Introduction
Neuroblastoma (NB) arises when early neuroblasts in
the fetus become cancerous causing widespread nervous system
impairments throughout the body including cognitive disabilities,
motor function impairment, and a compromised autonomic nervous
system (1,2). These cancerous neuroblasts originate
in the adrenal glands but can spread to other locations such as the
peripheral nervous system, spinal cord, and cortical regions of the
brain, leading to poor patient outcomes (3). Understanding the cellular mechanisms
underlying impaired differentiation and carcinogenesis in these
neuroblasts can improve future NB therapies.
The current treatments for NB include radiation
therapy and chemotherapy. However, these treatments are very toxic
and harmful (4). In addition, the
relapse rates are markedly high, usually leading to additional
chemotherapy or death (4). A
therapeutic approach that has minimized side effects is retinoic
acid (RA)-induced differentiation with RA analogues such as
all-trans RA. RA is a naturally-occurring retinoid
synthesized from vitamin A in embryos and adult vertebrates
(5–7). RA has been revealed to be very
effective for NB therapy in infants under 18 months with almost all
patients exhibiting complete remission from cancer (5). RA analogues promote differentiation of
neuroblasts to mature neurons. Since NB is a neuronal cancer of
immature neuronal cells, for the change to mature neurons, classic
neuronal characteristics are acquired including cell cycle arrest,
termination of aggressive cell movement, and formation of stronger
synapses compared to the immature cell (8).
The actions of RA are mediated by binding to nuclear
RA receptors (RARs), regulating the transcriptional activity of the
latter, as well as activating downstream signaling cascades
(9). Upon RA binding, activated RAR
binds to target DNA sequences [RA response elements (RAREs)]
(10). The downstream and
intermediate targets of RA-activated RAR, which contribute to
neuroblast differentiation are not well studied (11). Insight into the mechanism of
RA-induced differentiation via RAR would yield new targets to
develop improved differentiation therapies.
The purpose of the present study was to elucidate
the mechanism of RA-induced differentiation, providing insight to
key proteins that play an important regulatory role in this
process. In the present study, a novel mechanism of RA-induced
differentiation via the oncofetal receptor tyrosine kinase-like
orphan receptor 1 (ROR1) was described. Embryos and fetuses have
been revealed to highly express ROR1 in numerous tissues, including
epithelial and nervous tissue (12). There is minimal expression of ROR1
in normal adult tissues (12,13)
and data published from our laboratory has demonstrated a link
between overexpression of ROR1 and triple-negative breast cancer
cell migration and proliferation (14). Similarly, ROR1 levels have been
revealed to be abnormally high in NB (15) but only minimally expressed in
differentiated neurons. Notably, Wnt5a, the ligand for ROR1, is
underexpressed in NB and has previously been revealed to regulate
neuronal differentiation (16). RA
has been demonstrated to activate the PI3K/Akt signaling pathway
which is already known to be regulated by ROR1 (17). It was thus hypothesized that
differentiation therapy with RA may be mediated via the ROR1 and
Wnt5a signaling pathway. To corroborate this hypothesis, the
changes in ROR1 followed by RA treatment as well as ROR1-knockdown
experiments were investigated to reveal its role in RA-induced
differentiation.
For the present study, SK-N-SH neuronal epithelial
cells, which have been revealed to differentiate into mature
neurons that can be easily characterized by neurite outgrowth, were
used (18). This renders them
particularly useful for delineating signaling pathways involved in
neuronal differentiation. SK-N-SH has also been revealed to contain
RAR, specifically RAR-α, and has been previously studied in aspects
such as apoptosis (11).
Additionally, evidence of similar trends of ROR1 expression in
SH-SY5Y cells and patient primary cells from data collected from
the NCBI GEO datasets were provided.
The data from the present study provided further
insight into the molecular mechanism of RA-induced differentiation
and reiterated the potential of ROR1 and its downstream proteins
such as PI3K/AKT as a therapeutic target for poorly differentiated
neuroblastoma.
Materials and methods
Cell culture
SK-N-SH (HTB-11; ATCC) cells were cultured using
methods recommended by ATCC. Culture media was prepared using
DMEM/F-12 (Lonza Group, Ltd.) supplemented with fetal bovine serum
(10%) (cat. no. TMS-013-B; EMD Millipore), penicillin (100 U/ml),
and streptomycin (0.1 mg/ml) (cat. no. 516106; EMD Millipore).
Cells were maintained at 37°C with 5% CO2. Cell cultures
were validated and routinely checked for contaminants including
mycoplasma.
Differentiation of SK-N-SH cells using
RA
Neuroblastoma cells, SK-N-SH, were treated with 10
µM all-trans RA (CAS no. 302-79-4; Cayman Chemical Company)
(19,20) dissolved in DMSO and diluted in
culture media (final DMSO concentration, <0.1%) for up to 96 h
without the addition of new culture media. AGN 193109 (product no.
SML2034; EMD Millipore), a RAR inhibitor, was prepared by
dissolving in DMSO to a stock solution of 1 nM and diluted in
culture media to a final concentration of 10 µM. Cells were then
treated for 1 h before RA treatment and then treated in cell media
containing AGN 193109 and RA for up to 96 h without the addition of
new culture media.
Immunoblotting
Following appropriate treatments, cells were washed
with 1X PBS. To extract proteins, cells were scraped with a plastic
cell scraper and 1X PBS (cat. no. MT21040CV; Thermo Fisher
Scientific, Inc.) and centrifuged at 5,000 × g for 10 min. The cell
pellets were lysed in RIPA buffer (150 mM NaCl, 1.0% Triton X-100,
0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0) with 1 mM
PMSF. Protein concentrations from the supernatant were quantified
utilizing the Bradford Assay using Pierce 660 nm Protein Assay
reagent (Thermo Fisher Scientific, Inc.). The lysate was then
diluted in 2X Laemmli Buffer and 355 mM β-mercaptoethanol (CAS
60-24-2; EMD Millipore) to be boiled for 5 min. Protein samples
(10–15 µg) were loaded on a 4-15% gradient SDS-PAGE to ensure that
protein concentrations were equal amongst all lanes. Following
SDS-PAGE, separated proteins were transferred to nitrocellulose
membranes using electrophoresis. Membranes were then blocked in 5%
milk for 1 h at room temperature and subsequently washed with TBS-T
(20 mM Tris, 150 mM NaCl, 0.1% Tween-20). Primary antibody
incubations were performed overnight in 5% milk and then washed
with TBS-T. Secondary anti-mouse (product no. 7076; Cell Signaling
Technology, Inc.) or anti-rabbit IgGs (product no. 7074; Cell
Signaling Technology, Inc.) linked with horseradish peroxidase
diluted 1:1,000 were incubated at room temperature for 1 h and
washed with TBS-T. Protein bands were imaged via immunofluorescence
by SuperSignal™ West Dura Extended Duration Substrate (cat. no.
34076; Thermo Fisher Scientific, Inc.). The primary antibodies used
were as follows and diluted 1:1,000 unless specifically specified
differently under manufacturer recommendations: ROR1 (D6T8C;
product no. 16540; Cell Signaling Technology, Inc.), GAPDH (D16H11;
product no. 5174; Cell Signaling Technology, Inc.), p-AKT-ser473
(product no. 9271; Cell Signaling Technology, Inc.), AKT (C67E7;
product no. 4691; Cell Signaling Technology, Inc.), p-GSK3β-Ser9
(D17D2; product no. 8566; Cell Signaling Technology, Inc.), and
GSK3β (D75D3; product no. 5676; Cell Signaling Technology, Inc.),
β-catenin (cat. no. MA1-301; Invitrogen; Thermo Fisher Scientific,
Inc.), p-β-catenin-Ser33/37 (product no. 2009; Cell Signaling
Technology, Inc.), stabilized-β-catenin (product no. 9562; Cell
Signaling Technology, Inc.), Wnt-5a (cat. no. NBP2-24752SS; Novus
Biologicals, Ltd.), Synaptophysin (cat. no. 100298-T40; Sino
Biological, Inc.), β-tubulin (product code ab21057; Abcam) and
Lamin B1 (product code ab65986; Abcam).
Densitometric analysis
Densitometry of immunoblots was performed with
ImageJ (v1.52a; NIH) software utilizing established methods
(21,22). Blots were imported to ImageJ and
utilizing the ‘analyze gel’ feature, the areas under the curves
generated from the bands were obtained for further analysis.
MTT cell viability assay
Cell viability was determined using an MTT Assay
(Thermo Fisher Scientific, Inc.) according to a previously
published method (14). Cells were
seeded in a 96-well plate at 15,000 cells/well. A 0.3% vehicle
control group was used and after 96 h, treatment media was replaced
with media containing MTT dye and incubated at 37°C with 5%
CO2 for 3 h. Following incubation, DMSO was added and
absorbance was read using a spectrophotometer at 540 nm.
Immunofluorescence analysis
Following treatments with 10 µM RA, shROR1, 10 µM
RA+shROR1, or DMSO Control, cells were plated in Lab-Tek chamber
slides at a density of 10,000 cells/well. Cells were then fixed
with pre-chilled methanol (5 min, −20°C), washed with PBS and
blocked for 1 h at room temperature in 5% BSA/PBS (blocking
buffer). Primary antibody incubation was performed in blocking
buffer at 4°C overnight. Following three PBS washes, cells were
incubated with secondary antibodies Alexa-fluor-647 (Thermo Fisher
Scientific, Inc.) in blocking buffer for 1 h at room temperature.
The cells were then washed and stained with a 1-µg/ml DAPI/PBS
solution for 2 min at room temperature. Confocal fluorescence
microscopy at ×40 magnification was performed using a Leica DMi8
microscope. All the primary antibodies are mentioned in the
Immunoblotting section.
Analysis of neurite length/cell
size
The length of neurites or cell size was measured
using ImageJ considering the techniques published regarding simple
length analysis of neurons (23).
Images of the cells were imported and using the polygon shape tool,
manual tracing was performed with ~10–15 cells per image to obtain
cell size. The measurements were averaged to obtain the cell size
of the sample. Neurite length was obtained similarly but used the
line tool to manually measure the length between the middle of the
soma to the tip of the longest neurite. This was performed to 10–15
cells per image and averaged to obtain the neurite length of the
sample.
β-catenin luciferase reporter
assay
A construct encoding a TCF/LEF luciferase reporter
system (cat. no. 79787; SABiosciences) was transfected into cells
at 10,000 cells/well using Lipofectamine 3000 (cat. no. L3000008;
Thermo Fisher Scientific, Inc.). Expression of this construct is
modulated by the TCF/LEF transcription factor system specific to
the Wnt/β-catenin signaling pathway (24). Following transfection, cells were
treated with 10 µM RA up to 96 h. Following trypsinization of
treated cell cultures, the cells were collected using PBS and then
were pelleted by centrifugation at 5,000 × g for 5 min followed by
removal of the supernatant. Dual-Glo® Luciferase Assay
System reagent was added (100 µl; cat. no. E2920; Promega
Corporation). Following a 3-h incubation on ice, luminescence was
read at a 10-sec exposure per well. Luciferase measurements were
normalized to protein concentrations of 0 h Control group.
Luciferase experiments were performed as technical triplicates.
Analyzing the expression of Wnt5a and
ROR1 using real-time quantitative (RT-q)PCR
Following treatments with 10 µM RA for up to 96 h,
RNA extraction was performed using TRIzol (cat. no. 15596026;
Thermo Fisher Scientific, Inc.). cDNA synthesis from the extracted
RNA was performed using the SuperScript™ III First-Strand Synthesis
System (cat. no. 18080051; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Following a 1:50 dilution
of synthesized cDNA, RT-qPCR was performed with the CFX384 Touch
Real-Time PCR detection system (Bio-Rad Laboratories, Inc.) under
the following conditions: i) 95°C for 15 min; ii) 95°C for 10 sec;
iii) 60°C for 30 sec; and iv) repeat ii and iii for 39 more cycles.
The fluorophore used was SYBR Green (cat. no. 1725150; Bio-Rad
Laboratories, Inc.). The ROR1 (NCBI Gene ID: 4919) and Wnt5a (NCBI
Gene ID: 7474) primers used were as follows: ROR1 forward,
5′-TGTTTGTCAAGTTTGGCCCCC-3′ and reverse,
5′-AGGGAAGGAATGGCGAACTG-3′; and Wnt5a forward,
5′-CTTCAGCTCCGGTTCACTGC-3′ and reverse,
5′-TGGACTTCTTCATGGCGAGGG-3′. qPCR quantification utilized
2−ΔΔCT analysis (25).
Stand-alone qPCR experiments were conducted as biological
triplicates.
Chromatin-immunoprecipitation (ChIP)
assay to analyze the binding of a RAR to ROR1 promoter
Immunoprecipitation of RAR-bound chromatin was
performed using the Pierce Agarose ChIP Kit (cat. no. 26156; Thermo
Fisher Scientific, Inc.) according to manufacturer's instructions.
Briefly, 320,000 cells were plated in each well of 6-well plates
and treated with RA. Cells were then fixed with chilled 1%
paraformaldehyde solution at room temperature for 10 min and lysed.
Immunoprecipitation was performed using an anti-RAR-α antibody
(product no. 62294S; Cell Signal technology, Inc.) at a 1:50
dilution. RT-qPCR of Wnt5a and ROR1 promoter regions was performed
using the following ROR1 (NCBI Gene ID: 4919) and Wnt5a (NCBI Gene
ID: 7474) primers: ROR1 forward, 5′-TGTTTGTCAAGTTTGGCCCCC-3′ and
reverse, 5′-AGGGAAGGAATGGCGAACTG-3′; and Wnt5a forward,
5′-CTTCAGCTCCGGTTCACTGC-3′ and reverse,
5′-TGGACTTCTTCATGGCGAGGG-3′. ChIP assay experiments were conducted
as technical triplicates.
Analysis of gene expression data from
NCBI gene expression omnibus (GEO)
In dataset GSE58070 (26), SH-SY5Y cells were treated with 5 µM
RA for 30 min, 1, 3, 6 and 9 days. DMSO was used as a vehicle
control. For each treatment, total RNA was extracted using the
RNeasy Mini Kit (Qiagen) and gene expression was profiled using
NimbleGen 12×135 K Homo sapiens Expression Array (PROBE_ID version
100718_HG18_opt_expr) (NimbleGen-Roche). Expression data was
normalized to GAPDH using the 2−ΔΔCt method. ROR1 and
Wnt5a expression data was retrieved using ID NM_005012P02481 and
NM_003392P04985, respectively, in GEO2R. In dataset GSE45587
(27), BE(2)-C cells were treated
with 5 µM RA for 6, 24 and 72 h. DMSO was used as a vehicle
control. Genome-wide expression profiling was performed using
GeneChip Affymetrix U133A microarray (GeneChip). ROR1 and Wnt5a
expression data was retrieved in GEO2R using probe IDs 205805_s_at
and 205990_s_at respectively. In dataset GSE120920 (28), patient-derived xenograft cultures
were obtained from cerebral metastasis of a stage 4 neuroblastoma
patient (NB-PDX2) and a primary tumor in the adrenal gland of a
patient with stage 3 neuroblastoma (NB-PDX3). Both cell lines were
MYCN amplified. Both NB-PDX2 and NB-PDX3 were treated with RA for 6
and 24 h, with DMSO vehicle controls in both groups. Whole
transcriptome analysis was performed via DRUG-Seq (29). We visualized and analyzed the data
using the R2 Genomics Analysis and Visualization Platform
(r2.amc.nl) (30). ROR1 and Wnt5a
data was retrieved with probe IDs ENSG00000185483 and
ENSG00000114251 respectively. All data were expressed as the mean ±
standard error of mean and statistical analysis was conducted using
GraphPad Prism 7.04 (GraphPad Prism Software, Inc.).
ROR1 knockdown and overexpression
ROR1 expression was knocked down by shRNA (shROR1)
(plasmid DNA seq: 5′
CCGGCTTTACTAGGAGACGCCAATACTCGAGTATTGGCGTCTCCTAGTAAAGTTTTT 3′) (cat.
no. SHCLND-NM_005012; EMD Millipore) or overexpressed using an
ROR1-overexpressing plasmid (pROR1) containing a CMV promoter
(accession no. NM_005012; GenScript). Transfection of shRNA or
pROR1 was performed using the Lipofectamine 3000 Reagent (cat. no.
L3000008; Thermo Fisher Scientific, Inc.). Cells were plated at
300,000-400,000 cells per well in a 6-well plate. Twenty-four hours
after seeding, the cells were washed with PBS. Transfection was
performed in non-supplemented MEM with 1 µg DNA/well for shROR1,
0.1 µg DNA/well for pROR1, and/or 0.1 µg DNA/well for β-catenin
luciferase reporter plasmid. Cells were transfected for 24 h at
37°C then rescued in complete growth media for 24 h prior to
further experimentation.
Statistical analysis
All data were expressed as the mean ± standard
deviation unless otherwise specified. Statistically significant
differences between groups were analyzed using ANOVA with multiple
comparison Tukey's post hoc test to compare every condition with
each other condition in the experiment, unless otherwise indicated.
P-values <0.05 were considered to indicate a statistically
significant difference. All statistical analyses were performed
using GraphPad Prism 7.04 (GraphPad Software, Inc.).
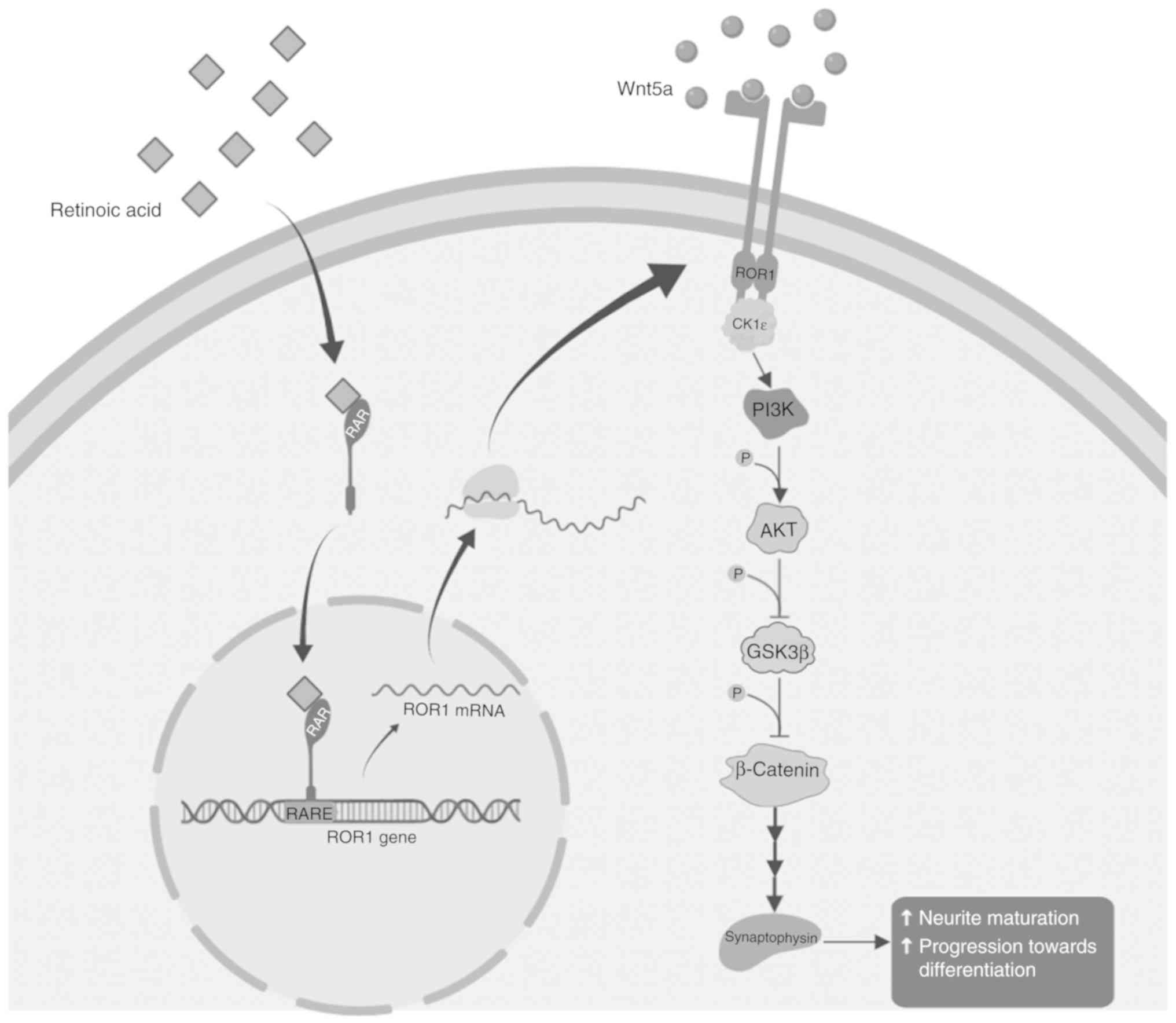
RA pathway model
The RA model graphic was created with BioRender.com
utilizing generic DNA, protein, cell membrane, and receptor images
available on the website.
Results
RA promotes neuroblastoma
differentiation and is mediated via ROR1
RA-mediated differentiation of SK-N-SH cells induced
a change in phenotype from an epithelial-like state to a more
typical neuronal-like state with distinct neurite processes.
Concurrently the increase in neurite processes and lengths led to
an increase in synaptic connections (Fig. 1C and D). Yellow arrows in phase
contrast images point to marked neurite extensions or clusters of
neurites that are suggestive of development (Fig. 1C). RA-induced differentiation was
monitored via immunoblotting assessing the levels of synaptophysin,
a ubiquitous synaptic marker (31).
We assume that an indication in increased neurite length paired
with an observation of increasing synaptophysin expression is
indicative of the formation of synapses, suggesting
differentiation. Furthermore, an increase in synaptophysin after 96
h of treatment, indicative of increased differentiation was
observed (Fig. 1A and B). Notably,
time-dependent modulation of ROR1 expression was also observed.
ROR1 levels increased initially from 0 to 72 h and then
significantly decreased at 96 h (Fig.
1A and B).
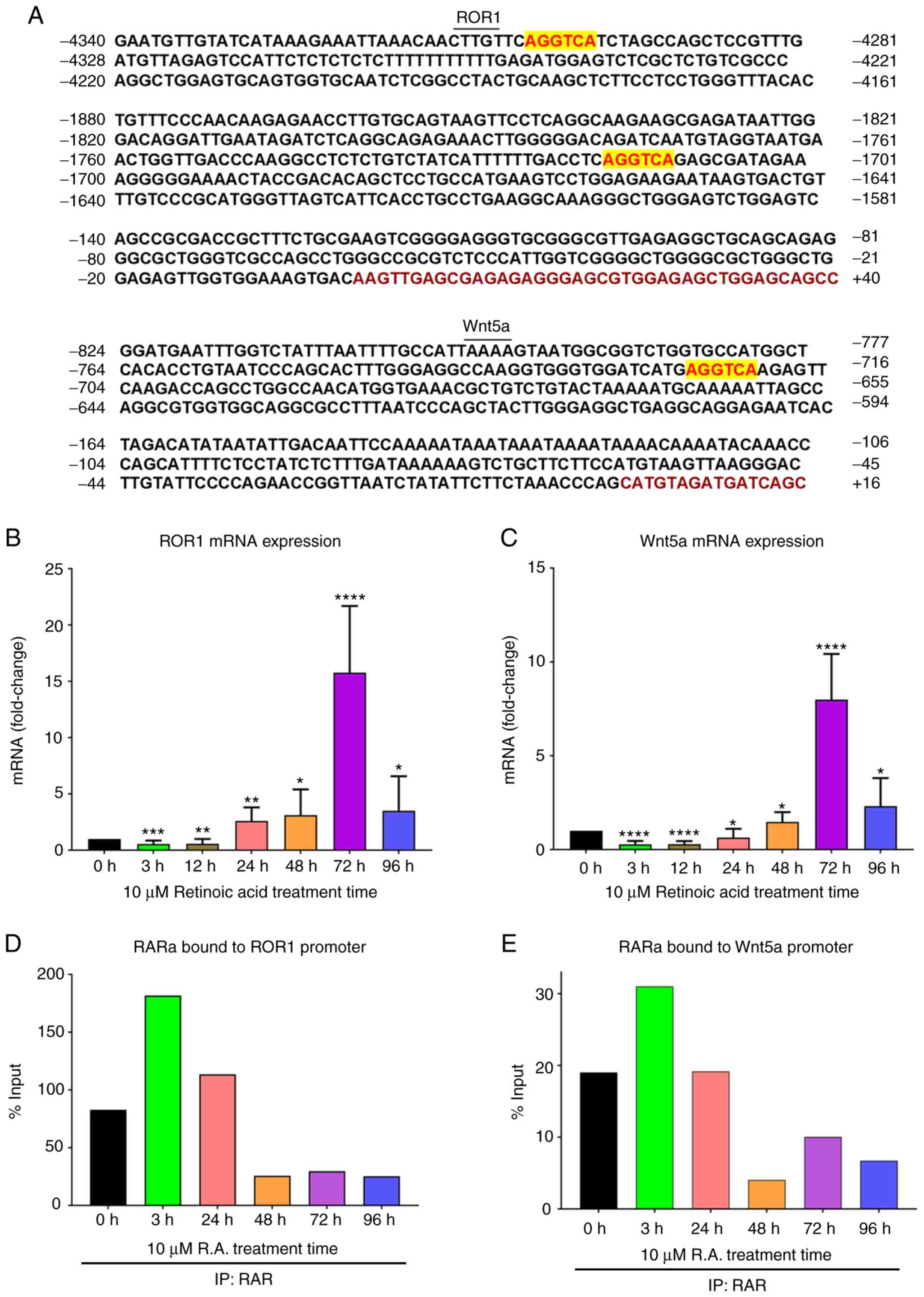
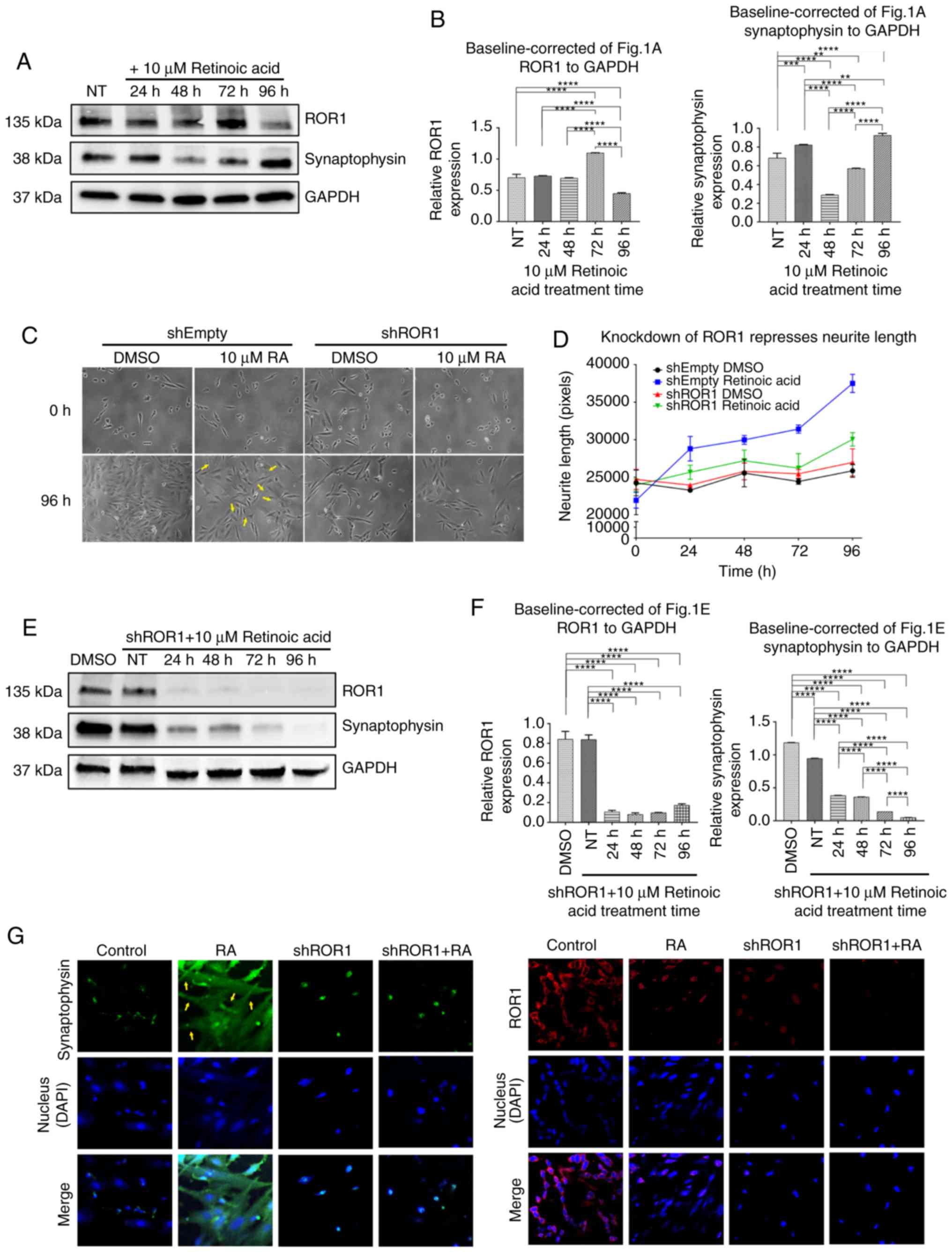 | Figure 1.RA promotes NB differentiation via
ROR1. (A) Immunoblotting revealed the changes in ROR1 and
synaptophysin levels under normal differentiation with RA. (B)
Densitometry of immunoblots from A, baseline-corrected to GAPDH
(ANOVA with Tukey's multiple comparison to all conditions, n=3).
**P<0.005, ***P<.0005, ****P<0.0001. (C) Phase contrast
images of SK-N-SH neuroblastoma cells undergoing RA or DMSO
treatment over 96 h under shRNA test Lipofectamine transfection,
knocking down ROR1 expression. Yellow arrows indicate marked
neurite extensions that suggest development. (D) Visualization of
the average neurite length over time following knockdown of ROR1
(n=3). (E) Immunoblots revealed the changes in ROR1 and
synaptophysin levels after Lipofectamine transfection, knocking
down ROR1 expression. (F) Densitometry of immunoblots from E,
baseline-corrected to GAPDH (ANOVA with Tukey's multiple comparison
test to compare each condition with every other condition in the
experiment, n=3). ****P<0.0001. (G) Immunofluorescence revealed
the effects of RA on synaptophysin (green) levels with or without
shRNA knockdown of ROR1 (red). DAPI (blue) highlights the nucleus
of the neurons. Yellow arrows indicate marked neurite extensions.
RA, retinoic acid; NB, neuroblastoma; ROR1, receptor tyrosine
kinase-like orphan receptor 1; NT, not treated. |
To investigate whether RA regulation of ROR1
expression was vital to the differentiation process, ROR1 was
knocked down via shRNA (shROR1) and differentiation was monitored.
A decrease in neurite length was observed in RA-treated cells with
ROR1 knockdown when compared to a vector control (Fig. 1E and F). The synaptophysin levels
were also monitored in shROR1-transfected cells following RA
treatment and a decrease in synaptophysin levels was observed
(Fig. 1E and F). Immunofluorescence
labelling synaptophysin in shROR1 and vector-transfected cells,
both treated with RA, similarly revealed impeded differentiation
(Fig. 1G).
RA regulation of ROR1 modulates the
PI3K/AKT/GSK3β pathway
ROR1 is a Wnt pathway receptor that regulates cell
migration, cell cycle progression, and apoptosis by promoting the
PI3K/AKT/GSK3β signaling axis (14–17).
To investigate the downstream effects of RA modulation of ROR1 the
PI3K/AKT/GSK3β pathway was monitored after RA treatment. An
increase in AKT phosphorylation on serine 473 was observed, which
is indicative of increased activation of the PI3K/AKT pathway
(Fig. 2A and B). This was
associated with increased phosphorylation and inhibition of GSK3β
until 72 h (Fig. 2A and B). GSK3β
phosphorylates β-catenin on Ser 33/37 inhibiting the activity of
the latter, tagging it for proteasomal degradation (32). Corroborating the increase in GSK3β
inhibition, a decrease in β-catenin phosphorylation was observed
(Fig. 2A and B). This would suggest
more active β-catenin. β-catenin activity was monitored using a
luciferase reporter system containing TCF/LEF responsive elements
and an increase in β-catenin activity after RA treatment from 0 to
96 h was observed (Fig. 2C). To
confirm that RA modulation of the PI3K/AKT/GSK3β pathway was
mediated via RA upregulation of ROR1, we knocked down ROR1 prior to
RA treatment and similarly monitored PI3K/AKT/GSK3β. A decrease in
AKT activation and an increase in phosphorylation of GSK3β until
the 72-h time-point, were observed. However, at 96 h, p-AKT and
p-GSK3β trends were reversed which is the suggested time of the end
of differentiation (Fig. 2D and E).
Fig. S2 depicts individual
proteins from Fig. 2A
baseline-corrected to the NT group. Fig. S3 depicts individual proteins from
Fig. 2D baseline-corrected to the
NT group.
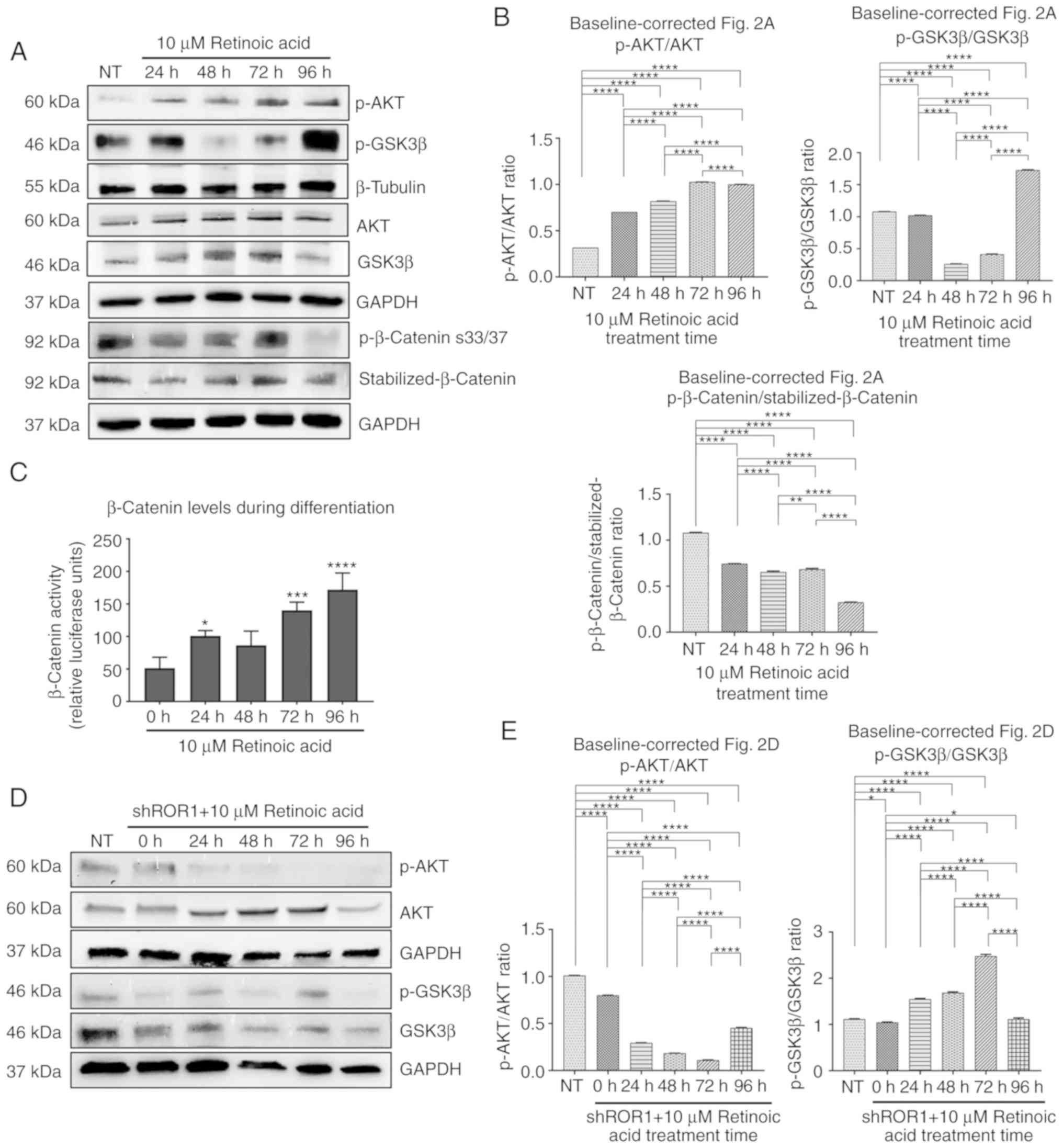 | Figure 2.RA modulation of ROR1 regulates
P13K/AKT/GSK3β. (A) Immunoblots of the ROR1 pathway under normal RA
treatment over 96 h. (B) Densitometry of ratioed baseline-corrected
proteins to their respective loading controls (ANOVA with Tukey's
multiple comparison test to all conditions, n=3). **P<0.005,
****P<0.0001. (C) Luciferase assay of total β-catenin levels
during differentiation over 96 h (ANOVA multiple comparisons to 0
h, n=3). *P<0.05, ***P<0.0005, ****P<0.0001. (D)
Immunoblots of the ROR1 pathway under RA treatment and knockdown of
ROR1 with shRNA over 96 h. (E) Densitometry of ratioed
baseline-corrected proteins to their respective loading controls
(ANOVA with Tukey's multiple comparison test to compare each
condition with every other condition in the experiment, n=3).
*P<0.05, ****P<0.0001. RA, retinoic acid; ROR1, receptor
tyrosine kinase-like orphan receptor 1; NT, not treated. |
RA regulates the transcription of
ROR1/Wnt5a
Subsequently, it was investigated how RA
mechanistically regulates ROR1 expression. RA typically binds to
and activates RAR, regulating the transcription of genes possessing
RARE sequences (5′-AGGTCA-3′) in the region upstream to their
promoter (9,10). ROR1 transcription was monitored
after RA treatment and an increase in ROR1 mRNA from 0–72 h was
observed, followed by a significant decrease at 96 h (Fig. 3B) mirroring our immunoblotting
results in Fig. 1A. The
transcription of the ligand of ROR1, Wnt5a, was also monitored
after RA treatment and a similar trend was observed: An increase in
Wnt5a mRNA from 0–72 h and then a significant decrease at 96 h
(Fig. 3C). Analysis of the sequence
upstream to the ROR1 promoter revealed the presence of two RARE
sequences. Similarly, one RARE sequence was found in the region
upstream to the Wnt5a promoter (Fig.
3A).
Next, it was investigated whether the presence of a
RARE sequence was associated with increased RARα binding to the
ROR1 and Wnt5a promoters after RA treatment. ChIP of RARα after RA
treatment was performed and ROR1 and Wnt5a bound to RARα were
assessed via qPCR. In both instances, an increase in RARα bound to
the ROR1 or Wnt5a promoter, 3 h after RA treatment followed by a
steady decrease until 96 h was observed (Fig. 3D and E).
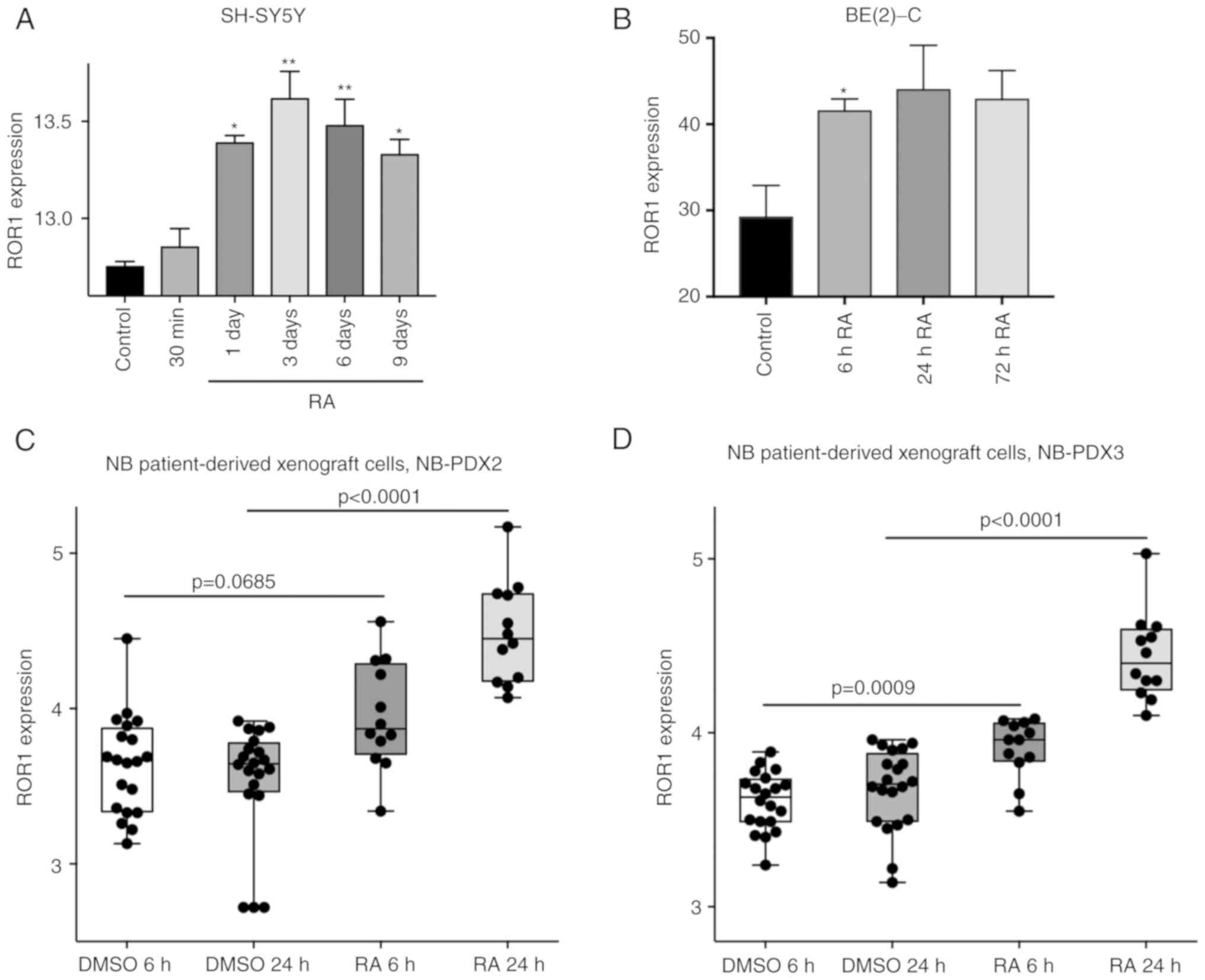
RA promotes ROR1 and Wnt5a
transcription in NB patient-derived cells and MYCN-amplified cell
lines
To corroborate the present findings and verify if
the RA-induced increase in ROR1 and Wnt5a transcription was
observed in other NB cell lines (both MYCN-amplified/non-amplified)
and patient-derived primary NB cells, publicly available gene
expression data were probed using GEO datasets from NCBI. In
dataset GSE58070 (26), SH-SY5Y
cells (MYCN-non-amplified) were treated with 5 µM RA and gene
expression, at various time-points, was profiled using NimbleGen
microarray. Analysis of ROR1 expression revealed an increase in
ROR1 mRNA starting at 30 min post-treatment, reaching a peak after
3 days of RA treatment (Fig. 4A).
ROR1 levels subsequently decreased on days 6 and 9 (Fig. 4A), mirroring our findings in SK-N-SH
cells (Fig. 3B). Wnt5a mRNA was
similarly increased after 1 day of RA treatment and exhibited a
downward trend in subsequent days (Fig. S1A). In another dataset, GSE45587
(27), BE(2)-C cells
(MYCN-amplified) were treated with 5 µM RA for 6, 24, and 72 h and
genome-wide expression profiling was performed using Affymetrix
U133A microarrays. ROR1 mRNA levels increased 6 h post-RA
treatment, reaching a peak at 24 h and decreasing at 72 h (Fig. 4B). Wnt5a mRNA levels initially
decreased at 6 h, and then increased until 72 h, however the data
was not statistically significant (Fig. S1B). In another dataset, GSE120920
(28), patient-derived xenograft
cultures from two patients with high-risk, MYCN-amplified NB
(NB-PDX2 and NB-PDX3), were treated with RA for 6 and 24 h and
whole transcriptome analysis was performed via DRUG-Seq. Similarly,
an increase in ROR1 mRNA in both cell cultures post-RA treatment
was observed (Fig. 4C and D). There
was no significant change in Wnt5a expression in the RA-treated
groups compared to the control (Fig.
S1C and D).
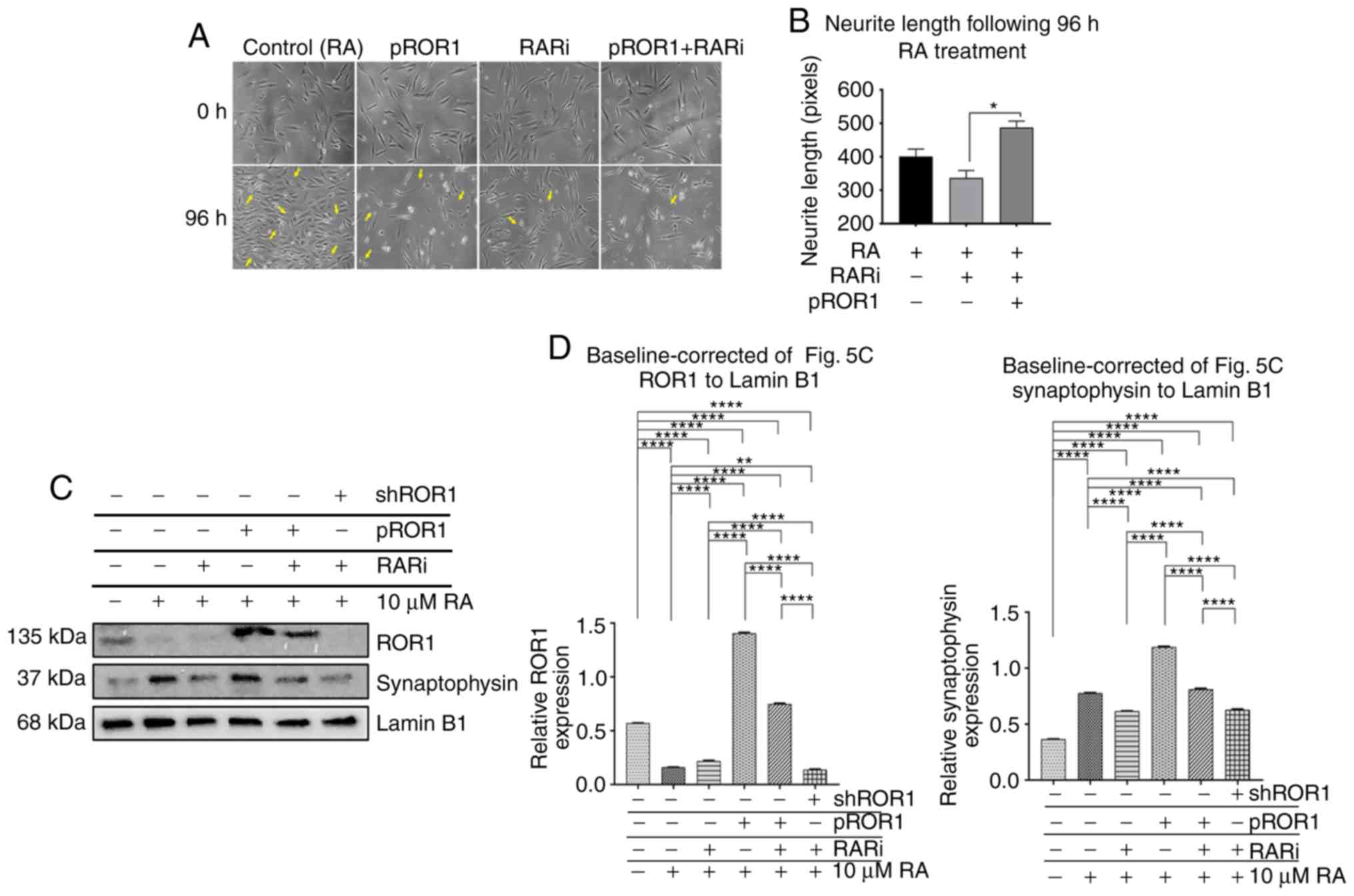
RA effect on differentiation and ROR1
is RAR-dependent
RA induces differentiation via activation of RAR
(33). However, RA has also been
revealed to bind to and induce several other cellular regulators
such as CRABP I and CRABP II which are involved in nervous system
development (34). To verify
whether RA-induced differentiation and upregulation of ROR1 was
RAR-dependent, a RAR was inhibited using a high affinity RAR
antagonist, AGN 193109 (RARi). Cells were pre-treated with RARi
prior to RA treatment. Then, differentiation was assessed by
measuring neurite length. As anticipated, neurite lengths were
decreased in the cells pre-treated with RARi compared to cells that
were not pretreated with RARi (Fig.
5A). RAR inhibition suppressed the effect of RA on ROR1 and
therefore it was investigated whether overexpressing ROR1 in RARi
pre-treated cells would rescue the differentiation phenotype. An
increase in neurite lengths in RARi pre-treated cells after ROR1
overexpression was observed that was comparable to the RA-only
group (Fig. 5A and B). To confirm
the present findings, synaptophysin protein levels after RA
treatment in cells with RAR inhibition and/or ROR1 overexpression
were assessed. Corroborating our phenotypic findings in Fig. 5A, a decrease in synaptophysin in the
RARi + RA group compared to the RA-only group was observed
(Fig. 5C and D). ROR1
overexpression in these RARi + RA cells rescued synaptophysin
expression (Fig. 5C and D).
Fig. S4 provides proof of
transfection based on RT-qPCR data for ROR1 expression due to the
transfection of pROR1.
Based on these findings, we propose a model where
treatment with RA leads to a cascade of downstream events initiated
by ROR1, resulting in differentiation of neuronal cells. The
ROR1-mediated effect is through the well-established PI3K/AKT/GSK3β
pathway (Fig. 6).
Discussion
The data from the present study demonstrated that
ROR1 regulated RA-induced differentiation by modulating RAR. In
some instances, spontaneous recovery has occurred in patients as a
result of differentiation of malignant tissue into normal, mature
neural tissue (3). Thus,
differentiation of malignant neuroblasts into normal mature neurons
remains a key therapeutic strategy for neuroblastoma. RA, a vitamin
A-derived morphogen, is a well-established differentiating agent
that has shown promise clinically for neuroblastoma and other
poorly-differentiated cancers, including lung, prostate, breast,
bladder, oral and skin cancers (5,35–38).
Understanding of the molecular mechanisms underlying RA-induced
differentiation remains limited. There are several genetic
heterogeneities in neuroblastomas which include amplification of
the MYCN oncogene, deletion of the distal short arm of chromosome 1
(1p), gain in the distal long arm in chromosome 17 (17q), and DNA
ploidy (39). Neuroblastoma
consists of tumorigenic ‘N’ type (neuronal) and non-tumorigenic ‘S’
type (Schwannian) cells. This cellular heterogeneity renders
prognosis difficult. NB aggressiveness and poor prognosis is mainly
associated with N-Myc amplification. Since the present study was
focused on understanding the cellular mechanisms that regulated
RA-induced differentiation, human neuroblastoma cell line SK-N-SH
which is markedly heterogeneous was selected (40). SK-N-SH cells present a suitable
model to study tumor-initiating neuroblasts as it has both cell
types, as well as the ‘I’ cell type which is an intermediate of S
and N types (41). Furthermore, the
time for in-vitro differentiation is significantly shorter
and possibly more reliable in this cell line than other
neuroblastoma cell lines, including the more studied SH-SY5Y cell
line (8,20). Hence, the data from the present
study, obtained in a highly heterogeneous cell line such as
SK-N-SH, is particularly relevant for highly heterogeneous tumors
such as the ones found in NB patients.
The present data revealed that RA treatment promoted
an initial increase in ROR1 protein and mRNA expression from 0–72 h
which was associated with activation of the P13K/AKT growth
pathway. The P13K/AKT pathway was previously revealed to play a
role in RA-induced differentiation through proliferation and cell
cycle progression (17,42). ROR1 is an upstream activator of
these pathways (14,15,43).
Downstream to the P13K/AKT pathway, activation of β-catenin
activity, another pathway known to promote differentiation, was
observed (32). ROR1 knockdown
suppressed RA-induced differentiation indicating ROR1 as a key
regulator of the process (12,13,15).
Notably, ROR1 levels markedly decreased after 96 h
of RA treatment. This corroborates previous findings suggesting
markedly minimal ROR1 levels in normal neurons and other
differentiated adult cells (15).
The mechanism of ROR1 sequestering post-differentiation remains
unknown. ROR1 can be rapidly degraded by the proteasome when Wnt5a
levels are high in certain cases (44). In the present study, an increase in
Wnt5a transcription following RA treatment from 0–72 h, followed by
a marked decrease at 96 h was observed. It is entirely plausible
that excessive Wnt5a levels after 72 h aid in the sequestering of
ROR1. This putative regulatory event would prevent ROR1-induced
proliferation and motility in mature neurons. Furthermore, this
suggests that the role of ROR1 is important for the process of
differentiation and is modulated to decrease once differentiation
is complete. This is consistent with the theory that ROR1 is
generally low in adult cells and is upregulated in cancers and
undifferentiated cells.
To investigate the mechanism of RA regulation of
ROR1 and Wnt5a, RAR activity was probed. RAR is a transcriptional
regulator induced by RA, responsible for most RA-induced molecular
events (9). RAR is found
consistently in neuroblastoma cell lines. It is a crucial protein
in regard to development and differentiation and thus, are useful
to target in complex diseases such as neuroblastoma (7,11).
Upon binding of RA, RAR bind to target DNA sequences containing a
RARE sequence (5′AGGTCA3′), regulating their transcription
(9). In fact, RAR may
heterodimerize with RXR depending on the target gene (7). Although this is a characteristic that
would be useful to characterize a transcription factor complex of a
gene, our focus was on observing and targeting RAR since it are
required regardless of heterodimerization with RXR. The downstream
and intermediate targets of RA-induced RAR that promote neuronal
differentiation has generally been studied inefficiently (11). However, we identified two RARE
sequences in the region upstream to the ROR1 promoter, and one in
the region upstream to the Wnt5a promoter. ChIP of RAR-bound
chromatin revealed a rapid increase in RAR accumulation at the ROR1
and Wnt5a promoters 3 h after RA treatment. This was associated
with an increase in ROR1 and Wnt5a transcription suggesting RAR as
a mediator of RA-induced regulation of both proteins. Corroborating
these findings, an increase in ROR1 and Wnt5a expression in other
MYCN non-amplified (SH-SY5Y) and amplified (BE(2)-C), as well as
primary NB patient-derived cells, was observed following RA
treatment.
Since RA can also bind to other cellular regulators
such as CRABP I and CRABP II (34),
we investigated whether the observed effects on ROR1 and
differentiation were RAR-dependent. Using a high-affinity RAR
inhibitor (AGN 193109), a decrease in RA-induced differentiation
was confirmed. Overexpression of ROR1 rescued the differentiation
phenotype suggesting a vital role for both the RAR and ROR1 in
RA-induced differentiation.
Based on the present results, we propose a model
where RA treatment induces ROR1 and Wnt5a transcription. Increased
ROR1 and Wnt5a activate MAPK/ERK and PI3K/AKT pathways to promote
differentiation as previously observed (42).
The implications of this research are several-fold.
Identifying ROR1 as a regulator of differentiation provides
valuable insight into the molecular mechanisms underlying
RA-induced differentiation. It also suggests that evaluating ROR1
levels prior to and during retinol differentiation therapy may have
prognostic value. As ROR1 is a highly active oncogenic receptor
(15), inducing its expression with
RA may lead to unwanted outcomes. ROR1 has recently been evaluated
as a therapeutic target for neuroblastoma (45,46).
In these ROR1 antagonist studies, concurrent efforts evaluating
neuroblast differentiation may shed more light on the implications
of ROR1 in neuroblast differentiation. However, these findings
underline the vital role for growth pathway regulators in
differentiation and further elucidate the molecular mechanisms
regulating RA-induced differentiation.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr. Zachary Klase
for support with research materials and instruments.
Funding
Funds for the present study, were provided through
the Milton Lev Faculty Research Fund, an internal grant awarded to
BP by The University of the Sciences.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on request.
Authors' contributions
All of the authors designed the study. AI and NF
performed the experiments. AI and NF wrote the manuscript. BP
revised the manuscript critically for important intellectual
content. All authors reviewed, read, and approved the manuscript
and agree to be accountable for all aspects of the research in
ensuring that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mitchell WG, Davalos-Gonzalez Y, Brumm VL,
Aller SK, Burger E, Turkel SB, Borchert MS, Hollar S and Padilla S:
Opsoclonus-ataxia caused by childhood neuroblastoma: Developmental
and neurologic sequelae. Pediatrics. 109:86–98. 2002.PubMed/NCBI
|
|
2
|
Yahya FS and Al-Shami HA: Posterior
mediastinal neuroblastoma masked as flaccid paraparesis in a 3 year
child. Neurosciences (Riyadh). 24:320–323. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Louis CU and Shohet JM: Neuroblastoma:
Molecular pathogenesis and therapy. Annu Rev Med. 66:49–63. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Habib EE, El-Kashef AT and Fahmy ES:
Management of neuroblastoma: A study of first-and second-line
chemotherapy responses, a single institution experience. Oncol Rev.
6:e32012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mandal A: Neuroblastoma treatment.
(online) News- Medical.net. Available at:
https://www.news-medical.net/health/Neuroblastoma-Treatment.aspx.
Feb 27–2019
|
|
6
|
Adamson PC, Matthay KK, O'Brien M, Reaman
GH, Sato JK and Balis FM: A phase 2 trial of all-trans-retinoic
acid in combination with interferon-alpha2a in children with
recurrent neuroblastoma or Wilms tumor: A pediatric oncology
branch, NCI and children's oncology group study. Pediatr Blood
Cancer. 49:661–665. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Reynolds CP, Matthay KK, Villablanca JG
and Maurer BJ: Retinoid therapy of high-risk neuroblastoma. Cancer
Lett. 197:185–192. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shipley MM, Mangold CA and Szpara ML:
Differentiation of the SH-SY5Y human neuroblastoma cell line. J Vis
Exp. 17:531932016.
|
|
9
|
Canon E, Cosgaya JM, Scsucova S and Aranda
A: Rapid effects of retinoic acid on CREB and ERK phosphorylation
in neuronal cells. Mol Biol Cell. 15:5583–5592. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Allenby G, Bocquel MT, Saunders M, Kazmer
S, Speck J, Rosenberger M, Lovey A, Kastner P, Grippo JF and
Chambon P: Retinoic acid receptors and retinoid X receptors:
Interactions with endogenous retinoic acids. Proc Natl Acad Sci
USA. 90:30–34. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nagai J, Yazawa T, Okudela K, Kigasawa H,
Kitamura H and Osaka H: Retinoic acid induces neuroblastoma cell
death by inhibiting proteasomal degradation of retinoic acid
receptor alpha. Cancer Res. 64:7910–7917. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Petrova IM, Malessy MJ, Verhaagen J,
Fradkin LG and Noordermeer JN: Wnt signaling through the Ror
receptor in the nervous system. Mol Neurobiol. 49:303–315. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rangel MC, Bertolette D, Castro NP,
Klauzinska M, Cuttitta F and Salomon DS: Developmental signaling
pathways regulating mammary stem cells and contributing to the
etiology of triple-negative breast cancer. Breast Cancer Res Treat.
156:211–226. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fultang N, Illendula A, Chen B, Wu C,
Jonnalagadda S, Baird N, Klase Z and Peethambaran B: Strictinin, a
novel ROR1-inhibitor, represses triple negative breast cancer
survival and migration via modulation of PI3K/AKT/GSK3ß activity.
PLoS One. 14:e02177892019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Borcherding N, Kusner D, Liu GH and Zhang
W: ROR1, an embryonic protein with an emerging role in cancer
biology. Protein Cell. 5:496–502. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Blanc E, Roux GL, Benard J and Raguenez G:
Low expression of Wnt-5a gene is associated with high-risk
neuroblastoma. Oncogene. 24:1277–1283. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bastien J, Plassat JL, Payrastre B and
Rochette-Egly C: The phosphoinositide 3-kinase/Akt pathway is
essential for the retinoic acid-induced differentiation of F9
cells. Oncogene. 25:2040–2047. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Akkuratov EE, Wu J, Sowa D, Shah ZA and
Liu L: Ouabain-induced signaling and cell survival in SK-N-SH
neuroblastoma cells differentiated by retinoic acid. CNS Neurol
Disord Drug Targets. 14:1343–1349. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hämmerle B, Yañez Y, Palanca S, Cañete A,
Burks DJ, Castel V and Font de Mora J: Targeting neuroblastoma stem
cells with retinoic acid and proteasome inhibitor. PLoS One.
8:e767612013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Niewiarowska-Sendo A, Patrzalek K, Kozik A
and Guevara-Lora I: The effect of differentiation agents on
inflammatory and oxidative responses of the human neuroblastoma
cell line SK-N-SH. Acta Biochim Pol. 62:435–443. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gassmann M, Grenacher B, Rohde B and Vogel
J: Quantifying Western blots: Pitfalls of densitometry.
Electrophoresis. 30:1845–1855. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tan HY and Ng TW: Accurate step wedge
calibration for densitometry of electrophoresis gels. Opt Commun.
281:3013–3017. 2008. View Article : Google Scholar
|
|
23
|
Pemberton K, Mersman B and Xu F: Using
ImageJ to assess neurite outgrowth in mammalian cell cultures:
Research data quantification exercises in undergraduate
neuroscience lab. J Undergrad Neurosci Educ. 16:A186–A194.
2008.
|
|
24
|
Clevers H: Wnt/beta-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Y, Chakravarty P, Ranes M, Kelly G,
Brooks PJ, Neilan E, Stewart A, Schiavo G and Svejstrup JQ:
Dysregulation of gene expression as a cause of cockayne syndrome
neurological disease. Proc Natl Acad Sci USA. 111:14454–14459.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Frumm SM, Fan ZP, Ross KN, Duvall JR,
Gupta S, VerPlank L, Suh BC, Holson E, Wagner FF, Smith WB, et al:
Selective HDAC1/HDAC2 inhibitors induce neuroblastoma
differentiation. Chem Biol. 20:713–725. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Almstedt E, Elgendy R, Hekmati N, Rosén E,
Wärn C, Olsen TK, Dyberg C, Doroszko M, Larsson I, Sundström A, et
al: Integrative discovery of treatments for high-risk
neuroblastoma. Nat Commun. 11:712020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ye C, Ho DJ, Neri M, Yang C, Kulkarni T,
Randhawa R, Henault M, Mostacci N, Farmer P, Renner S, et al:
DRUG-seq for miniaturized high-throughput transcriptome profiling
in drug discovery. Nat Commun. 9:43072018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Koster J, Volckmann R, Zwijnenburg D,
Molenaar P and Versteeg R: R2: Genomics analysis and visualization
platform. Cancer Res. 24902019.
|
|
31
|
Tarsa L and Goda Y: Synaptophysin
regulates activity-dependent synapse formation in cultured
hippocampal neurons. Proc Natl Acad Sci USA. 99:1012–1016. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rogers HA, Ward JH, Miller S, Lowe J,
Coyle B and Grundy RG: The role of the WNT/ß-catenin pathway in
central nervous system primitive neuroectodermal tumours (CNS
PNETs). Br J Cancer. 108:2130–2141. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kam RK, Deng Y, Chen Y and Zhao H:
Retinoic acid synthesis and functions in early embryonic
development. Cell Biosci. 2:112012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Donovan M, Olofsson B, Gustafson AL,
Dencker L and Eriksson U: The cellular retinoic acid binding
proteins. J Steroid Biochem Mol Biol. 53:459–465. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lindley D, Goranov B, Ponthan F and
Redfern C: Abstract# 5280: The role of retinoic acid receptors in
differentiation, gene expression and apoptosis of neuroblastoma.
Cancer Res. 69 (9 Suppl):S52802009.
|
|
36
|
Kedishvili NY: Retinoic acid synthesis and
degradation. Subcell Biochem. 81:127–161. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Peinemann F, van Dalen EC, Enk H and
Berthold F: Retinoic acid postconsolidation therapy for high-risk
neuroblastoma patients treated with autologous haematopoietic stem
cell transplantation. Cochrane Database Syst Rev.
8:CD0106852017.PubMed/NCBI
|
|
38
|
Chen MC, Hsu SL, Lin H and Yang TY:
Retinoic acid and cancer treatment. Biomedicine (Taipei). 4:222014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bown N: Neuroblastoma tumour genetics:
Clinical and biological aspects. J Clin Pathol. 54:897–910. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Boeva V, Louis-Brennetot C, Peltier A,
Durand S, Pierre-Eugene C, Raynal V, Etchevers HC, Thomas S,
Lermine A, Daudigeos-Dubus E, et al: Heterogeneity of neuroblastoma
cell identity defined by transcriptional circuitries. Nat Genet.
49:1408–1413. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ross RA, Biedler JL and Spengler BA: A
role for distinct cell types in determining malignancy in human
neuroblastoma cell lines and tumors. Cancer Lett. 197:35–39. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Qiao J, Paul P, Lee S, Qiao L, Josifi E,
Tiao JR and Chung DH: PI3K/AKT and ERK regulate retinoic
acid-induced neuroblastoma cellular differentiation. Biochem
Biophys Res Commun. 424:421–426. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fultang N, Illendula A, Lin J, Pandey MK,
Klase Z and Peethambaran B: ROR1 regulates chemoresistance in
Breast Cancer via modulation of drug efflux pump ABCB1. Sci Rep.
10:18212020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
O'Connell MP, Marchbank K, Webster MR,
Valiga AA, Kaur A, Vultur A, Li L, Herlyn M, Villanueva J, Liu Q,
et al: Hypoxia induces phenotypic plasticity and therapy resistance
in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer
Discov. 3:1378–1393. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Park H, Awasthi A, Ayello J, Chu Y,
Riddell SR, Rosenblum J, Lee DA and Cairo MS: ROR1-specific
chimeric antigen receptor (CAR) NK cell immunotherapy for high risk
neuroblastomas and sarcomas. Biol Blood Marrow Transplant.
23:S136–S137. 2017. View Article : Google Scholar
|
|
46
|
Dave H, Butcher D, Anver M and Bollard CM:
ROR1 and ROR2-novel targets for neuroblastoma. Pediatr Hematol
Oncol. 36:352–364. 2019. View Article : Google Scholar : PubMed/NCBI
|