
Proliferation
Exponentially growing SW480 CRC cells were labeled
with the dye eFluor® 670 (eBioscience; Thermo Fisher
Scientific, Inc.) according to the manufacturers' instructions.
Then, the labeled cells were seeded into 12-well plates at a
density of 1×105 cells/well. Four hours later, the cells
were infected with Ad.Spred2 or Ad.Null at a multiplicity of
infection (MOI) of 50. At 0, 24, 48, 72 and 96 h after infection,
the cells were collected. The fluorescence intensity of the eFluor
670 dye was detected by flow cytometry, and the proliferation index
was calculated.
Apoptosis
Exponentially growing SW480 cells were plated into a
6-well plate at a density of 5.0×105 cells/well.
Twenty-four hours later, the cells were infected with Ad.Spred2 or
Ad.Null at an MOI of 50. After 4 h of incubation, the culture media
were replaced with fresh RPMI-1640 medium plus 10% FCS. At 24 and
48 h after infection, the cells were collected, labeled with APC
conjugated Annexin-V and PI (Sungene Biotech Co., Ltd.), and
analyzed by flow cytometry using CellQuest Pro software version 5.1
(FACSCalibur; BD Biosciences).
Migration assay
For the wound healing assay, exponentially growing
SW480 cells were seeded in a 6-well plate at a density of
5.0×105 cells/well. Twenty-four hours later, the cells
were infected by Ad.Spred2 or Ad.Null at an MOI of 50, or treated
with 10 ng/ml recombinant human TGF-β (PeproTech, Inc.). After
another 24 h of incubation, confluent cells were scratched to
generate wounds. At 0 and 24 h after the scratch, live cell images
(×100) were obtained at indicated positions using an inverted
microscope and charge-coupled device (Olympus Corporation). The
distances between the two margins of the wounds were measured, and
the percentage of wound healing was calculated as previously
described (19).
For the Transwell assay, exponentially growing SW480
cells were infected with Ad.Spred2 or Ad.Null at an MOI of 50.
Twenty-four hours later, the cells were collected and resuspended
in FCS-free RPMI-1640 medium at a density of 5.0×105
cells/ml. Then, a Transwell insert was added to the well by
inserting the bottom of the insert into the medium (RPMI-1640
medium plus 2% FCS) in the lower compartment. Then, 200 µl of cells
was added to the Transwell insert. After 8 h of incubation, the
migrated cells were fixed by 800 µl methanol for 30 min and stained
with 0.1% crystal violet for 20 min at room temperature. The cells
were observed by inverted microscope and counted in 5 randomly
selected views using a magnification of ×100.
Giemsa staining
Exponentially growing SW480 cells were seeded into
6-well plates at a density of 5×105 cells/well.
Twenty-four hours later, 10 ng/ml TGF-β was added and incubation
continued for another 48 h. Cells were stained with Giemsa staining
buffer (Beijing Solarbio Science & Technology Co., Ltd.) for
15–30 min at room temperature. Cell images were obtained by
inverted microscope and charge-coupled device using a magnification
of ×400.
Rhodamine-phalloidin staining
A total of 1×105 exponentially growing
SW480 cells and HCT116 cells were seeded into a 35-mm cell culture
dish with a glass bottom. Twenty-four hours later, the cells were
treated with Ad.Spred2, Ad.Null, or 10 ng/ml TGF-β. After another
48 h of incubation, the cells were stained with
rhodamine-phalloidin for 1 h at room temperature (Thermo Fisher
Scientific, Inc.) according to the manufacturers' instructions. The
results were analyzed by confocal microscopy (Perkin Elmer).
Immunofluorescence
Exponentially growing SW480 cells were plated on a
35-mm cell culture dish as aforementioned. Twenty-four hours later,
the cells were infected with Ad.Spred2 or Ad.Null at a MOI of 50,
or treated by 10 ng/ml TGF-β. Forty-eight hours later, the
expression of E-cadherin, N-cadherin and vimentin was analyzed by
immunofluorescence. Briefly, cells were incubated with rabbit
anti-human E-cadherin antibody (product code ab40772), rabbit
anti-human N-cadherin antibody (product code ab18203) or mouse
anti-human vimentin antibody (product code ab20346) (all from
Abcam) with a dilution of 1:500 at 4°C overnight. The following
day, corresponding secondary antibodies, including FITC-conjugated
goat anti-rabbit IgG H&L (cat. no. ZF0311) and goat anti-mouse
IgG H&L (cat. no. ZF0312) (ZSGB-Bio; OriGene Technologies,
Inc.), or TRITC conjugated goat anti-rabbit IgG H&L (cat. no.
BA1090) and goat anti-mouse IgG H&L (cat. no. BA1089) (Boster
Biological Technology, Ltd.) were added at a dilution of 1:2,000
and incubated for 1 h at room temperature. The results were
observed by confocal microscopy.
Western blotting
Twenty-four and forty-eight hours after infection
with Ad.Spred2 and Ad.Null, SW480 and HCT116 cells were collected
and the protein was extracted by direct lysis buffer (DLB; Beijing
Yangguang Yingrui Biotech, Inc.). At the indicated time-points, the
expression of Spred2, ERK, phosphorylated ERK (p-ERK), E-cadherin,
N-cadherin, vimentin, SMAD2/3, phosphorylated SMAD2/3 (p-SMAD2/3)
and SMAD4 was analyzed by western-blotting using rabbit anti-human
Spred2 antibody (cat. no. PRS4849) (Sigma-Aldrich; Merck KGaA),
rabbit anti-MAPK/ERK antibody (cat. no. 4695), rabbit
anti-phospho-Erk1/2 (Thr202/Tyr204) antibody (cat. no. 4370) (all
from Cell Signaling Technology Inc.), rabbit anti-human E-cadherin
antibody (product code ab40772), rabbit anti-human N-cadherin
antibody (product code ab18203), mouse anti-human vimentin antibody
(product code ab20346) (all from Abcam), rabbit anti-Slug antibody
(cat. no. 9585), rabbit anti-Snail antibody (cat. no. 3879), rabbit
anti-SMAD2/3 antibody (cat. no. 8685), rabbit anti-phospho-Smad2
(Ser465/467)/Smad3 (Ser423/425) antibody (cat. no. 8828) and rabbit
anti-SMAD4 antibody (cat. no. 9515) (all from Cell Signaling
Technology Inc.), respectively. Briefly, the concentration of total
proteins was determined using Bicinchoninic Acid (BCA) method.
Then, 40 µg total proteins were loaded and separated by 10–15%
SDS-polyacrylamide gel. After electroblotting onto a polyvinylidene
difluoride membrane (EMD Millipore), the proteins were blocked by
Tris-buffered saline containing 7.5% non-fat dry milk for 1 h at
room temperature and labeled by corresponding primary antibodies at
a dilution of 1:500 for 2 h at room temperature. Then, the membrane
was incubated with corresponding horseradish peroxidase
(HRP)-conjugated goat anti-rabbit antibody (cat. no. ZDR-5306) and
HRP-conjugated goat anti-mouse antibody (cat. no. ZDR-5307)
(ZSGB-Bio; OriGene Technologies, Inc.) at a dilution of 1:5,000.
Then, the polyvinylidene difluoride membrane was visualized by
using an ECL Plus Western Blotting Substrate (Pierce; Thermo Fisher
Scientific, Inc.) and scanned by MultiImage II system (Alpha
Innotech). The densitometry was analyzed by AlphaEase®
Image Analysis software version 4.0 (Alpha Innotech). The relative
expression of each indicated protein was normalized to GAPDH
expression.
Real-time RT-PCR
Total RNA was isolated from tumor samples,
corresponding normal tissues, and adenoviral vector-transfected
SW480 cells. Then, cDNA was synthesized using RevertAid First
Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Inc.). The
mRNA expression of Spred1 and Spred2 was quantified using
SYBR® Premix Ex Taq™ (Tli RNaseH Plus) (Takara Bio,
Inc.) on a 7500 Fast real-time PCR system (Applied Biosystems/Life
Technologies; Thermo Fisher Scientific, Inc.). The optimal
thermocycling conditions included: Pre-denaturation at 95°C for 2
min; denaturation at 95°C for 30 sec, then annealing and extension
at 60°C for 1 min, repeated for 40 cycles. The relative expression
levels were calculated by the 2−ΔΔCq method, using
β-actin as the control (20). The
following primers were used, Spred1 sense,
5′-ggaagcactagaaactggcattatt-3′ and antisense,
5′-cacctggctgctaggcaaac-3′; Spred2 sense,
5′-ctcatccatggtgaacgacagaa-3′ and antisense,
5′-tgtcaaaggctcgggcatc-3′; and β-actin sense,
5′-gggacctgactgactacctc-3′ and antisense,
5′-cttaatgtcacgcacgattt-3′.
Statistical analysis
Data are presented as the mean ± SEM and were
statistically analyzed by using GraphPad Prism software version 5
(GraphPad Software, Inc.). Paired t-tests were used to analyze
Spred2 and Spred1 expression in the tumor tissues of CRC patients,
and unpaired t-tests were used to analyze differences in other data
between two groups. One-way analysis of variance (ANOVA) followed
by the Bonferroni's post hoc test was performed to analyze multiple
groups. Differences were considered statistically significant with
a two-sided P<0.05.
Results
Spred2 inhibits the growth and
migration of CRC cells, but promotes cellular apoptosis
Spred1 and Spred2, two important members of the
Spred family, were revealed to be frequently downregulated in
various cancers, such as hepatocellular carcinoma and prostate
cancer (9,13). In the present study, Spred1 and
Spred2 expression in the tumor tissues of CRC patients were
analyzed. The expression of Spred2, but not Spred1, was
significantly downregulated in tumor tissues, compared to
corresponding distal normal tissues (Fig. 1A and B). Moreover, it was also
demonstrated that Spred2 mRNA expression was decreased in CRC cell
lines, compared to normal tissues (Fig.
1C). These results indicated that Spred2 plays pivotal roles in
regulating tumor initiation and progression.
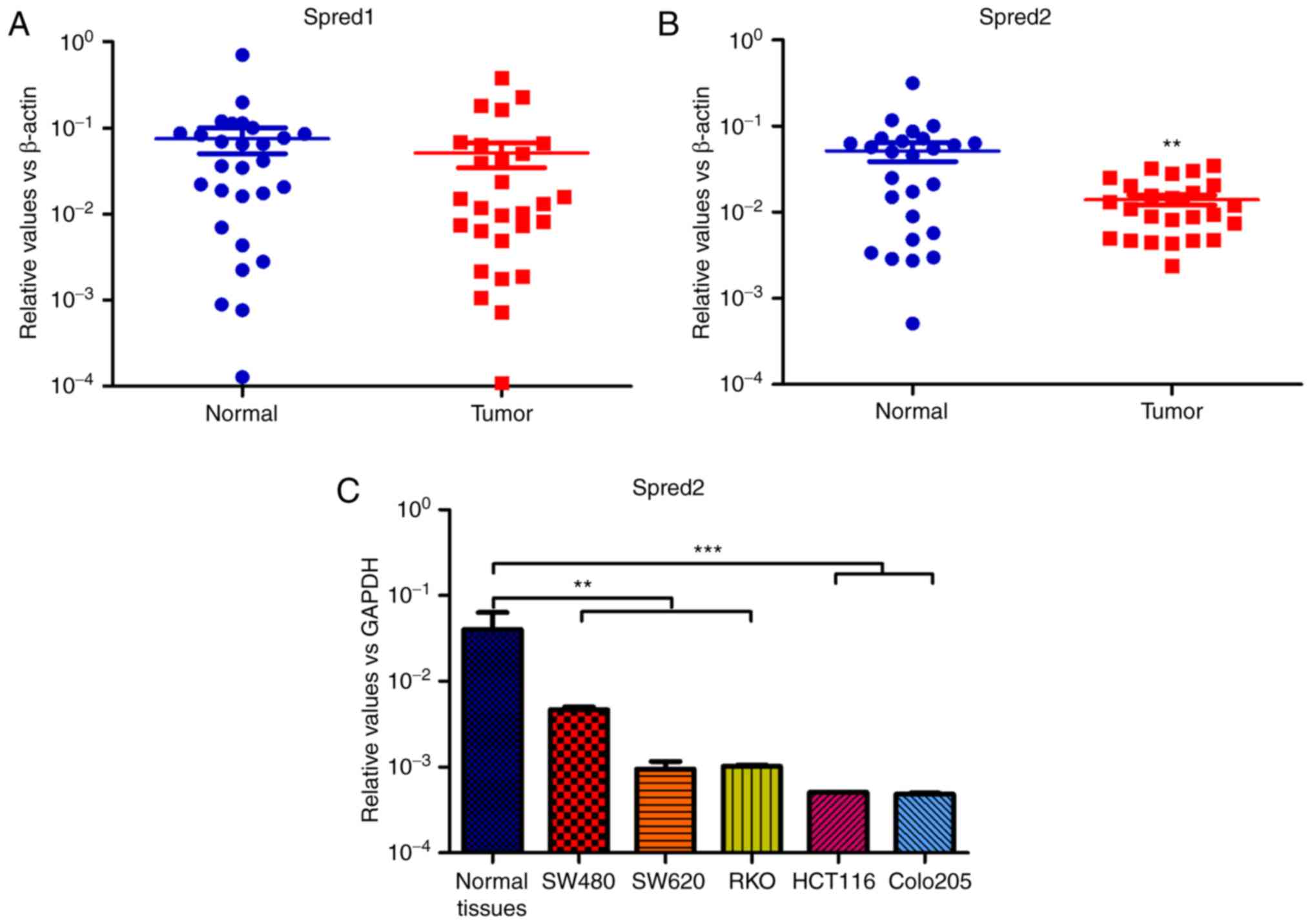 | Figure 1.Spred1 and Spred2 expression in the
tumor lesions of CRC patients. Surgical specimens were obtained
from 26 CRC patients, and were divided into tumor tissues,
paracancerous tissues and distal normal tissues according to the
results of the clinical pathological examination. The total RNA was
isolated from tumor lesions and corresponding distal normal
tissues. After cDNA synthesis, the mRNA expression of (A) Spred1
and (B) Spred2 was detected by RT-PCR, and normalized to β-actin.
Moreover, the mRNA expression in CRC cell lines and distal normal
tissues aforementioned, was also detected by real-time RT-PCR, and,
the relative expression level of Spred2 was normalized to GAPDH.
(C) Data are presented as the mean ± SEM. **P<0.01 and
***P<0.001, compared to the expression in corresponding normal
tissues. Spred2, sprouty-related EVH1 domain protein 2; CRC,
colorectal cancer; RT-PCR, real-time reverse transcription
polymerase chain reaction. |
To investigate the roles and mechanisms of Spred2 in
regulating the biological characteristics of CRC cells, a
fiber-modified adenoviral vector expressing Spred2, Ad.Spred2, was
prepared. Colon cancer cell lines, including SW480 cells,
transfected with Ad.Spred2 effectively expressed Spred2 (Fig. 2A and B). In SW480 cells, Spred2
overexpression inhibited proliferation and promoted cellular
apoptosis (Fig. 2C and D).
Moreover, Spred2 expression also significantly decreased migration
(Fig. 2E and F). These results
indicated that Spred2 is a pivotal molecule in modulating tumor
growth, progression and metastasis.
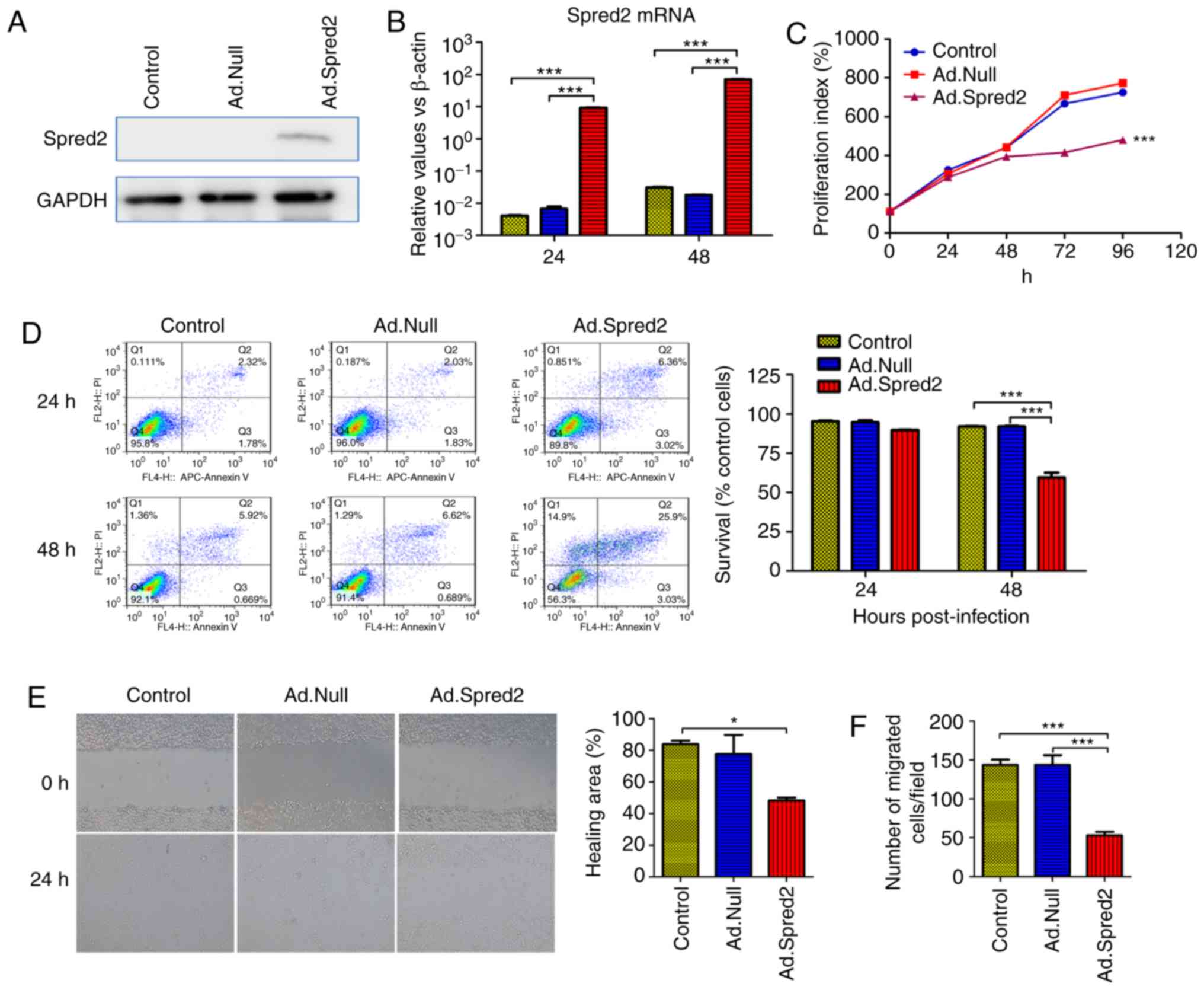 | Figure 2.Ad.Spred2 transduction reduces
proliferation, induces apoptosis and inhibits migration in SW480
cells. Exponentially growing SW480 cells were infected with
Ad.Spred2, an adenoviral vector expressing Spred2, at an MOI of 50.
(A and B) Ad.Spred2 mediated Spred2 protein and mRNA expression.
(A) Forty-eight hours later, the cells were collected and Spred2
protein expression was analyzed by western blotting. (B) In
addition, at 24 and 48 h after infection, Spred2 mRNA expression
was also detected by real-time RT-PCR (B) (C-F) The effects of
Spred2 on the biological characteristics of SW480 cells were
analyzed. (C) Cellular proliferation at 24, 48, 72 and 96 h after
infection was analyzed by flow cytometry using Fluor 670 dye
staining. The relative proliferation index was calculated. (D) At
24 and 48 h after infection, cells were collected and labeled with
Annexin V/APC and PI. Cellular apoptosis was analyzed by flow
cytometry, and the survival rate was calculated. (E) Twenty-four
hours after adenoviral infection, confluent SW480 cells were
scratched to generate wounds, and the percentage of the wounded
area filled was analyzed and is presented. (F) Furthermore, the
migratory ability was also analyzed by Transwell assay, and the
cells that passed through the membrane were counted. All the data
were obtained from at least three independent experiments, and data
in B-F are presented as the mean ± SEM. *P<0.05 and
***P<0.001, vs. the corresponding control group. Spred2,
sprouty-related EVH1 domain protein 2. |
Ad.Spred2 inhibits the EMT of CRC
cells in vitro
As aforementioned, Spred2 significantly impaired the
migratory ability of CRC cells in vitro. Therefore, the
mechanisms involved in Spred2 regulation of tumor metastasis, were
explored. In SW480 and HCT116 cells, decreased filamentous actin
(F-actin) expression and aggregation indicated that Spred2 may play
pivotal roles in regulating EMT, an essential event during tumor
metastasis to distant sites (Fig.
3A).
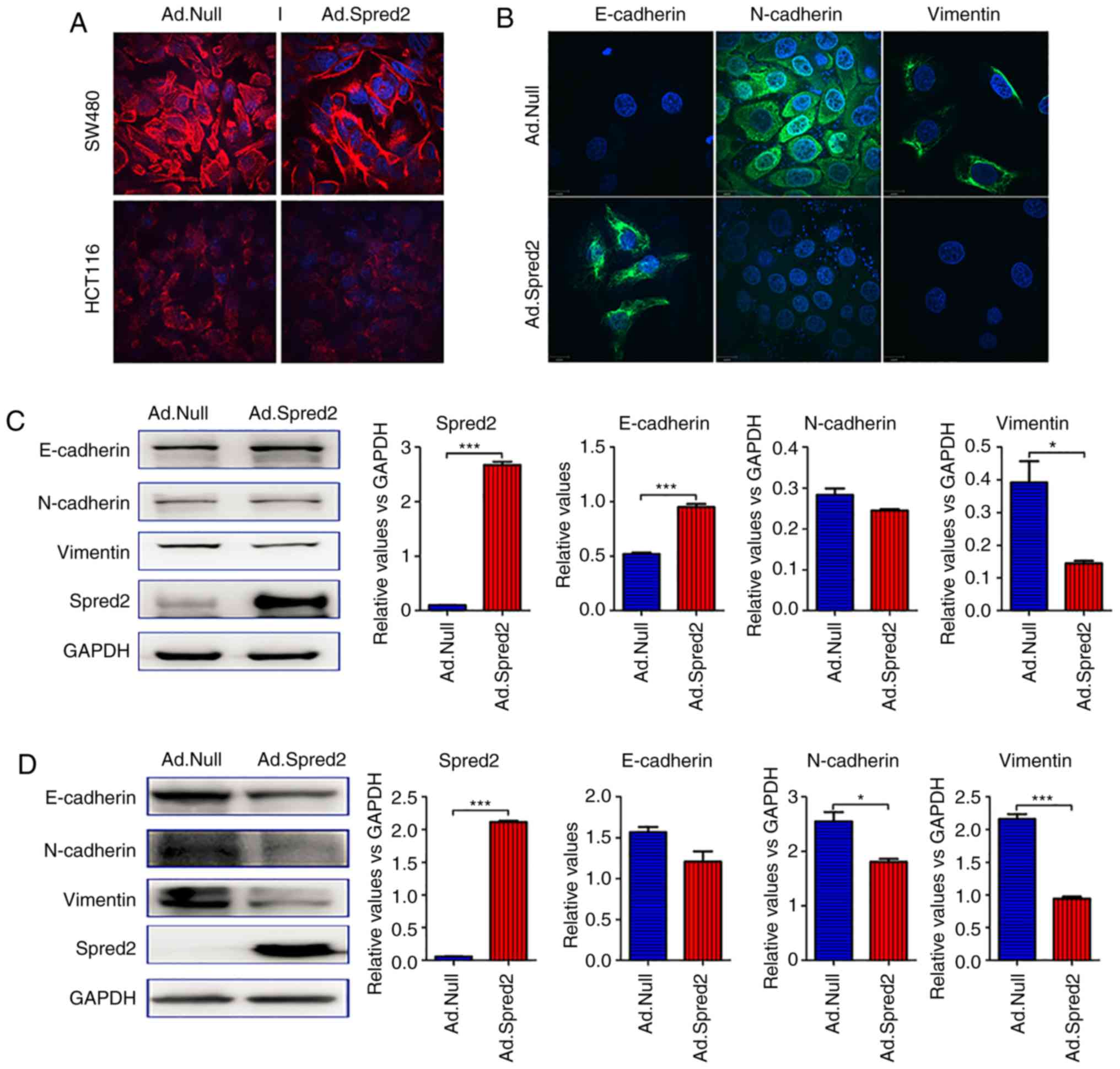 | Figure 3.Ad.Spred2 inhibits EMT of CRC cells.
(A) Exponentially growing SW480 cells and HCT116 cells were
infected with Ad.Spred2 at an MOI of 50. At 48 h after infection,
the expression and intercellular distribution of F-actin, an
important indicator of EMT, was analyzed by rhodamine-phalloidin
staining and observed by confocal microscopy. (B) Then, the protein
expression and intercellular location of the epithelial marker
E-cadherin, and mesenchymal markers N-cadherin and vimentin in
SW480 cells were analyzed by immunofluorescence staining and
observed by confocal microscopy. In addition, Spred2, E-cadherin,
N-cadherin and vimentin protein expression in (C) SW480 cells and
(D) HCT116 cells was also detected by western blotting. The results
of semi-quantitative analysis are also presented in C and D. All
the data were obtained from at least three independent experiments.
Data in C and D are presented as the mean ± SEM. *P<0.05 and
***P<0.001, vs. Ad.Null-treated cells. Spred2, sprouty-related
EVH1 domain protein 2; EMT, epithelial-mesenchymal transition; CRC,
colorectal cancer. |
During EMT, cells lose epithelial markers, such as
E-cadherin, but acquire mesenchymal markers, such as N-cadherin and
vimentin. In the present study, it was determined that Spred2
overexpression increased E-cadherin expression and decreased
N-cadherin and vimentin protein in SW480 cells (Fig. 3B and C). However, in HCT116 cells,
Spred2 only significantly downregulated the mesenchymal markers,
N-cadherin and vimentin, but did not affect E-cadherin (Fig. 3D). The results indicated that Spred2
can impair EMT in CRC cells.
TGF-β activates SMAD signaling and
induces EMT in CRC cells
TGF-β, a multifunctional cytokine, can induce EMT in
tumor cells via both SMAD-dependent and SMAD-independent activation
of EMT-related transcription factors (21,22).
Firstly, it was confirmed that 10 ng/ml TGF-β could activate
classic SMAD signaling in CRC cells. In fact, it enhanced the
phosphorylation of SMAD2/3 and increased SMAD4 levels in SW480
cells (Fig. 4A).
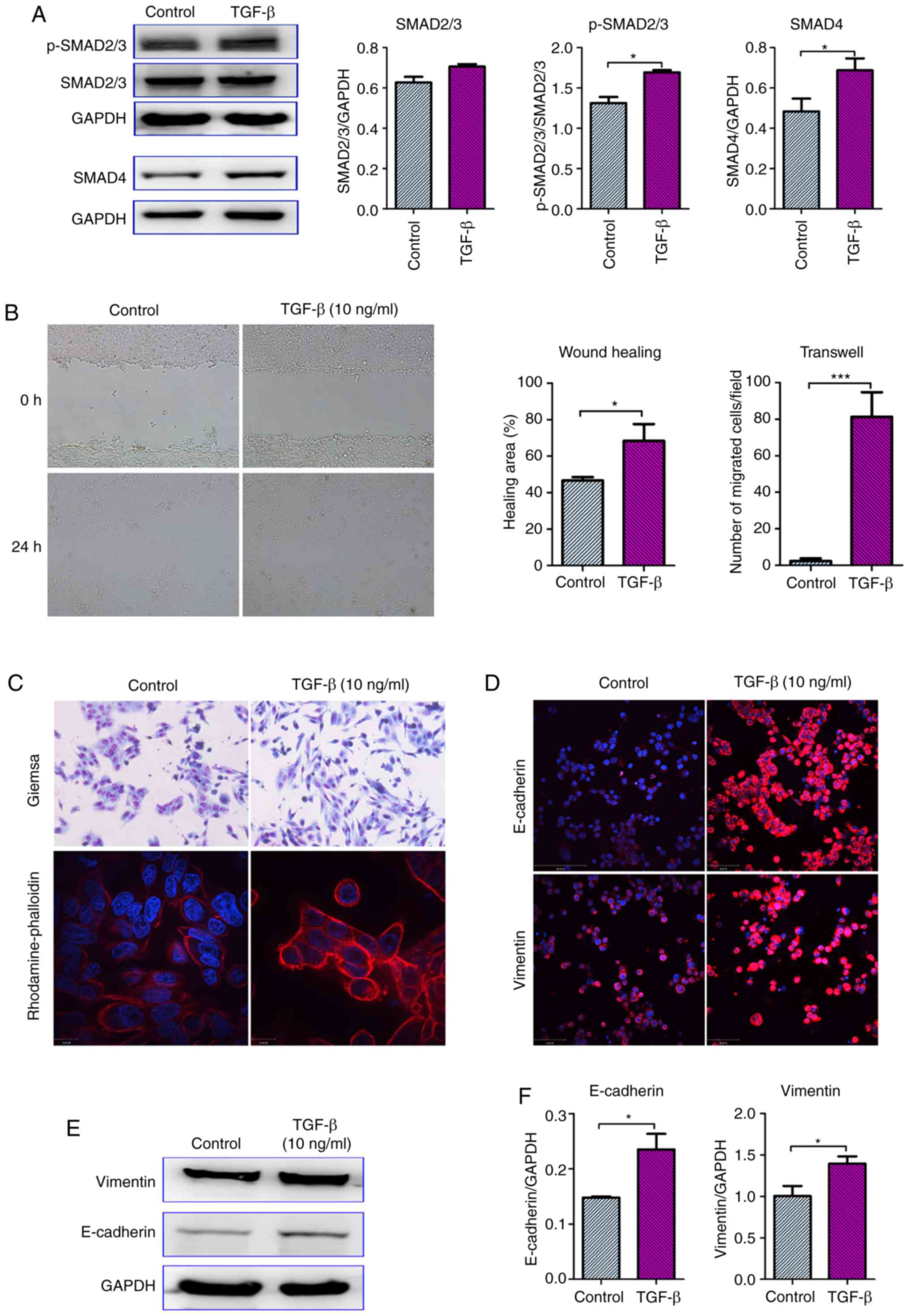 | Figure 4.TGF-β activates SMAD signaling and
induces EMT in SW480 cells. Exponentially growing SW480 cells were
stimulated with 10 ng/ml TGF-β. At 48 h after treatment, the
expression of pivotal molecules in the SMAD signaling pathway,
SMAD2/3, phosphorylated SMAD2/3 and SMAD4, was detected by western
blotting. Representative images and the results of
semi-quantitative analysis are presented in A. (B) The effects of
TGF-β on the migratory ability of SW480 cells were analyzed by
wound healing and Transwell assays. For the wound healing assay,
confluent SW480 cells were scratched to generate wounds and
stimulated with 10 ng/ml TGF-β for 24 h. For the Transwell assay,
10 ng/ml TGF-β was added in the lower compartment, while cells were
seeded in the insert compartment. (C) The cellular morphology was
observed after Giemsa staining, while the F-actin distribution was
detected by rhodamine-phalloidin staining. Moreover, the expression
of E-cadherin and vimentin was detected by (D) immunofluorescence
staining and (E and F) western blotting and quantitative analysis.
All the data were obtained from at least three independent
experiments. Data in A, B and F are presented as the mean ± SEM.
*P<0.05 and ***P<0.001, vs. the control (TGF-β untreated
cells). TGF-β, transforming growth factor-β; EMT,
epithelial-mesenchymal transition. |
Consistent with previous studies, it was
demonstrated that TGF-β treatment significantly enhanced the
migration of SW480 cells (Fig. 4B)
(23,24). Then, the effects of TGF-β on EMT
were analyzed. Untreated SW480 cells exhibited a typical epithelial
morphology, but TGF-β treatment increased the number and percentage
of fibroblast-like spindle-shaped cells. Moreover,
rhodamine-phalloidin staining was used to show the distribution of
F-actin, which has also emerged as an important marker of EMT in
tumor cells. As expected, F-actin filaments showed a pericellular
plasma membrane distribution in untreated SW480 cells. However,
TGF-β treatment enhanced rhodamine-phalloidin staining, and F-actin
was assembled into thick parallel bundles (Fig. 4C). Furthermore, the expression of
E-cadherin and vimentin was also analyzed. Notably, TGF-β increased
the protein expression of both vimentin and E-cadherin (Fig. 4D and E). It was hypothesized that
the ratio of vimentin and E-cadherin, but not their absolute
expression determines the EMT phenotype of tumor cells.
Spred2 inhibits EMT in CRC cells in an
ERK signaling-dependent manner
Spred2 is a negative regulator of growth
factor-induced RAS-ERK activation. It was revealed that Spred2
overexpression significantly inhibited the phosphorylation of ERK
in SW480 cells (Fig. 5A). Then, ERK
signaling was blocked by using a specific inhibitor, PD98059, and
it was revealed that the level of phosphorylated ERK was
significantly downregulated. Similar to Spred2, PD98059 increased
E-cadherin expression, but reduced vimentin protein levels in SW480
cells (Fig. 5B), indicating that
ERK signaling plays pivotal roles in regulating EMT in SW480
cells.
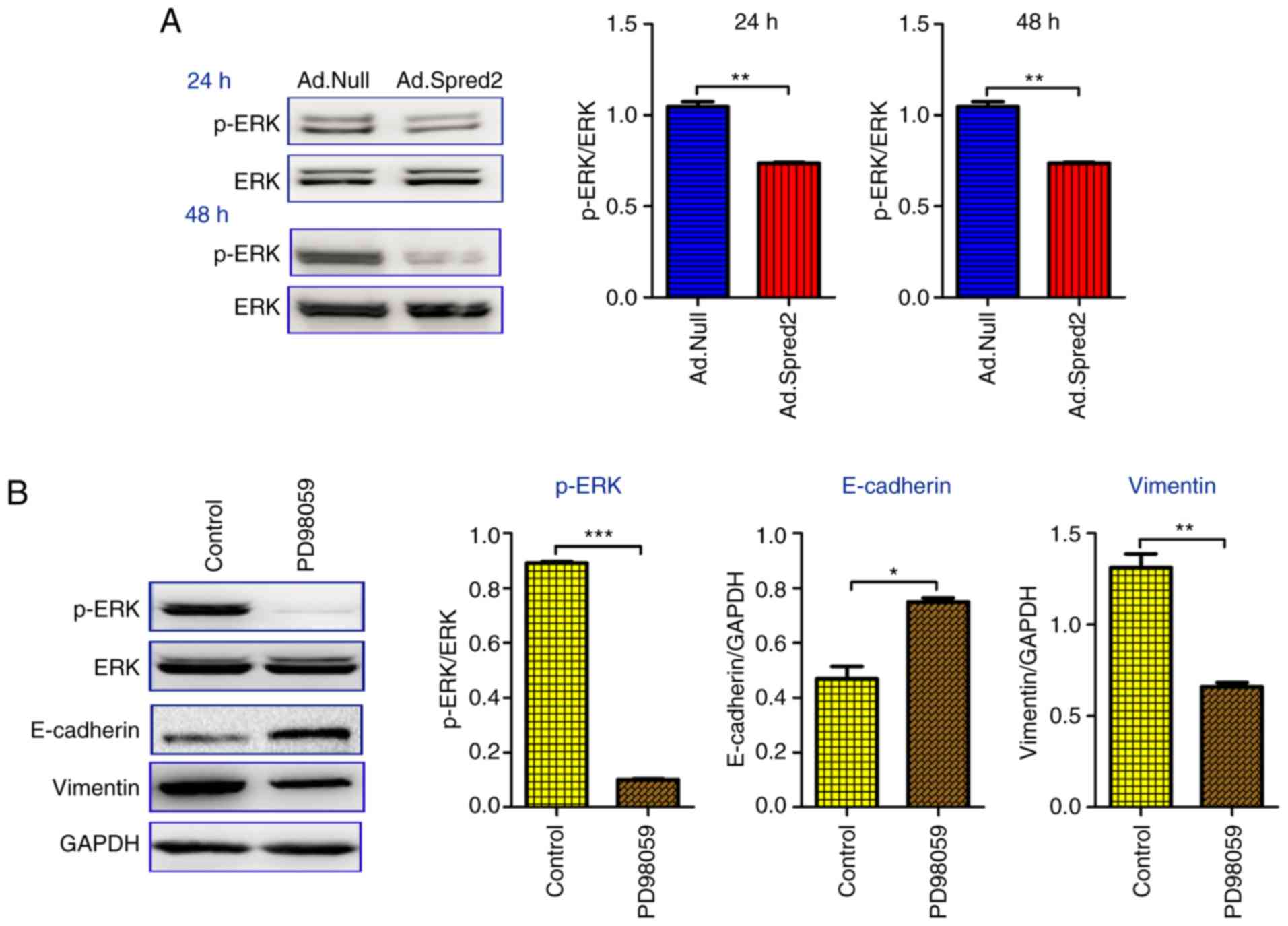 | Figure 5.Ad.Spred2 impairs ERK signaling, but
not SMAD signaling to inhibit EMT in SW480 cells. Exponentially
growing SW480 cells were infected with Ad.Spred2 and Ad.Null at an
MOI of 50. (A) Then, the phosphorylated ERK expression was detected
at the indicated time-points and normalized to ERK expression. ERK
specific inhibitor, PD98059, was used to inhibit ERK
phosphorylation, which was confirmed by western blotting at 24 h
after treatment. The (B) EMT markers, E-cadherin, N-cadherin and
vimentin, and the (C) SMAD signaling molecules SMAD2/3,
phosphorylated SMAD2/3 and SMAD4 were analyzed by western blotting
48 h after PD98059 treatment. (D) Moreover, the SMAD2/3 and SMAD4
protein expression was also analyzed 48 h after adenoviral
transduction. All the data are obtained from at least three
independent experiments, and presented as the mean ± SEM.
*P<0.05, **P<0.01 and ***P<0.001, vs. the corresponding
group. ERK, extracellular-regulated kinase; EMT,
epithelial-mesenchymal transition. |
As aforementioned, the TGF-β/SMAD signaling pathway
plays important roles in regulating EMT in colon cancer cells.
Therefore, it was investigated whether Spred2 could block
TGF-β/SMAD signaling to inhibit EMT processes. SW480 cells were
first treated with PD98059, and then stimulated with 10 ng/ml
TGF-β. The expression of SMAD2/3 and SMAD4, and the level of
phosphorylated SMAD2/3 were not affected by PD98059 (Fig. 5C), which suggested that ERK
signaling blockade can inhibit EMT in SW480 cells in a
SMAD-independent manner. It was also demonstrated that Ad.Spred2
had no significant effect on the expression of SMADs (Fig. 5D). The results indicated that Spred2
inhibited EMT in SW480 cells by regulating ERK signaling, but not
TGF-β/SMAD signaling.
In HCT116 cells, phosphorylated ERK was also
significantly downregulated by Ad.Spred2 at 48 h after transduction
(Fig. 6A). It was determined that
neither PD98059 nor Ad.Spred2 had a marked effect on E-cadherin
expression, but both reduced vimentin protein levels (Fig. 6B), suggesting that Spred2 reduced
EMT in HCT116 cells by inhibiting ERK signaling. Moreover, SMAD2/3
and SMAD4 expression after treatment with Ad.Spred2 and PD98059 was
also analyzed. Notably, Ad.Spred2 and PD98059 had opposite effects
on the expression of SMAD2/3 and SMAD4. PD98059 increased SMAD4
expression, while Ad.Spred2 reduced SMAD2/3 and SMAD4 levels. These
results indicated that Spred2 regulated TGF-β/SMAD signaling via an
ERK signaling-independent manner. Unlike its roles in SW480 cells,
Ad.Spred2 may also participate in the regulation of EMT by
inhibiting TGF-β signaling in HCT116 cells. Therefore, it was
concluded that Ad.Spred2 can inhibit EMT in CRC cells in an ERK
signaling-dependent manner.
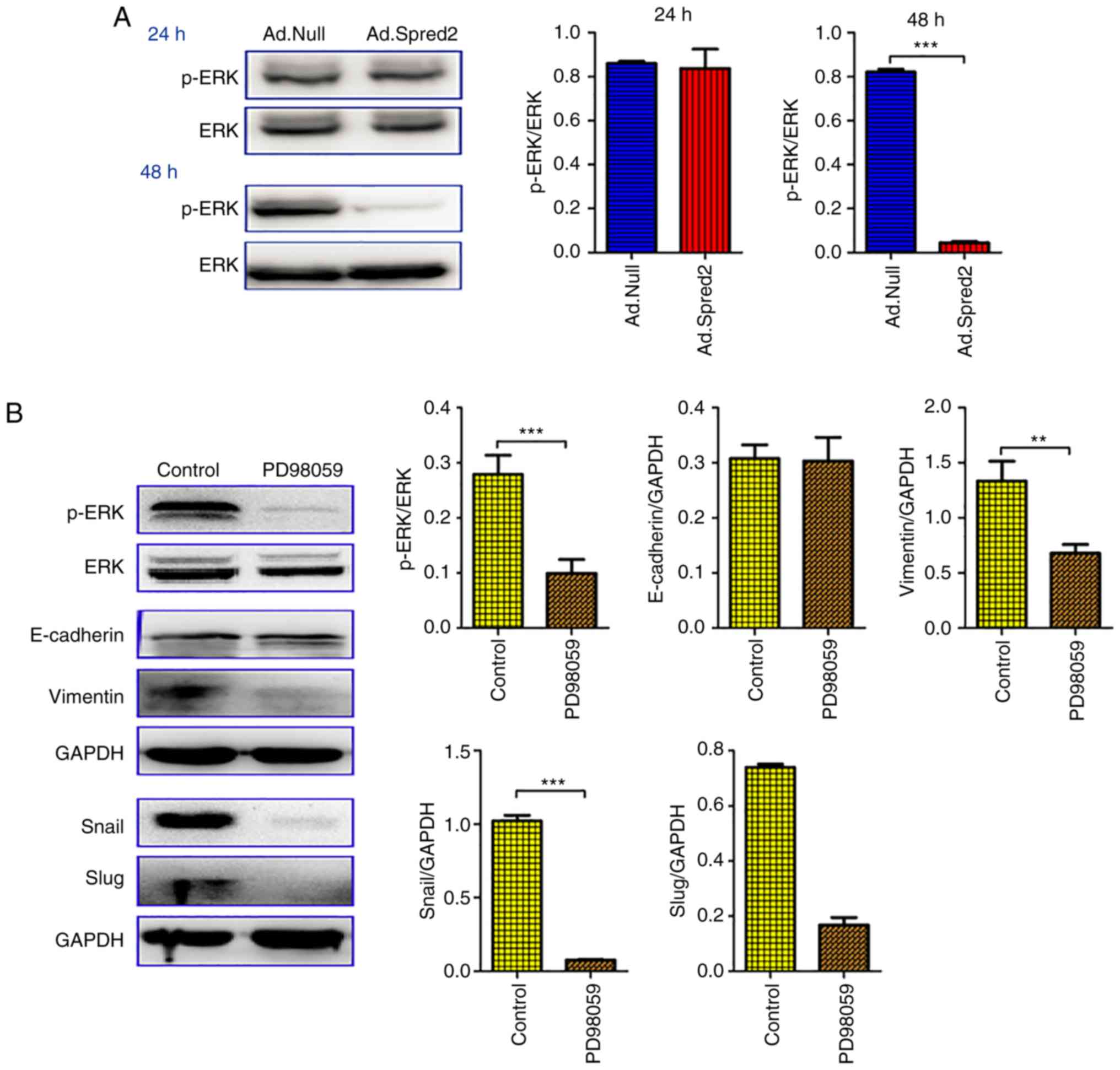 | Figure 6.Ad.Spred2 impairs ERK signaling and
SMAD signaling to inhibit EMT in HCT116 cells. (A) Exponentially
growing HCT116 cells were infected by Ad.Spred2 or Ad.Null at an
MOI of 50, and the ERK and phosphorylated ERK levels were detected
at 24 and 48 h after adenoviral infection. (B) The phosphorylated
ERK and ERK levels were analyzed at 24 h after PD98059 treatment.
Moreover, the EMT markers E-cadherin, N-cadherin and Vimentin, and
the EMT-related transcription factors Snail and Slug, were analyzed
at 48 h after PD98059 treatment. In addition, at 48 h after the
indicated treatments, the protein expression of SMAD2/3 and SMAD4
was analyzed in both (C) PD98059-treated cells and (D)
adenoviruses-infected cells. All the data were obtained from at
least three independent experiments, and are presented as the mean
± SEM. *P<0.05, **P<0.01, ***P<0.001, vs. the
corresponding group. ERK, extracellular-regulated kinase; EMT,
epithelial-mesenchymal transition. |
Discussion
CRC is the fourth leading cause of cancer-related
deaths worldwide, and there are still no effective approaches to
treat advanced and metastatic CRC (1,25,26).
Exploring the mechanisms of CRC initiation and progression will be
beneficial for discovering novel therapeutic targets. The
Ras/Raf-1/ERK signaling pathway is always aberrantly activated in
various tumors (27–30). Moreover, Spred1 and Spred2, two
important members of the Spred family, are frequently downregulated
in various cancers, such as hepatocellular carcinoma and prostate
cancer (13). Notably, the protein
level of Spred is closely associated with tumor progression and
metastasis. In the present study, it was revealed that Spred2, but
not Spred1, was markedly decreased in the tumor tissues of CRC
patients and CRC cell lines. Moreover, Spred2 restoration inhibited
the proliferation, migration and survival of CRC cells. Notably, it
was revealed that Spred2 mRNA expression increased 5–10 times from
24 to 48 h after culture, in both control and rAd.Null-treated
groups. Spred2 is a natural inhibitor of ERK signaling, which plays
important roles in regulating cellular apoptosis. Therefore, it was
surmised that upregulation of Spred2 at 48 h may be associated with
cellular apoptosis caused by nutrient deficiencies after long-time
culture. The investigation of the mechanisms is of interest for
future studies. These results indicated that Spred2 is a pivotal
molecule in CRC development and progression.
EMT, which plays crucial roles in both embryonic
development and tumor metastasis, is characterized by the loss of
epithelial markers and acquisition of mesenchymal markers (31–33).
TGF-β, a pleiotropic cytokine, is a pivotal molecule in the EMT
processing of tumor cells and can activate EMT-related
transcription factors in both SMAD-dependent and SMAD-independent
manners (21,22,34).
It was confirmed that 10 ng/ml TGF-β could activate SMAD signaling
and increase cellular migration in CRC cells by promoting
reorganization of the actin cytoskeleton and upregulating vimentin
expression. However, the epithelial marker E-cadherin was also
reduced by TGF-β. It was speculated that TGF-β regulates the EMT of
SW480 cells by modulating the ratio of mesenchymal markers to
epithelial markers, but not their absolute expression.
Spred proteins negatively regulate TGFβ-induced EMT
(35–36). In keratinocytes, Spred2 was revealed
to inhibit TGFβ-induced EMT by restoring E-cadherin levels,
reducing vimentin expression, and preventing reorganization of the
actin cytoskeleton. Ad.Spred2-mediated Spred2 overexpression
significantly inhibited EMT in both SW480 and HCT116 cells. In
addition, Ad.Spred2 both increased E-cadherin levels and reduced
vimentin levels in SW480 cells, however, it only decreased
mesenchymal markers in HCT116 cells. It was speculated that
Ad.Spred2 regulated EMT in CRC cells with various genetic
backgrounds in different ways. Combining immunofluorescence and
confocal microscopy is a widely used tool to analyze the expression
and distribution of proteins. In the present study, E-cadherin and
vimentin expression was detected by immunofluorescence in SW480
cells, but not in HCT116 cells. To further clarify the mechanisms
underlying Spred2-regulated EMT, the distribution of EMT markers
will be investigated in CRC cells with various genetic backgrounds.
Notably, it was revealed that SMAD2/3 and SMAD4 expression was
downregulated by Ad.Spred2 in HCT116 cells but not in SW480 cells,
which may be attributed to the different regulatory mechanisms of
various tumor cells. However, the mechanisms underlying these
different responses to Ad.Spred2 were not investigated in this
study. The distinct sensitivity of CRCs to TGF-β may be a reason
for these discrepancies. Therefore, study of the effects of
treatment with TGF-β at various doses will be necessary to clarify
the exact mechanisms. Data from our studies and others have
suggested that Spred2 is a negative regulator of EMT in epithelial
cells, including normal and tumor cells (14,35,36).
However, the roles of Spred2 in mesenchymal cells are still largely
unexplored. Notably, investigation of whether Spred2 regulates
mesenchymal-epithelial transition (MET) in human mesenchymal stem
cells is of great interest.
Numerous studies have demonstrated that
mitogen-activated protein kinase (MAPK)/ERK signaling participates
in multiple cellular processes during tumor progression and
metastasis, including growth, proliferation, differentiation and
migration (7,8,28–30).
Notably, aberrant activation of MAPK/ERK signaling is always
closely associated with the EMT of tumor cells in various cancers.
In CRC cells, the carboxyl terminus of Hsc70-interacting protein
(CHIP) was revealed to promote tumor metastasis by activating MAPK
signaling and reducing E-cadherin expression (37). Moreover, miR-27b could induce EMT in
gastric cancer by inhibiting Sprouty2, which plays a suppressive
role in the MAPK/ERK signaling pathway (38). Spred2, another important negative
regulator of MAPK/ERK signaling, could markedly inhibit the level
of phosphorylated ERK in CRC cells. Further analysis revealed that
Ad.Spred2 and the ERK-specific inhibitor, PD98059, had similar
effects on the expression of EMT markers in both SW480 cells and
HCT116 cells. These results indicated that ERK signaling inhibition
is a pivotal mechanism underlying Spred2-mediated impairment of EMT
in CRC cells. Furthermore, neither Ad.Spred2 nor PD98059 could
regulate SMAD2/3 and SMAD4 levels in SW480 cells, indicating that
Spred2 modulates EMT in a SMAD-independent manner. However,
Ad.Spred2 reduced intercellular SMAD2/3 and SMAD4 levels, while
PD98059 increased SMAD4 expression in HCT116 cells. It was
speculated that Ad.Spred2 can inhibit SMAD signaling in an ERK
signaling-independent manner. Therefore, Ad.Spred2 may regulate EMT
in both SMAD-dependent and SMAD-independent manners.
In conclusion, the expression of negative regulator
of MAPK/ERK signaling, Spred2, was markedly downregulated in the
tumor tissues of CRC patients. In vitro restoration of
Spred2 significantly inhibited the growth, survival and migration
of CRC cells. Moreover, Spred2 could inhibit the migration of tumor
cells by impairing the EMT of CRC cells, as it downregulated
E-cadherin and upregulated vimentin. Furthermore, Ad.Spred2
inhibited EMT by impairing ERK signaling, with or without reduced
TGF-β/SMAD signaling (Fig. S1).
Therefore, the introduction of the clinical application of Spred2
has great potential for development as a gene therapy approach for
CRC.
Supplementary Material
Supporting Data
Acknowledgements
We are thankful to the National Clinical Research
Center for Digestive Disease, Department of General Surgery,
Beijing Friendship Hospital for providing clinical samples in this
study.
Funding
The present study was supported by the National
Natural Science Foundation of China (no. 81402558&81472396),
and the National High Technology Research and Development Program
of China (863 Program) (SS2014AA020515). These funding agencies had
no role in the study design, data collection and analysis, decision
to publish, or preparation of the manuscript.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
SL and LW conceived and designed the experiments.
HW, FK, FX, YL, SZ and DH performed the experiments. HW and YY
analyzed the data. FX contributed to the collection of
reagents/materials/analytical tools. YY wrote the paper. All
authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
All the procedures were approved by the Ethics
Committee of Beijing Friendship Hospital. All patients provided
written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD, Fedewa SA, Ahnen DJ,
Meester RGS, Barzi A and Jemal A: Colorectal cancer statistics,
2017. CA Cancer J Clin. 67:177–193. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gonzalez-Pons M and Cruz-Correa M:
Colorectal cancer bio-markers: Where are we now? Biomed Res Int.
2015:1490142015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brenner H, Stock C and Hoffmeister M:
Colorectal cancer screening: The time to act is now. BMC Med.
13:2622015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wrobel P and Ahmed S: Current status of
immunotherapy in metastatic colorectal cancer. Int J Colorectal
Dis. 34:13–25. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Van der Jeught K, Xu HC, Li YJ, Lu XB and
Ji G: Drug resistance and new therapies in colorectal cancer. World
J Gastroenterol. 24:3834–3848. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li Y, Zhang M, Dorfman RG, Pan Y, Tang D,
Xu L, Zhao Z, Zhou Q, Zhou L, Wang Y, et al: SIRT2 promotes the
migration and invasion of gastric cancer through RAS/ERK/JNK/MMP-9
pathway by increasing PEPCK1-related metabolism. Neoplasia.
20:745–756. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Qiu XY, Hu DX, Chen WQ, Chen RQ, Qian SR,
Li CY, Li YJ, Xiong XX, Liu D, Pan F, et al: PD-L1 confers
glioblastoma multiforme malignancy via Ras binding and Ras/Erk/EMT
activation. Biochim Biophys Acta Mol Basis Dis. 1864:1754–1769.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu D, Zhao B, Qi X, Peng F, Fu H, Chi X,
Miao QR and Shao S: Nogo-B receptor promotes epithelial-mesenchymal
transition in non-small cell lung cancer cells through the
Ras/ERK/Snail1 pathway. Cancer Lett. 418:135–146. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hu YC, Wang AM, Lu JK, Cen R and Liu LL:
Long noncoding RNA HOXD-AS1 regulates proliferation of cervical
cancer cells by activating Ras/ERK signaling pathway. Eur Rev Med
Pharmacol Sci. 21:5049–5055. 2017.PubMed/NCBI
|
|
11
|
Wakioka T, Sasaki A, Kato R, Shouda T,
Matsumoto A, Miyoshi K, Tsuneoka M, Komiya S, Baron R and Yoshimura
A: Spred is a sprouty-related suppressor of ras signalling. Nature.
412:647–651. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nobuhisa I, Kato R, Inoue H, Takizawa M,
Okita K, Yoshimura A and Taga T: Spred-2 suppresses
aorta-gonad-mesonephros hematopoiesis by inhibiting MAP kinase
activation. J Exp Med. 199:737–742. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kachroo N, Valencia T, Warren AY and
Gnanapragasam VJ: Evidence for downregulation of the negative
regulator SPRED2 in clinical prostate cancer. Br J Cancer.
108:597–601. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma XN, Liu XY, Yang YF, Xiao FJ, Li QF,
Yan J, Zhang QW, Wang LS, Li XY and Wang H: Regulation of human
hepatocellular carcinoma cells by Spred2 and correlative studies on
its mechanism. Biochem Biophys Res Commun. 410:803–808. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Momeny M, Khorramizadeh MR, Ghaffari SH,
Yousefi M, Yekaninejad MS, Esmaeili R, Jahanshiri Z and Nooridaloii
MR: Effects of silibinin on cell growth and invasive properties of
a human hepatocellular carcinoma cell line, HepG-2, through
inhibition of extracellular signal-regulated kinase 1/2
phosphorylation. Eur J Pharmacol. 591:13–20. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yoshida T, Hisamoto T, Akiba J, Koga H,
Nakamura K, Tokunaga Y, Hanada S, Kumemura H, Maeyama M, Harada M,
et al: Spreds, inhibitors of the Ras/ERK signal transduction, are
dysregulated in human hepatocellular carcinoma and linked to the
malignant phenotype of tumors. Oncogene. 25:6056–6066. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu XY, Yang YF, Wu CT, Xiao FJ, Zhang QW,
Ma XN, Li QF, Yan J, Wang H and Wang LS: Spred2 is involved in
imatinib-induced cytotoxicity in chronic myeloid leukemia cells.
Biochem Biophys Res Commun. 393:637–642. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lu ZZ, Ni F, Hu ZB, Wang L, Wang H, Zhang
QW, Huang WR, Wu CT and Wang LS: Efficient gene transfer into
hematopoietic cells by a retargeting adenoviral vector system with
a chimeric fiber of adenovirus serotype 5 and 11p. Exp Hematol.
34:1171–1182. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu W, Neill T, Yang Y, Hu Z, Cleveland E,
Wu Y, Hutten R, Xiao X, Stock SR, Shevrin D, et al: The systemic
delivery of an oncolytic adenovirus expressing decorin inhibits
bone metastasis in a mouse model of human prostate cancer. Gene
Ther. 22:247–256. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang Y, Li G, Wang K, Mu Z, Xie Q, Qu H,
Lv H and Hu B: Collagen type VI alpha 3 chain promotes
epithelial-mesenchymal transition in bladder cancer cells via
transforming growth factor β (TGF-β)/Smad pathway. Med Sci Monit.
24:5346–5354. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thakur AK, Nigri J, Lac S, Leca J, Bressy
C, Berthezene P, Bartholin L, Chan P, Calvo E, Iovanna JL, et al:
TAp73 loss favors Smad-independent TGF-β signaling that drives EMT
in pancreatic ductal adenocarcinoma. Cell Death Differ.
23:1358–1370. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Han S, Bui NT, Ho MT, Kim YM, Cho M and
Shin DB: Dexamethasone inhibits TGF-β1-induced cell migration by
regulating the ERK and AKT pathways in human colon cancer cells via
CYR61. Cancer Res Treat. 48:1141–1153. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lampropoulos P, Zizi-Sermpetzoglou A,
Rizos S, Kostakis A, Nikiteas N and Papavassiliou AG: TGF-β
signalling in colon carcinogenesis. Cancer Lett. 314:1–7. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Elez E, Argiles G and Tabernero J:
First-line treatment of metastatic colorectal cancer: Interpreting
FIRE-3, PEAK, and CALGB/SWOG 80405. Curr Treat Options Oncol.
16:522015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fakih MG: Metastatic colorectal cancer:
Current state and future directions. J Clin Oncol. 33:1809–1824.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cakir M and Grossman AB: Targeting MAPK
(Ras/ERK) and PI3K/Akt pathways in pituitary tumorigenesis. Expert
Opin Ther Targets. 13:1121–1134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Khavari TA and Rinn J: Ras/Erk MAPK
signaling in epidermal homeostasis and neoplasia. Cell Cycle.
6:2928–2931. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Torii S, Yamamoto T, Tsuchiya Y and
Nishida E: ERK MAP kinase in G cell cycle progression and cancer.
Cancer Sci. 97:697–702. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Samatar AA and Poulikakos PI: Targeting
RAS-ERK signalling in cancer: Promises and challenges. Nat Rev Drug
Discovery. 13:928–942. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pastushenko I and Blanpain C: EMT
transition states during tumor progression and metastasis. Trends
Cell Biol. 29:212–226. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen T, You Y, Jiang H and Wang ZZ:
Epithelial-mesenchymal transition (EMT): A biological process in
the development, stem cell differentiation, and tumorigenesis. J
Cell Physiol. 232:3261–3272. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim DH, Xing T, Yang Z, Dudek R, Lu Q and
Chen YH: Epithelial mesenchymal transition in embryonic
development, tissue repair and cancer: A comprehensive overview. J
Clin Med. 7:E12017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tan QY and Cheng ZS: TGFβ1-smad signaling
pathway participates in interleukin-33 induced
epithelial-to-mesenchymal transition of A549 cells. Cell Physiol
Biochem. 50:757–767. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhao G, Wojciechowski MC, Jee S, Boros J,
McAvoy JW and Lovicu FJ: Negative regulation of TGFβ-induced lens
epithelial to mesenchymal transition (EMT) by RTK antagonists. Exp
Eye Res. 132:9–16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Villar V, Kocic J and Santibanez JF:
Spred2 inhibits TGF-beta1-induced urokinase type plasminogen
activator expression, cell motility and epithelial mesenchymal
transition. Int J Cancer. 127:77–85. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xu J, Zhou J, Dai H, Liu F, Li W, Wang W
and Guo F: CHIP functions as an oncogene by promoting colorectal
cancer metastasis via activation of MAPK and AKT signaling and
suppression of E-cadherin. J Transl Med. 16:1692018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jiang J, Yi B, Qin C, Ding S and Cao W:
Upregulation of microRNA27b contributes to the migration and
invasion of gastric cancer cells via the inhibition of
sprouty2-mediated ERK signaling. Mol Med Rep. 13:2267–2272. 2016.
View Article : Google Scholar : PubMed/NCBI
|














