Introduction
From the 1960s to the 1980s, survival rates for
patients with osteosarcoma significantly increased due to
neoadjuvant and postsurgery chemotherapy (1). However, the outcome remains poor for
the majority of patients with metastatic or recurrent osteosarcoma
(2). Therefore, novel insights into
the mechanisms underlying tumorigenesis and metastasis are urgently
required to further improve overall survival in patients with
osteosarcoma.
MicroRNAs (miRNAs/miRs) have important roles in the
initiation and development of tumors (3,4).
Various miRNAs, which have been reported to act as either tumor
promoters or tumor suppressors, have been identified in
osteosarcoma (5–7). In our previous study, the miRNA
expression spectrum of two osteosarcoma cell lines with different
metastatic potentials was compared and a group of differentially
expressed miRNAs was identified (8). Among the identified miRNAs, miR-193b
revealed a ~2-fold difference between the high metastatic subline
(F5M2) and the low metastatic subline (F4). Although the
tumor-suppressive role of miR-193b has been demonstrated in several
types of cancer (9–13), the involvement of miR-193b in
osteosarcoma has rarely been discussed. Therefore, the purpose of
the present study was to investigate the effects of miR-193b on the
malignant behaviors of osteosarcoma, and to clarify its underlying
mechanism by exploring its upstream regulator and downstream
targets. KRAS is one of the front-line sensors that initiate the
activation of an array of signaling molecules, thus affecting cell
differentiation, growth, chemotaxis and apoptosis (14). Stathmin 1 (STMN1) regulates cell
division by destabilizing microtubules in a
phosphorylation-dependent manner, and acts as an important
regulator of cell cycle progression, proliferation, invasion and
survival (15). KRAS and STMN1 have
been predicted as downstream effectors of miR-193b, and they have
been reported to be upregulated in several human tumors, including
osteosarcoma (16,17). Thus, the present study focused on
KRAS and STMN1 as downstream effectors of miR-193b to clarify the
mechanism by which miR-193b may modulate malignant behaviors of
osteosarcoma. As for the upstream regulator of miR-193b, this study
focused on the well-recognized oncogene MYC, since it may be able
to bind to the promoter region of miR-193b as predicted by
bioinformatics tools (18).
The present study reported that miR-193b suppressed
the malignant behaviors of osteosarcoma cells through the direct
inhibition of its targets; namely, KRAS and STMN1. In addition,
miR-193b and MYC exerted mutual suppressive effects against each
other. Such mutual suppressive effects may lead to sustained MYC
activation, miR-193b inhibition, and the eventual overexpression of
KRAS and STMN1.
Materials and methods
Cell culture
F4 and F5M2 osteosarcoma cell lines with different
metastatic potential, screened and established from the human
osteosarcoma cell line SOSP-9607, were acquired from the Department
of Orthopedics, Tangdu Hospital, Fourth Military Medical University
(Xi'an, China) (8). 293A cells were
purchased from the Institutes for Biological Sciences at the
Chinese Academy of Sciences. Osteosarcoma cells and 293A cells were
separately cultured in RPMI (HyClone; GE Healthcare Life Sciences)
or DMEM (Gibco; Thermo Fisher Scientific, Inc.), respectively,
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Sciences, Inc.) at 37°C in a humidified incubator with 5%
CO2.
Oligonucleotides and plasmids
Synthetic miR-193b mimic, miR-193b inhibitor,
negative controls (NC and inhibitor NC), silencer Select Negative
Control (siNC), and KRAS, STMN1 and MYC small interfering RNAs
(siRNAs) were obtained from Shanghai GenePharma Co., Ltd.
Lentiviral particles containing miR-193b (LV-193b), miR-193b
inhibitor (LV-193b inhibitor) and negative controls (LV-NC and
LV-inhibitor-NC) were purchased from GeneCopoeia, Inc. The coding
sequences of KRAS and STMN1 were PCR-amplified from F5M2 cell cDNA
using primers listed in Table I and
were cloned into the BgIII and XhoI sites of pMSCV
(Addgene), yielding the pMSCV-KRAS and pMSCV-STMN1 overexpression
vectors (19); empty pMSCV was used
as the control. MYC overexpression vector and its control vector
(pBabe-MYC and pBabe) were kindly provided by Professor Rui Zhang
(Departments of Immunology, Fourth Military Medical University,
Xi'an, China). The sequences of oligonucleotides and plasmids are
presented in Table II.
 | Table I.Primers used in the study. |
Table I.
Primers used in the study.
| Gene | Sequence |
|---|
| Primers for
construction of KRAS/STMN1 expression vector |
|
|
KRAS-F |
5′-CAGATCTATGACTGAATATAAACTTGTGG-3′ |
|
KRAS-R |
5′-GCCTCGAGTTACATAATTACACACTTTGTC-3′ |
|
STMN1-F |
5′-CAGATCTATGGCTTCTTCTGATATCCAGG-3′ |
|
STMN1-R |
5′-GCCTCGAGTTAGTCAGCTTCAGTC-3′ |
| Primers for
RT-qPCR |
|
|
Hsa-miR-193b-F |
5′-AACTGGCCCTCAAAGTCCCGCT-3′ |
|
KRAS-F |
5′-CGGTCATCCAGTGTTGTCAT-3′ |
|
KRAS-R |
5′-AATGCTCTTGATTTGTCAGCAG-3′ |
|
STMN1-F |
5′-CCTCTGTTTGGCGCTTTTGTGCG-3′ |
|
STMN1-R |
5′-GGCACGCTTCTCCAGTTCTTTCACC-3′ |
|
Actin-F |
5′-TGGCATCCACGAAACTACC-3′ |
|
Actin-R |
5′-GTGTTGGCGTACAGGTCTT-3′ |
|
U6-F |
5′-CTCGCTTCGGCAGCACA-3′ |
|
U6-R |
5′-AACGCTTCACGAATTTGCGT-3′ |
| Primers used in
construction of dual-luciferase reporters |
|
| (wild
type) KRAS-1F |
5′-CGGAATTCGGTGTTGATGATGCCTTC-3′ |
| (wild
type) KRAS-1R |
5′-GCTGCAGATCATCATCAGGAAGCCC-3′ |
| (wild
type) KRAS-2F |
5′-CGGAATTCAGGCAGACCCAGTATGAA-3′ |
| (wild
type) KRAS-2R |
5′-GCTGCAGAATGTCTTGGCACACCACCA-3′ |
|
(mutant) KRAS-1F |
5′-CGGAATTCGAACTAGCAATGCCTGTG-3′ |
|
(mutant) KRAS-1R |
5′-GCTGCAGATCATCATCAGGAAGCCC-3′ |
|
(mutant) KRAS-2F |
5′-GCGAATTCAGAGACCAAGGTTGCAAG-3′ |
|
(mutant) KRAS-2R |
5′-GCTGCAGAATGTCTTGGCACACCACCA-3′ |
| (wild
type) STMN1-F |
5′-GCGGAATTCTTGTTCTGAGAACTGACTTTC-3′ |
| (wild
type) STMN1-R |
5′-GCTGCAGGTCATTTGTGCGTTGGGTAT-3′ |
|
(mutant) STMN1-F |
5′-GCGGAATTCAATGGCTAGTACTGTATTGG-3′ |
|
(mutant) STMN1-R |
5′-GCTGCAGGTCATTTGTGCGTTGGGTAT-3′ |
| Primers used in
construction of promoter reportersa,b |
|
| t1
fragment |
|
193b-5′-UTR-1F |
5′-GAGCTCTCGAGTTGGCTTTAGCTTCTGC-3′ |
|
193b-5′-UTR-1R |
5′-AGATCTACAGCCTCCAAAAGCCTC-3′ |
| t2
fragment |
|
|
193b-5′-UTR-2F |
5′-TTTGAGCTCACGCTTGTCTGGGCTGCGATT-3′ |
|
193b-5′-UTR-2R |
5′-CTCGAGGCAGAAGCTAAAGCCAAC-3′ |
| Primers for ChIP
assay |
|
|
c193b-F1 |
5′-TCCGTGCCCCCTGTTTGAA-3′ |
|
c193b-R1 |
5′-CAGAGAGGGCGAGAGCCTGGAA-3′ |
|
c193b-F2 |
5′-TGAGTGCTCCCCTTCTTCC-3′ |
|
c193b-R2 |
5′-AATCGCAGCCCAGACAAGCGT-3′ |
|
c193b-F3 |
5′-CCTGGGCTTGGAAATTGAC-3′ |
|
c193b-R3 |
5′-GGTAACTCTCTGGGGACGGT-3′ |
|
c193b-F4 |
5′-GGCGCGCAGCAAATTTGACT-3′ |
|
c193b-R4 |
5′-TTGATCCCGGGGTGTCTCTT-3′ |
|
c193b-F5 |
5′-GGAGGAGAAAGTACATTCCC-3′ |
|
c193b-R5 |
5′-TGTTGCAATTCCAGGTGGAAGC-3′ |
| Table II.Oligonucleotides sequences used for
transfection. |
Table II.
Oligonucleotides sequences used for
transfection.
|
Oligonucleotide | Sequence |
|---|
| miR-193b mimic | F:
5′-AACUGGCCCUCAAAGUCCCGCU-3′ |
|
| R:
5′-CGGGACUUUGAGGGCCAGUUUU-3′ |
| NC | F:
5′-UUCUCCGAACGUGUCACGUTT-3′ |
|
| R:
5′-ACGUGACACGUUCGGAGAATT-3′ |
| miR-193b
inhibitor |
5′-AGCGGGACUUUGAGGGCCAGUU-3′ |
| Inhibitor NC |
5′-CAGUACUUUUGUGUAGUACAA-3′ |
| siNC | F:
5′-UUCUCCGAACGUGUCACGUTT-3′ |
|
| R:
5′-ACGUGACACGUUCGGAGAATT-3′ |
| siKRAS | F:
5′-GGGCUUUCUUUGUGUAUUUTT-3′ |
|
| R:
5′-AAAUACACAAAGAAAGCCCTT-3′ |
| siSTMN1 | F:
5′-GAUGUUUAUUUGCAAACAACC-3′ |
|
| R:
5′-UUGUUUGCAAAUAAACAUCUG-3′ |
| siMYC | F:
5′-AACGUUAGCUUCACCAACAUU-3′ |
|
| R:
5′-UGUUGGUGAAGCUAACGUUUU-3′ |
Transfection assay
F4 and F5M2 osteosarcoma cells were plated in 6-well
plates at a density of 4×105 cells/well, cultured
overnight and transfected with 50 nmol/l oligonucleotides using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) for 8 h at 37°C. The cells were continuously
cultured for 48 h before the commencement of other experiments.
Lentiviral infection was performed to stably
increase or reduce intracellular levels of miR-193b in F5M2 or F4
cells, respectively. Briefly, 2×105 cells were plated in
a 6-well plate 24 h prior to infection. Fresh culture media
containing 8 µg/ml polybrene was added to the cells when they
reached 60–70% confluence. Lentivirus (multiplicity of infection,
5) was then added to the culture media. After 24 h, viral
supernatant was removed and fresh culture medium was added. After
incubation for 72 h, the cells were subcultured into 2×100 mm
dishes and 1.0 µg/ml puromycin was added for selection. After
antibiotic selection for 2 weeks, the stable clones were
obtained.
Cell proliferation assay
F4 cells transfected with the miR-193b inhibitor or
infected with LV-miR-193b inhibitor, and F5M2 cells transfected
with miR-193b or infected with LV-193b were maintained in 96-well
plates at a density of 2×103 cells/well. After 48 h of
culture, cells were treated with 0.5% MTT solution for 4 h at 37°C.
After formazan crystals were dissolved in dimethyl sulfoxide, the
proliferation rate was measured as the optical density at an
absorbance of 570 nm. The experiment was repeated three times with
five replicates.
Colony formation assay
A total of 200 cells/well were plated in a 6-well
plate and cultured for 2 weeks. Colonies were counted after they
were fixed with 100% methanol for 20 min and stained with 0.1%
crystal violet in 20% methanol for 30 min at room temperature.
After the cells were washed and dried, visible cell colonies were
counted and images were captured. Colony-forming efficiency was
calculated using the following equation: Total number of visible
cell colonies/200×100. The experiment was repeated three times with
five replicates.
Cell cycle analysis
Cell cycle analysis was conducted using a cell cycle
detection kit (Beyotime Institution of Biotechnology). Cells were
collected and fixed in 75% ethanol at 4°C overnight. After washing
with PBS, the cells were treated with 1 mg/ml RNase at 37°C for 40
min. Subsequently, the cells were stained with 50 µg/ml propidium
iodide (PI) for 30 min at 4°C in the dark. Cell cycle distribution
was analyzed on a COULTER FC500 MCL flow cytometer (Beckman
Coulter, Inc.) using MultiCycle for Windows 32-bit software
(Beckman Coulter, Inc.). The experiment was repeated three times
with five replicates.
Cell apoptosis analysis
Cells (1×103 cells/ml) were harvested and
washed, then stained with FITC-conjugated Annexin V (Beyotime
Institute of Biotechnology) and PI (Beyotime Biotechnology) at room
temperature in the dark for 15 min. Cell apoptosis was measured on
a COULTER FC500 MCL flow cytometer (Beckman Coulter, Inc.) using
EXPO32 ADC software (Beckman Coulter, Inc.). Apoptotic index was
calculated by adding the proportion of Annexin V(+)/PI(−) early
apoptotic cells to that of Annexin V(+)/PI(+) late apoptotic cells.
The experiment was repeated three times with five replicates.
Wound-healing assay
F4 and F5M2 cells at a density of 4×105
cells/well were seeded on 6-well plates and cultured in RPMI medium
supplemented with 10% fetal bovine serum. When the cells reached
100% confluence after 24 h of culture, a wound-healing assay was
performed. A 20 µl pipette tip was used to make three parallel
wounds. After the cells were cultured in serum-free medium for 48
or 96 h, images were captured under a light microscope and analyzed
with ImageJ (version 1.8.0; National Institutes of Health). The
experiment was repeated three times with three replicates.
Transwell cell invasion assay
The upper chamber of Transwell culture plates (8 µm;
Corning, Inc.) was coated with 0.4 mg/ml Matrigel (BD Biosciences)
and incubated for 24 h at 4°C. Cells were seeded into the upper
chamber of Transwell culture plates at a density of
2×105 cells/well. The lower chamber was filled with RPMI
supplemented with 10% fetal bovine serum. After 36 h, cells on the
upper membrane that did not migrate were wiped with a cotton swab,
and the membrane in the lower chamber was fixed with absolute ethyl
alcohol and stained with 0.1% crystal violet for 30 min at room
temperature. The number of migrated cells was then counted under a
light microscope in at least five fields. The experiment was
repeated three times with three replicates.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA (containing small RNAs) was extracted
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and reversed transcribed into cDNA. For miR-193b,
RT was conducted at 37°C for 1 h, followed by 85°C for 5 sec using
the PrimeScript miRNA qPCR Starter kit ver. 2.0 (Takara
Biotechnology Co., Ltd.). The forward primer for miR-193b was
5′-AACTGGCCCTCAAAGTCCCGCT-3′ as described in Table I, while the reverse primer was a
universal primer from the PrimeScript™ miRNA qPCR Starter kit. The
expression levels of miR-193b were quantified by qPCR in a
real-time PCR detector (Bio-Rad Laboratories, Inc.) using the
PrimeScript™ miRNA qPCR Starter kit ver. 2.0. For the quantitative
detection of KRAS, STMN1 and MYC mRNA, RT was conducted at 37°C for
15 min, followed by 85°C for 5 sec with a PrimeScript RT reagent
kit (Bio-Rad Laboratories, Inc.). Subsequently, qPCR was conducted
using the SYBR Green PCR Master Mix kit (Applied Biosystems; Thermo
Fisher Scientific, Inc.). Two-step PCR amplification was conducted
at 95°C for 180 sec, followed by 40 cycles at 95°C for 10 sec and
57°C for 30 sec. Final extension was performed at 72°C for 5 min.
U6 and GAPDH served as internal controls for miRNA and mRNA
expression levels, respectively. Relative expression levels were
calculated using the 2−ΔΔCq method (20). All primers used for RT-qPCR analysis
are listed in Table I. The
experiment was carried out in triplicate and repeated three
times.
Western blot analysis
After cells were lysed with RIPA lysis buffer
(Beyotime Institute of Biotechnology), the supernatant was
harvested by centrifugation at 14,000 × g for 30 min at 4°C.
Protein quantification was performed using the BCA method and 30 µg
protein was loaded on SDS-polyacrylamide gels for SDS-PAGE.
Proteins were resolved on a 12% precast gradient gel and were then
transferred to a nitrocellulose membrane. The membranes were
blocked with 5% (w/v) non-fat dry milk in TBS-0.05% Tween (Beyotime
Institute of Biotechnology) for 1 h at room temperature and were
then incubated with primary antibodies overnight at 4°C, followed
by incubation with a secondary antibody (1:3,000; cat. nos. 7076 or
5127; Cell Signaling Technology, Inc.) for 1 h at room temperature.
To visualize protein expression, enhanced chemiluminescence (ECL)
was performed with the Pierce™ ECL Western Blotting Substrate kit
(Thermo Fisher Scientific, Inc.). The gray value of the target band
was analyzed using Image Lab (Version 3.1; Bio-Rad Laboratories,
Inc.). Antibodies against KRAS (1:250; F234; cat. no. sc-30; Santa
Cruz Biotechnology, Inc.), STMN1 (1:1,000; cat. no. ab131481;
Abcam), MYC (1:1,000; cat. no. ab32072; Abcam), and actin (1:2,000;
cat. no. A5441; Sigma-Aldrich; Merck KGaA) were used. The
experiment was performed in triplicate.
Dual-luciferase reporter assay
Human KRAS-3′-untranslated region (3′-UTR) and
STMN1-3′-UTR reporter constructs containing the putative binding
site of miR-193b, and their identical sequences with a mutation in
the seed sequence, were amplified by PCR using the primers listed
in Table I as described previously
(21), and inserted between the
EcoRI and PstI restriction sites of the PGL3-basic
reporter vector (Promega Corporation). There are two binding sites
for miR-193b on KRAS 3′UTR. Thus, to clarify whether the two
binding sites are both effective, two reporter vectors for KRAS
with the corresponding binding sites were constructed. 293A cells
at a density of 2×105 cells/well were co-transfected
with 50 nM miR-193b mimic or NC mimic and 1 mg reporter vectors
containing wild-type or mutant 3′-UTR of KRAS or STMN1 according to
the aforementioned transfection protocol. At 6 h post-transfection,
the medium was replaced with complete medium and at 48 h
post-transfection, cells were lysed and luciferase activity was
detected using the Dual-Luciferase Assay kit (Promega Corporation).
The primers used to amplify the wild-type and mutant 3′-UTRs by PCR
are listed in Table I. Each sample
was tested in triplicate, and the experiment was repeated three
times.
Promoter reporter assay
A 1.0 kb wild-type promoter fragment of miR-193b
with a putative MYC binding site, or a truncated promoter fragment
of miR-193b without a MYC binding site (synthesized by Sangon
Biotech Co., Ltd.) was PCR-amplified and then cloned into the
SacI and BglII sites of a pGL3-Basic vector (Promega
Corporation). 293A cells were seeded in 24-well at a density of
2×105 cells/well and were cultured until confluence
reached 80%. Subsequently, the cells were co-transfected with 400
ng wild-type or truncated reporter vector together with 400 ng
pBabe or pBabe-MYC plasmid. The pTK-luc (Renilla) luciferase
vector (20 ng; Thermo Fisher Scientific, Inc.) was transfected into
293A cells together with the reporter vectors and pBabe plasmids
for normalization. The luciferase activity was measured 48 h
post-transfection using the Dual-Luciferase Reporter Assay system
(Promega Corporation) according to the manufacturer's
instructions.
Chromatin immunoprecipitation
(ChIP)-qPCR
Cells (3×107) were fixed with 1.5%
formaldehyde for 10 min at room temperature and quenched with
glycine. After cell lysis with RIPA buffer (Beyotime Institute of
Biotechnology), chromatin was fragmented into 100–500 bp fragments
using an ultrasonic cell breaker (DH92-IIN; Lawson Scientific Ltd.)
with a cycle of 1 sec on/1 sec off for 8 min (frequency, 20 kHz).
Protein-DNA complexes were immunoprecipitated with MYC antibody
(cat. no. ab32072; Abcam) or anti-IgG antibody (cat. no. ab171870;
Abcam) conjugated with Dynabeads Protein G (cat. no. 10007D;
Invitrogen; Thermo Fisher Scientific, Inc.) as described previously
(22). After being eluted from the
beads using ChIP Elution Buffer (cat. no. 714231S; Cell Signaling
Technology, Inc.) for 30 min at 65°C, the antibody-protein-DNA
complex was reverse cross-linked by incubation at 65°C with 200 mM
NaCl. Six primers were designed according to the predicted binding
sites on miR-193b (sequences are listed in Table I), and quantification of
immunoprecipitated DNA was performed by RT-qPCR. Each sample was
tested in triplicate, and the experiment was repeated three
times.
In vivo tumorigenicity and metastasis
analysis
Female BALB/c nude-mice (age, 6 weeks; weight, 20 g;
n=5 mice/group) were obtained and maintained at the Qualified
Animal Care Facility, The Fourth Military Medical University.
During the study, animal welfare was seriously taken into
consideration. All animal handling and experimental procedures were
approved by the Animal Ethics Committees of The Fourth Military
Medical University and were in accordance with the guidelines of
the China Council of Animal Care (Regulation for the Administration
of Affairs Concerning Experimental Animal approved by Decree No. 2
of the State Science and Technology Commission on November
14th, 1988). All mice were housed in polycarbonate
cages, provided with free access to food and water, and maintained
under a 12 h light-dark cycle at 22±2°C with 60±5% humidity. Humane
endpoints and clinical signs used in all experiments to determine
when to euthanize the mice include: Tumors 2.0 cm diameter, weight
loss >20%, tumor ulceration, abnormal posture, quick weight
loss, loss of appetite for >3 days, and too weak to access water
and food. However, no experimental mice met these criteria and were
euthanized before the end of our experiments. On the indicated time
of sacrifice, mice were anesthetized with isoflurane (4%) for ~1
min. Animals with no heartbeat for 30 sec accompanied by no
response to the toe pinch reflex were considered to be dead.
Subsequently, cervical dislocation was performed to verify
euthanasia. In the tumor formation in vivo experiment, the
maximum tumor burden was <4% animal weight.
For the tumorigenicity study, female BALB/c
nude-mice (age, 6 weeks; weight, 20 g; n=5 mice/group) were
anesthetized with isoflurane after initial induction in a chamber
provided with oxygen (2 l/min) containing 2% isoflurane, in order
to minimize suffering and pain during cell injection. F5M2 cells
stably transfected with LV-miR-193b or LV-NC were subcutaneously
injected into the dorsal flank of the mice. When tumors were
palpable on day 4, the tumor diameter was measured and documented
every 5 days until day 35. Tumor volume was calculated as follow:
Volume = width2 × length ×0.5 (23). The animals were sacrificed on day
35, and the tumors were collected, weighed and photographed. For
the metastasis study, F5M2 cells transfected with LV-NC or
LV-miR-193b were injected into the tail vein of mice. A total of 6
weeks after tumor cell implantation, mice were euthanized, and the
lungs of the experimental animals were collected, photographed and
weighed. Subsequently, the number of tumor nodules formed on the
lung surfaces was counted. Metastatic lungs were fixed in 4%
paraformaldehyde (Beyotime Institute of Biotechnology) for 24 h at
room temperature, and then embedded with paraffin. Serial sections
of lungs were obtained and stained with hematoxylin and eosin to
confirm the pulmonary metastatic lesions under a light
microscope.
Immunofluorescence staining
Paraffin-embedded sections were dewaxed and
endogenous peroxidase was removed using 3%
H2O2 for 10 min at room temperature.
Heat-mediated antigen retrieval was performed by immersing the
sections in citrate buffer (Abcam) and boiling them in a microwave
oven for 10 min. Subsequently, the sections were cooled, washed and
blocked in PBS containing 2% goat serum (Thermo Fisher Scientific,
Inc.) and 0.1% Triton X-100 for 60 min at room temperature.
Subsequently, the sections were incubated with primary antibodies
against KRAS (1:200; cat. no. sc-30; Santa Cruz Biotechnology,
Inc.), STMN1 (1:200; cat. no. ab221017; Abcam) or MYC (1:200; cat.
no. ab39688; Abcam) overnight at 4°C. The sections were then washed
and incubated with Alexa Fluor® 488-conjugated goat
anti-mouse IgG antibody (1:500; cat. no. ab150113; Abcam) for 1 h
at room temperature. After washing, DAPI nuclear staining was
performed. The stained sections were observed under an Olympus IX53
inverted fluorescence microscope (Olympus Corporation). The
experiment was performed in triplicate.
Bioinformatics analysis
The target genes of miR-193b were predicted by
computer-aided algorithms using TargetScan (http://www.targetscan.org/, release 7.1: June 2016)
and Microcosm Targets (http://www.mirbase.org/, release 21: 2014). The
transcription factors that could bind to the promoter region of
miR-193b were predicted using Genome Browser from National Center
for Biotechnology Information (http://www.ncbi.nlm.nih.gov/), University of
California Santa Cruz (http://www.genome.ucsc.edu/), and Ensembl (http://www.ensembl.org/index.html).
Statistical analysis
Each experiment was repeated at least three times.
The data are presented as the mean ± standard deviation. SPSS V18.0
software (SPSS, Inc.) was used for statistical analysis. The
normality of data from various groups was tested using the
Kolmogorov-Smirnov test and homogeneity was tested using the Levene
test. The statistical significance of differences between two
groups was evaluated using unpaired Student's t-test (two-tailed).
For comparisons of datasets containing multiple groups, one-way
ANOVA and post-hoc Tukey test was performed. P<0.05 was
considered to indicate a statistically significant difference.
Results
miR-193b inhibits osteosarcoma growth
in vitro and in vivo
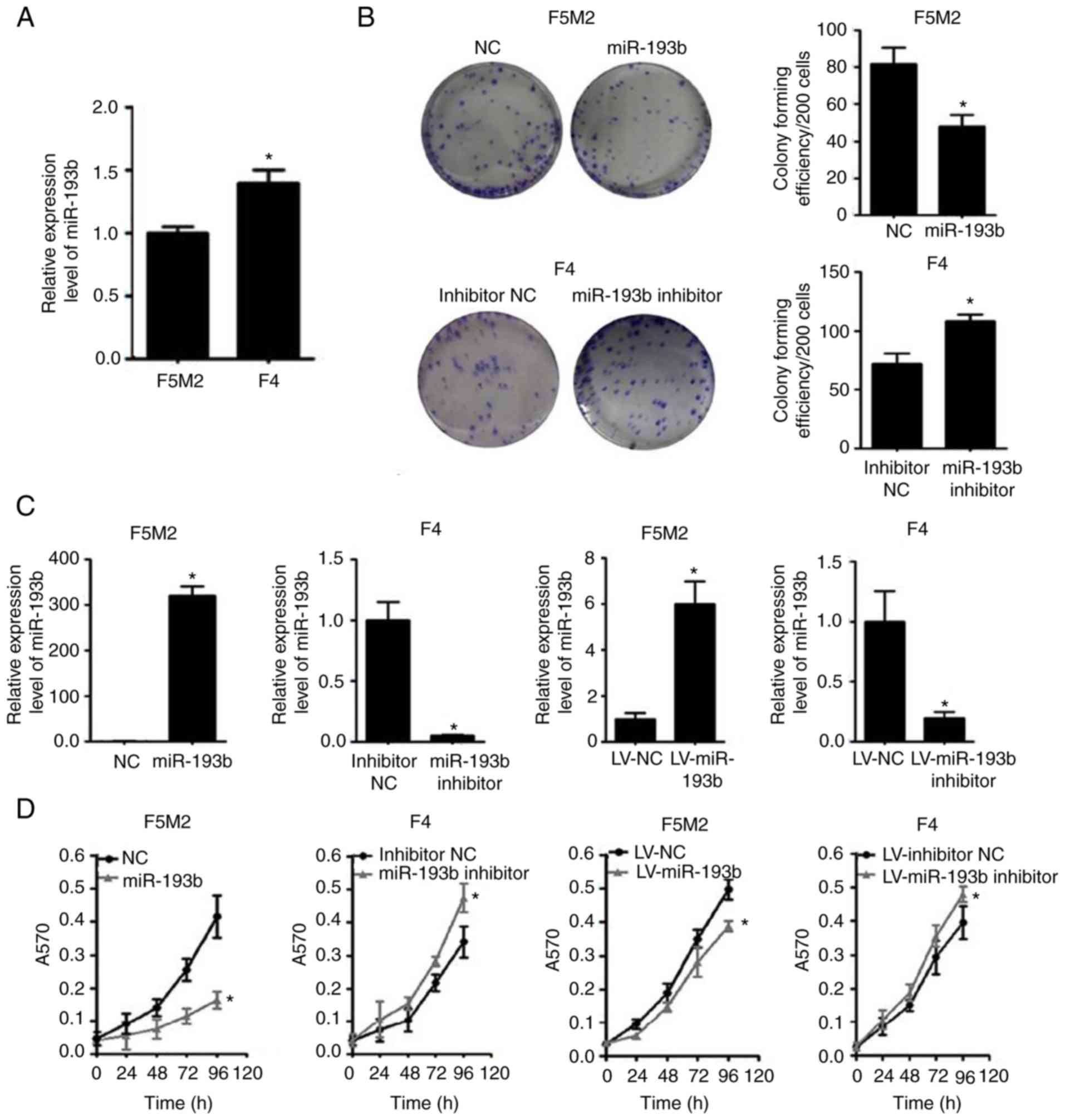
When the expression of miR-193b was compared between
the two cell lines, F5M2 cells, which have a relatively high
metastatic potential, exhibited lower miR-193b expression than F4
cells, which have lower metastatic potential (Fig. 1A).
Subsequently, the expression of miR-193b was
transiently or stably altered (Fig.
1C), and the effects of miR-193b on proliferation and
colony-forming capacities were investigated. The results revealed
that the miR-193b mimic reduced the proliferation and
colony-forming capacity of F5M2 cells, whereas the miR-193b
inhibitor enhanced these abilities in F4 cells (Fig. 1B and D). Similar results were found
in F5M2 and F4 cells with stably altered miR-193b expression
(Fig. 1B and D).
As shown in Fig.
1E-G, the xenograft tumor formation assay revealed that the
tumor volume and weight were significantly higher in the group
injected with wild-type F5M2 cells compared with those injected
with F5M2 cells with stably upregulated miR-193b. Detailed
information about animal weight, tumor dimension and tumor weight
for each experimental animal is provided in Table SI.
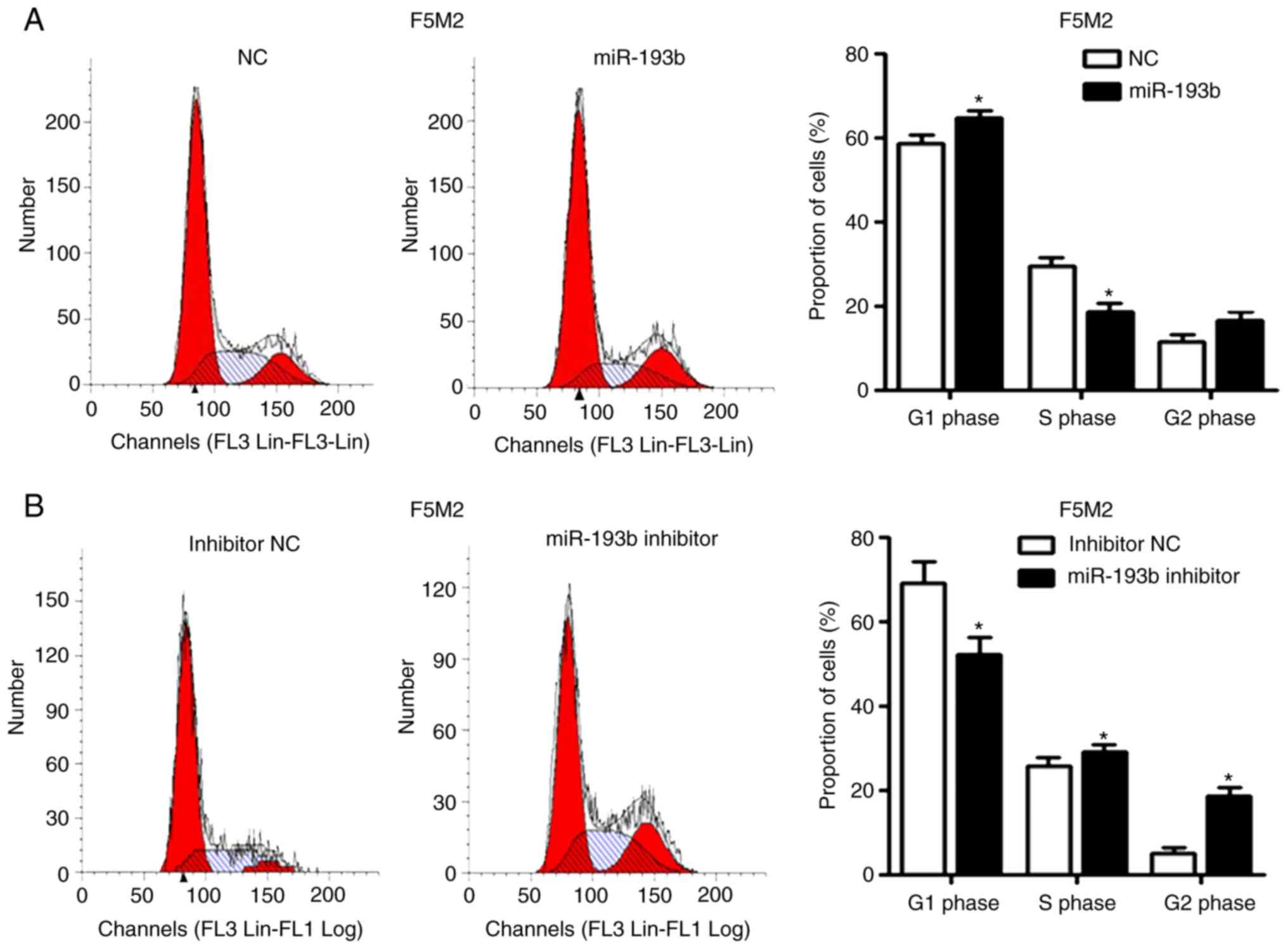
miR-193b induces cell cycle arrest and
apoptosis
Increasing the expression levels of miR-193b in F5M2
cells induced cell cycle arrest in the G1 phase
(Fig. 2A) and significantly
increased the percentage of apoptotic cells (Fig. 2C). After knockdown of miR-193b in F4
cells, the percentage of cells in G1 phase decreased
(Fig. 2B), whereas the proportion
of cells in S and G2 phases was increased. The
proportion of apoptotic cells decreased in
miR-193b-inhibitor-transfected F4 cells (Fig. 2D).
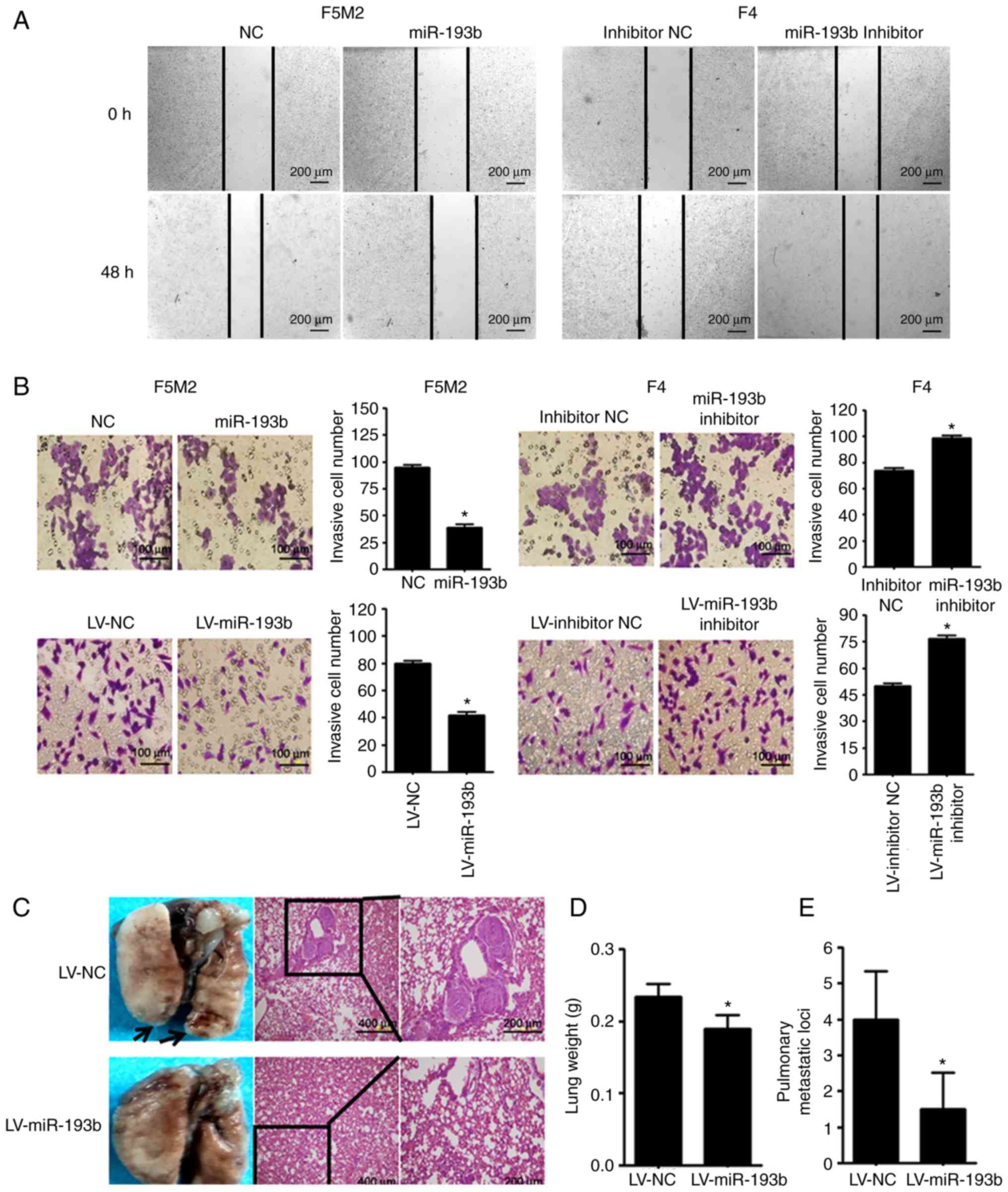
miR-193b suppresses the migratory and
invasive ability of osteosarcoma cells
As shown in Fig. 3A and
B, wound-healing assay and Transwell cell invasion assay
revealed that transient upregulation of miR-193b reduced the
migration and invasion abilities of F5M2 cells, whereas
downregulation of miR-193b in F4 cells exerted opposite effects. In
addition, when a Transwell cell invasion assay was performed after
miR-193b was stably altered, results were consistent with those
aforementioned.
The in vivo study revealed that mice injected
with miR-193b-upregulated F5M2 cells had significantly lower lung
weights and fewer pulmonary metastatic nodules compared with mice
injected with wild-type F5M2 cells (Fig. 3C-E).
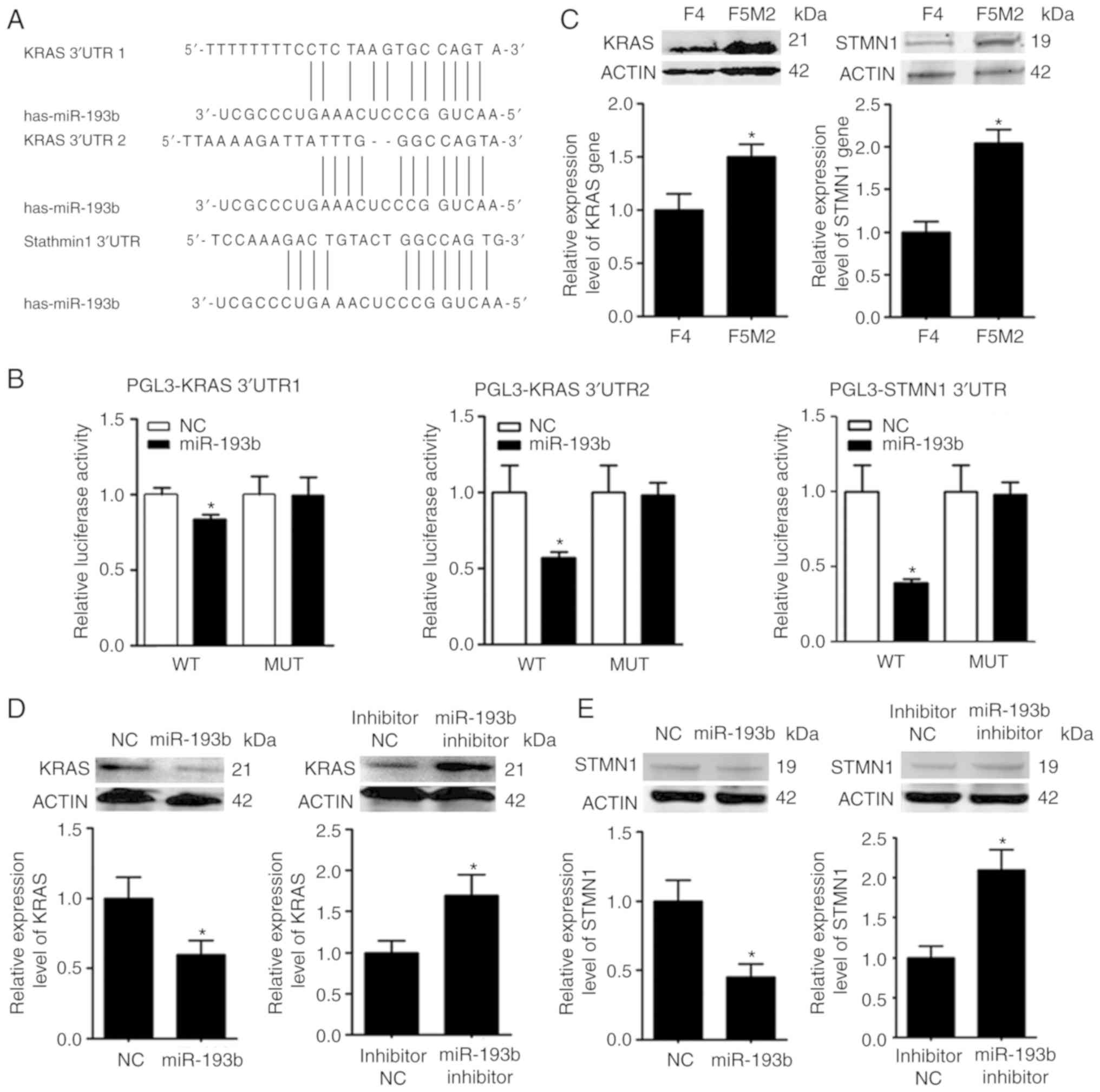
KRAS and STMN1 are targets of
miR-193b
The target molecules of miR-193b were predicted
using numerous miRNA target gene prediction websites. KRAS and
STMN1 were selected from the predicted targets (Fig. 4A), since they have been associated
with the malignant behavior of tumors (16,17).
The expression levels of KRAS and STMN1 were higher in F5M2 cells
than in F4 cells (Fig. 4B), showing
an inverse trend compared with miR-193b. The prediction was further
validated using a luciferase reporter assay, which revealed that
miR-193b markedly reduced the luciferase activities of the reporter
vectors containing wild-type constructs of KRAS and STMN1, but not
vectors containing mutant constructs (Fig. 4C). In addition, upregulation of
miR-193b in F5M2 cells induced a decrease in KRAS and STMN1
expression (Fig. 4D), whereas the
downregulation of miR-193b in F4 cells exerted opposite effects
(Fig. 4E). Immunofluorescence
staining for KRAS and STMN1 in xenograft tumor sections also
revealed that upregulation of miR-193b in osteosarcoma cells
reduced the expression of KRAS and STMN1 in vivo (Fig. 4F).
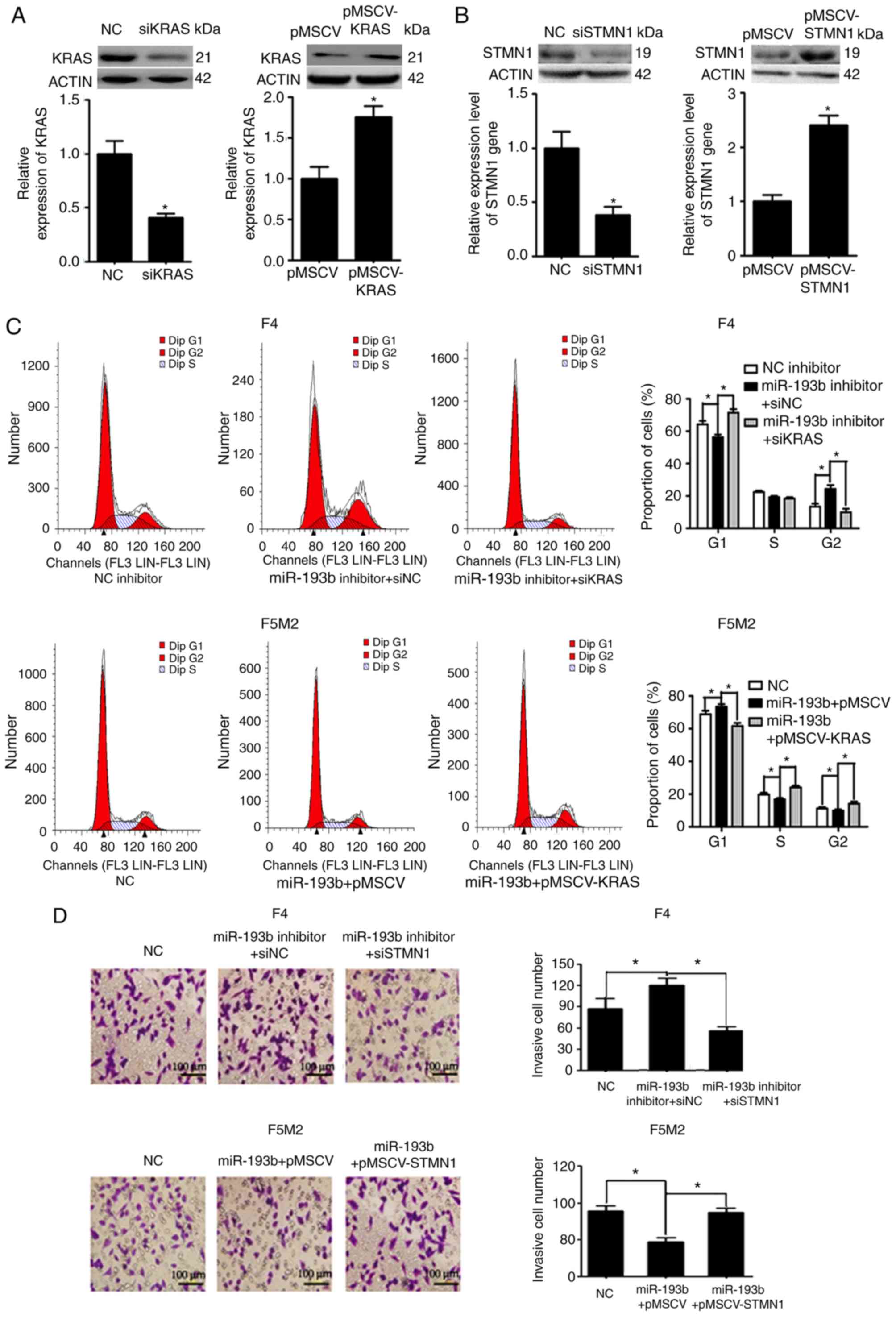
miR-193b inhibits proliferation via
KRAS and suppresses migration and invasion via STMN1
Expression levels of KRAS and STMN1 were
deliberately enhanced or reduced using pMSCV-KRAS/pMSCV-STMN1 or
KRAS siRNA (siKRAS)/STMN1 siRNA (siSTMN1). Transfection efficiency
was validated using western blotting and RT-qPCR (Fig. 5A and B). F5M2 cells were transfected
with miR-193b mimic plus empty pMSCV or miR-193b mimic plus
pMSCV-KRAS. F4 cells were transfected with miR-193b inhibitor plus
siNC or miR-193b inhibitor plus siKRAS. The cells were then
subjected to cell cycle analysis. As shown in Fig. 5C, pMSCV-KRAS reversed the cell cycle
arrest induced by miR-193b in F5M2 cells. Similarly siKRAS
abolished the miR-193b inhibitor-induced increase in the percentage
of cells in G2 phase in F4 cells (Fig. 5C). To study the involvement of STMN1
in miR-193b-mediated regulation of invasion and migration, F5M2
cells were transfected with miR-193b mimic plus empty pMSCV or
miR-193b mimic plus pMSCV-STMN1, and F4 cells were transfected with
miR-193b inhibitor plus siNC or miR-193b inhibitor plus siKRAS.
Subsequently, Transwell cell invasion assay and wound-healing
assays were performed. As can be seen from the results of Transwell
cell invasion assay, co-transfection of F5M2 cells with the
miR-193b mimic and pMSCV-STMN1 reversed miR-193b-induced inhibition
of invasion, whereas co-transfection of F4 cells with the miR-193b
inhibitor and siSTMN1 abolished miR-193b inhibitor-induced
enhancement of tumor cell invasion (Fig. 5D). Similarly, wound-healing assay
results revealed that pMSCV-STMN1 reversed miR-193b-induced
inhibition of migration in F5M2 cells, whereas siSTMN1 abolished
miR-193b inhibitor-induced enhancement of tumor cell migration in
F4 cells (Fig. 5E).
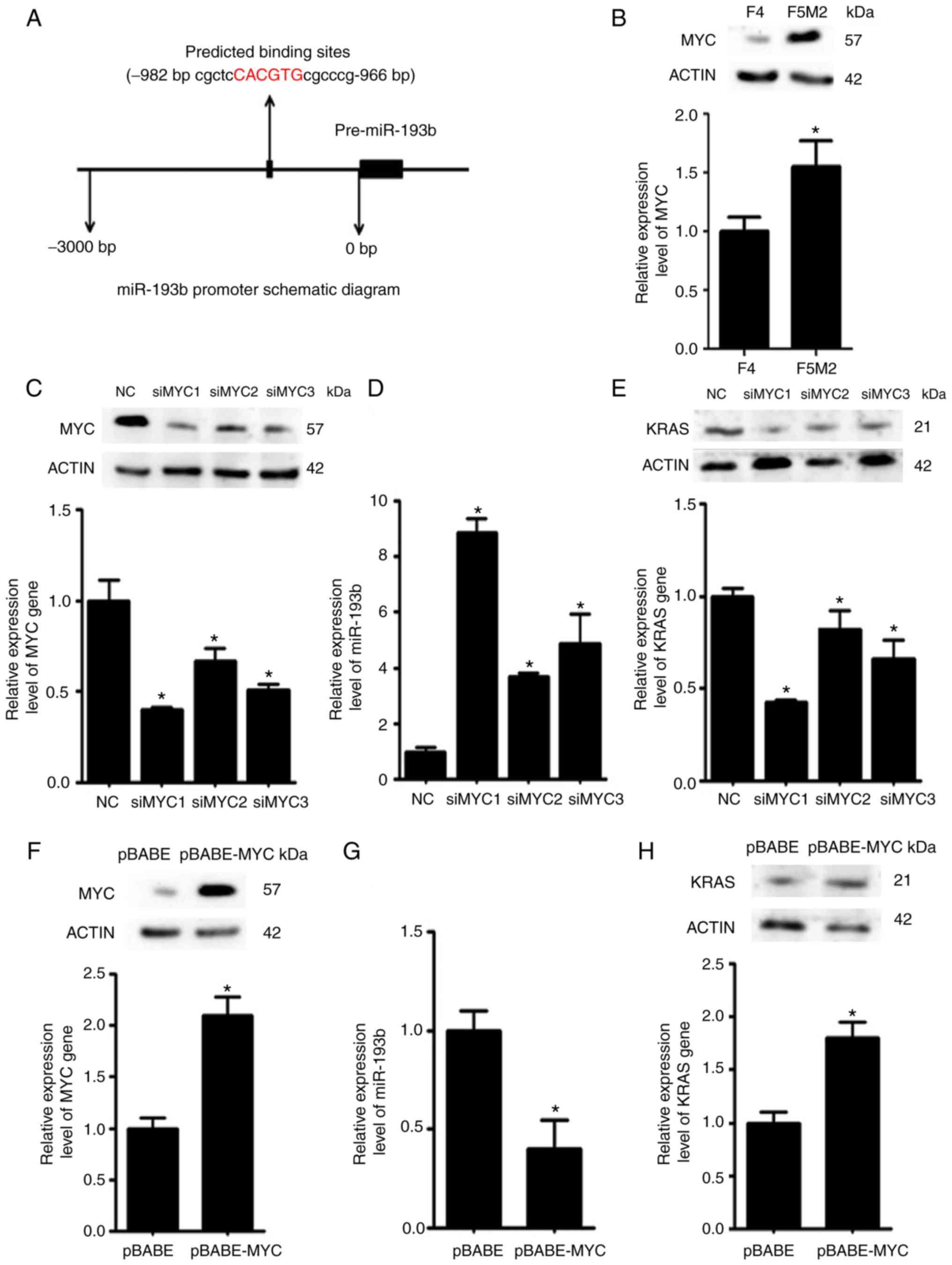
MYC downregulates miR-193b
expression
Using bioinformatics tools, it was predicted that
the transcription factor MYC may bind to the promoter region of
miR-193b and regulate its expression (Fig. 6A). Western blotting and RT-qPCR
revealed that the expression levels of MYC were higher in F5M2
cells than in F4 cells (Fig. 6B).
To validate the regulatory effects of MYC on miR-193b, the
expression levels of miR-193b were examined after the expression of
MYC was deliberately reduced in F5M2 cells (Fig. 6C) or enhanced in F4 cells (Fig. 6F). The expression of miR-193b was
increased when MYC was downregulated in F5M2 cells (Fig. 6D). Conversely, miR-193b expression
was decreased when MYC was overexpressed in F4 cells (Fig. 6G). In addition, the expression of
KRAS, which is a target of miR-193b, was reduced after MYC was
downregulated in F5M2 cells (Fig.
6E) and enhanced after MYC was upregulated in F4 cells
(Fig. 6H).
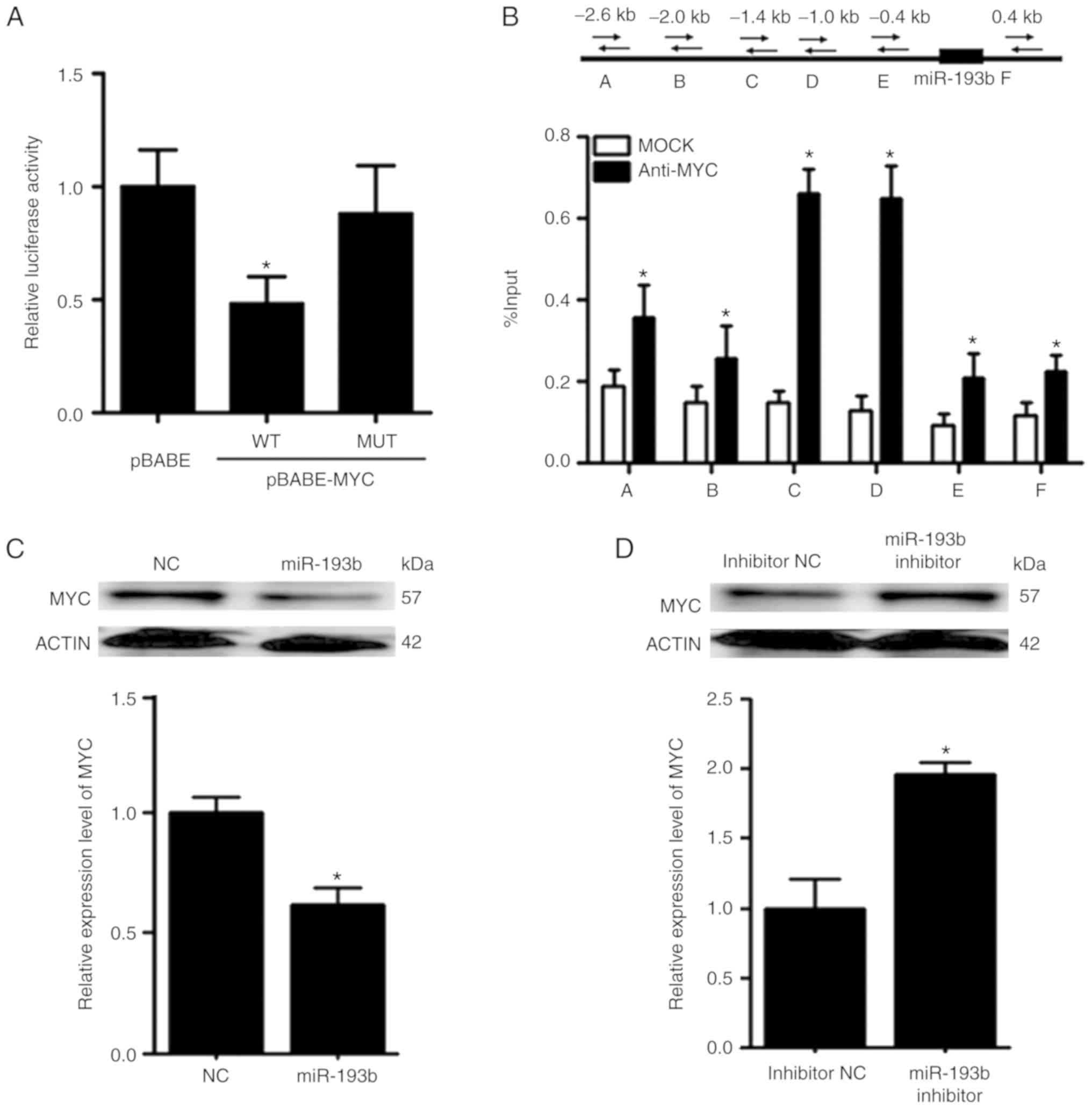
MYC and miR-193b exert mutual
suppressive effects against each other
The promoter reporter assay revealed that MYC could
reduce the luciferase activity of the reporter construct harboring
the wild-type promoter region of miR-193b (Fig. 7A). To further validate the binding
between MYC and miR-193b, ChIP-qPCR was performed. In the ChIP-qPCR
assay, six primers were designed according to the predicted binding
sites on miR-193b. It was revealed that MYC was able to bind to the
promoter region of miR-193b, and the relative enrichment levels of
the precipitated DNA for anti-MYC groups were 1.5–5-fold greater
than those for the IgG MOCK control groups (Fig. 7B).
Following overexpression of miR-193b in F5M2 cells,
RT-qPCR and western blot analysis revealed that the expression of
MYC was decreased (Fig. 7C). In
contrast, the expression of MYC was increased after miR-193b
expression was downregulated in F4 cells (Fig. 7D). Furthermore, when xenograft tumor
sections were subjected to immunofluorescence staining for MYC,
tumor sections from mice injected with F5M2 cells with upregulated
miR-193b exhibited relatively lower MYC expression than those from
mice in the control group. These results indicated that miR-193b
had a negative effect on MYC expression. However, through exploring
various target gene prediction websites, no direct binding site for
miR-193b was detected on the 3′-UTR of MYC, indicating that
miR-193b may negatively modulate the expression of MYC through some
unknown mechanism. Since miR-193b has many direct targets, it is
possible that one or several of these direct targets may
subsequently influence the expression of MYC. Further research is
required to clarify the mechanism underlying negative regulation of
MYC by miR-193b.
Discussion
Due to their critical role in tumor generation and
progression, miRNAs have been recognized as potential therapeutic
targets for cancer treatment. By comparing the miRNA expression of
two primary osteosarcoma cell lines with different metastatic
potentials, miR-193b was identified as a potential regulator of
malignant behaviors in osteosarcoma. Notably, miR-193b has been
reported as a tumor suppressor that may be downregulated in various
types of cancer, such as gastric cancer (24), Ewing sarcoma (25), colorectal cancer (26), pancreatic cancer (27), glioma (28), lung cancer (13), hepatocellular carcinoma (11), prostate cancer (10) and melanoma (29). Although the majority of reports have
indicated that miR-193b may serve a protective role against various
types of cancer, in head and neck squamous cell carcinoma, miR-193b
functioned as a tumor promoter via the negative regulation of
neurofibromin 1 (30). The
underlying reason for this discrepancy remains unclear, but it
might be due in part to organ-specific actions and the different
cellular contexts of tumors (31).
To the best of our knowledge, the role of miR-193b in the malignant
behavior of osteosarcoma remains to be elucidated. Thus, a thorough
investigation was performed in the present study to reveal the role
of miR-193b in the generation and development of osteosarcoma.
Compared with the highly metastatic F5M2 cell line,
F4 cells with a low metastatic capacity exhibited relatively high
expression levels of miR-193b. This result indicated that, in
accordance with the majority of published data, miR-193b may act as
a tumor suppressor in osteosarcoma. To confirm this hypothesis,
gain-and-loss-of-function studies were performed. As expected,
upregulation of miR-193b in F5M2 cells significantly inhibited cell
proliferation, cell cycle progression, cell migration and cell
invasion, and markedly promoted apoptosis. Conversely, knockdown of
miR-193b expression using inhibitors in F4 cells exerted the
opposite effects. Furthermore, in vivo studies demonstrated
that, compared with mice injected with wild-type F5M2 cells, nude
mice injected with F5M2 cells with increased miR-193b levels
exhibited reduced tumor mass and fewer lung metastatic nodules.
Based on the aforementioned evidence, it was suggested that
miR-193b may have a tumor-suppressive role in osteosarcoma.
Published studies have demonstrated that the
tumor-suppressive role of miR-193b is mainly caused by inhibition
of the expression of its target genes (9–13). To
investigate the molecular mechanism underlying the
tumor-suppressive role of miR-193b in osteosarcoma, numerous miRNA
target gene prediction websites were used, and KRAS and STMN1 were
identified as two potential targets for miR-193b. The expression
levels of KRAS and STMN1 were further evaluated and it was revealed
that they were negatively associated with the levels of miR-193b.
The upregulation of miR-193b in F5M2 cells inhibited the expression
of KRAS and STMN1, whereas the downregulation of miR-193b in F4
cells enhanced their expression, demonstrating negative influences
of miR-193b on KRAS and STMN1 expression. To further verify whether
miR-193b influenced the expression of KRAS and STMN1 by directly
targeting their promoter regions, a luciferase reporter assay was
performed. It was revealed that miR-193b reduced the luciferase
activity of the reporter gene with the wild-type construct but not
with the mutant 3′-UTR, demonstrating that miR-193b could indeed
reduce KRAS and STMN1 expression by directly binding to their
3′-UTRs.
STMN1 encodes a cytosolic phosphoprotein, which is
involved in the formation and function of the mitotic spindle. By
acting as a microtubule destabilizer, STMN1 participates in various
cellular biological processes, such as cell division, motility and
differentiation (32). Recent
studies have demonstrated that STMN1 may act as an oncogene, which
was upregulated in numerous types of malignant tumors, including
lung cancer (33), esophageal
carcinoma (34), breast cancer
(35), hepatocellular carcinoma
(36), gastric cancer (37), pancreatic cancer (38) and colorectal cancer (39). STMN1 may promote the occurrence and
development of tumors by interfering with microtubule dynamics,
enhancing tumor cell proliferation and motility, and inhibiting
apoptosis (40). The oncogenic role
of STMN1 in osteosarcoma has also been revealed by recent
publications. Jiang et al (41) reported that the inhibition of STMN1
expression using small interfering RNA induced growth inhibition,
cell cycle arrest and apoptosis. Other researchers have reported
that STMN1 may be involved in the drug resistance of osteosarcoma,
and inhibition of STMN1 enhanced the chemosensitivity of
osteosarcoma cells (42–44). In the present study, pMSCV-STMN1
reversed miR-193b-induced inhibition of migration and invasion in
F5M2 cells, indicating that miR-193b may inhibit migration and
invasion of osteosarcoma through STMN1.
The KRAS oncogene, which encodes a GTPase signaling
protein, is a key driver of tumorigenesis (45). An oncogenic role of KRAS in
osteosarcoma has been reported in recent years. Sun et al
(46) investigated the differences
in gene expression levels between metastatic and nonmetastatic
osteosarcoma, and demonstrated that KRAS had the potential to be
used as a biomarker for highly metastatic osteosarcoma. Zhang et
al (47) reported that KRAS was
a target of let-7a that may interfere with the viability,
invasiveness and migration of osteosarcoma cells. Additionally,
quercetin, a potential anticancer agent, enhanced the cisplatin
sensitivity of osteosarcoma by modulating the miR-217-KRAS axis
(48). In the present study,
pMSCV-KRAS reversed miR-193b-induced cell cycle arrest at
G1 phase, indicating that miR-193b inhibited the
proliferation of osteosarcoma cells through KRAS.
Although a substantial number of publications have
discussed the roles of miRNAs and their targets in human cancer,
only a small number of them concern the upstream molecules that
regulate miRNAs. MYC, which is a key basic helix-loop-helix leucine
zipper transcription factor, acts as an important regulator of
several cellular processes (49–51).
Moreover, MYC has been recognized as an oncogene that is
overexpressed in several types of human cancer (52,53),
including osteosarcoma (54). In
recent years, increasing evidence has indicated that there is a
mutual regulatory effect between miRNAs and MYC. This study
investigated the involvement of MYC in the miRNA-regulated
malignant behaviors of osteosarcoma cells. In contrast to miR-193b,
highly metastatic F5M2 cells had a higher expression level of MYC
than F4 cells with low metastatic potential. Furthermore, the
downregulation of MYC in F5M2 cells significantly enhanced the
expression of miR-193b and subsequently reduced the expression of
KRAS. Conversely, the upregulation of MYC in F4 cells revealed
opposite results. Taken together, these findings indicated that MYC
may act as an upstream negative regulator of miR-193b. To test this
hypothesis, several online databases were searched and it was
revealed that a potential binding site for MYC is present in the
promoter region of miR-193b. The direct binding of MYC to the
miR-193b promoter region was validated using a ChIP-qPCR assay and
luciferase reporter assay. Considering the recently published data
regarding the mutual regulatory effects between MYC and miRNAs,
this study aimed to assess whether the expression of MYC was
subjected to regulation by miR-193b. Overexpression of miR-193b
induced a reduction in MYC expression, whereas downregulation of
miR-193b caused an increase in MYC expression. The negative
regulation of MYC by miR-193b was also supported by in vivo
studies, which revealed that MYC expression was relatively lower in
tumor tissues from nude mice injected with miR-193b-upregulated
F5M2 cells compared with the control group. To determine whether
miR-193b regulated MYC through directly targeting the MYC 3′-UTR,
various target gene prediction websites were explored. However, no
direct binding site for miR-193b was identified. Since one miRNA
can have a great number of targets, it may be hypothesized that
miR-193b influences the expression of MYC indirectly through
targeting other MYC-regulatory proteins or miRNAs.
In conclusion, the evidence from this study
indicated that MYC and miR-193b exhibited mutual negative
interactions with each other. Such a mutual interaction could lead
to sustained MYC activation, miR-193b downregulation, STMN1 and
KRAS upregulation, and may eventually promote development and
metastasis of osteosarcoma.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by the Natural Science
Foundation of China (grant nos. 30901784, 81172289 and
81472633).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contribution
LQS and TW designed the study and helped interpret
the data. JJG performed the experiments, acquired the data and
wrote the manuscript. FY, XC, WW, JPZ and YFL performed the
experiments and acquired the data. SM performed data analysis,
helped write the manuscript and revised the manuscript. All authors
read and approved the final manuscript, and agreed to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethic approval and consent to
participate
All animal handling and experimental procedures were
approved by the Animal Ethics Committees of The Fourth Military
Medical University and were in accordance with the guidelines of
the China Council of Animal Care.
Patient consent for publication
Not applicable.
Competing interest
The authors declare that they have no competing
interests.
References
|
1
|
Ottaviani G and Jaffe N: The epidemiology
of osteosarcoma. Cancer Treat Res. 152:3–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marina N, Gebhardt M, Teot L and Gorlick
R: Biology and therapeutic advances for pediatric osteosarcoma.
Oncologist. 9:422–441. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Esquela-Kerscher A and Slack FJ:
Oncomirs-microRNAs with a role in cancer. Nat Rev Cancer.
6:259–269. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gao J, Yang TT, Qiu XC, Yu B, Han JW, Fan
QY and Ma BA: Cloning and identification of microRNA from human
osteosarcoma cell line SOSP-9607. Ai Zheng. 26:561–565. 2007.(In
Chinese). PubMed/NCBI
|
|
6
|
Palmini G, Marini F and Brandi ML: What is
new in the mirna world regarding osteosarcoma and chondrosarcoma?
Molecules. 7:4172017. View Article : Google Scholar
|
|
7
|
Leichter AL, Sullivan MJ, Eccles MR and
Chatterjee A: MicroRNA expression patterns and signalling pathways
in the development and progression of childhood solid tumours. Mol
Cancer. 16:152017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen X, Yang TT, Qiu XC, Ji ZG, Li CX,
Long H, Zhou Y, Ma BA, Ma Q, Zhang X and Fan QY: Gene expression
profiles of human osteosarcoma cell sublines with different
pulmonary metastatic potentials. Cancer Biol Ther. 11:287–292.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu W, Lin Z, Zhuang Z and Liang X:
Expression profile of mammalian microRNAs in endometrioid
adenocarcinoma. Eur J Cancer Prev. 18:50–55. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rauhala HE, Jalava SE, Isotalo J, Bracken
H, Lehmusvaara S, Tammela TL, Oja H and Visakorpi T: MiR-193b is an
epigenetically regulated putative tumor suppressor in prostate
cancer. Int J Cancer. 127:1363–1372. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu C, Liu S, Fu H, Li S, Tie Y, Zhu J,
Xing R, Jin Y, Sun Z and Zheng X: MicroRNA-193b regulates
proliferation, migration and invasion in human hepatocellular
carcinoma cells. Eur J Cancer. 46:2828–2836. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li XF, Yan PJ and Shao ZM: Downregulation
of miR-193b contributes to enhance urokinase-type plasminogen
activator (uPA) expression and tumor progression and invasion in
human breast cancer. Oncogene. 28:3937–3948. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hu H, Li S, Liu J and Ni B: MicroRNA-193b
modulates proliferation, migration, and invasion of non-small cell
lung cancer cells. Acta Biochim Biophys Sin (Shanghai). 44:424–430.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jancik S, Drabek J, Radzioch D and Hajduch
M: Clinical relevance of KRAS in human cancers. J Biomed
Biotechnol. 2010:1509602010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mao Q, Chen Z, Wang K, Xu R, Lu H and He
X: Prognostic role of high stathmin 1 expression in patients with
solid tumors: Evidence from a meta-analysis. Cell Physiol Biochem.
50:66–78. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao C, Li H, Wang L and Sun W: An
immunohistochemical study of stathmin 1 expression in osteosarcoma
shows an association with metastases and poor patient prognosis.
Med Sci Monit. 31:6070–6078. 2018. View Article : Google Scholar
|
|
17
|
Zhang H, He QY, Wang GC, Tong DK, Wang RK,
Ding WB, Li C, Wei Q, Ding C, Liu PZ, et al: MiR-422a inhibits
osteosarcoma proliferation by targeting BCL2L2 and KRAS. Biosci
Rep. 21:382018.
|
|
18
|
Swier L, Dzikiewicz-Krawczyk A, Winkle M,
van den Berg A and Kluiver J: Intricate crosstalk between MYC and
non-coding RNAs regulates hallmarks of cancer. Mol Oncol. 13:26–45.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang N, Wang X, Huo Q, Sun M, Cai C, Liu
Z, Hu G and Yang Q: MicroRNA-30a suppresses breast tumor growth and
metastasis by targeting metadherin. Oncogene. 33:3119–3128. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bai JX, Yan B, Zhao ZN, Xiao X, Qin WW,
Zhang R, Jia LT, Meng YL, Jin BQ, Fan DM, et al: Tamoxifen
represses miR-200 microRNAs and promotes epithelial-to-mesenchymal
transition by up-regulating c-Myc in endometrial carcinoma cell
lines. Endocrinology. 154:635–645. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang W, Xiao X, Chen X, Huo Y, Xi WJ, Lin
ZF, Zhang D, Li YF, Yang F, Wen WH, et al: Tumor-suppressive
miR-145 co-repressed by TCF4-β-catenin and PRC2 complexes forms
double-negative regulation loops with its negative regulators in
colorectal cancer. Int J Cancer. 142:308–321. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang YQ, Jiang DM, Hu SS, Zhao L, Wang L,
Yang MH, Ai ML, Jiang HJ, Han Y, Ding YQ and Wang S: SATB2-AS1
suppresses colorectal carcinoma aggressiveness by inhibiting
SATB2-Dependent snail transcription and epithelial-mesenchymal
transition. Cancer Res. 79:3542–3556. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang L, Zhang Y, Zhao L, Liu S, Yu S, Ma Y
and Sun G: MicroRNA-193b inhibits the proliferation, migration and
invasion of gastric cancer cells via targeting cyclin D1. Acta
Histochem. 118:323–330. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Moore C, Parrish JK and Jedlicka P:
MiR-193b, downregulated in Ewing Sarcoma, targets the ErbB4
oncogene to inhibit anchorage-independent growth. PLoS One.
12:e01780282017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guo F, Luo Y, Mu YF, Qin SL, Qi Y, Qiu YE
and Zhong M: MiR-193b directly targets STMN1 and inhibits the
malignant phenotype in colorectal cancer. Am J Cancer Res.
6:2463–2475. 2016.PubMed/NCBI
|
|
27
|
Jin X, Sun Y, Yang H, Li J, Yu S, Chang X,
Lu Z and Chen J: Deregulation of the MiR-193b-KRAS axis contributes
to impaired cell growth in pancreatic cancer. PLoS One.
10:e01255152015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhong Q, Wang T, Lu P, Zhang R, Zou J and
Yuan S: MiR-193b promotes cell proliferation by targeting Smad3 in
human glioma. J Neurosci Res. 92:619–626. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen J, Zhang X, Lentz C, Abi-Daoud M,
Paré GC, Yang X, Feilotter HE and Tron VA: MiR-193b regulates Mcl-1
in melanoma. Am J Pathol. 179:2162–2168. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lenarduzzi M, Hui AB, Alajez NM, Shi W,
Williams J, Yue S, O'Sullivan B and Liu FF: MicroRNA-193b enhances
tumor progression via down regulation of neurofibromin 1. PLoS One.
8:e537652013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sampson VB, Yoo S, Kumar A, Vetter NS and
Kolb EA: MicroRNAs and potential targets in osteosarcoma: Review.
Front Pediatr. 3:692015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rubin CI and Atweh GF: The role of
stathmin in the regulation of the cell cycle. J Cell Biochem.
93:242–250. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nie W, Xu MD, Gan L, Huang H, Xiu Q and Li
B: Overexpression of stathmin 1 is a poor prognostic biomarker in
non-small cell lung cancer. Lab Invest. 95:56–64. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Akhtar J, Wang Z, Jiang WP, Bi MM and
Zhang ZP: Stathmin overexpression identifies high risk for
lymphatic metastatic recurrence in pN0 esophageal squamous cell
carcinoma patients. J Gastroenterol Hepatol. 29:944–950. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Baquero MT, Hanna JA, Neumeister V, Cheng
H, Molinaro AM, Harris LN and Rimm DL: Stathmin expression and its
relationship to microtubule-associated protein tau and outcome in
breast cancer. Cancer. 118:4660–4669. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen YL, Uen YH, Li CF, Horng KC, Chen LR,
Wu WR, Tseng HY, Huang HY, Wu LC and Shiue YL: The E2F
transcription factor 1 transactives stathmin 1 in hepatocellular
carcinoma. Ann Surg Oncol. 20:4041–4054. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kang W, Tong JH, Chan AW, Lung RW, Chau
SL, Wong QW, Wong N, Yu J, Cheng AS and To KF: Stathmin1 plays
oncogenic role and is a target of microRNA-223 in gastric cancer.
PLoS One. 7:e339192012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lu Y, Liu C, Cheng H, Xu Y, Jiang J, Xu J,
Long J, Liu L and Yu X: Stathmin, interacting with Nf-κB, promotes
tumor growth and predicts poor prognosis of pancreatic cancer. Curr
Mol Med. 14:328–339. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tan HT, Wu W, Ng YZ, Zhang X, Yan B, Ong
CW, Tan S, Salto-Tellez M, Hooi SC and Chung MC: Proteomic analysis
of colorectal cancer metastasis: Stathmin-1 revealed as a player in
cancer cell migration and prognostic marker. J Proteome Res.
11:1433–1445. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Biaoxue R, Xiguang C, Hua L and Shuanying
Y: Stathmin-Dependent molecular targeting therapy for malignant
tumor: The latest 5 years' discoveries and developments. J Transl
Med. 14:2792016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang HZ, Wang Y, Gao P, Lin F, Liu L, Yu
B, Ren JH, Zhao H and Wang R: Silencing stathmin gene expression by
survivin promoter-driven siRNA vector to reverse malignant
phenotype of tumor cells. Cancer Biol Ther. 5:1457–1461. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang Z, He R, Xia H, Wei Y and Wu S:
Knockdown of STMN1 enhances osteosarcoma cell chemosensitivity
through inhibition of autophagy. Oncol Lett. 13:3465–3470. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Feng T, Qiao G, Feng L, Qi W, Huang Y, Yao
Y and Shen Z: Stathmin is key in reversion of doxorubicin
resistance by arsenic trioxide in osteosarcoma cells. Mol Med Rep.
10:2985–2992. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang R, Dong K, Lin F, Wang X, Gao P, Wei
SH, Cheng SY and Zhang HZ: Inhibiting proliferation and enhancing
chemosensitivity to taxanes in osteosarcoma cells by RNA
interference-mediated downregulation of stathmin expression. Mol
Med. 13:567–575. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Karnoub AE and Weinberg RA: Ras oncogenes:
Split personalities. Nat Rev Mol Cell Biol. 9:517–531. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sun B, Wang F, Li M and Yang M:
Identifications of genetic differences between metastatic and
non-metastatic osteosarcoma samples based on bioinformatics
analysis. Med Oncol. 32:1532015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang S, Hou C, Li G, Zhong Y, Zhang J,
Guo X, Li B, Bi Z and Shao M: A single nucleotide polymorphism in
the 3′-untranslated region of the KRAS gene disrupts the
interaction with let-7a and enhances the metastatic potential of
osteosarcoma cells. Int J Mol Med. 38:919–926. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang X, Guo Q, Chen J and Chen Z:
Quercetin enhances cisplatin sensitivity of human osteosarcoma
cells by modulating microRNA-217-KRAS axis. Mol Cells. 38:638–642.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Li Y, Liu H, Lai C, Du X, Su Z and Gao S:
The Lin28/let-7a/c-Myc pathway plays a role in non-muscle invasive
bladder cancer. Cell Tissue Res. 354:533–541. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Akinyeke T, Matsumura S, Wang X, Wu Y,
Schalfer ED, Saxena A, Yan W, Logan SK and Li X: Metformin targets
c-MYC oncogene to prevent prostate cancer. Carcinogenesis.
34:2823–2832. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Dueck AC, Reinholz MM, Geiger XJ, Tenner
K, Ballman K, Jenkins RB, Riehle D, Chen B, McCullough AE, Davidson
NE, et al: Impact of c-MYC protein expression on outcome of
patients with early-stage HER2+ breast cancer treated with adjuvant
trastuzumab NCCTG (alliance) N9831. Clin Cancer Res. 19:5798–5807.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Li K, Chen MK, Situ J, Huang WT, Su ZL, He
D and Gao X: Role of co-expression of c-Myc, EZH2 and p27 in
prognosis of prostate cancer patients after surgery. Chin Med J
(Engl). 126:82–87. 2013.PubMed/NCBI
|
|
53
|
Liu Z, Jiang Y, Hou Y, Hu Y, Cao X, Tao Y,
Xu C, Liu S, Wang S, Wang L, et al: The IkB family member Bcl-3
stabilizes c-Myc in colorectal cancer. J Mol Cell Biol. 5:280–282.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Pello OM and Andres V: Role of C-MYC in
tumor-associated macrophages and cancer progression.
Oncoimmunology. 2:e229842013. View Article : Google Scholar : PubMed/NCBI
|