Introduction
Among the most prevalent tumor types, colorectal
carcinoma (CRC) is third most common, and is the second leading
cause of cancer-associated deaths worldwide (1). Although the 5-year overall survival
rate of patients with CRC is 64%, this rate decreases to ≤10% in
patients who have developed metastases, and several patients with
CRC will develop local or distant relapses or metastasis (2). It is suggested that only the
application of more advanced drugs directed at novel targets may
improve the survival rate of patients with CRC (3). However, the pathogenetic mechanisms of
CRC are insufficiently understood, hampering drug development.
Thus, identifying the molecular mechanism underlying development
and/or progression of CRC may facilitate the discovery of
potentially novel therapeutic targets.
Previously, it was shown that chemo-prevention is a
suitable and effective treatment against cancer (4). Ellagic acid [EA;
2,3,7,8-tetrahydroxy-chromeno (5,4,3-cde) chromene-5,10-dione;
International Union of Pure and Applied Chemistry] is a
polyphenolic compound abundantly found in woody plants, berries,
grapes and nuts (5). EA is
considered a potential chemo-preventive agent, and has been shown
to inhibit proliferation in a variety of cancer types (5,6). In
previous studies, both in vitro and in vivo, EA
exhibited notable inhibitory effects against CRC, suggesting its
anti-tumor role against CRC (7–9).
However, the molecular mechanisms associated with the cellular
responses induced by EA, particularly the regulatory mechanisms
involved in altered transcription and protein interactions, have
not been determined, to the best of our knowledge. Additionally,
previous studies have not assessed the involvement of any relevant
pathways. Therefore, the aim of the present study was to identify
the molecular targets of EA which underlie the effects of EA on
HCT-116 CRC cells, and thus provide a theoretical basis for
precision therapy in CRC.
In our previous study, using cDNA microarray
analysis, it was shown that treatment with EA reduced proliferation
of CRC cells. Furthermore, a total of 4,738 genes were shown to be
significantly differentially expressed (1.2 fold change) after 72 h
of treatment with EA (10). EA was
shown to be associated with G0/G1 cell cycle arrest in HCT-116
cells and thereby induced apoptosis. Several genes involved in the
TGF-β1 and Smad3 signaling pathways were upregulated by treatment
with EA. Additionally, in another of our previous studies, it was
shown that EA can regulate breast cancer cell cycle arrest in
vitro via TGF-β/Smad signaling (11), although this has not been shown in
CRC yet, to the best of our knowledge. Following on from our
previous study, in the present study, it was shown that EA induced
cell cycle arrest in HCT-116 CRC cells by enhancing TGF-β1-induced
phosphorylation of Smad3, thereby inducing subsequent
apoptosis.
Materials and methods
Reagents and cell culture
EA was purchased from Sigma-Aldrich (Merck KGaA) and
diluted to a working concentration in DMSO (<1%). The solution
was sterilized using a 0.22 µm filter and stored at −20°C. HCT-116
cells were purchased from The Cell Bank of Type Culture Collection
of the Chinese Academy of Sciences, and were cultured in DMEM
supplemented with 10% FBS and 1% penicillin streptomycin solution,
and incubated in a humidified incubator at 37°C and 5%
CO2, as described previously (10). All standard reagents for cell
culture were purchased from Gibco (Thermo Fisher Scientific,
Inc.).
Treatment with EA
Cells were cultured in DMEM supplemented with FBS
initially. After incubation for 24 h, cells were cultured in DMEM
without FBS, as described previously (10). Cells were plated in a T25 flask at a
density of 5×105 cells/ml and reached a confluence of
50–60% after incubation for 6 h. As cells had been grown in
serum-free media, the cellular growth had been synchronized
(10). After a further 6 h
incubation in supplemented media, cells were treated EA with 0, 25,
50, 100 or 150 µM. DMSO diluted to <0.1% was used to treat the
negative control cells. After treatment with EA or negative control
at 37°C for 24 or 72 h, the cells were harvested, washed in
ice-cold PBS and fixed in 70% ethanol at 4°C for at least 12 h. The
samples were subsequently adjusted to a density of 1×106
cells/ml, and then stained with 80 mg/ml RNase A and 50 µg/ml
propidium iodide for 30 min at room temperature. The distribution
of the cells in the different phases of the cell cycle was detected
using a FACScan cytometer (Becton, Dickinson and Company) as
described previously. Flow cytometry were analyzed using BD Cell
Quest™ Pro version 3.2 (Becton, Dickinson and Company) (10,11).
Apoptosis analysis
The rate of apoptotic cells was analyzed using an
Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide kit
(Becton, Dickinson and Company) according to the manufacturer's
protocol. Briefly, upon following treatment with various
concentration of EA for 24 h, the cells were digested with 0.25%
trypsin and harvested. The samples were washed three times using
ice-cold PBS and re-suspended in binding buffer (500 µl) at a
density of 1×106 cells/ml, from which 500 µl was
transferred to a flow cytometry tube, and 5 µl each Annexin V-FITC
(50 µg/ml) and propidium iodide (50 µg/ml) was added. Cells were
left to stain in the dark for 30 min at room temperature. Using
flow cytometry, the proportion of apoptotic cells per 10,000 cells
was detected to calculate the apoptotic rate (11).
Cell cycle analysis
HCT-116 cells (5×105) were seeded in T25
culture flasks and grown for 6 h to a confluence of 50–60%. Cells
were starved in serum-free medium for 24 h to achieve
synchronization. Cells were subsequently grown for a further 6 h in
supplemented media for 6 h, and treated with EA as described above.
After treatment for 24 h at 37°C, floating and adherent cells were
collected, washed with ice-cold PBS and fixed with 70% ethanol for
at least 12 h at 4°C. The cells were then treated with 80 mg/ml
RNase A and 50 µg/ml PI at a density of 1×106 cells/ml
for 30 min, and the stained cells were analyzed using a FACScan
cytometer (Becton, Dickinson, Company).
Reverse transcription-quantitative
(RT-qPCR)
After 24 h of treatment with EA, total RNA was
extracted using TRIzol® reagent (Thermo Fisher
Scientific, Inc.) and an RNeasy kit (Qiagen GmbH) according to the
manufacturer's protocol. Specifically, for ribosomal RNA, purity
and integrity were further evaluated as described previously
(10,11).
Total RNA from HCT-116 cells treated with 100 µM EA,
an optimal concentration of EA determined in our previous study
(10), was used for transcriptomics
analysis of the selected target genes using RT-qPCR. RNA (2 µg) was
reverse transcribed to cDNA using oligo(dT) primers and SuperScript
II reverse transcriptase kit (Thermo Fisher Scientific, Inc.). The
thermocycling conditions used were: 30 sec at 95°C; followed by 40
cycles of 5 sec at 95°C and 34 sec at 60°C. A melt curve was
plotted between 60–95°C. Primers were purchased from Sangon Biotech
Co., Ltd. The sequences of the primers are stated in Table I. qPCR was performed using an ABI
Prism 7900HT sequence detection system (Applied Biosystems; Thermo
Fisher Scientific, Inc.). Data were analyzed using the comparative
2−ΔΔCq method (12). The
normalization of results was based on β-actin levels respectively
(10,11).
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Gene | Primer
sequence |
|---|
| β-actin |
CTCACCATGGATGATGATATCGC |
| Forward |
CTCACCATGGATGATGATATCGC |
| Reverse |
AGGAATCCTTCTGACCCATGC |
| TGFβ1 |
|
| Forward |
TGGAAACCCACAACGAAATCTATG |
| Reverse |
GCTAAGGCGAAAGCCCTCA |
| Smad3 |
|
| Forward |
ATGGCCGGTTGCAGGTGTC |
| Reverse |
GGTTCATCTGGTGGTCACTGGTTTC |
| P15 |
|
| Forward | TGGTGGC
TACGAATCTTCCG |
| Reverse |
TCGTCGCTTGCACATCCTC |
Western blotting analysis
To analyze protein expression, total protein was
extracted using lysis buffer (Beyotime Institute of Biotechnology)
and western blotting was performed. Briefly, 20 µg protein was
loaded on a 10–15% SDS gel, resolved using SDS-PAGE and transferred
to a PVDF membrane. The membrane was blocked with 5% skimmed milk
in PBS-Tween for 1 h at room temperature with shaking and
subsequently incubated with the primary antibody at 4°C overnight.
The antibodies used were: TGF-β1 (cat. no. 3709), Smad3 (cat. no.
9513), p-Smad3 (cat. no. 9520), β-Actin (cat. no. 4967) and p15
(cat. no. 4138) were obtained from Cell Signaling Technology, Inc.
All antibodies were used at a 1:1,000 dilution. Signals were
developed using an Enhanced Chemiluminescence Plus Detection kit
(Pierce; Thermo Fisher Scientific, Inc.). Signals were visualized
using a digital camera and densitometry analysis was performed
using ImageJ version 1.41q (National Institutes of Health). Protein
expression was normalized to β-actin levels.
Small interfering (si)RNA transfection
and specific inhibitors
si-TGF-β1 (5-CACUGCAAGUGGACAUCAATT-3 and
5-UUGAUGUCCACUUGCAGUGTT-3) and a negative control scrambled siRNA
(5′-GCCTAACTGTGTCAGAAGGAA-3′) were purchased from Shanghai
GenePharma, Co., Ltd. and 20 nM transfected into HCT-116 cells
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). CRC cells were pretreated with 10 ng/ml TGF-β1
siRNAs for 16 h, prior to treatment with EA (100 µM) for 24 h. To
inhibit Smad3, cells were pretreated with 3 µmol/l of the specific
inhibitor SIS3 for 6 h, prior to a 24 h EA treatment.
Statistical analysis
Data are presented as the mean ± the standard error
of the mean. Data were analyzed using GraphPad Prism version 5.0
(GraphPad Software, Inc.). The statistical differences between
>2 groups were determined using one-way ANOVA followed by a
Tukey's post-hoc test or a two-way ANOVA followed by a Bonferroni
post-hoc test. Statistical differences between 2 groups were
determined using a Student's t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
EA reduces cell proliferation and
results in G0/G1 cell cycle arrest
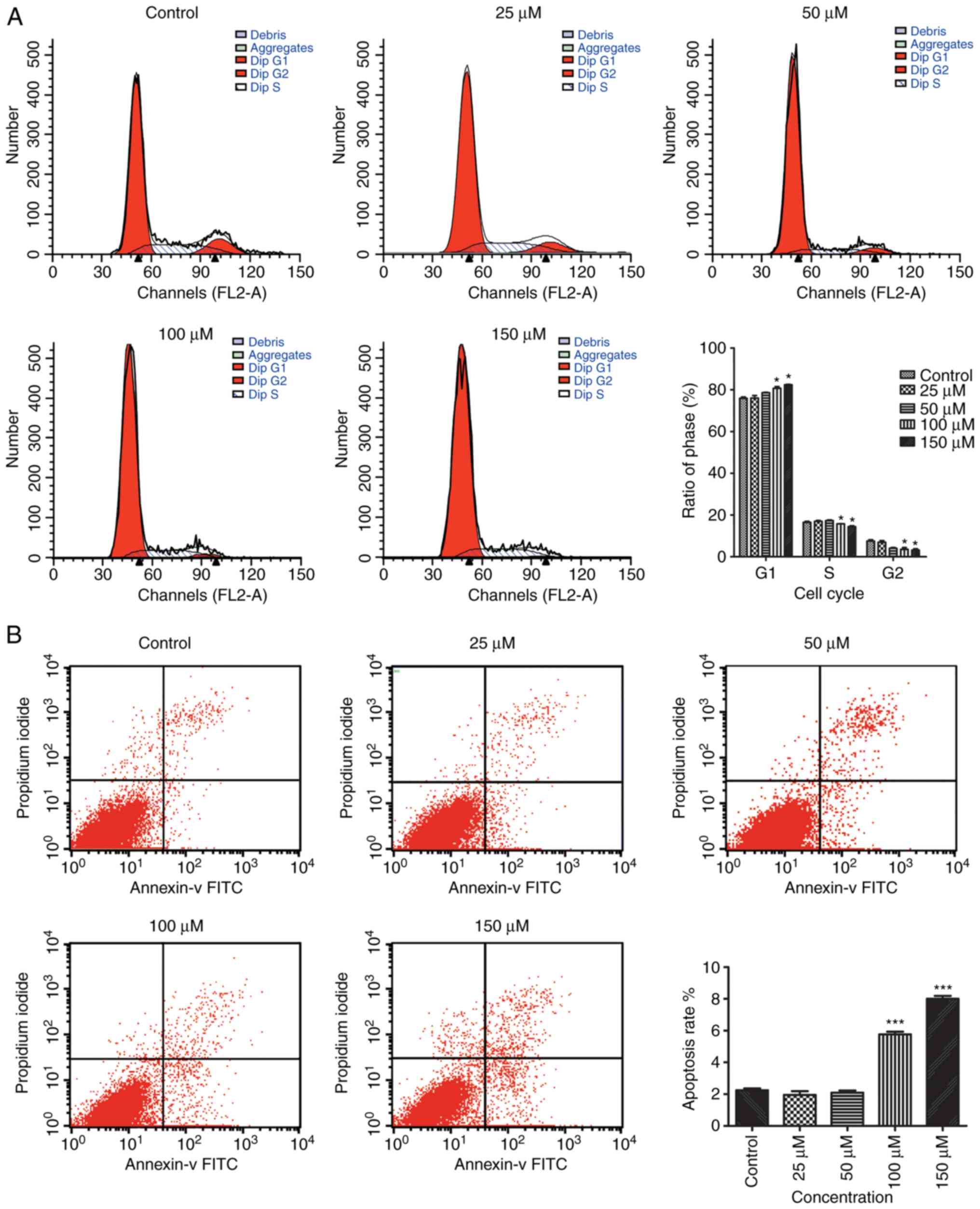
The effect of EA treatment on the cell cycle was
investigated using flow cytometry to determine whether the
EA-related decrease in the quantity of cells was the result of cell
cycle arrest. Treatment with 100 and 150 µM EA resulted in a
significant increase in the proportion of G0/G1 cells, and a
significant decrease in the proportion of cells in the S and G2/M
phase (Fig. 1A). These results
suggest that EA treatment was associated with G0/G1 arrest in
HCT-116 cells.
EA induces apoptosis in HCT-116
cells
Flow cytometry was used to assess the effects of EA
treatment on HCT-116 with regards to apoptosis. The results showed
that treatment with 100 and 150 µM EA for 24 h increased apoptosis
in a dose-dependent manner. These results showed that EA was
associated with the activation of apoptosis in HCT-116 cells, and
this effect was positively associated with concentration of EA
(Fig. 1B).
TGF-β/Smad signaling is involved in
the inhibitory effects of EA
Based on the further analysis of the microarray
data, the TGF-β1/Smad3 pathway was considered to be associated with
the induction of G0/G1 cell cycle arrest as well as reduction of
HCT-116 cell proliferation.
Subsequently, the expression levels of five
differentially expressed genes (DEGs) (TGF-β1, Smad3, E2F4, E2F5
and p15), which were enriched in the TGF-β/Smad signaling pathway,
were assessed using RT-qPCR. These findings were consistent with
those of the microarray analysis, indicating the 100% accuracy of
the array data (Table II).
| Table II.Relative expression changes in the
expression of five genes associated with the TGF-β1/Smad3 signaling
pathway in HCT-116 cells treated with 100 µM EA for 24 or 72 h,
determined using Affymetrix microarray analysis (72 h) and RT-qPCR
(24 h). |
Table II.
Relative expression changes in the
expression of five genes associated with the TGF-β1/Smad3 signaling
pathway in HCT-116 cells treated with 100 µM EA for 24 or 72 h,
determined using Affymetrix microarray analysis (72 h) and RT-qPCR
(24 h).
| Gene | Fold change,
Microarray | P-value,
Microarray | Fold change,
RT-qPCR | P-value,
RT-qPCR |
|---|
| TGFB1 | 1.503268 |
0.034882a | 3.122347 |
0.003244b |
| SMAD3 | 1.437719 |
0.006965b | 2.256482 |
0.007712b |
| E2F4 | −1.553536 |
0.007042b | −1.78425 |
0.003536b |
| E2F5 | −1.626492 |
0.002574b | −1.85462 | 0.006491 |
| CDKN2B/p15 | 1.740624 |
0.003070b | 2.10236 |
0.012624a |
TGF-β1 is a key factor involved in
EA-mediated regulation of cell cycle arrest and apoptosis
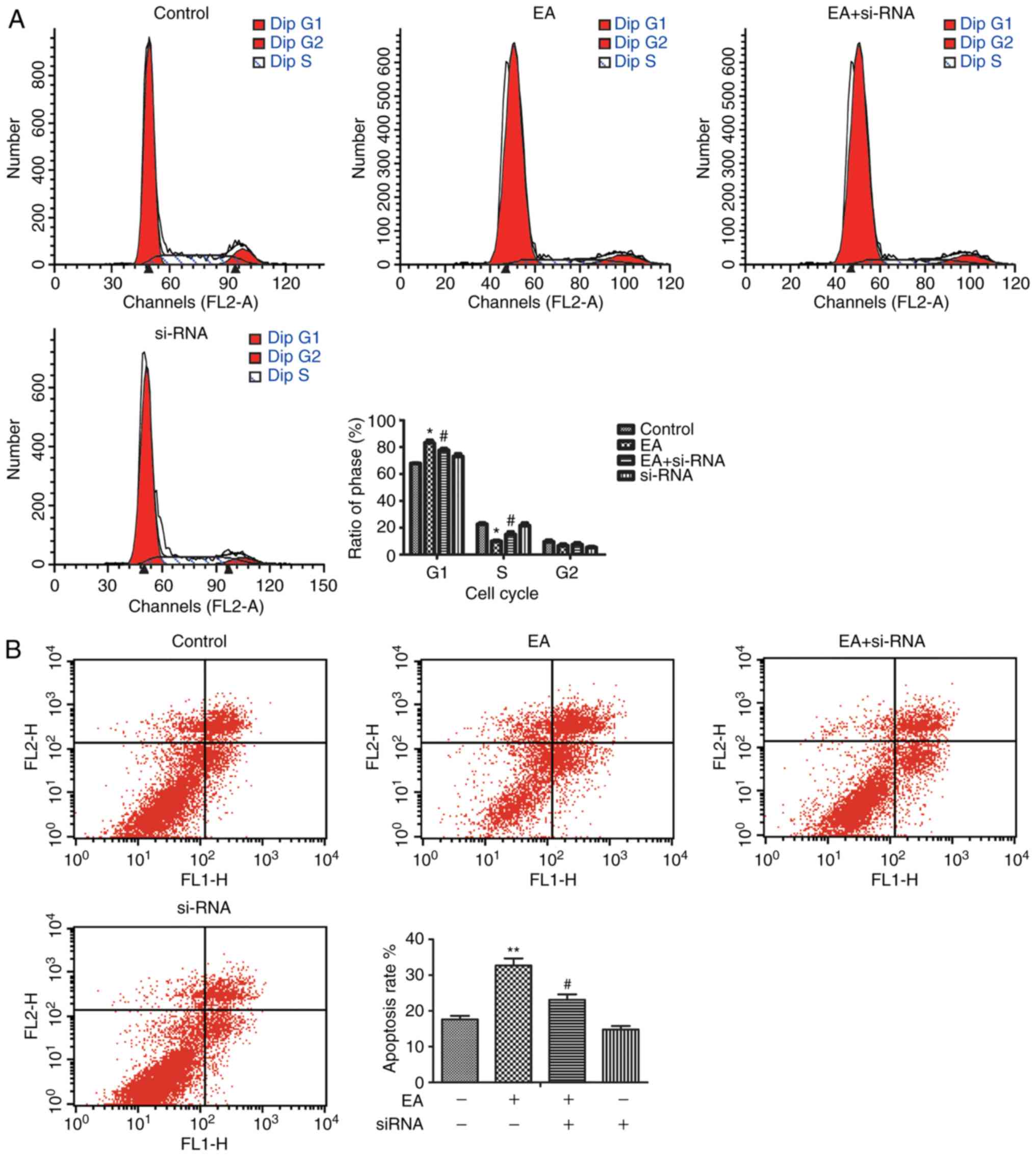
si-TGF-β1 were transfected into HCT-116 cells, to
determine the role of TGF-β1 in the EA-mediated effects on cell
behavior. The results showed that, si-TGF-β1 effectively reduced
TGF-β1 mRNA and protein expression levels. Additionally, si-TGF-β1
reduced the expression of Smad3 and P15 (Fig. 2). si-TGF-β1 abrogated the effects of
EA on cell cycle arrest and apoptosis (Fig. 3). Thus, it was demonstrated that
TGF-β1 was a crucial factor involved in the EA-mediated effects on
HCT-116 cells.
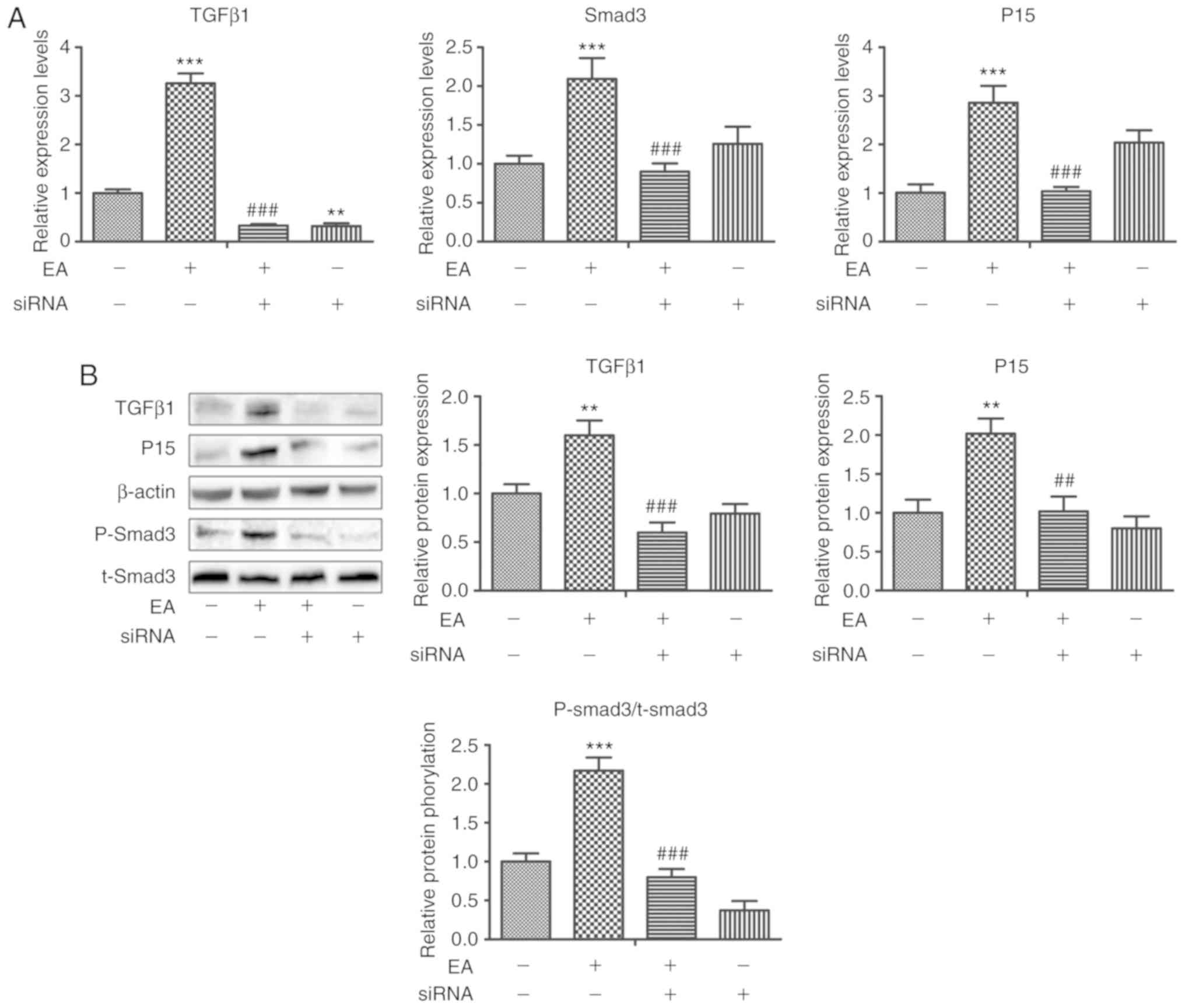 | Figure 2.Changes in the expression of genes
and proteins associated with TGF-β/Smad signaling pathways in
HCT-116 cells treated with EA and with TGF-β1 expression knocked
down. (A) Relative mRNA expression levels of TGF-β1, Smad3 and p15
in HCT-116 cells treated with DMSO+scrambleRNA, EA+scrambleRNA,
EA+TGF-β1 siRNA, or DMSO+TGF-β1 siRNA for 24 h. **P<0.01,
***P<0.001 vs. DMSO+scramble; ###P<0.001 vs.
EA+scrambleRNA (B) Western blotting showed that TGF-β1, Smad3, and
p15 expression patterns were altered in HCT-116 cells treated with
DMSO+scrambleRNA, EA+scrambleRNA, EA+TGF-β1 siRNA, or DMSO+TGF-β1
siRNA for 24 h. **P<0.01, vs. ***P<0.001 vs. DMSO+scramble;
##P<0.01, ###P<0.001 vs.
EA+scrambleRNA. EA, ellagic acid; si, small interfering; p-,
phospho; t-, total. |
Smad3 phosphorylation is involved in
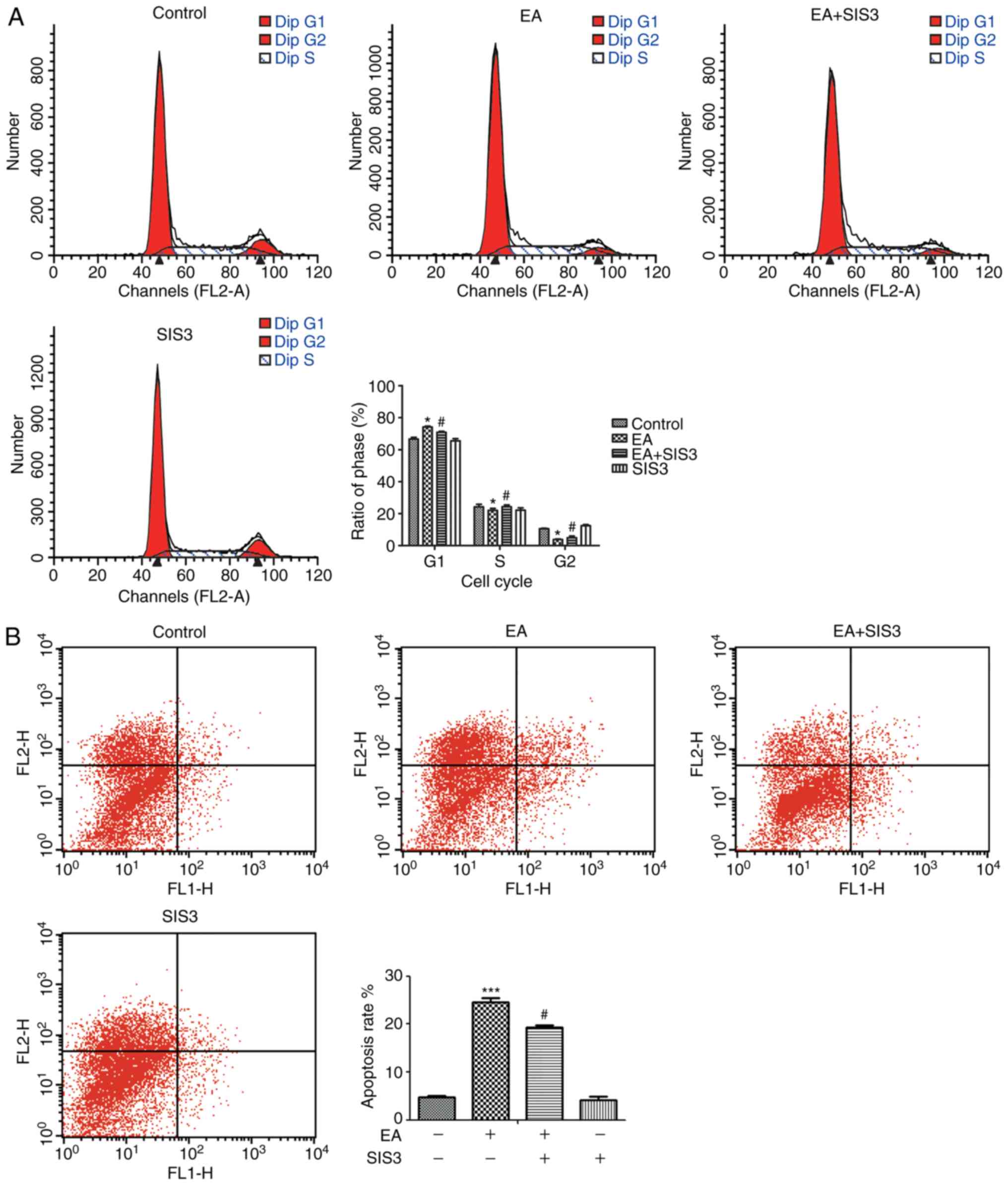
EA-mediated regulation of cell cycle arrest and apoptosis
The specific inhibitor SIS3 was used to treat CRC
cells, which had been pretreated with EA (100 µM) for 6 h. After 24
h of treatment, SIS3 effectively reduced the mRNA and protein
expression levels of P15 and the levels of phospho-Smad3 (Fig. 4). In addition, the regulatory
function of EA on the cell cycle and apoptosis were reduced
(Fig. 5). Therefore, Smad3 may be
crucial in the EA-mediated effects on HCT-116 cells.
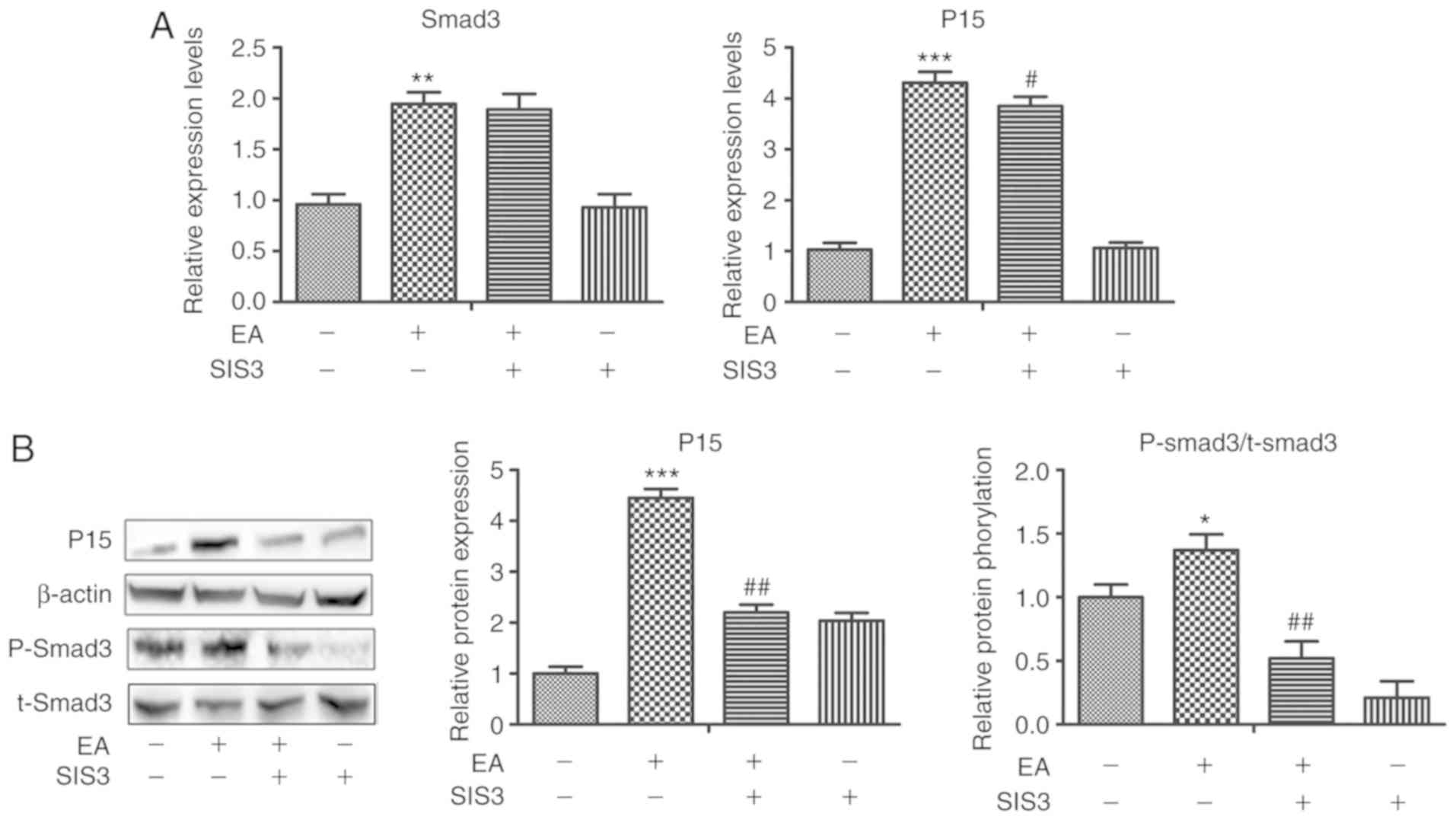 | Figure 4.Changes in the expression of genes
and proteins associated with the TGF-β/Smad signaling pathway in
HCT-116 cells treated with DMSO+scrambleRNA, EA+scrambleRNA,
EA+SIS3 or DMSO+SIS3 for 24 h. (A) Relative mRNA expression levels
of TGF-β1, Smad3 and p15 in HCT-116 cells treated with
DMSO+scrambleRNA, EA+scrambleRNA, EA+SIS3 or DMSO+SIS3 for 24 h.
**P<0.01, ***P<0.001 vs. DMSO+scramble; #P<0.05
vs. EA+scrambleRNA. (B) Altered protein expression levels
associated with the TGF-β/Smad signaling pathway in HCT-116 cells
treated with DMSO+scrambleRNA, EA+scrambleRNA, EA+SIS3 or DMSO+SIS3
treatment for 24 h. SIS3 rescued the levels of p15 in cells
co-treated with EA and SIS3 compared with cells treated with EA
alone. Error bars represent the standard error of the means based
on three independent experiments. *P<0.05, **P<0.001 vs.
DMSO+scramble; ##P<0.01 vs. EA+scrambleRNA. EA,
ellagic acid; si, small interfering; p-, phospho; t-, total. |
Discussion
CRC is a serious malignant disease, ranking fifth
and second among the most common causes of cancer-associated death
in China (13) and western
countries (14), respectively.
Although effective therapeutic strategies have been developed over
the previous decades, the 5-year overall survival of patients with
CRC has remained unsatisfactory, owing to limitations of currently
available prognostic factors (such as, vascular and neural
invasion, a low lymphocyte-to-monocyte ratio and tumor stage
III/IV) (3). Chemo-prevention is an
effective method for the inhibition of cancer cell growth. However,
our understanding regarding CRC pathogenesis remains limited. Thus,
it is crucial to further determine the molecular mechanisms
underlying CRC, with the aim of facilitating the discovery of novel
therapeutic targets.
EA may be a potential chemo-preventive agent, which
has been shown to inhibit proliferation in various types of cancer
(15). Based on in vitro and
in vivo experiments, EA was shown to significantly delay
progression of CRC, suggesting that it may serve an anti-tumor role
in CRC (8,16,17).
However, the relevant molecular pathways underlying the cellular
response to EA, especially those associated with transcriptional
regulation and protein production, have not been determined.
Therefore, it is clinically significant to determine the molecular
mechanisms and targets of EA for the inhibition of growth in
HCT-116 CRC cells. Although several studies have hypothesized the
involvement of various pathways in this process, few of these have
investigated these pathways further (7,8,16).
Microarray profiling was used to investigate the
molecular mechanisms underlying EA-induced inhibition for HCT-116
CRC cell proliferation. In our previous study, it was shown that EA
reduced cancer cell proliferation. Furthermore, the cDNA microarray
analysis showed that, after 72-h EA treatment, a total of 4,738
genes exhibited a >1.2-fold change in their expression levels
(10). Based on the present study,
EA was shown to arrest the HCT-116 cell cycle in the G0/G1 phase,
thus resulting in apoptosis. Furthermore, among the DEGs identified
in the cDNA microarray analysis, the changes in TGF-β1, Smad3,
E2F4, E2F5 and p15, involved in the TGF-β1/Smad3 pathway, were
verified using RT-qPCR. Treatment with si-TGF-β1 and a Smad3
inhibitor were used to assess the function of TGF-β1 and Smad3,
respectively, the corresponding regulatory functions of EA were
abrogated in HCT-116 cells, and the expression patterns of
downstream DEGs in TGF-β1/Smad3 pathway, including the
cyclin-dependent kinase inhibitor 2B (CDKN2B) (also known as p15),
were altered.
TGF-β is a family of multifunctional polypeptides
that promotes differentiation and inhibits growth and proliferation
in most epithelial cell types in vitro as well as in
vivo (18). TGF-β1 is one of
the most predominant cytokines which participates in various
biological pathways in critical physiological functions (19). Following hetero-oligomerization of
the type I and type II transmembrane TGF-β receptors, signals are
transmitted by TGF-β from the cellular membrane to the nuclear
targets via a signaling cascade involving Smad proteins (20). Smad3, one mediator involved in the
signaling cascade, is critically responsible for the transduction
of TGF-β signals to its nuclear targets, thereby inhibiting
cellular growth. Once a TGF-β signal is activated, the
carboxyl-terminal serine amino acids of two important downstream
targets, Smad2 and Smad3, are phosphorylated allowing them to bind
to Smad4 to form heteromeric complexes. Smad2/3/4 complexes are
translocated to the nucleus to regulate transcription of target
genes, such as CDKN2B (21).
TGF-β induces G1 cell cycle arrest via its
regulatory function on CDKN2B (21). TGF-β also induces apoptosis in
several types of cancer cells through multiple mechanisms (21,22).
Based on in vitro experiments using breast cancer cells, our
previous study showed that EA induced cell cycle arrest
predominantly via modulation of the TGF-β/Smad signaling pathway
(11). Although the role of EA on
regulating TGF-β/Smad3 pathway has been demonstrated in several
types of tumor (9,11,15,23),
to the best of our knowledge, the present study is the first to
report the anti-tumor role of EA by regulating TGF-β1/Smad3
signaling pathway in CRC.
CDKN2B regulates cell growth control during the G1
phase (24). The cyclin-dependent
kinase inhibitor encoded by CDKN2B targets CDK4 or CDK6, binding
with them, to prevent CDK activation. Therefore, the protein
product of CDKN2B is a regulator of cell growth, specifically
controlling cell cycle progression to the G1 phase (25). CDKN2B expression was detected in
multiple breast cancer cell lines and epithelial cells of normal
breast tissue (26). Furthermore,
it was shown to be associated with cell aging and is hypothesized
to be a tumor suppressor gene (27). In an in vivo melanoma model,
loss of p15 promoted the transition from benign nevus to melanoma,
demonstrating its importance in this process (28).
Binding of CDK inhibitors (p15, p21 and p27) to the
corresponding cyclin/CDK complexes results in the inactivation of
cyclin/CDK complexes, and thus the subsequent restriction of cell
growth (29). In HeLa cells, the
growth-inhibitory effect of
1-(2-hydroxy-5-methylphenyl)-3-phenyl-1, 3-propanedione may be
induced by blocking the G1/S transition, through upregulation of
these CDK inhibitors (30). In our
previous study, EA was shown to upregulate TGF-β1 and Smad3
expression levels and promote the phosphorylation of Smad3
(23). phospho-Smad3 bound to Smad4
in the nucleus, thereby regulating the expression of the p15 target
gene (31,32). In the present study, EA inhibited
HCT-116 cell proliferation, to a certain extent, via the
TGF-β1/Smad3 pathway.
Based on the results of the present study, EA caused
G0/G1 cell cycle arrest in HCT-116 cells, thereby inducing
apoptosis in vitro. EA was predicted to regulate
TGF-β1/Smad3 signaling based on microarray profiling analysis.
Furthermore, it was shown that EA regulated TGF-β1/Smad3/CDKN2B
signaling via phosphorylation of Smad3, resulting in increased
transcriptional activity of CDKN2B. These observations show the
relationship between TGF-β1/Smad3 signaling and EA treatment which
resulted in reduced cancer cell growth. In the microarray analysis
the protein markers of the cell cycle, including Cyclin E, D and B,
and apoptosis markers, including caspase-3, were not differentially
expressed genes in our previous study (10). Collectively, the present data
demonstrated that EA inhibited CRC cell cycle progression via
upregulation of CDKN2B, a cell cycle inhibitor.
There are some limitations in the present study.
First, only a single cell line was used for in vitro
experiments, and in vivo experiments were not performed.
Thus, additional studies using different CRC cell lines and in
vivo models of CRC are required to verify the role and
mechanism of EA further. Secondly, in-depth mechanisms underlying
the properties of EA were not explored. Thirdly, the results of
present study have not been verified in clinical specimen. Finally,
TGF-β1/Smad3 signaling is known to be associated with epithelial to
mesenchymal transition. Whether EA affects epithelial to
mesenchymal transition in colon cancer cells will be explored in
future studies.
In conclusion, the present study provides
preliminary evidence showing the anti-growth function of EA in CRC
cells. The results suggested that EA treatment may alter
TGF-β1/Smad3 signaling pathway and thus upregulate the cell cycle
inhibitor, CDKN2B. Therefore, the present study highlights the
therapeutic potential of EA for treatment of CRC. However, further
research is required to develop a suitable clinical approach for
use of EA in the treatment of cancer.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (gran no. 81372612), Excellent Youth
Project of the Fourth Affiliated Hospital of Harbin Medical
University (grant no. HYDSYYXQN202006), The Youth Project of
Science and Technology Innovation Project of Heilongjiang Academy
of Traditional Chinese Medicine (grant no. ZHY19-080), Outstanding
Youth Training Foundation of Academician Yu-Weihan in Harbin
Medical University, and the Science Foundation for Key Project of
the Fourth Affiliated Hospital of Harbin Medical University (grant
no. HYDSYJQ201602).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
ML, GL and JZ conceived and designed the study. JW,
SD and XY analyzed the microarray data. JZ, LD, ZZ, HZ and CS
performed the experiments. JZ, GL and HC wrote the article. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jermal A, Siegel R, Ward E, Murray T, Xu J
and Thun MJ: Cancer statistics, 2007. CA Cancer J Clin. 57:43–66.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wu Q, Hu T, Zheng E, Deng X and Wang Z:
Prognostic role of the lymphocyte-to-monocyte ratio in colorectal
cancer: An up-to-date meta-analysis. Medicine (Baltimore).
96:e70512017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cragg GM and Newman DJ: Plants as a source
of anti-cancer agents. J Ethnopharmacol. 100:72–79. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Whitley AC, Stoner GD, Darby MV and Walle
T: Intestinal epithelial cell accumulation of the cancer preventive
polyphenol Ellagic acid-extensive binding to protein and DNA.
Biochem. Pharmacol. 66:907–915. 2003.
|
|
6
|
Aiyer HS, Vadhanam MV, Stoyanova R, Caprio
GD, Clapper ML and Gupta RC: Dietary berries and Ellagic acid
prevent oxidative DNA damage and modulate expression of DNA repair
genes. Int J Mol Sci. 9:327–341. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cho H, Jung H, Lee H, Yi HC, Kwak HK and
Hwang KT: Chemopreventive activity of ellagitannins and their
derivatives from black raspberry seeds on HT-29 colon cancer cells.
Food Funct. 6:28612015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mertens-Talcott SU, Lee JH, Percival SS
and Talcott ST: Induction of cell death in Caco-2 human colon
carcinoma cells by Ellagic acid rich fractions from muscadine
grapes. J Agric Food Chem. 54:5336–5343. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li LW, Na C, Tian SY, Chen J, Ma R, Gao Y
and Lou G: Ellagic acid induces HeLa cell apoptosis via regulating
signal transducer and activator of transcription 3 signaling. Exp
Ther Med. 16:29–36. 2018.PubMed/NCBI
|
|
10
|
Zhao JL, Li GD, Bo WL, Zhou YH, Dang SW,
Wei JF, Li XL and Liu M: Multiple effects of Ellagic acid on human
colorectal carcinoma cells identified by gene expression profile
analysis. Int J oncol. 50:613–621. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen HS, Bai MH, Zhang T, Li GD and Liu M:
Ellagic acid induces cell cycle arrest and apoptosis through
TGF-β/Smad3 signaling pathway in human breast cancer MCF-7 cells.
Int J Oncol. 46:1730–1738. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sung JJ, Lau JY, Young GP, Sano Y, Chiu
HM, Byeon JS, Yeoh KG, Goh KL, Sollano J, Rerknimitr R, et al: Asia
Pacific consensus recommendations for colorectal cancer screening.
Gut. 57:1166–1176. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ferlay J, Colombet M, Soerjomataram I,
Dyba T, Randi G, Bettio M, Gavin A, Visser O and Bray F: Cancer
incidence and mortality patterns in Europe: Estimates for 40
countries and 25 major cancers in 2018. Eur. J Cancer. 103:356–387.
2018.
|
|
15
|
Ceci C, Lacal PM, Tentori L, De Martino
MG, Miano R and Graziani G: Experimental Evidence of the antitumor,
antimetastatic and antiangiogenic activity of Ellagic acid.
Nutrients. 10(pii): E17562018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Umesalma S and Sudhandiran G: Differential
inhibitory effects of the polyphenol Ellagic acid on inflammatory
mediators NF-kappaB, iNOS, COX-2, TNF-alpha, and IL-6 in
1,2-dimethylhydrazine-induced rat colon carcinogenesis. Basic Clin
Pharmacol Toxicol. 107:650–655. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kong X, Ding X and Yang Q: Identification
of multi-target effects of Huaier aqueous extract via microarray
profling in triple-negative breast cancer cells. Int J Oncol.
46:2047–2056. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Heldin CH, Miyazono K and ten Dijke P:
TGF-beta signaling from cell membrane to nucleus through SMAD
proteins. Nature. 390:465–471. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Blobe GC, Schiemann WP and Lodish HF: Role
of transforming growth factor beta in human disease. N Engl J Med.
342:1350–1358. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Siegel PM and Massagué J: Cytostatic and
apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev
Cancer. 3:807–821. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pardali K and Moustakas A: Actions of
TGF-beta as tumor suppressor and pro-metastatic factor in human
cancer. Biochim Biophys Acta. 1775:21–62. 2007.PubMed/NCBI
|
|
22
|
Ikushima H and Miyazono K: TGFbeta
signaling: A complex web in cancer progression. Nat Rev Cancer.
10:415–424. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang T, Chen HS, Wang LF, Bai MH, Wang
YC, Jiang XF and Liu M: Ellagic acid exerts anti-proliferation
effects via modulation of TGF-β/Smad3 signaling in MCF-7 breast
cancer cells. Asian Pac J Cancer Prev. 15:273–276. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou X, Suzuki H, Shimada Y, Imamura M,
Yin J, Jiang HY, Tarmin L, Abraham JM and Meltzer S: Genomic DNA
and messenger RNA expression alterations of the CDKN2B and CDKN2
genes in esophageal squamous carcinoma cell lines. Genes
Chromosomes Cancer. 13:285–290. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hannon G and Beach D: p15INK4B is a
potential effector of TGF-beta-induced cell cycle arrest. Nature.
371:257–261. 1994. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Musgrove EA, Lilischkis R, Cornish AL, Lee
CS, Setlur V, Seshadri R and Sutherland RL: Expression of the
Cyclin-dependent kinase inhibitors p16INK4, p15INK4B and
p21WAF1/CIP1 in human breast cancer. Int J Cancer. 63:584–591.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Erickson S, Sangfelt O, Heyman M, Castro
J, Einhorn S and Grandér D: Involvement of the Ink4 proteins p16
and p15 in T-lymphocyte senescence. Oncogene. 17:595–602. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McNeal AS, Liu K, Nakhate V, Natale CA,
Duperret EK, Capell BC, Dentchev T, Berger SL, Herlyn M, Seykora JT
and Ridky TW: CDKN2B loss promotes progression from benign
melanocytic nevus to melanoma. Cancer Discov. 5:1072–1085. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Malumbres M and Barbacid M: Mammalian
cyclin-dependent kinases. Trends Biochem. Sci. 30:630–641.
2005.
|
|
30
|
Tsai JH, Hsu LS, Huang HC, Lin CL, Pan MH,
Hong HM and Chen WJ: 1-(2-Hydroxy-5-methylphenyl)-3-phenyl-1,
3-propanedione Induces G1 cell cycle arrest and autophagy in HeLa
cervical cancer cells. Int J Mol Sci. 17(pii): E12742016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Derynck R, Zhang Y and Feng XH: Smads:
Transcriptional activators of TGF-beta responses. Cell. 95:737–740.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Miyazono K, ten Dijke P and Heldin CH:
TGF-beta signaling by Smad proteins. Adv Immunol. 75:115–157. 2000.
View Article : Google Scholar : PubMed/NCBI
|