Introduction
Prostate cancer (PCa) is the most common cancer in
men and the second leading cause of cancer-related mortality in men
worldwide (1). Because of the
limited treatment options available for PCa, patients may
experience disease relapse and are often treated with anti-androgen
therapy (2). However, advanced PCa
results from the emergence of androgen-independent PCa and
eventually must be treated with chemotherapy. Although
docetaxel-based therapy is mostly used in advanced PCa, patients
show low survival rates when taking docetaxel, limiting the
treatment options available for advanced PCa (3). Therefore, targeting PCa independently
with chemotherapy is crucial. p53 is a transcription factor known
as the guardian of the genome because it is involved in the
transcription of regulatory genes that control cell apoptosis, cell
cycle arrest, and DNA repair (4).
p53 causes cell-cycle arrest by activating the cyclin-dependent
kinase inhibitor p21 (4). In
addition, p53 induces apoptosis by activating the mitochondrial
pro-apoptotic proteins p53 upregulated modulator of apoptosis
(PUMA) [also known as Bcl-2-binding component 3 (BBC3)] and
Phorbol-12-myristate-13-acetate-induced protein 1 (NOXA) (5). p53 undergoes post-translation
modification (PTM) at several sites in response to DNA damage and
oncogene activation in a cell (4,6). The
p53 PTM enables it to be activated and stabilized after DNA damage
(4). It was previously shown that
phosphorylation and acetylation of p53 enhance the transcription of
its target genes (7–9). On the other hand, p53 also undergoes
ubiquitination and sumoylation, which are associated with the
nuclear export of p53 and with the inhibition of p53
transcriptional activities (10–12).
The TP53 gene is mutated in about 50% of cancers (13,14).
On the other hand, p53 still retains its wild-type status in about
50% of carcinomas, but its activity is diminished. Murine double
minute-2 (MDM2), an E3 ligase enzyme, is the major negative
regulator of p53 that is overexpressed in half of the cancers,
including PCa (15). MDM2 is also a
target gene for p53, helping to establish an autoregulatory
feedback loop in which p53 promotes the transcription of its
negative regulator (16). MDM2
regulates p53 by three main mechanisms, as follows: It inhibits p53
transcriptional activity, exports p53 from the nucleus, and
ubiquitinates p53 and degrades it in the proteasome (17,18).
Thus, MDM2 becomes a promising target for cancer therapy in cancers
that are harboring wild-type p53 (19). Although MDM2 is the main negative
regular of p53, it also regulates other proteins independently of
p53 (16). Among these proteins is
X-linked inhibitor of apoptosis protein (XIAP) protein that is
known for its inhibition to caspase 3 (18).
Epidemiological studies, including PCa cell lines
and in vivo xenograft models, suggest that the consumption
of a selected variety of fruits and vegetables rich in polyphenolic
compounds is effective against several types of cancer (20, 21).
The pomegranate, a widely consumed fruit, suppresses the growth of
several cancers, including PCa (22,23).
The effect of the pomegranate on cancers is attributed to its
polyphenolic compounds, particularly ellagic acid (EA) and
urolithin A (UA), which are potent antioxidants with anticancer
activity (24,25). Previous studies have shown that EA
causes PCa cell inhibition, cell cycle arrest, and apoptosis in
22RV1, DU-145, PC3, and LNCaP PCa cell lines (23,26–28).
In addition, EA has been shown to inhibit vascular endothelial
growth factor in LNCaP cells, which is responsible for cancer
angiogenesis (29). In addition to
the in vitro studies, some investigators have extended the
in vitro observation to in vivo. For example, using
immunodeficient murine models of PCa, the authors found EA reduced
angiogenesis and metastasis formation (30). However, these effects were not by EA
alone since EA was given in combination with other polyphenols,
luteolin, and punicic acid (30).
Another study showed that EA inhibited PCa carcinogenesis in
vivo (25). Thus, EA interferes
with multiple biological processes involved in PCa initiation,
angiogenesis, and metastasis (31).
Although these and other investigations have confirmed the
anticancer effect of EA on PCa, the mechanisms by which EA
influences the p53/MDM2 pathway in PCa remain incompletely
understood. We have recently demonstrated that EA's metabolite, UA,
increased p53 expression, and inhibited MDM2-mediated p53
polyubiquitination (32). The aim
of the present study was to investigate the influence of EA on the
p53/MDM2 signaling pathway in PCa cells. Here, we confirmed the
effect of the parent compound, EA, on the p53/MDM2 pathway by
downregulating MDM2 and upregulating p53. Our data suggest that EA
suppresses PCa progression partly via targeting the p53/MDM2
pathway.
Materials and methods
Cell cultures
Human PCa LNCaP, 22RV1, and PC3 cell lines were
obtained from the American Type Culture Collection (ATCC). Both
LNCaP and 22RV1 cell lines were grown using RPMI-1640 media (ATCC)
containing 10% fetal bovine serum (FBS), penicillin (100 mg/ml) and
streptomycin (100 mg/ml). PC3 cells were grown using F12 knight
media (ATCC) containing 10% FBS, penicillin (100 mg/ml) and
streptomycin (100 mg/ml). Mouse embryonic fibroblast (MEF) cells
possessing double knockouts of p53 and MDM2 (p53−/−
MDM2−/−) were obtained from Professor Guillermina Lozano
(MD Anderson Cancer Center, University of Texas, Austin, TX, USA).
Wild-type MEF cells were obtained from ATCC. Both wild-type MEF and
MEF (p53−/− MDM2−/−) were grown using DMEM
media (ATCC) containing 10% fetal bovine serum (FBS), penicillin
(100 mg/ml) and streptomycin (100 mg/ml). All cell lines were grown
in a 37°C incubator with 5% CO2 according to ATCC
protocols.
Reagents and antibodies
EA was purchased from Santa Cruz Biotechnology, Inc.
Anti-cleaved PARP (cat. no. 5625), anti-cleaved caspase-3 (cat. no.
9661), anti-phospho-MDM2-ser166 (cat. no. 3521), anti-p14ARF (cat.
no. 2407) anti-PUMA (cat. no. 12450), anti-NOXA (cat. no. 14766),
anti-phospho-p53-ser15 (cat. no. 9284), anti-phospho-p53-ser 20
(cat. no. 9287), anti-XIAP (2042T), anti-GAPDH (cat. no. 2118) and
anti-β-actin (cat. no. 3700) antibodies were purchased from Cell
Signaling Biotechnology, Inc. Anti-MDM2 antibody (cat. no. 556353)
was purchased from BD Biosciences. Anti-p53 (sc-126) and anti-p21
(sc-6246) antibodies were purchased from Santa Cruz Biotechnology,
Inc.
Cell viability assay
All cells were incubated in 96-well plates overnight
at a concentration of 120,000 cells/ml to allow them to adhere and
to reach 70% confluency before treatment. Cells were then treated
with EA at concentrations of 5, 10, 20, 40, 80 and 160 µM. DMSO was
used to dissolve EA and was used as a control (CTRL) at a final
concentration of 0.08%, while staurosporine (ST) was used as a
positive control. Cell viability was measured using the
CellTiter-Glo® luminescent assay (Promega Corp.) after
24 and 48 h of EA treatment according to the manufacturer's
protocol.
Immunoblotting (IB)
Immunoblotting was conducted as described previously
(33). Cells were cultured in
100-mm plates at a concentration of 120,000 cells/ml and incubated
for 24 h in a 37°C incubator. Following incubation, cells were
treated for 24 or 48 h with either EA (20, 40 and 80 µM) or vehicle
control prepared in serum-free medium. Protein lysates were
extracted using 1X cell lysis buffer containing 20 mM Tris-HCl (pH
7.5) 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton,
2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM
Na3VO4 and 1 µg/ml leupeptin (Cell Signaling
Technology, Inc.). The dishes were then scraped, and the lysate was
collected in a microcentrifuge tube and placed on ice for 30 min.
The lysate was then passed through a 21-gauge needle to break up
the cell aggregates. The cell lysate was centrifuged at 14,000 × g
for 10 min and was quantified by BCA reagent (Thermo Fisher
Scientific, Inc.). An amount 30 µg of protein lysate for each
sample was loaded equally onto SDS-PAGE for separation using
gradient (4-20%) gels (Bio-Rad Laboratories). The gel was then
transferred to a nitrocellulose membrane (Bio-Rad Laboratories)
using Trans-Blot Turbo Transfer System (Bio-Rad Laboratories) with
transfer buffer (containing 230 mM glycine, 25 mM Tris, 0.7 mM SDS,
20% methanol). The membrane was then blocked using Odyssey blocking
buffer (LI-COR Biosciences) for 1 h at room temperature to block
the nonspecific binding sites on the membrane. The membrane was
then incubated at 4°C overnight with blocking buffer containing p53
(1:1,000), p-p53-ser 15 (1:1,000), p-p53-ser20 (1:1,000), MDM2
(1:500), p-MDM2 ser166 (1:1,000), p21 (1:500), cleaved caspase 3
(1:1,000), cleaved PARP (1:1,000), p14ARF (1:500), NOXA (1:500),
PUMA (1:1,000), XIAP (1:1,000), GAPDH (1:1,000) and β-actin
(1:1,000). The membrane then was washed three times with
Tris-buffered saline with 0.1% Tween 20 (TBST), and then incubated
for 1 h at room temperature with a blocking buffer containing the
appropriate secondary antibody (1:15,000). Protein bands were
visualized using the LI-COR Odyssey CLx imaging system (LI-COR
Biosciences). The loading controls used for western blotting were
GAPDH and β-actin. The densitometry for each band was measured
using ImageJ 1.5k software (National Institutes of Health,
Bethesda). Each band was normalized to its corresponding loading
control as shown on the y-axis for each quantitative analysis of
the western blot.
Quantitative real-time polymerase
chain reaction (RT-qPCR)
PCa cells were treated with 40 and 80 µM of EA for
24 h for 22RV1 and LNCaP cell lines, respectively, and 20 µM of EA
for PC3 cells also for 24 h. Total RNA was extracted and purified
from the cell lines using the miRNeasy Mini kit (Qiagen) according
to the manufacturer's guidelines. The complementary DNA was
generated from the total RNA by using the iScript cDNA Synthesis
kit (Bio-Rad Laboratories). The quantitative real-time polymerase
chain reaction was performed with a real-time thermal cycler
(Bio-Rad Laboratories) using SsoAdvanced™ Universal
SYBR® Green Supermix (Bio-Rad Laboratories). RT-qPCR
reactions were conducted for 40 cycles. Each cycle included
denaturation (95°C for 39 sec), annealing (57°C for 30 sec), and
extension (60°C for 30 sec). Specific primers were used as follows:
For human P21 forward (CTGAGACTCTCAGGGTCGAA) and reverse
(CGGCGTTTGGAGTGGTAGAA); for human MDM2 forward
(TGGCGTGCCAAGCTTCTCTGT) and reverse (ACCTGAGTCCGATGATTCCTGCT); for
human GAPDH forward (CAGCCTCAAGATCATCAGCA) and reverse
(GTCTTCTGGGTGGCAGTGAT). The mRNA expressions of MDM2 and
P21 were calculated using the 2−ΔΔCq method
(34), with the GAPDH as an
internal control, and data were represented as fold change.
Statistical analysis
All data are representative of three or more
independent experiments. Data are presented as mean ± standard
error of the mean (SEM). Unpaired student's T-test was used to
compare two groups. One-way ANOVA, followed by Dunnett's multiple
comparison tests were performed to compare three groups. Data were
generated using GraphPad Prism v7.04 (GraphPad Software, Inc.).
P<0.05 was considered to indicate a statistically significant
difference.
Results
EA induces apoptosis in PCa cells
Different concentrations of EA were used for cell
viability assay ranging from 5 to 160 µM for 24 and 48 h. For 22RV1
and LNCaP cell lines, EA did not promote any significant decrease
in cell viability after 24 h of treatment compared with the vehicle
control (Fig. 1A). Separately, for
PC3 cells, EA significantly inhibited PC3 cell viability after 24
h, starting at 20 µM. On the other hand, PCa cell viability was
decreased in all cell lines after 48 h compared with the vehicle
control (Fig. 1A; lower panel). We
further confirmed the apoptotic effect at 24 h at 40 and 80 µM EA
in the LNCaP and 22RV1 cell lines (Fig.
1B). Although EA did not affect cell viability at 24 h, it
enhanced the expression of cleaved PARP in the LNCaP and 22RV1 cell
lines compared with the vehicle control, suggesting apoptosis at 24
h following treatment with 40 and 80 µM EA (Fig. 1B). For PC3 cells, cleaved caspase-3
was significantly increased after treatment with 20 µM of EA for 24
h when compared with the vehicle control (Fig. 1B; right panel). Since we aimed to
ascertain the effect of EA on MDM2 and p53 at the same
concentrations that caused apoptosis, we continued to use 40 and 80
µM for subsequent experiments.
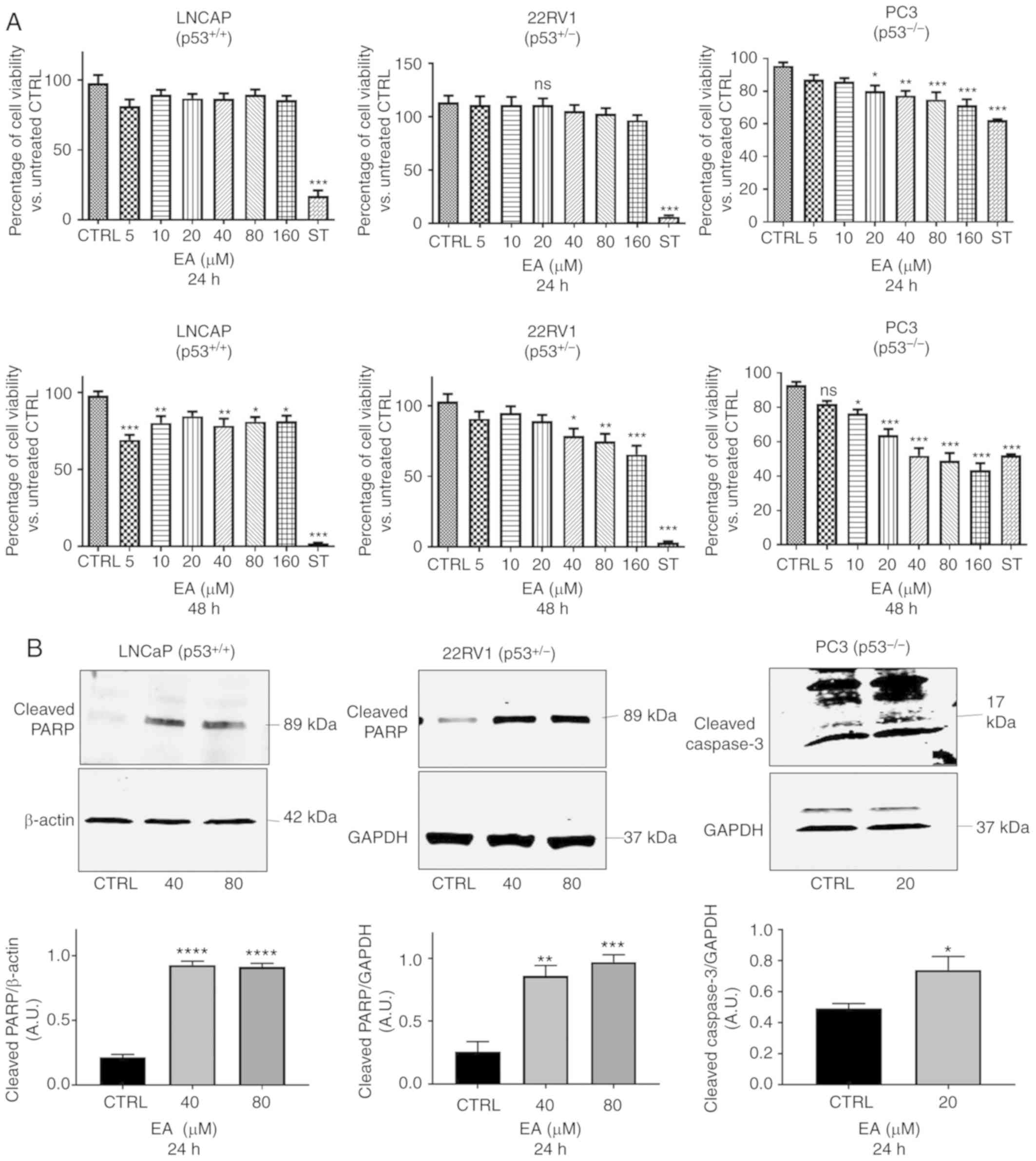 | Figure 1.(A) Cell viability assay for the
effect of EA on PCa cell lines. Cell Titer-Glo assays were
performed to determine the cell viability of LNCaP, 22RV1 and PC3
cells following treatment with EA at concentrations of 0–160 µM for
24 h (top panels) and 48 h (lower panels). The percentages of
viable cells were determined relevant to untreated cells as
compared to vehicle control (CTRL). One-way ANOVA of three
measurements in triplicate (mean ± SEM) was performed. *P<0.05,
**P<0.01, ***P<0.001 significant results of different
concentrations. of EA vs. CTRL (vehicle control); ns, not
significant; ST, staurosporine. (B) Representative western blotting
of cleaved PARP protein expression in PCa LNCaP and 22RV1 cells
with their corresponding quantitative analyses. EA significantly
increased the protein expression of cleaved PARP in LNCaP (40 and
80 µM, P≤0.0001) and in 22RV1 cells (40 µM, P=0.023; 80 µM,
P=0.0009). Also, EA significantly increased the protein expression
of cleaved caspase 3 in PC3 cells (20 µM, P=0.0476). Results are
expressed as arbitrary units (A.U.) and represent the means ± SEM
of 3 experiments *P<0.05, **P<0.01,
***P<0.001,****P<0.0001 significant results of 40 and 80 µM
EA vs. CTRL (vehicle control). PCa, prostate cancer; EA, ellagic
acid; PARP, poly(ADP-ribose) polymerase. |
EA increases p53 protein expression
and enhances the expression of p53 target proteins
Following the results of apoptosis, we sought to
investigate the effect of EA on p53 expression. The p53 protein
level was significantly increased in 22RV1 and LNCaP with 40 and 80
µM of EA at 24 h after treatment compared with the vehicle control
(Fig. 2A and B). Previous studies
have shown that many polyphenols increase p53 expression by
inducing DNA damage (35); thus we
investigated the effects of EA on p53 PTM, particularly
phosphorylation. We found that EA increased phosphorylated p53
(p-p53) at ser15 and ser20 in the case of 22RV1 (Fig. 2A). On the other hand, the increase
in phosphorylated p53 at ser15 was not significant in LNCaP
following treatment with EA at both concentrations used, and no
expression of phosphorylated p53 at ser20 was detected in LNCaP
cells (Fig. 2B).
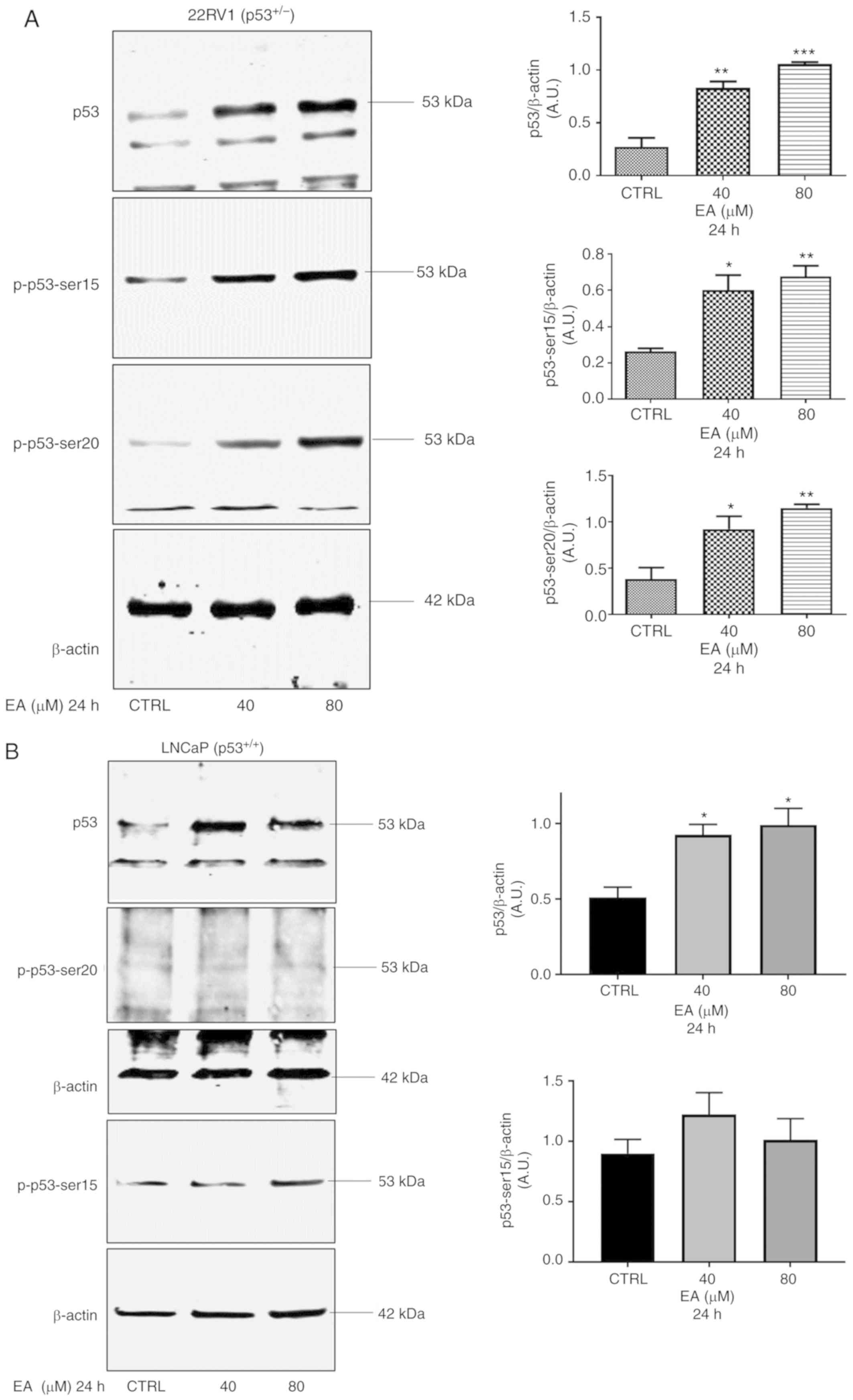 | Figure 2.Representative western blotting of
p53, p-p53-ser15, and p-p53-ser20 protein expression in PCa (A)
22RV1 and (B) LNCaP cells with their corresponding quantitative
analyses. (A) In 22RV1 cells, EA significantly increased the
protein expression of p53 (40 µM, P=0.0011; 80 µM P=0.0002), p-p53
ser 15 (40 µM, P=0.0122; 80 µM, P=0.0047) and p-p53-ser 20 (40 µM,
P=0.0231; 80 µM, P=0.0046). (B) In addition, EA significantly
increased the protein expression of p53 (40 µM, P=0.0164; 80 µM,
P=0.0110) in LNCaP cells. Results are expressed as arbitrary units
(A.U.) and represent the means ± SEM of 3 experiments. *P<0.05,
**P<0.01, ***P<0.001, significant result of 40 and 80 µM EA
vs. CTRL (vehicle control). PCa, prostate cancer; EA, ellagic acid;
p-, phosphorylated. |
Next, we investigated the effects of EA on p53 main
target proteins, MDM2, and p21. As expected, the protein level of
p21 was increased by 40 and 80 µM EA in both 22RV1 and LNCaP cell
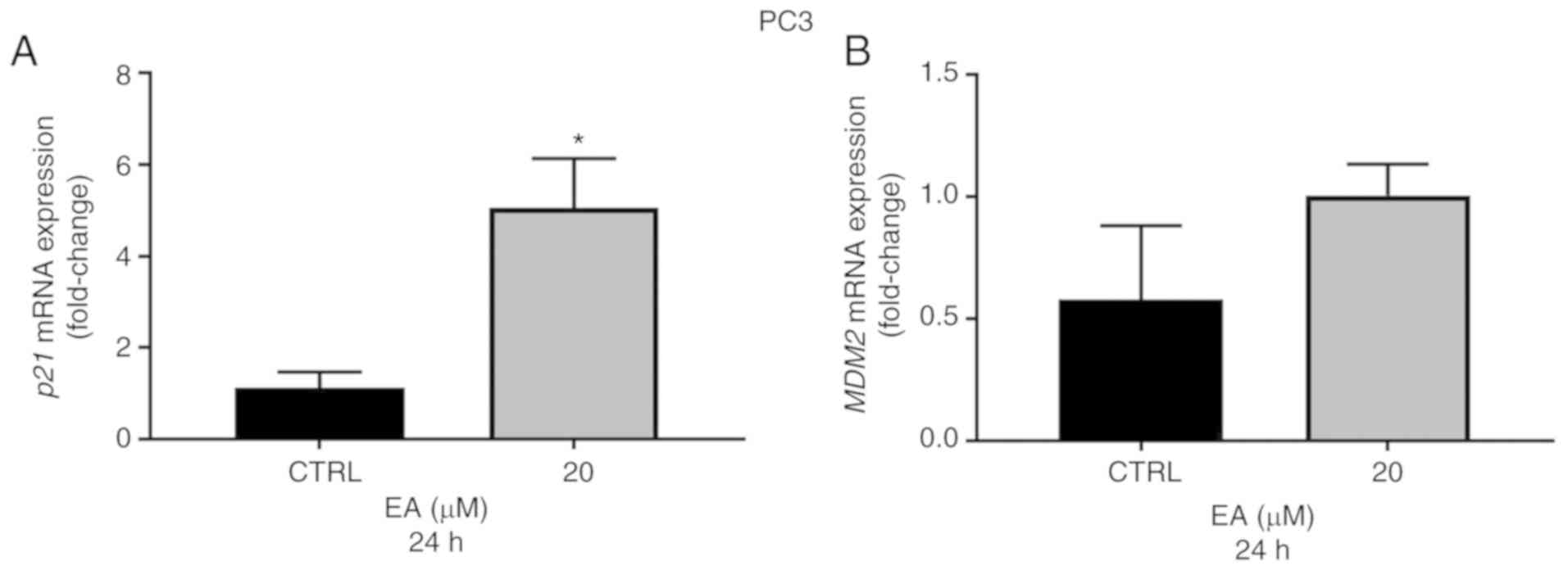
lines at 24 h compared with the vehicle control (Fig. 3A and C). Importantly, the p21 gene
and protein expressions were also significantly elevated in PC3
(p53−/−) cells following 20 µM EA at 24 h when compared
with the vehicle control (Figs. 3E
and 4A). Of note, although MDM2 is
also a target gene for p53, the protein expression of MDM2 was
significantly decreased in all cell lines examined in this study
when compared with the vehicle control (Fig. 3B, D and E). Moreover, EA at 40 and
80 µM downregulated phosphorylated MDM2 (p-MDM2) at ser166 in 22RV1
and LNCaP cells at 24 h when compared with the vehicle control
(Fig. 3B and D). To further
understand the nature of the MDM2 protein downregulation promoted
by EA, we investigated the effects of EA on MDM2 mRNA
expression. As expected, 40 and 80 µM EA caused the downregulation
of MDM2 gene expression in both 22RV1 and LNCaP cell lines
at 24 h when compared with the vehicle control (Fig. 4C and D) although the decrease was
not significant at 80 µM in 22RV1 cells. However, the gene
expression of MDM2 was not significantly changed in PC3
cells following treatment with 20 µM EA at 24 h when compared with
the vehicle control (Fig. 4B). To
further understand the MDM2 downregulation by EA, we also
investigated the effects of EA on the MDM2/p14ARF pathway in PCa
cells. Interestingly, p14ARF was markedly increased in 22RV1 and
PC3 cells (Fig. 3A and E) but not
in LNCaP cells (Fig. 3C).
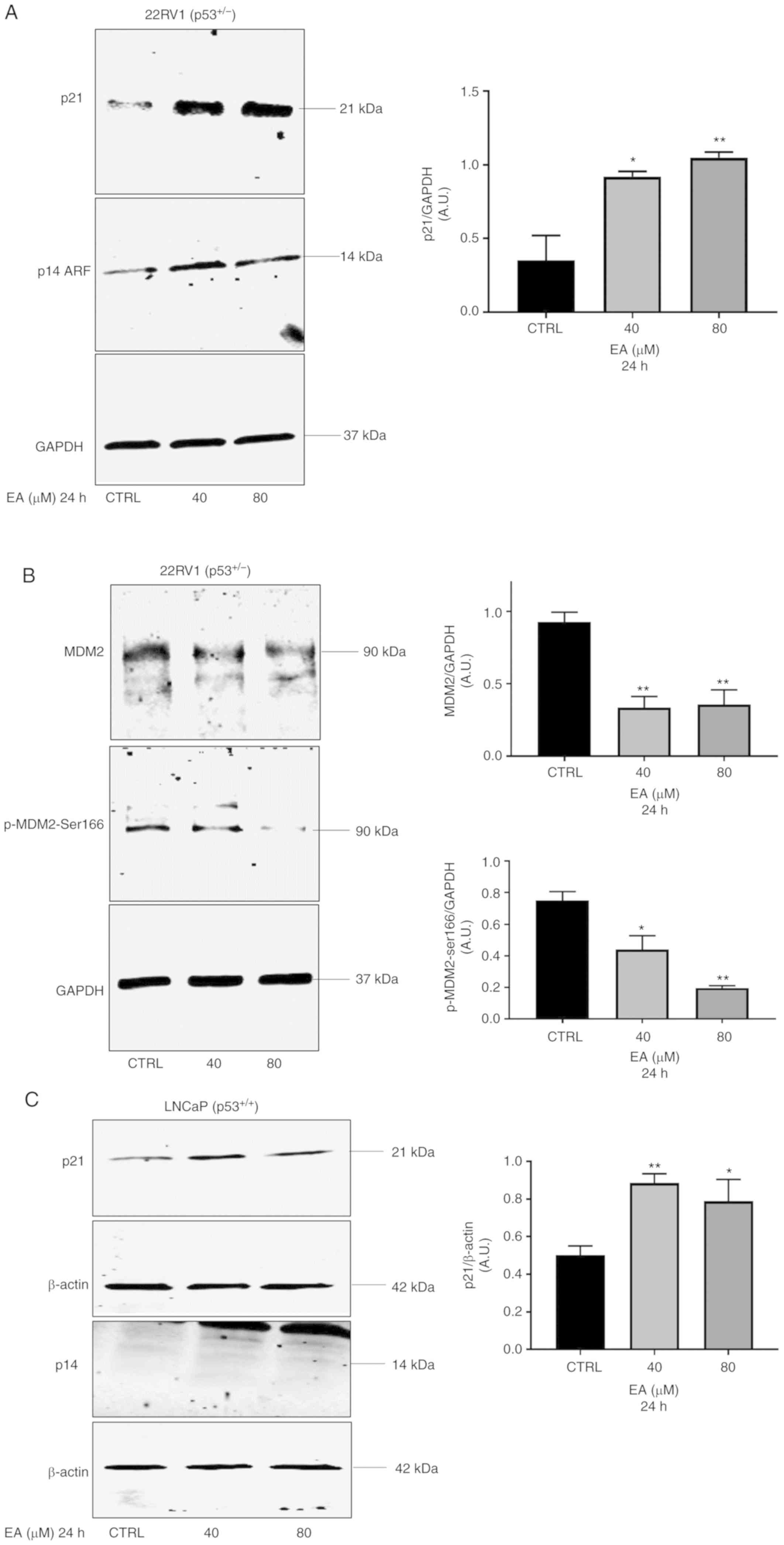 | Figure 3.(A-D) Representative western blotting
of MDM2, p-MDM2-ser166, p21, and p14ARF protein expression in PCa
22RV1, LNCaP, and PC3 cells with their corresponding quantitative
analyses. EA significantly increased the protein expression of p21
in (A) 22RV1 cells (40 µM, P=0.00137; 80 µM, P=0.0053), (C) LNCaP
cells (40 µM, P=0.0057; 80 µM, P=0.0363) and (E) PC3 cells (20 µM,
P=0.0025). (B) In 22RV1 cells, EA significantly decreased protein
expression of MDM2 (40 µM, P=0.044; 80 µM, P=0.0083), and p-MDM2 at
ser166 (40 µM, P=0.0212; 80 µM, P=0.0013). (D) In LNCaP cells, EA
significantly decreased MDM2 protein expression (40 µM, P=0.0282;
80 µM, P=0.0121), and p-MDM2 at ser166 (80 µM, P=0.0221). (E) In
PC3 cells, EA significantly decreased protein expression of MDM2
(20 µM, P=0.0131) and significantly increased protein expression of
p14ARF (20 µM, P=0.0328). Results are expressed as arbitrary units
(A.U.) and represent the means ± SEM of 3 experiments. *P<0.05,
**P<0.01, significant result of 20, 40 and 80 µM EA vs. CTRL
(vehicle control). PCa, prostate cancer; EA, ellagic acid; p-,
phosphorylated; MDM2, murine double minute-2. |
EA induces apoptosis in a
p53-dependent and -independent manner
p53 provokes apoptosis mainly through the induction
of the pro-apoptotic proteins PUMA and NOXA (36). We therefore examined the effects of
EA on expression levels of these proteins. As expected, EA
increased the levels of PUMA and NOXA in 22RV1 and LNCaP cells at
40 and 80 µM at 24 h when compared with the vehicle control
(Fig. 5A and B). To validate the
apoptotic effect in PC3 cells in the absence of p53, we separately
found that EA significantly downregulated XIAP protein at 20 µM at
24 h compared with the vehicle control, leading to increase cleaved
caspase-3 (Fig. 5C). These data
suggest that EA induces apoptosis in a p53-dependent and
-independent manner by downregulating MDM2.
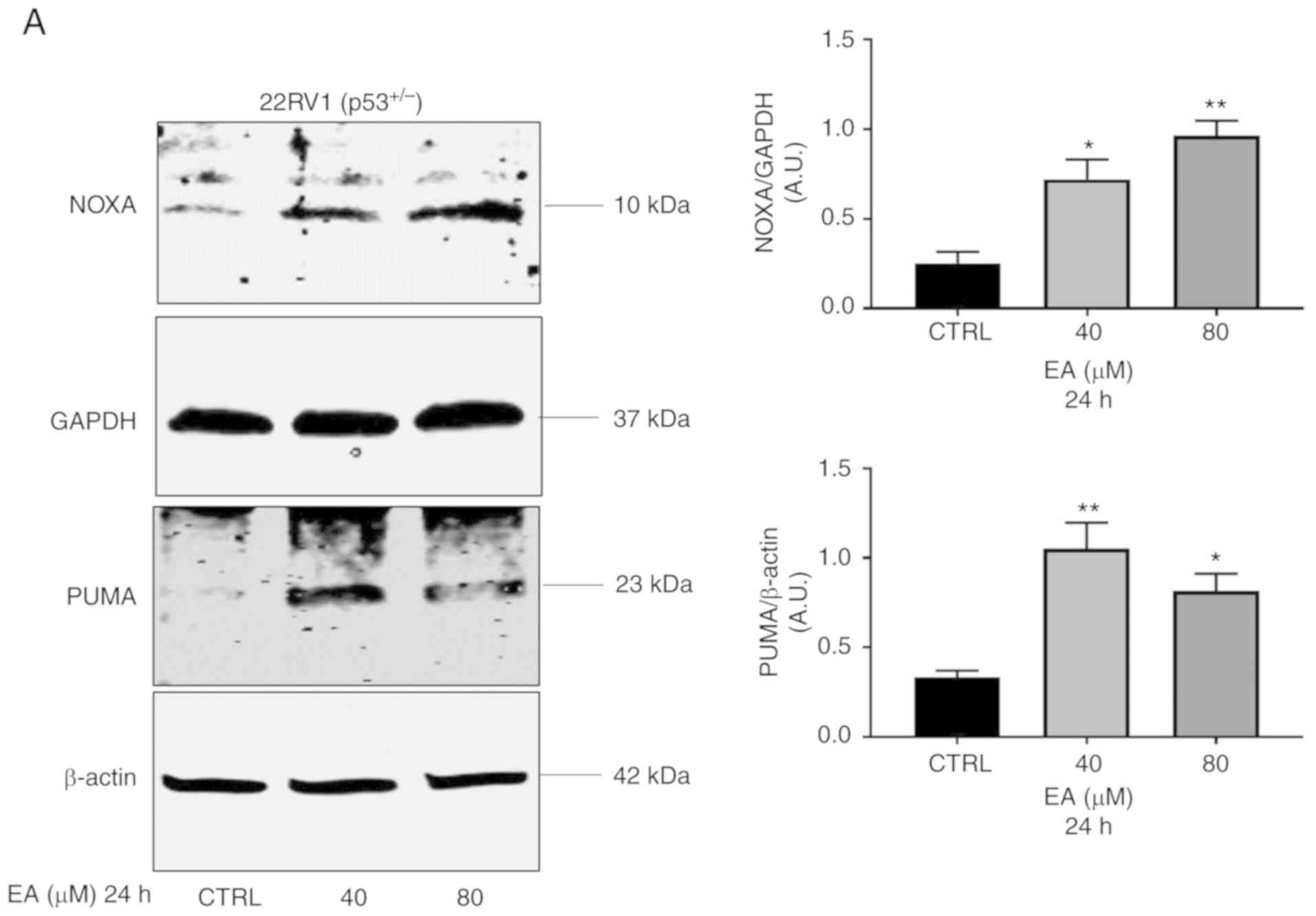 | Figure 5.Representative western blotting of
NOXA, and PUMA protein expression in PCa (A) 22RV1 and (B) LNCaP
cells with their corresponding quantitative analyses. (A) EA
significantly increased the protein expression of PUMA in 22RV1 (40
µM, P=0.0052; 80 µM, P=0.0173) and (B) LNCaP cells (80 µM,
P=0.0362). EA also increased the level of NOXA protein expression
in (A) 22RV1 (40 µM, P=0.0172; 80 µM, P=0.0028), and (B) LNCaP
cells (40 µM, P=0.0432). (C) Representative western blotting of
MDM2, XIAP and cleaved caspase 3 protein expression in PC3 cells.
EA significantly increased the protein expression of XIAP (20 µM,
P=0.0436). Results are expressed as arbitrary units (A.U.) and
represent the means ± SEM of 3 experiments. *P<0.05,
**P<0.01, significant result of 20, 40 and 80 µM EA vs. vs. CTRL
(vehicle control). PCa, prostate cancer; EA, ellagic acid; MDM2,
murine double minute-2; PUMA, p53 upregulated modulator of
apoptosis [also known as Bcl-2-binding component 3 (BBC3)]; NOXA,
Phorbol-12-myristate-13-acetate-induced protein 1; XIAP, X-linked
inhibitor of apoptosis protein. |
Effects of EA are independent of p53
and MDM2
The above results showed that EA induces PCa
suppression via targeting MDM2 and activating p53. To further
examine the effect of EA on the status of p53 and MDM2, we used MEF
cells that have double knockouts for both p53 and MDM2
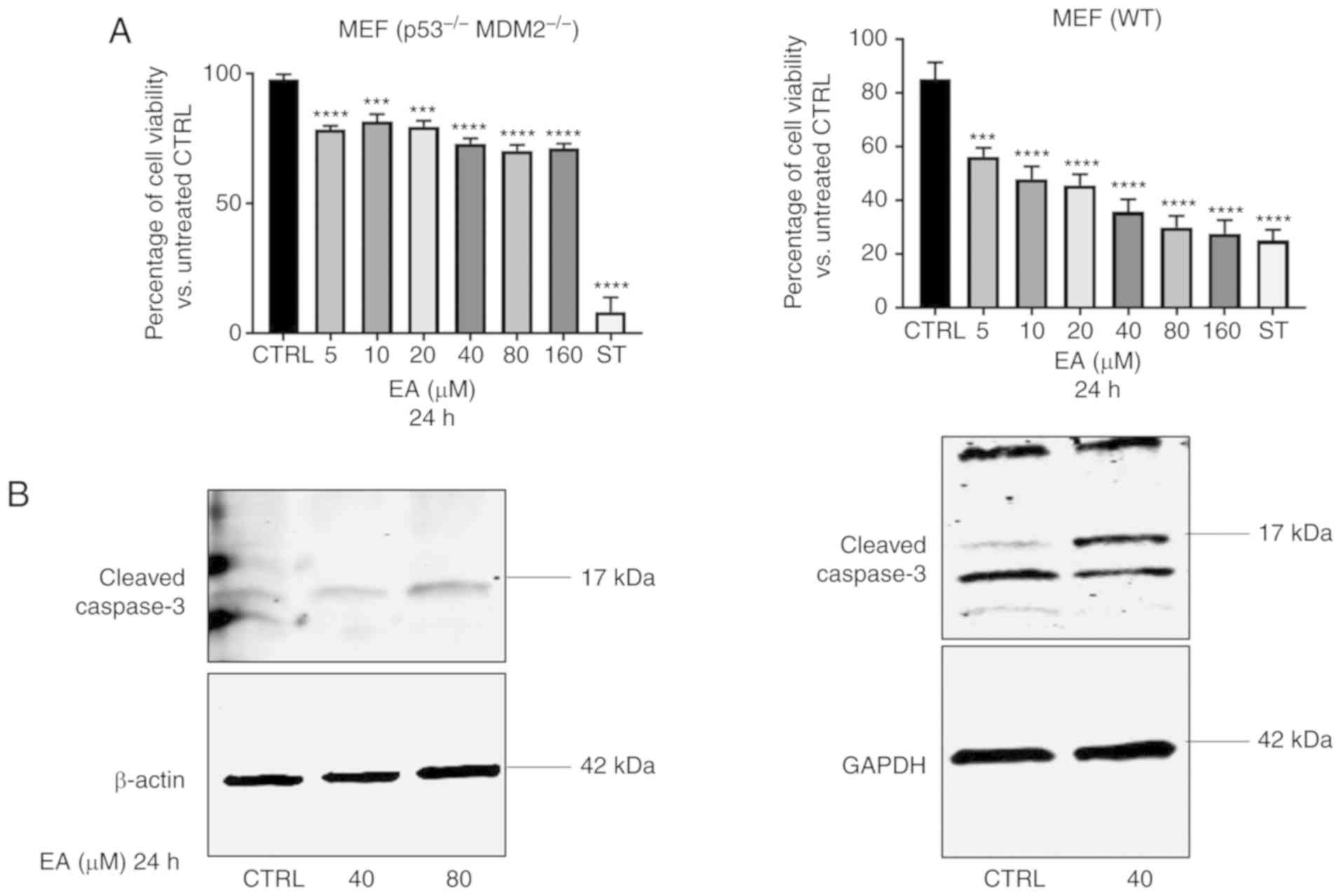
(p53−/− MDM2−/−). Cell viability assay showed
that EA did inhibit MEF cell viability at the concentration range
of from 5 to 160 µM at 24 h in both the WT and double knockout MEF
cells (Fig. 6A; upper panel).
Moreover, EA induced cleaved caspase-3 expression In both the WT
and double knockout MEF cells, suggesting apoptosis was induced
independently of p53 and MDM2 (Fig.
6B; lower panel).
Discussion
EA is a naturally occurring polyphenolic compound
that is derived from punicalagins (23). Although EA is known for its ability
to induce cytotoxic effects in several types of cancer, including
PCa (37), the influence of EA on
MDM2 and p53 in PCa is not yet fully understood. We confirmed that
EA induced apoptosis by increasing the expression of cleaved PARP
in LNCaP, 22RV1, and PC3 cell lines, although no significant effect
was found on the viability of PCa cells after 24 h. We assumed this
discrepancy between cell viability assay and apoptosis after 24 h
is that apoptosis is an early event in cell death, while cell
viability is reduced at the late stage of apoptosis. We further
confirmed this by analyzing the viability after 48 h, finding that
EA suppressed PCa cell viability. Moreover, we chose 40 and 80 µM
as these two concentrations caused a significant effect on
apoptosis after 24 h. Since we aimed to ascertain the effect of EA
on MDM2 and p53 at the same concentrations that caused apoptosis,
we used 40 and 80 µM for all other experiments. To note, EA reduced
the LNCaP cell viability at a low concentration. In a separate
experiment, we found that EA induced apoptosis at a low
concentration but without affecting the p53 and MDM2 at low
concentration (data not shown). We conclude that EA can induce
apoptosis at a low concentration in a pathway that is independent
of the p53-MDM2 pathway; such a pathway requires a high
concentration of EA to get stimulated. Vanella et al
(28) showed that EA induced LNCaP
cell death by targeting the mTOR pathway and lowering the
intracellular level of β-catenin. Moreover, their study also showed
that EA increased the expression of p21 in LNCaP cells (28).
In the present study, we demonstrated the influences
of EA on the p53/MDM2 pathway in PCa in vitro using three
models of PCa that each harbor a distinct TP53 gene. Our
western blot data indicated that EA increased the p53 protein
level. p53 activation is mainly regulated by phosphorylation and
acetylation, which are part of the post-translation modification
(PTM) of p53 (6). Phosphorylation
of p53 at ser15 occurred as a response to DNA damage that resulted
from a single- or double-strand break (38). Previous research has shown that
other polyphenols induce p53 expression by inducing DNA damage,
which triggers the ATM/CHK kinases that cause phosphorylation to
the p53 (35). Here, we found that
EA induced phosphorylation of p53 at ser15 in 22RV1 cells,
indicating a DNA damage mechanism induced by EA in 22RV1 cells.
However, p53-ser 15 was not significantly increased after EA
treatment in LNCaP cells, suggesting that EA does not produce DNA
damage effects in LNCaP as it does in 22RV1 cells. Prior research
showed that phosphorylation of p53 at ser20 weakens the interaction
between p53 and MDM2 (19). In our
study, we found EA only induces the phosphorylation of p53 at ser20
in 22RV1 cells, but not in LNCaP cells. Moreover, the increased
level of p53 protein achieved by EA was accompanied by increases in
p53′s main target protein, p21, in both 22RV1 and LNCaP cells,
suggesting cell-cycle arrest of PCa by EA in these cells. This
finding is in agreement with those previously reported by Vanella
et al (28,29). p53 is negatively regulated by MDM2,
which itself is a target gene of p53, forming an autoregulatory
feedback loop. We recently found that a metabolite of EA, urolithin
A (UA), increased p53 protein expression, and that this increase
was accompanied by increases in MDM2 gene and protein expression,
forming an autoregulatory feedback loop (32). Interestingly, although the level of
p53 was increased by EA, we found that EA did not increase MDM2
protein expression as a feedback loop but did downregulate it. We
speculated the MDM2 downregulation by EA may be occurring at the
transcriptional level or via other MDM2 regulators. p14ARF, another
tumor suppressor, is known to inhibit the p53–MDM2 interaction and
MDM2 ligase activity (39). A
previous study revealed that apigenin downregulated MDM2 by
increasing p14ARF in 22RV1 cells (40). In 22RV1 and PC3 cells treated with
EA, we found that p14ARF was increased, suggesting a possible
mechanism of MDM2 downregulation by EA. However, p14AFR was not
increased in LNCAP cells, indicating that EA may affect either the
gene expression of MDM2 or affect MDM2 at the protein level. Our
data showed that EA downregulated MDM2 mRNA in LNCaP and
22RV1 cells, further explaining the MDM2 protein downregulation. We
concluded that the downregulation of MDM2 by EA suggested that EA
inhibited the ligase activity of MDM2, preventing p53
ubiquitination and degradation. On the other hand, the MDM2
mRNA level was not altered in PC3 cells, suggesting that EA
downregulates MDM2 protein expression by inducing p14ARF or that EA
may affect MDM2 protein directly. Interestingly, the gene and
protein expressions of p21 were also increased in PC3 cells
independently of p53. These results are in agreement with those of
a previous study that confirmed p21 expression in the absence of
p53 (41). Moreover, MDM2 itself
can negatively regulate p21 in a p53-independent manner (42). Therefore, we speculate that the MDM2
downregulation by EA in PC3 cells may aid in the increased in
protein expression of p21. It is known that p53 produces apoptosis
when the p53 protein level reaches the apoptotic threshold
(43). Upon reaching the apoptotic
threshold, p53 induces the expression of two main pro-apoptotic
proteins, p53 upregulated modulator of apoptosis (PUMA) [also known
as Bcl-2-binding component 3 (BBC3)] and
Phorbol-12-myristate-13-acetate-induced protein 1 (NOXA). (36). PUMA and NOXA can bind the
mitochondrial antiapoptotic proteins Bcl-2, Bcl-X, and MCL-1
(36), producing an intrinsic
apoptotic effect. In the present study, the increased level of p53
by EA was accompanied by increases in PUMA and NOXA, suggesting
p53-dependent apoptosis by EA.
MDM2 is phosphorylated by AKT at ser166, enabling it
to enter the nucleus, where it binds and inhibits the
transcriptional activity of p53 (44). In the present study, we found that
EA downregulated phosphorylated MDM2 in LNCaP and 22RV1 cells at
ser166, restoring the activity of p53 and its target genes. Our
data on mouse embryonic fibroblasts (MEFs) (p53−/−,
MDM2−/−) suggest that the influence of EA on the
p53-MDM2 pathway may be partly contributed to the cytotoxic effect
of EA on PCa.
All these mechanisms of EA on the p53-MDM2 pathway
demonstrate its effectiveness in suppressing PCa through this
pathway in vitro. However, EA has low bioavailability after
consumption of pomegranate or other dietary sources in other fruits
(45). The poor bioavailability of
EA will eventually limit the metabolically active urolithins,
limiting the therapeutic effectiveness of these compounds after
consumption of pomegranate (46).
In the current study, we selected 40 and 80 µM
concentrations of EA and UA because they showed significant
apoptotic effect after a 24-h treatment and a significant effect on
p53 and its target genes and proteins in vitro. We
acknowledge that these concentrations are not bioavailable after
consumption of pomegranate. Therefore, the current research
suggests that EA can be useful to treat PCa if both are extracted
from pomegranate or other dietary sources. Furthermore, there are
recent efforts by researchers to improve the bioavailability of EA
(47,48), which will focus on improving EA drug
delivery to cancer sites, including PCa.
In conclusion, this study demonstrated the role of
the natural polyphenol EA in the suppression of PCa cell growth by
inhibiting the oncogene MDM2 and inducing p53 protein
expression and its target proteins. However, further experiments
are required to validate these data; for example, using luciferase
reporter assay for p53 before and after EA treatment in PCa cells
will be useful to validate the gene expression by p53. Moreover,
RNA-seq analysis will be useful to validate the alteration in gene
expression by p53. Additionally, it may be useful to observe the
effect of EA on AKT to validate the EA effect on the phosphorylated
MDM2 at ser-166. Furthermore, Annexin V apoptotic assay using flow
cytometry will be helpful in validating the apoptotic effect by EA.
A major problem with MDM2 inhibitors is that they produce p53
accumulation in normal cells, causing toxicity to normal cells.
Therefore, further research is needed to examine the concept of p53
accumulation in a normal prostate cell line provoked by EA.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Department of
Pharmacology and Toxicology at East Carolina University
(Greenville, NC, USA).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
YIMS and MS conceptualized the study. YIMS designed
the study and performed all the experiments and analyzed the data.
YIMS wrote the manuscript, and MS revised the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wirth MP, Hakenberg OW and Froehner M:
Antiandrogens in the treatment of prostate cancer. Eur Urol.
51:306–313, discussion 314. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Logan IR, McNeill HV, Cook S, Lu X, Lunec
J and Robson CN: Analysis of the MDM2 antagonist nutlin-3 in human
prostate cancer cells. Prostate. 67:900–906. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Toufektchan E and Toledo F: The guardian
of the genome revisited: p53 downregulates genes required for
telomere maintenance, DNA repair, and centromere structure. Cancers
(Basel). 10:1352018. View Article : Google Scholar
|
|
5
|
Brady CA and Attardi LD: p53 at a glance.
J Cell Sci. 123:2527–2532. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jin L, Li C, Xu Y, Wang L, Liu J, Wang D,
Hong C, Jiang Z, Ma Y, Chen Q and Yu F: Epigallocatechin gallate
promotes p53 accumulation and activity via the inhibition of
MDM2-mediated p53 ubiquitination in human lung cancer cells. Oncol
Rep. 29:1983–1990. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kruse JP and Gu W: SnapShot: p53
posttranslational modifications. Cell. 133:930–30.e1. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Muñoz-Fontela C, González D, Marcos-Villar
L, Campagna M, Gallego P, González-Santamaría J, Herranz D, Gu W,
Serrano M, Aaronson SA and Rivas C: Acetylation is indispensable
for p53 antiviral activity. Cell Cycle. 10:3701–3705. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sakaguchi K, Herrera JE, Saito S, Miki T,
Bustin M, Vassilev A, Anderson CW and Appella E: DNA damage
activates p53 through a phosphorylation-acetylation cascade. Genes
Dev. 12:2831–2841. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jimenez GS, Khan SH, Stommel JM and Wahl
GM: p53 regulation by post-translational modification and nuclear
retention in response to diverse stresses. Oncogene. 18:7656–7665.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee JT and Gu W: The multiple levels of
regulation by p53 ubiquitination. Cell Death Differ. 17:86–92.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yuan J, Luo K, Zhang L, Cheville JC and
Lou Z: USP10 regulates p53 localization and stability by
deubiquitinating p53. Cell. 140:384–396. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Olivier M, Hollstein M and Hainaut P: TP53
mutations in human cancers: Origins, consequences, and clinical
use. Cold Spring Harb Perspect Biol. 2:a0010082010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Robles AI and Harris CC: Clinical outcomes
and correlates of TP53 mutations and cancer. Cold Spring Harb
Perspect Biol. 2:a0010162010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Leite KR, Franco MF, Srougi M, Nesrallah
LJ, Nesrallah A, Bevilacqua RG, Darini E, Carvalho CM, Meirelles
MI, Santana I and Camara-Lopes LH: Abnormal expression of MDM2 in
prostate carcinoma. Mod Pathol. 14:428–436. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bohlman S and Manfredi JJ: p53-independent
effects of Mdm2. Subcell Biochem. 85:235–246. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Alarcon-Vargas D and Ronai Z: p53-Mdm2 -
the affair that never ends. Carcinogenesis. 23:541–547. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nag S, Qin J, Srivenugopal KS, Wang M and
Zhang R: The MDM2-p53 pathway revisited. J Biomed Res. 27:254–271.
2013.PubMed/NCBI
|
|
19
|
Moll UM and Petrenko O: The MDM2-p53
interaction. Mol Cancer Res. 1:1001–1008. 2003.PubMed/NCBI
|
|
20
|
Khan N, Bharali DJ, Adhami VM, Siddiqui
IA, Cui H, Shabana SM, Mousa SA and Mukhtar H: Oral administration
of naturally occurring chitosan-based nanoformulated green tea
polyphenol EGCG effectively inhibits prostate cancer cell growth in
a xenograft model. Carcinogenesis. 35:415–423. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Robson CH, Ganapathy M, Swanson GP,
Natarajan M, Papanikolaou N, Hanes MA, Yeh IT, Ghosh R and Kumar
AP: Phellodendron amurense bark extract enhances radiosensitivity
by inhibition of nf-kappa B in transgenic adenocarcinoma of mouse
prostate model and human prostate cancer cells. J Urol. 181((4S)):
4792009. View Article : Google Scholar
|
|
22
|
Paller CJ, Pantuck A and Carducci MA: A
review of pomegranate in prostate cancer. Prostate Cancer Prostatic
Dis. 20:265–270. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Seeram NP, Adams LS, Henning SM, Niu Y,
Zhang Y, Nair MG and Heber D: In vitro antiproliferative, apoptotic
and antioxidant activities of punicalagin, ellagic acid and a total
pomegranate tannin extract are enhanced in combination with other
polyphenols as found in pomegranate juice. J Nutr Biochem.
16:360–367. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Han DH, Lee MJ and Kim JH: Antioxidant and
apoptosis-inducing activities of ellagic acid. Anticancer Res.
26:3601–3606. 2006.PubMed/NCBI
|
|
25
|
Naiki-Ito A, Chewonarin T, Tang M,
Pitchakarn P, Kuno T, Ogawa K, Asamoto M, Shirai T and Takahashi S:
Ellagic acid, a component of pomegranate fruit juice, suppresses
androgen-dependent prostate carcinogenesis via induction of
apoptosis. Prostate. 75:151–160. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Losso JN, Bansode RR, Trappey A II, Bawadi
HA and Truax R: In vitro anti-proliferative activities of ellagic
acid. J Nutr Biochem. 15:672–678. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Malik A, Afaq S, Shahid M, Akhtar K and
Assiri A: Influence of ellagic acid on prostate cancer cell
proliferation: A caspase-dependent pathway. Asian Pac J Trop Med.
4:550–555. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vanella L, Di Giacomo C, Acquaviva R,
Barbagallo I, Cardile V, Kim DH, Abraham NG and Sorrenti V:
Apoptotic markers in a prostate cancer cell line: Effect of ellagic
acid. Oncol Rep. 30:2804–2810. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vanella L, Di Giacomo C, Acquaviva R,
Barbagallo I, Li Volti G, Cardile V, Abraham NG and Sorrenti V:
Effects of ellagic acid on angiogenic factors in prostate cancer
cells. Cancers (Basel). 5:726–738. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang L, Li W, Lin M, Garcia M, Mulholland
D, Lilly M and Martins-Green M: Luteolin, ellagic acid and punicic
acid are natural products that inhibit prostate cancer metastasis.
Carcinogenesis. 35:2321–2330. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang HM, Zhao L, Li H, Xu H, Chen WW and
Tao L: Research progress on the anticarcinogenic actions and
mechanisms of ellagic acid. Cancer Biol Med. 11:92–100.
2014.PubMed/NCBI
|
|
32
|
Mohammed Saleem YI, Albassam H and Selim
M: Urolithin A induces prostate cancer cell death in p53-dependent
and in p53-independent manner. Eur J Nutr. 59:1607–1618. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Van Dross RT: Metabolism of anandamide by
COX-2 is necessary for endocannabinoid-induced cell death in
tumorigenic keratinocytes. Mol Carcinog. 48:724–732. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nelly Etienne-Selloum ID, Tanveer Sharif
CAa and Schini-Kerth VB: Polyphenolic Compounds Targeting
p53-Family Tumor Suppressors. Current Progress and Challenges.
2013.
|
|
36
|
Khoo KH, Verma CS and Lane DP: Drugging
the p53 pathway: Understanding the route to clinical efficacy. Nat
Rev Drug Discov. 13:217–236. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ceci C, Lacal PM, Tentori L, De Martino
MG, Miano R and Graziani G: Experimental evidence of the antitumor,
antimetastatic and antiangiogenic activity of ellagic acid.
Nutrients. 10:17562018. View Article : Google Scholar
|
|
38
|
Loughery J, Cox M, Smith LM and Meek DW:
Critical role for p53-serine 15 phosphorylation in stimulating
transactivation at p53-responsive promoters. Nucleic Acids Res.
42:7666–7680. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Agrawal A, Yang J, Murphy RF and Agrawal
DK: Regulation of the p14ARF-Mdm2-p53 pathway: An overview in
breast cancer. Exp Mol Pathol. 81:115–122. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shukla S and Gupta S: Apigenin-induced
prostate cancer cell death is initiated by reactive oxygen species
and p53 activation. Free Radic Biol Med. 44:1833–1845. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Aliouat-Denis CM, Dendouga N, Van den
Wyngaert I, Goehlmann H, Steller U, van de Weyer I, Van Slycken N,
Andries L, Kass S, Luyten W, et al: p53-independent regulation of
p21Waf1/Cip1 expression and senescence by Chk2. Mol Cancer Res.
3:627–634. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang Z, Wang H, Li M, Agrawal S, Chen X
and Zhang R: MDM2 is a negative regulator of p21WAF1/CIP1,
independent of p53. J Biol Chem. 279:16000–16006. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kracikova M, Akiri G, George A,
Sachidanandam R and Aaronson SA: A threshold mechanism mediates p53
cell fate decision between growth arrest and apoptosis. Cell Death
Differ. 20:576–588. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Meek DW and Knippschild U:
Posttranslational modification of MDM2. Mol Cancer Res.
1:1017–1026. 2003.PubMed/NCBI
|
|
45
|
Seeram NP, Lee R and Heber D:
Bioavailability of ellagic acid in human plasma after consumption
of ellagitannins from pomegranate (Punica granatum L.)
juice. Clin Chim Acta. 348:63–68. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bell C and Hawthorne S: Ellagic acid,
pomegranate and prostate cancer-a mini review. J Pharm Pharmacol.
60:139–144. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bala I, Bhardwaj V, Hariharan S, Kharade
SV, Roy N and Ravi Kumar MN: Sustained release nanoparticulate
formulation containing antioxidant-ellagic acid as potential
prophylaxis system for oral administration. J Drug Target.
14:27–34. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jeong YI, Prasad Yv R, Ohno T, Yoshikawa
Y, Shibata N, Kato S, Takeuchi K and Takada K: Application of
Eudragit P-4135F for the delivery of ellagic acid to the rat lower
small intestine. J Pharm Pharmacol. 53:1079–1085. 2001. View Article : Google Scholar : PubMed/NCBI
|