Introduction
Protein S (PS) is a naturally occurring
anticoagulant. Primarily produced by the liver, PS circulates in
the plasma at a concentration of 350 nM and mediates its
anticoagulant function by either direct binding to Factor IXa or as
a cofactor of activated protein C (APC) and tissue factor pathway
inhibitor (TFPI) (1–3). PS also has a variety of
non-anticoagulant functions (4). PS
binds and activates a family of receptor tyrosine kinases
collectively known as TAM (Tryo3, Axl, and Mer) receptors (5,6).
PS-TAM receptor interaction regulates a number of physiological
processes, such as cell proliferation, survival, apoptosis,
vasculogenesis, and inflammatory cytokine release (7–10).
Growth arrest specific 6 (GAS6) protein is another
TAM receptor ligand (5). Heart,
lungs, kidneys, endothelial, and muscle cells produce GAS6. Despite
the structural homology (~42%) shared with PS, GAS6 functions only
in activating TAM receptors (11–13).
Both PS and GAS6 bind the different TAM receptors with variable
affinities (14). GAS6 affinity for
the TAMs are Axl > Tyro3 >> Mer; PS activates Mer and
Tyro3, although its binding with Axl is still debated (12). Small molecule inhibitors and
knockdown of TAM receptors are approaches that have been
investigated as therapies to inhibit proliferation of various types
of tumors; however, these studies have had limited success in
inhibition of proliferation (15).
Paradoxically, although TAM receptor inhibition restricts tumor
progression, the downstream TAM signaling necessary for
phagocytosis and other antitumor effector functions are also
inhibited (15–17). Furthermore, because TAM receptors
and their ligands are implicated in multiple pathways, therapies
that target these receptors must take into account the possibility
of adverse responses (14).
Pancreatic ductal adenocarcinoma (PDAC) is estimated
to be the fourth most lethal form of cancer in the United States
(18). The systemic nature of the
disease confers poor prognosis, with an overall median survival of
5–8 months and with <5% of patients surviving more than 5 years
(19). Cancer patients are at an
increased risk of venous thromboembolism (VTE), the highest
incidence occurring among individuals with pancreatic and brain
cancers (20–22). Lindahl et al reported that,
during disease progression, patients with pancreatic cancer
exhibited a significant decrease in plasma anticoagulants such as
antithrombin, protein C, and free PS (23). However, the effect of the decrease
in free PS on TAM signaling has not been investigated.
In the present study, the ratio of PS and GAS6 was
revealed to be associated to a function of the aggressiveness (time
of replication) of the two cell lines, PANC-1 and MIA PaCa-2. MIA
PaCa-2 is currently used as an in vitro model of PDAC
carcinogenesis (24). We then
modulated the PS/GAS6 ratio to confirm that the balance between the
two proteins determines the cell survival and proliferation rate.
GAS6 overexpression enhanced survival and proliferation of PANC-1
and MIA PaCa-2 cells. Conversely, either overexpression of PS or
knocking down of GAS6 inhibited cell proliferation and promoted
apoptosis. Notably, the degree of inhibition of proliferation by
either PS overexpression or GAS6 knockdown was comparable to that
achieved with a Mer-specific inhibitory drug. It was concluded that
PS functioned as a natural promoter of apoptosis in pancreatic
cancer cells. In addition, the present study demonstrated that
increased expression of PS could be a strategy for reducing
aggressiveness of PDAC without targeting essential TAM receptor
signaling pathways.
Materials and methods
Cell culture
Human pancreatic cancer cell lines (PANC-1, MIA
PaCa-2, and BxPC-3) were obtained from ATCC. PANC-1 cells were
grown in high glucose DMEM supplemented with 10% fetal bovine serum
(FBS) (Life Technologies; Thermo Fisher Scientific, Inc.) and (1X)
antibiotic-antimycotic solution (Life Technologies; Thermo Fisher
Scientific, Inc.) at 37°C with 5% CO2. MIA PaCa-2
received the same complete medium with supplementation of horse
serum (Life Technologies; Thermo Fisher Scientific, Inc.) to a
final concentration of 2.5%. BxPC-3 cells were grown in RPMI-1640
medium (Life Technologies; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS and antibiotic-antimycotic solution.
These cell lines were selected based on the variations in the cell
doubling times which were referred to as the aggressiveness of the
cell. MIA PaCa-2 has been revealed to have the shortest doubling
time (40 h), PANC-1 a moderate doubling time (52 h) and BxPC-3 the
highest doubling time (72 h) (25,26).
BxPC-3 cells were not used in our PS/Gas6 overexpression studies
because the cells grow too slowly. We could not use healthy
pancreatic duct epithelial cells due to their unavailability.
Regarding other non-cancerous control cells, such as H6C7, which
are commercially available, since these cell lines are immortalized
and their phenotypes are not the same as the primary cells
(27,28) they were not used. Instead,
respective empty vectors were used as controls since we
overexpressed or knocked down the genes in our experiments.
Transfection and lentiviral
infection
PANC-1 and MIA PaCa-2 cells were separately seeded
in 6-well plates at 1.5×106 cells/well and transfected
with pcDNA-V5-His containing Pros1 (protein S) insert, a
gift from Dr Rezende (29). The
transfection was performed with 4 µg of plasmid DNA and
Lipofectamine 2000 (Life Technologies; Thermo Fisher Scientific,
Inc.) as previously described (30). The cells were subjected to 10 µg/ml
Blasticidin (Sigma-Aldrich; Merck KGaA) antibiotic selection to
obtain stable expression clones. For GAS6 overexpression and
knockdown studies, we obtained from Dr Sonja Loges (University
Medical Center Hamburg-Eppendorf, Hamburg, Germany) LeGo-iG2-Puro
(GFP-expressing) vector containing the GAS6 insert and LeGO-C-Zeo
(mCherry expressing) containing shGAS6 insert, which we verified by
sequencing prior to use. The production of lentiviral particles and
the lentiviral gene transfer into PANC-1 and MIA PaCa-2 cells were
performed as outlined by Weber et al (31). For detailed protocols and vector
maps, please refer to http://www.lentigo-vectors.de. Generation of virus
particles and titration were performed as previously described
(32). The GAS6-overexpressing
cells were subsequently sorted by FACS analysis for GFP-positive
cells and cultured to generate stable cell lines. The cells with
GAS6 knocked down were stably selected with 500 µg/ml Zeocin
(Thermo Fisher Scientific, Inc.).
RT-qPCR assessment of gene
expression
Cells were seeded in 6-well plates at a density of
3×105 cells/well. RNA was isolated using RNeasy Plus
Mini Kit (Qiagen, Inc.), according to the manufacturer's
instructions. The RNA concentration was measured via NanoDrop
(Thermo Fisher Scientific, Inc.). Two µg of RNA was used to
synthesize cDNA. RT-qPCR was performed with qScript®
cDNA Supermix (Quanta BioSciences, Inc.), according to the
manufacturer's instructions. The quantification of the genes of
interest was performed with PerfecCTa SYBR Green FastMix (Quanta
BioSciences, Inc.) according to the manufacturer's instructions.
The following primers were used: 5′-TCATGAAGATCCTCACCGAG-3′
(forward) and 5′-TTGCCAATGGTGATGACCTG-3′ (reverse) for β-Actin;
5′-ATCAAGGTCAACAGGGATGC-3′ (forward) and 5′-CTTCTCCGTTCAGCCAGTTC-3′
(reverse) for GAS6; 5′-CCTAGTGCTTCCCGTCTCAG-3′ (forward) and
5′-TTTCCGGGTCATTTTCAAAG-3′ (reverse) for Pros1;
5′-ACACCCCAGAGGTGCTAATG-3′ (forward) and 5′-ACGAGAAGGCAGGAGTTGAA-3′
(reverse) for Axl; 5′-TGCCCTGGGAATGGAGTATC-3′ (forward) and
5′-ATCTTAGCAATGCGGCCTTG-3′ (reverse) for Mertk;
5′-GTGGCTGACTTCGGACTGTC-3′ (forward) and 5′-AGCTGTCATGATCTCCCACA-3′
(reverse) for Tyro3. The qPCR conditions included initial
denaturation at 95°C for 5 min, denaturation at 95°C for 15 sec
annealing and extension at 60°C for 1 min with 40 cycles. To
analyze the expression levels, the average of each sample was used
according to its primer and the following calculations were
performed to determine the fold change: ΔCq=(PROS1-actin),
ΔCq=(GAS6-actin)→ΔΔCq=(ΔCqsample-ΔCqreference)→Fold
Change=2−ΔΔCq (33).
Flow cytometry
PANC-1 and MIA PaCa-2 stable cell lines
overexpressing control vector (pCDNA6-V5-His) or PS were produced
as aforementioned. These stable cell lines were synchronized by
serum starvation for 60 h. After synchronization, the cells were
washed with PBS and collected by treatment with Accutase (cat. no.
AT-104; Innovative Cell Technologies, Inc.) digestion performed at
room temperature for 5 min. The Accutase was neutralized by regular
growth medium containing 10% FBS. Cells were stained for Annexin-V
and 7AAD using BD Biosciences (cat. no. 559763) and analyzed for
Annexin-V and 7AAD binding using FACSCalibur. We were unable to
perform flow cytometric experiments with the cells that were
transfected with Gas-6-knockdown construct since the constructs
contained GFP as internal control.
Protein expression via
immunoblotting
PANC-1 and MIA PaCa-2 cells [empty vector (EV)
control], PS overexpressing, GAS6 overexpressing and knocked down)
were separately plated in 6-well plates at a density of
3×105 cells/ml and grown for 48 h. The cells were lysed
in RIPA buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM Na2 EDTA,
1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM
Na4P2O7, 1 mM
Na3VO4), and total lysate protein was
assessed by BCA assay (Thermo Fisher Scientific, Inc.). SDS-PAGE
(10%) and immunoblotting were performed with 40 µg of protein from
each sample. With the exception of sheep PS antibody (dilution
1:1,000; cat. no. PAHPS-S; Haematologic Technologies, LLC), the
following primary antibodies used for probing were obtained from
Cell Signaling Technology (CST), Inc.: Mouse phospho-p53 antibody
(dilution 1:1,000; product no. 9286) and rabbit phospho-HSP27
antibody (dilution 1:1,000; product no. 9709). All blots were
normalized with rabbit β-actin antibody (dilution 1:1,000; product
no. 4967S). Blots were incubated with primary antibodies at 4°C
overnight. After washing, the blots were incubated with the
appropriate horseradish peroxidase-conjugated secondary antibody at
room temperature for 30 min. The secondary antibodies obtained from
Cell Signaling Technology, Inc. were the following: Anti-rabbit
IgG, HRP-linked antibody (dilution 1:5,000; product no. 7074) and
anti-mouse IgG, HRP-linked antibody (dilution 1:5,000; product no.
7076) was obtained from Cell Signaling Technology, Inc. For PS
detection, anti-sheep IgG, HRP-linked secondary antibody (dilution
1:5,000; cat. no. 81–8620) was obtained from Thermo Fisher
Scientific, Inc. Images were developed using ECL reagent (cat. no.
32109; Thermo Fisher Scientific, Inc.) with Amersham Imager 600 (GE
Healthcare). All immunoblots were repeated thrice, and the band
densities were quantitated by ImageJ V1.51 software (National
Institutes of Health).
Cell viability assay using MTT
dye
Cells were plated in 96-well plates at a density of
5×103 cells/well in 90 µl of complete medium. After one
and five days of incubation, 10 µl of 5 mg/ml MTT (Sigma-Aldrich;
Merck KGaA) was added to the cells and further incubated for 4 h.
The supernatants were discarded, 100 µl of DMSO was added per well,
and the crystals were dissolved by agitating for 5 min. The
absorbance value was measured by a microplate reader at 570 nm. The
relative fold change of absorbance between PS/GAS6-overexpressing
cells compared with control cells that contained the empty vector
was calculated for the subsequent days, with the day 1 absorbance
value as the baseline. At least three independent experiments were
performed. For experiments involving drug treatments, UNC2025
(MedChemExpress) was added to cells at variable concentrations
alongside a similar volume of vehicle control. Treatment was
performed 8 h after cell seeding and readings were obtained on days
1, 3, and 5.
Cell proliferation assay using crystal
violet
To assess the aggressiveness of single cells with
differing genotypes to grow into a colony, PANC-1 and MIA PaCa-2
cells (EV control, PS-overexpressing, GAS6-overexpressing) were
plated at a low density (1,000 cells/well) in 6-well plates and
allowed to generate single colonies for 14 days. The medium was
replenished every three days. The colonies were washed twice in PBS
and stained with 0.5% (v/v) crystal violet for 20 min at room
temperature as previously described (34). The plates were imaged under white
light with Amersham Imager 600 and colonies with a minimum of 50
cells in each, were compared using Clono-Counter software (35).
Statistical analysis
Data from PS, Gas6 or Gas6 shRNA or overexpressing
cells were compared against an empty vector or a scramble shRNA
vector control. The test group data were analyzed against the
control group using one-way ANOVA. A Bonferroni correction was
applied to the P-values obtained from ANOVA to control for multiple
comparisons, and this was performed separately for each of the two
cell lines PANC-1 and MIA PaCa-2. The data for the colony
formation, MTT assays were expressed as the mean ± standard
deviation (SD) and error bars in the figures represent the SD.
Statistical analysis was performed using GraphPad Prism 7 software
(GraphPad Software, Inc.). P-values of <0.05 were considered to
indicate a statistically significant difference.
Results
PS/GAS6 ratio is directly associated
with the aggressiveness of pancreatic cancer cell lines
Since the selected cell lines were similar in
origin, it was investigated whether cell growth rate was related to
the basal levels of PS and GAS6. MIA PaCa-2, PANC-1, and BXPC-3
with population doubling times of 40, 52 and 72 h respectively (per
information from ATCC) were used. The basal levels of the three TAM
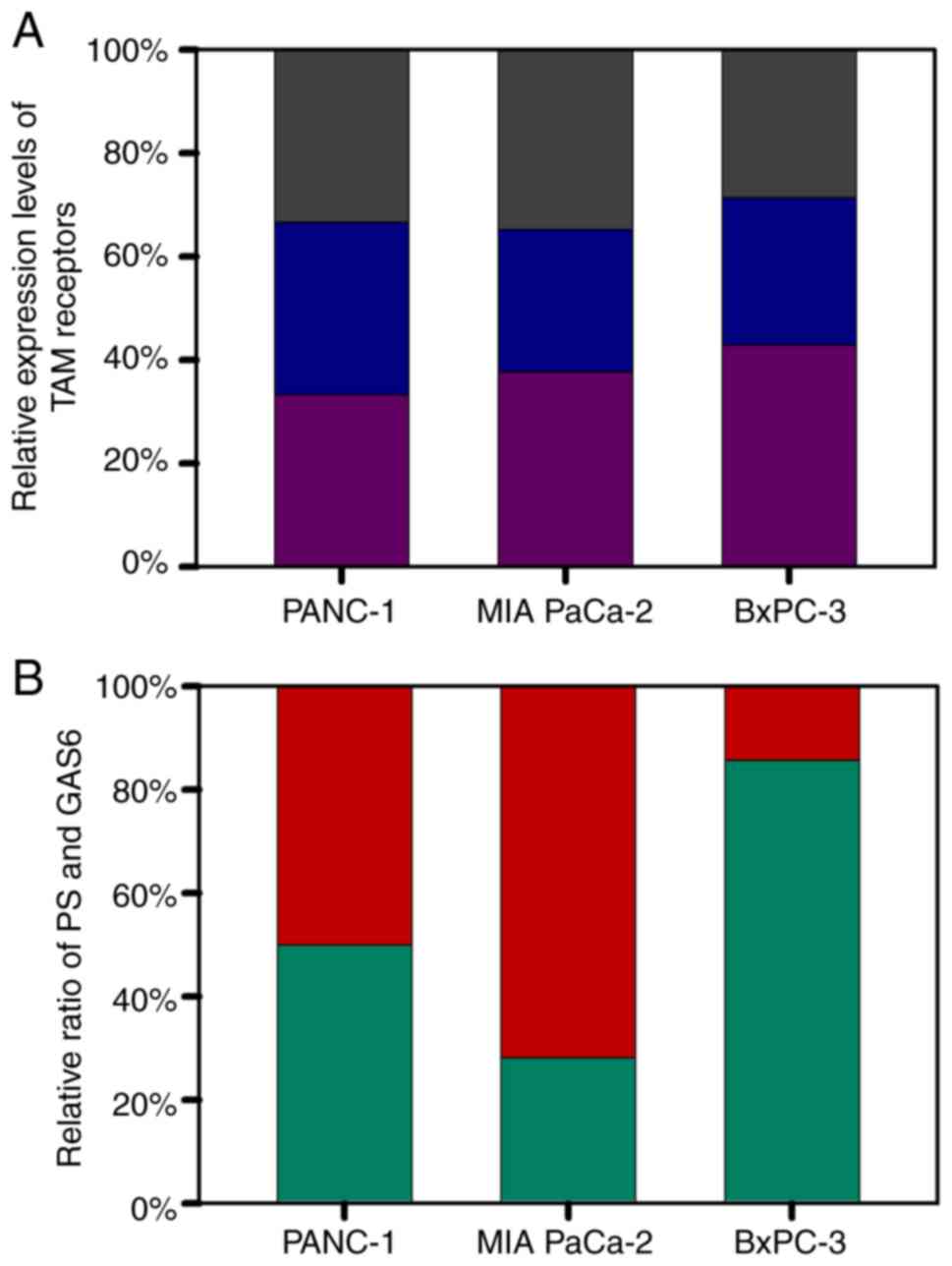
receptors were relatively uniform among the three cell lines, i.e.,
less than 5% difference in the expression level (Fig. 1A). Notably, although PANC-1 cells,
with an intermediate degree of aggressiveness, had a PS/GAS6 ratio
of 1, MIA PaCa-2 cells had a significantly lower PS/GAS6 ratio
(2.183:5.588) (indicating that the most aggressive cell line had
more Gas6) and BXPC-3 cells, the least aggressive cell line, had a
markedly higher PS/GAS6 ratio (12.817:2.151) (Fig. 1B). These data associated the degree
of aggressiveness of the cell lines to the PS/GAS6 ratio, and the
data revealed that a lower PS/GAS6 ratio was associated with
increased proliferative potential, whereas a higher ratio was
associated with decreased proliferative ability.
PS overexpression reduces the survival
and proliferation of PANC-1 and MIA PaCa-2 cells
Since the relative levels of PS and GAS6 were
associated with the aggressiveness of pancreatic cancer cell lines,
the PS/GAS6 ratios were altered to confirm their importance in
determining proliferative potential. The expression of
apoptotic/survival markers of PANC-1 and MIA PaCa-2 cells stably
overexpressing PS was first assessed by immunoblotting and was
compared with the corresponding EV control cells. Subsequently
these data were associated with the aggressiveness of cell growth
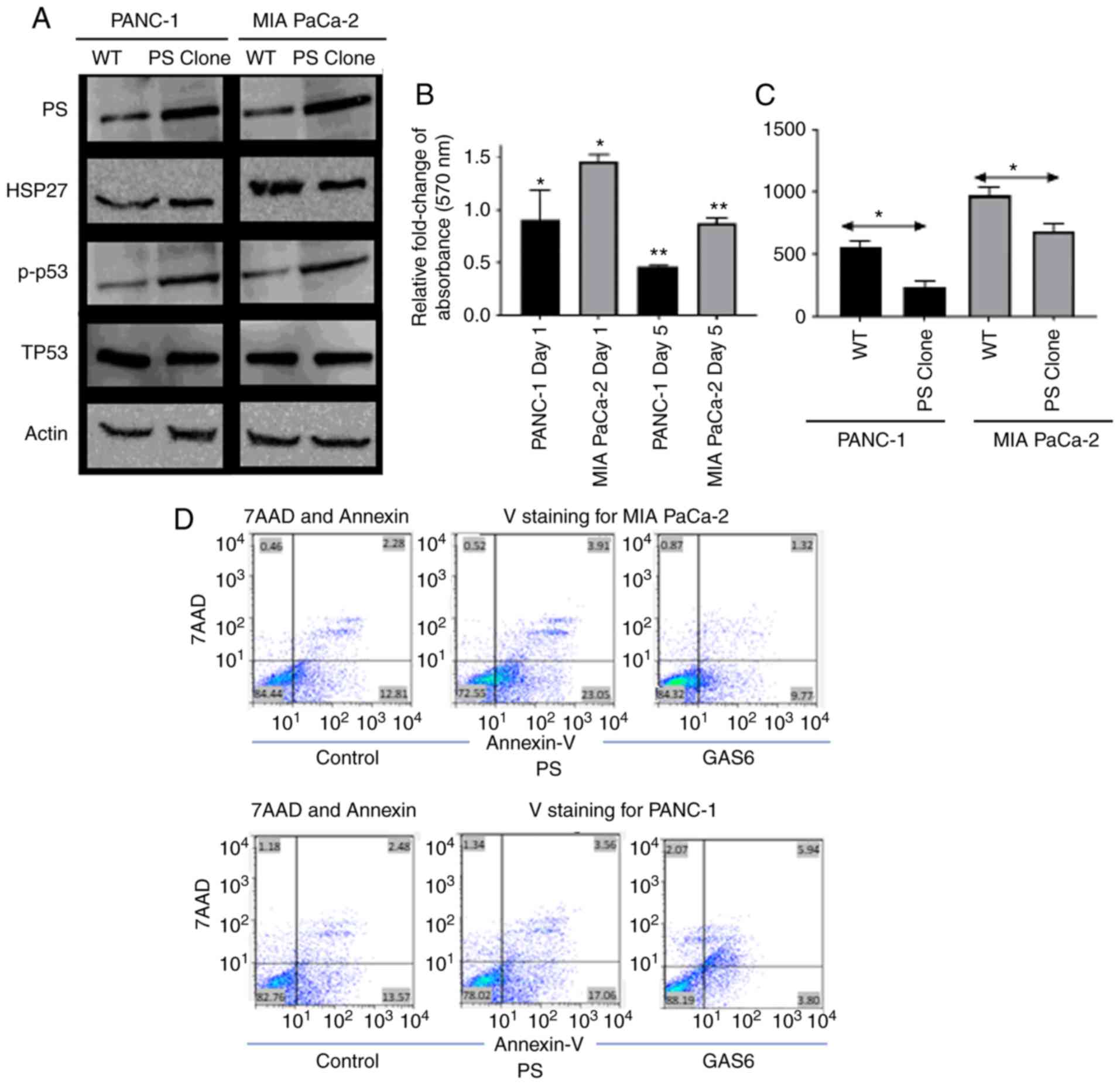
and proliferation rate. In both cell lines, overexpression of PS
was associated with increased expression of the apoptotic marker
phospho-p53 and decreased expression of the survival marker HSP27
(25 and 15% reduction in immunoblot band intensities for PANC-1 and
MIA PaCa-2 cells, respectively; Fig.
2A). PS overexpression increased apoptosis (Fig. 2D) and caused a decrease in the
survival of both cell lines, as assessed by the MTT assay (Fig. 2B). PS-overexpressing PANC-1 and MIA
PaCa-2 cells exhibited 0.5-fold and 0.8-fold reductions in cell
survival on Day 5 compared with the corresponding EV control cells
(relative absorbance of the EV control was normalized to 1 and the
data is not presented in the figure). PANC-1 cells exhibited a
0.9-fold reduction in cell survival on day 1; however, MIA PaCa-2
cells did not exhibit any decrease but a significant increase in
the number of cells on day 1. For both cell lines, this further
resulted in a decreased number of colonies being formed during a
14-day period, compared with the EV control cells (Fig. 2C); PS overexpression had no
significant effect on endogenous Gas6 expression (data not shown).
The difference in aggressiveness in the growth rates of PANC-1 and
MIA PaCa-2 was clearly evident. MIA PaCa-2 cells formed 808/438 (EV
control/PS-overexpressing) colonies compared with 492/251 (EV
control/PS-overexpressing) colonies for PANC-1 cells. The data are
an average of the number of colonies calculated from three
independent experiments.
To further confirm that PS overexpression caused an
increase in apoptosis of pancreatic cells, flow cytometric data was
generated using Annexin V/7-Aminoactinomycin D (7AAD) staining.
Despite the considerable difference in the aggressiveness in growth
between the two cell lines, PS overexpression resulted in increased
Annexin binding for both cell lines. For example, 82±0.43% control
PANC-1 cells were double negative for Annexin V and 7AAD, 13±0.37%
were Annexin V-positive, 1.28±0.06% were 7AAD positive, and
3.06±0.32 were double positive. PANC1 cells that overexpressed PS
were 77.33±0.47 double negative, 18±0.53% Annexin V-positive,
1.24±0.16 7AAD-positive, and 3.34±0.11% double positive (Fig. 2D). Conversely, MIA PaCa-2 cells
transfected with control vector were 84.4±0.4% double negative,
12.3±0.67% Annexin V-positive, 0.8±0.17% 7AAD-positive, and
2.3±0.25% double positive. MIA PaCa-2 cells overexpressing PS were
72.79±1.48% double negative, 23.35±1.3% Annexin V-positive,
0.57±0.13% 7AAD-positive, and 3.2±0.33% double positive. Therefore,
the flow cytometric data (Fig. 2D)
revealed that overexpression of PS in PANC-1 increased the
apoptotic population of cells by 5%, whereas overexpression of PS
in MIA PaCa-2 cells increased the apoptotic population by 13%. The
difference in the effect of PS overexpression in PANC-1 compared to
MIA PaCa-2 may be attributed to the difference in the basal level
of endogenous PS in the cells.
GAS6 overexpression enhances the
survival and proliferation of PANC-1 and MIA PaCa-2 cells
GAS6 has been reported to promote prostate cancer
cell survival by inhibition of apoptotic pathways (36). It was investigated whether this
phenomenon occurred in pancreatic cancer cells. The comparative
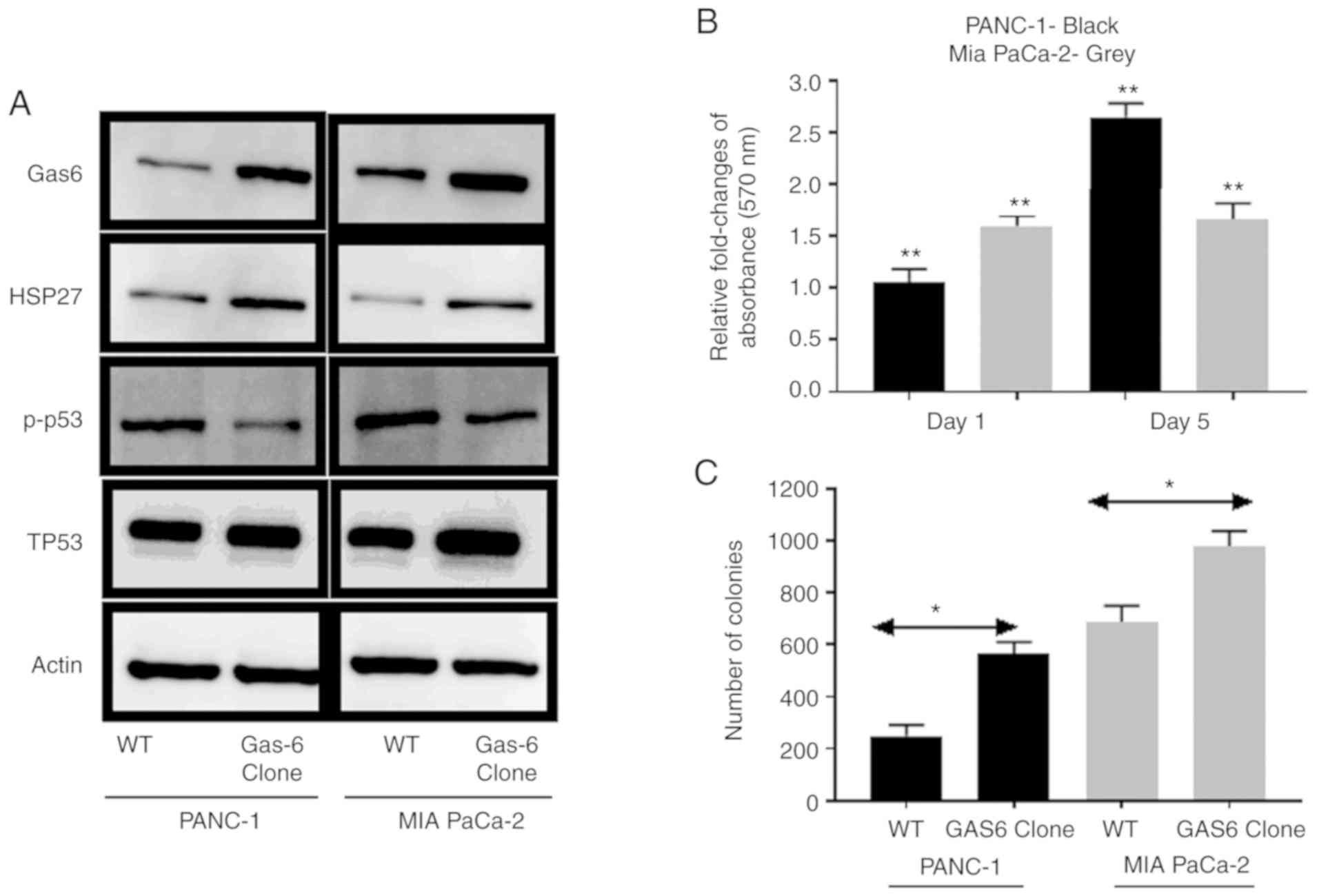
analysis of molecular markers revealed that stable overexpression
of GAS6 was associated with reduced signaling in apoptotic
phosphorylated (p)-p53 marker and enhanced survival signaling in
HSP27 compared with the EV control cells (Fig. 3A). The direct effect of
GAS6-mediated apoptotic inhibition was pronounced in PANC-1 cells,
in which GAS6 overexpression caused ~2.7-fold increase in relative
MTT assay absorbance on Day 5 compared with the EV control cells
(relative absorbance of the EV control was normalized to 1 and the
data is not presented in the figure), whereas in MIA PaCa-2 cells,
the increase was ~1.7-fold (Fig.
3B). Furthermore, the number of colonies formed by both cell
types was higher in GAS6-overexpressing cells compared with the EV
control cells (Fig. 3C). MIA PaCa-2
cells formed 689/977 (EV control/GAS6-overexpressing) colonies
compared with 249/563 (EV control/GAS6-overexpressing) colonies for
the PANC-1 cells. Exogenous overexpression of Gas6 had no
significant effect on the endogenous PS level (data not shown).
GAS6 knockdown reduces the survival
and proliferation of PANC-1 and MIA PaCa-2 cells
Since GAS6 overexpression was associated with
increased survival, it was anticipated that knockdown of GAS6 would
reduce growth and proliferation (37). This was confirmed from the
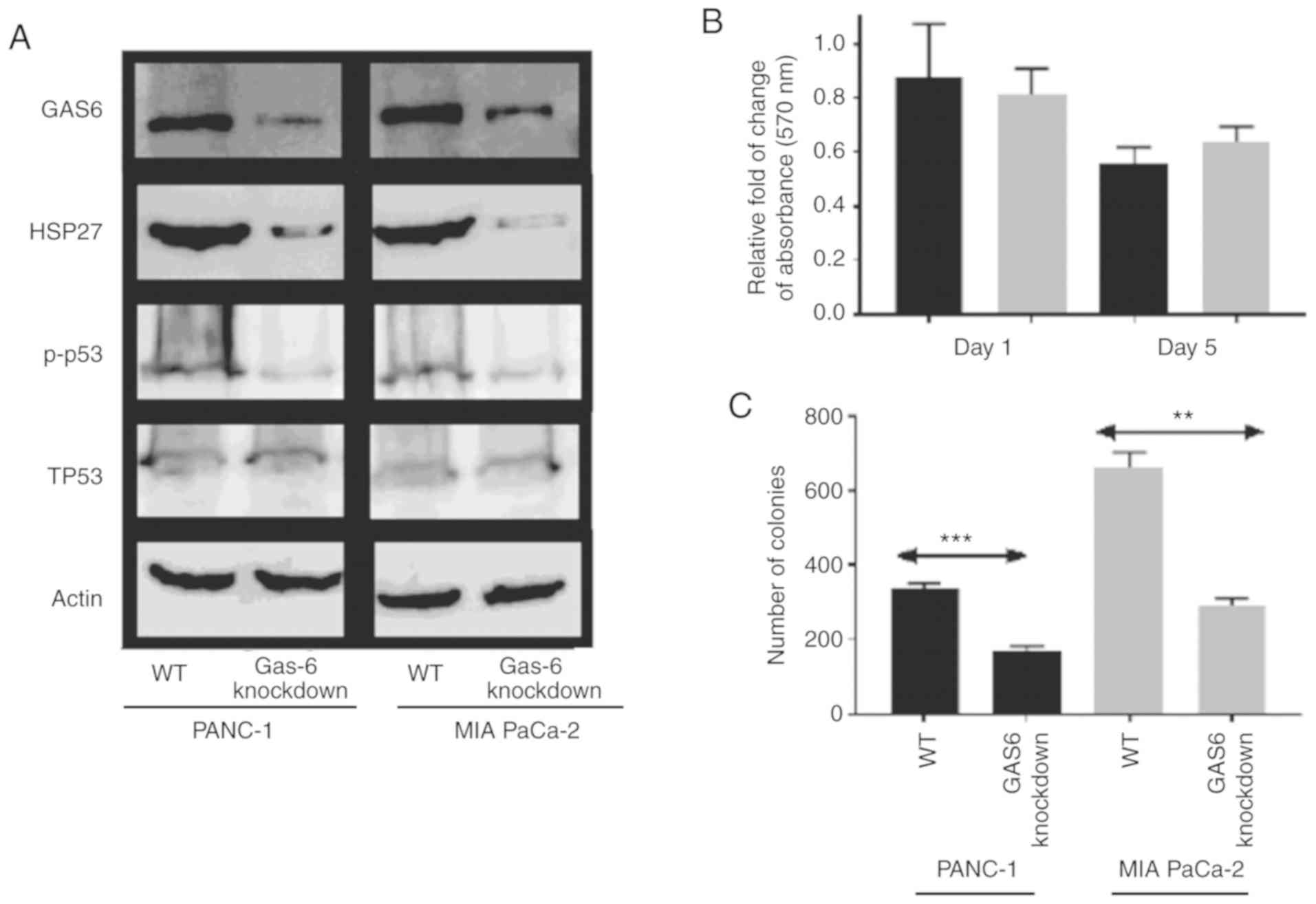
expression profiles of the apoptotic/survival markers, in which
p-p53 levels were reduced in GAS6-knockdown clones compared with
the EV control clones (Fig. 4A).
The reduced HSP27 signal in the knockdown clones indicated an
induction in apoptosis over survival, which, consequently, resulted
in a decrease in the relative change of MTT assay absorbance on Day
5 in PANC-1 and MIA PaCa-2 cells compared with the EV control cells
(relative absorbance of the EV control was normalized to 1 and the
data is not presented in the figure) (Fig. 4B). Colony formation assays revealed
a consistent, significantly higher number of colonies, 337 of
PANC-1 EV control cells compared with their respective
GAS6-knockdown clones, which produced 170 colonies (Fig. 4C). Conversely, MIA PaCa-2 cells
produced an average of 661 colonies for EV control clones compared
with 291 colonies for the GAS6-knockdown clones.
PS overexpression produces effects
similar to the Mer-inhibitor UNC2025 in PANC1 and MIA PaCa-2
cells
PS is an important ligand for MER in vivo;
Mer overexpression in cancerous cells has been revealed to activate
survival and proliferative signaling pathways (38,39).
Consequently, Mer is a significant target for therapeutic
manipulation. A Mer-specific small molecule inhibitor, UNC2025, was
used as a potential agent for treatment of glioblastoma (40,41).
To determine whether UNC2025 initiated an apoptotic response, a
dose-dependent assay using UNC2025 was performed and it was
observed that 75 nM was an optimal concentration (data not shown).
Therefore, 75 nM was applied to PANC-1 and MIA PaCa-2 EV control
cells. An MTT assay revealed a marked reduction in cell survival
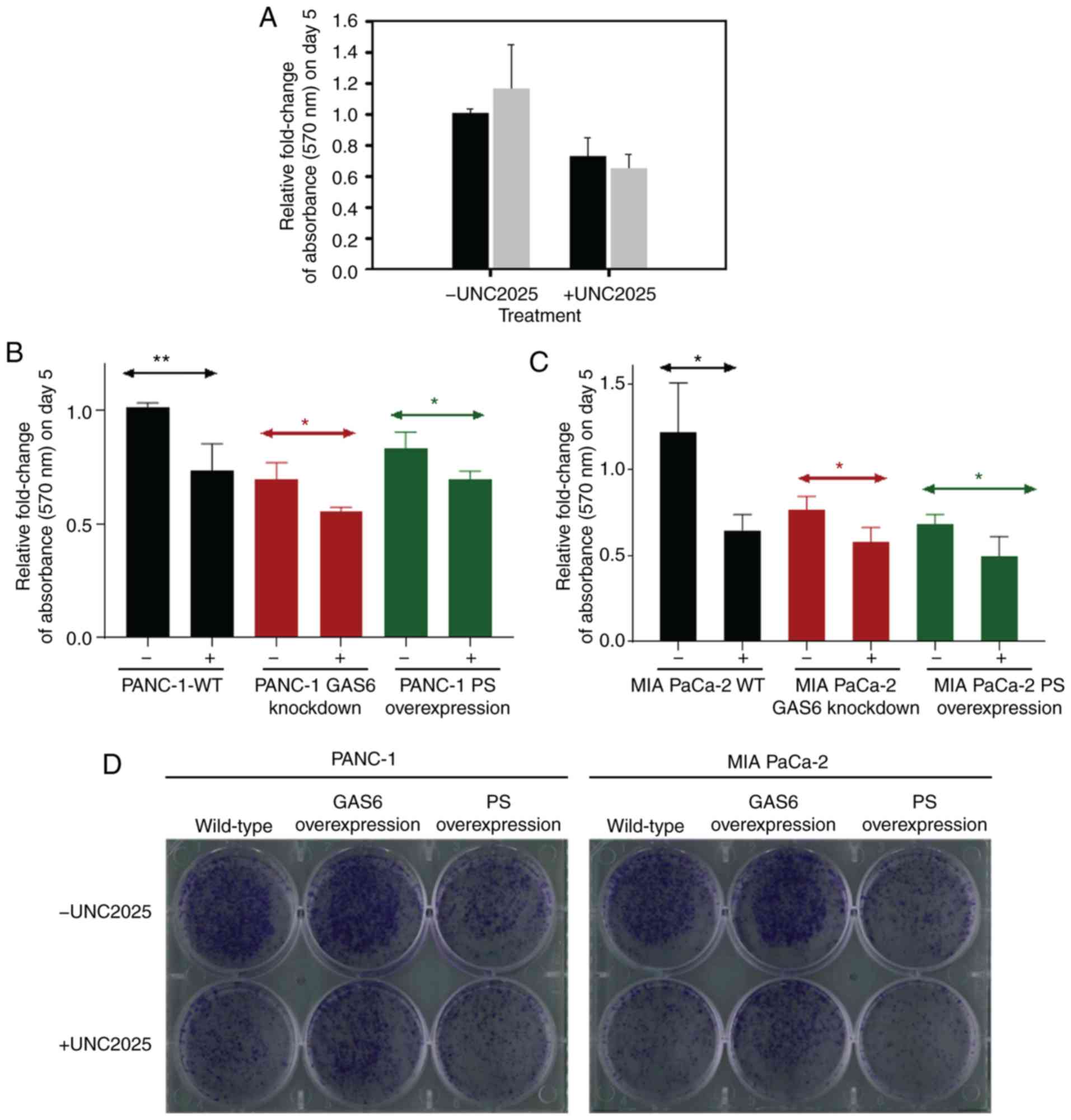
for the drug-treated cells (Fig.
5A). The differences in relative fold change of absorbance for
PANC-1 and MIA PaCa-2 cells subjected to drug treatment compared
with cells that received vehicle control were ~0.3-fold and
~0.5-fold, respectively, during the five days of treatment.
However, this assay did not distinguish whether apoptosis following
inhibition of Mer receptor signaling was due to the absence of
interaction by either GAS6, PS, or both (12,16).
Thus, once the anti-proliferative effect of the drug was
established, the effect of drug-induced apoptotic signaling and the
natural apoptosis signaling initiated by PS overexpression in the
pancreatic cancer cell lines was compared. A 0.69-fold relative
change of absorbance was observed following UNC2025 treatment in
PANC-1 GAS6-knockdown cells (Fig.
5B). This change was comparable to the PANC-1 PS overexpression
clones that exhibited a 0.67-fold relative change of absorbance
following treatment with the vehicle control. Further treatment
with UNC2025 further decreased this change to 0.56-fold in the
PS-overexpressing clones. MIA PaCa-2 GAS6-knockdown cells exhibited
a milder, 0.59-fold relative change of absorbance following UNC2025
treatment compared with the MIA PaCa-2 PS overexpression clones
that exhibited 0.64-fold relative change of absorbance when treated
with the vehicle (Fig. 5C).
However, the 0.65-fold relative change of absorbance following
UNC2025 treatment of MIA PaCa-2 EV control cells was exactly the
same as that of PS-overexpressing clones under vehicle treatment,
with UNC2025 further decreasing the change to 0.49-fold.
A colony formation assay was used to assess the
long-term efficacy of this PS-mediated decrease of cell
proliferation. PS-overexpressing PANC-1 and MIA PaCa-2 cells were
plated alongside the corresponding EV control cells and
GAS6-overexpressing cells, each receiving either 75 nM UNC2025 or
vehicle control. PS overexpression clones without drug treatment
had decreased cell proliferation and colony formation to extents
comparable to those of EV control cells and GAS6-overexpressing
clones treated with the drug (Fig.
5D). These results provided conclusive evidence that PS
overexpression achieves the same effect as that of the
Mer-inhibitory drug UNC2025 in arresting cell proliferation. As
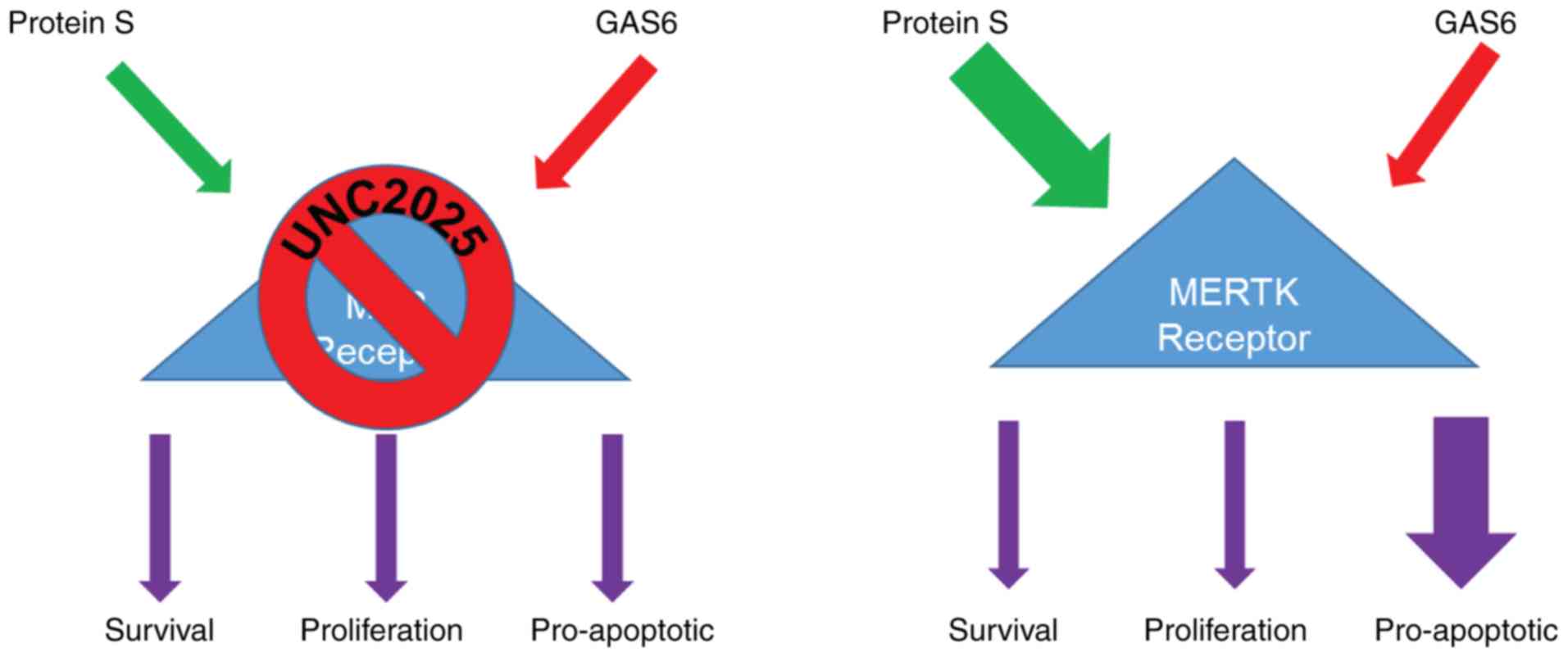
elucidated in Fig. 6, it was
confirmed that the PS/GAS6 ratio is crucial in terms of TAM
receptor signaling, and a change in this ratio can drive cancer
cells toward proliferation (GAS6 overexpression) or growth arrest
(PS overexpression/GAS6 knockdown).
Discussion
PS is a natural anticoagulant, whose plasma
concentration is reportedly reduced during the progression of PDAC
(23). However, its properties as a
signaling molecule in the context of cancer progression and
cellular aggressiveness have not been studied in-depth. In the
present study, two PDAC cell lines with variable degrees of
aggressiveness in growth were used. The expression levels of the
individual TAM receptors exhibited less than 1-fold variation
between the cell lines, whereas PS and GAS6 levels were
significantly different. With the PS and GAS6 levels of the
moderately aggressive cell line PANC-1 as a baseline, a higher GAS6
level was associated with the highly aggressive cell line MIA
PaCa-2, whereas significantly higher PS levels were exhibited by
the least aggressive BxPC-3 cell line. To associate PS and GAS6
signaling to a specific phenotype in the proliferative potential of
cells, overexpression studies on PANC-1 and MIA-PaCa-2 cells were
performed. Immunoblot analysis further revealed enhanced expression
of the apoptotic marker p-p53 and reduced expression of the
survival marker protein HSP27. Using MTT and colony formation
assays, both the short-term and the long-term pro-apoptotic effects
of PS overexpression were confirmed. These assays confirmed that PS
and GAS6 play vital roles in regulating the aggressiveness of
pancreatic cancer cell lines; however, the present study has a
limitation in elucidating the roles of PS and GAS-6 in the motility
of PANC-1 and MIA-PaCa-2. In general, aggressiveness of a cell line
is assessed by invasion and migration assays; however,
aggressiveness is also described in terms of cell doubling time and
thereby, we did not perform invasion or migration assays. We
studied the ability of the cell doubling time by colony formation
assay.
GAS6 overexpression was achieved by lentiviral
transduction using a LeGo-iG2-Puro plasmid containing the GAS6
gene. Overexpression of PS increased Annexin V binding compared
with the basal Annexin V of control cells. Similar results were
observed for MIA PaCa-2 cells. PS overexpression increased Annexin
V staining compared with cells carrying the control vector. Once
verified, PANC-1 and MIA PaCa-2 cells overexpressing GAS6 were
subjected to the same assays. The upregulation of survival protein
HSP27 was associated with an increased relative fold change of
absorbance as assessed by MTT assay, and the increased cell number
exhibited a proliferative phenotype caused by GAS6 overexpression.
GAS6 knockdown produced the opposite effect and the trend was
pro-apoptotic, similar to the effect of PS-overexpression. Both
plasmids (GAS6 overexpression/knockdown) have been used
successfully for lentivirus-mediated transduction, and the methods
were adhered to without any modification (42). By blocking the Mer receptor, the
drug UNC2025 effectively inhibited any downstream signaling that
results from GAS6-Mer and PS-Mer interaction. The present MTT
results revealed a growth inhibitory effect of UNC2025 on PANC-1
and MIA PaCa-2 cells. The efficacy of the PS-overexpression,
however, was comparable to the drug action. Thus, the present study
confirmed that overexpression of PS from a plasmid effectively
decreased aggressiveness of tumor cells, a phenomenon that could be
exploited to circumvent the side effects of drug-mediated blocking
of TAM receptors.
Globally, cancer-associated thrombosis (CAT) is a
significant risk factor in cancer treatment and therapy, but little
is known about its mechanistic basis (43–45).
Iodice et al examined a number of population studies and
inferred that patients with VTE have a six-fold higher risk of
developing pancreatic cancer than the general population (46). As reviewed by Sohail and Saif,
multiple events can result in hypercoagulability in pancreatic
cancer (47). Such events include
retroperitoneal location of tumors, tumor specific increase in
procoagulant factors (tissue factor, thrombin and fibrin), and
decreases in anticoagulants such as APC, TFPI, and PS. Initially
identified as a cofactor of APC (48,49),
PS was later found to mediate an APC-independent anticoagulant
function as a cofactor of TFPI (2,50).
Recently, we demonstrated a more direct function of PS in the
coagulation cascade, whereby PS directly binds to activated Factor
IX in an APC-independent manner and inhibits the formation of the
intrinsic tenase complex required for thrombus formation (51,52).
Incidentally, patients with heterozygous PS-deficiency exhibited an
increased risk of VTE (53). An
initial screening of the players in the coagulation pathway
revealed an interesting participant in PS, a well-known
anticoagulant with diverse functions, including serving as a
signaling molecule (7,43).
PS and GAS6 are TAM receptor family ligands
(54,55), with variable affinities for the
individual receptors (56). The
expression levels of the TAM receptors vary across cell and tissue
type and are generally upregulated in tumors (57). As a result, the downstream effects
of TAM signaling, such as proliferation, efferocytosis, and
anti-inflammatory activity, become dysregulated (39,58–60).
The overexpression of GAS6 and Axl has been reported in pancreatic
cancer, both in vitro and in vivo (61–63).
Shen et al have extensively elucidated the scope of
targeting TAM receptors for designing drugs intended to inhibit
cancer progression (64). They
reported that, despite considerable effort, application of
TAM-inhibitory drugs has had minimal success. The pathological
complexity of cancer and the global output of TAM-dependent
signaling pathways are significant obstacles for use of these drugs
because they cause severe side effects and are associated with drug
resistance. A screening of the common anticoagulants during the
progression of PDAC in a group of patients revealed an increase in
the plasma level of TFPI, whereas the level of free PS exhibited a
statistically significant decrease (23). A similar association of decreased PS
and increased GAS6 levels has been reported in patients with
systemic lupus erythromatosus, resulting in widespread autoimmune
disorders mediated by an imbalance in TAM signaling (65,66).
The main goal of the present study was to
demonstrate that overexpressing PS could be a strategy for reducing
aggressiveness of PDAC without targeting essential TAM receptor
signaling pathways. Thrombotic complications and
hypercoagulopathies are commonly associated with the progression of
PDAC. The amount of PS, a known anticoagulant, has been reported to
decrease in patients during the progression of PDAC, possibly
contributing to the hypercoagulopathies. PS has also been
identified as an important signaling molecule that binds a family
of tyrosine kinase receptors known as TAM receptors, thereby
triggering multiple downstream functions, such as cell survival,
proliferation, efferocytosis, and apoptosis. However, the
properties of PS as a signaling molecule in the context of cancer
progression and cellular aggressiveness have not been studied
adequately. In the present study, it was demonstrated that PS-TAM
interaction produced a pro-apoptotic effect, whereas GAS6-mediated
TAM signaling promoted proliferation and survival in select PDAC
cell lines. Both long-term and short-term effects of natural PS
overexpression were comparable with treatment of the cell lines
with UNC2025, a drug that inhibits the Mer-receptor. We intend to
study the thrombotic effects of PS in pancreatic cancer in the near
future.
The physiological importance of the present study
lies in the fact that a drug action was emulated by overexpressing
PS, a natural protein that is already reduced in pancreatic cancer
and whose deficiency is strongly associated with the severe
thrombotic complications that are synchronous with PDAC
progression. The present study can provide the basis for developing
newer strategies of PDAC treatment and concurrently improving the
quality of life of patients with cancer-associated thrombosis.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Louisiana
State University Health Sciences Center Special Appropriation Award
(0101500039) and the National Heart, Lung, and Blood Institute,
National Institutes of Health grant no. 5R01HL118557-02.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
VSP performed the experiments, analyzed the data,
and assisted in revising the manuscript by collecting new data. ADa
performed the experiments, analyzed the data, and wrote the initial
version of the manuscript. ADo maintained the cells, analyzed the
data, and reviewed the manuscript. BL collected flow cytometric
data and provided resources for the flow cytometric assessment. RM
designed the study, performed the statistical analysis, and
reviewed and edited the final manuscript. All authors read and
approved the final manuscript and agree to be accountable for all
aspects of the research in ensuring that the accuracy and integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
PS
|
protein S
|
|
GAS6
|
growth arrest specific protein 6
|
|
PDAC
|
pancreatic ductal adenocarcinoma
|
|
TAM
|
Tyro3, Axl and Mer
|
References
|
1
|
Arnljots B and Dahlbäck B: Protein S as an
in vivo cofactor to activated protein C in prevention of
microarterial thrombosis in rabbits. J Clin Invest. 95:1987–1993.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hackeng TM, Seré KM, Tans G and Rosing J:
Protein S stimulates inhibition of the tissue factor pathway by
tissue factor pathway inhibitor. Proc Natl Acad Sci USA.
103:3106–3111. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rosing J, Maurissen LF, Tchaikovski SN,
Tans G and Hackeng TM: Protein S is a cofactor for tissue factor
pathway inhibitor. Thromb Res. 122 (Suppl 1):S60–S63. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Suleiman L, Négrier C and Boukerche H:
Protein S: A multifunctional anticoagulant vitamin K-dependent
protein at the crossroads of coagulation, inflammation,
angiogenesis, and cancer. Crit Rev Oncol Hematol. 88:637–654. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lemke G: Biology of the TAM receptors.
Cold Spring Harb Perspect Biol. 5:a0090762013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lemke G and Rothlin CV: Immunobiology of
the TAM receptors. Nat Rev Immunol. 8:327–336. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Anderson HA, Maylock CA, Williams JA,
Paweletz CP, Shu H and Shacter E: Serum-derived protein S binds to
phosphatidylserine and stimulates the phagocytosis of apoptotic
cells. Nat Immunol. 4:87–91. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Burstyn-Cohen T: TAM receptor signaling in
development. Int J Dev Biol. 61:215–224. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Burstyn-Cohen T, Heeb MJ and Lemke G: Lack
of protein S in mice causes embryonic lethal coagulopathy and
vascular dysgenesis. J Clin Invest. 119:2942–2953. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fraineau S, Monvoisin A, Clarhaut J,
Talbot J, Simonneau C, Kanthou C, Kanse SM, Philippe M and
Benzakour O: The vitamin K-dependent anticoagulant factor, protein
S, inhibits multiple VEGF-A-induced angiogenesis events in a Mer-
and SHP2-dependent manner. Blood. 120:5073–5083. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schneider C, King RM and Philipson L:
Genes specifically expressed at growth arrest of mammalian cells.
Cell. 54:787–793. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nagata K, Ohashi K, Nakano T, Arita H,
Zong C, Hanafusa H and Mizuno K: Identification of the product of
growth Arrest-specific Gene 6 as a common ligand for Axl, Sky, and
mer receptor tyrosine kinases. J Biol Chem. 271:30022–30027. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mark MR, Chen J, Hammonds RG, Sadick M and
Godowsk PJ: Characterization of Gas6, a member of the superfamily
of G domain-containing proteins, as a ligand for rse and Axl. J
Biol Chem. 271:9785–9789. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
van der Meer JH, van der Poll T and van't
Veer C: TAM receptors, Gas6, and protein S: Roles in inflammation
and hemostasis. Blood. 123:2460–2469. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zagórska A, Través PG, Lew ED, Dransfield
I and Lemke G: Diversification of TAM receptor tyrosine kinase
function. Nat Immunol. 15:920–928. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dransfield I and Farnworth S: Axl and mer
receptor tyrosine kinases: Distinct and nonoverlapping roles in
inflammation and cancer? Adv Exp Med Biol. 930:113–132. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bosurgi L, Bernink JH, Delgado Cuevas V,
Gagliani N, Joannas L, Schmid ET, Booth CJ, Ghosh S and Rothlin CV:
Paradoxical role of the proto-oncogene Axl and Mer receptor
tyrosine kinases in colon cancer. Proc Natl Acad Sci USA.
110:13091–13096. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bilimoria KY, Bentrem DJ, Ko CY, Stewart
AK, Winchester DP and Talamonti MS: National failure to operate on
early stage pancreatic cancer. Ann Surg. 246:173–180. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cronin-Fenton DP, Søndergaard F, Pedersen
LA, Fryzek JP, Cetin K, Acquavella J, Baron JA and Sørensen HT:
Hospitalisation for venous thromboembolism in cancer patients and
the general population: a population-based cohort study in Denmark,
1997–2006. Br J Cancer. 103:947–953. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Walker AJ, Card TR, West J, Crooks C and
Grainge MJ: Incidence of venous thromboembolism in patients with
cancer-a cohort study using linked United Kingdom databases. Eur J
Cancer. 49:1404–1413. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Horsted F, West J and Grainge MJ: Risk of
venous thromboembolism in patients with cancer: A systematic review
and meta-analysis. PLoS Med. 9:e10012752012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lindahl AK, Odegaard OR, Sandset PM and
Harbitz TB: Coagulation inhibition and activation in pancreatic
cancer. Changes during progress of disease. Cancer. 70:2067–2072.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gradiz R, Silva HC, Carvalho L, Botelho MF
and Mota-Pinto A: MIA PaCa-2 and PANC-1-pancreas ductal
adenocarcinoma cell lines with neuroendocrine differentiation and
somatostatin receptors. Sci Rep. 6:216482016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tan MH, Nowak NJ, Loor R, Ochi H, Sandberg
AA, Lopez C, Pickren JW, Berjian R, Douglass HO Jr and Chu TM:
Characterization of a new primary human pancreatic tumor line.
Cancer Invest. 4:15–23. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yunis AA, Arimura GK and Russin DJ: Human
pancreatic carcinoma (MIA PaCa-2) in continuous culture:
Sensitivity to asparaginase. Int J Cancer. 19:128–135. 1977.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tai MH, Olson LK, Madhukar BV, Linning KD,
Van Camp L, Tsao MS and Trosko JE: Characterization of gap
junctional intercellular communication in immortalized human
pancreatic ductal epithelial cells with stem cell characteristics.
Pancreas. 26:e18–e26. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Qian J, Niu J, Li M, Chiao PJ and Tsao MS:
In vitro modeling of human pancreatic duct epithelial cell
transformation defines gene expression changes induced by K-ras
oncogenic activation in pancreatic carcinogenesis. Cancer Res.
65:5045–5053. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rezende SM, Razzari C and Simmonds RE: In
vitro high level protein S expression after modification of protein
S cDNA. Thromb Haemost. 90:1214–1215. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Levkovitz Y and Baraban JM: A dominant
negative inhibitor of the Egr family of transcription regulatory
factors suppresses cerebellar granule cell apoptosis by blocking
c-Jun activation. J Neurosci. 21:5893–5901. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Weber K, Bartsch U, Stocking C and Fehse
B: A multicolor panel of novel lentiviral ‘gene ontology’ (LeGO)
vectors for functional gene analysis. Mol Ther. 16:698–706. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Weber K, Mock U, Petrowitz B, Bartsch U
and Fehse B: Lentiviral gene ontology (LeGO) vectors equipped with
novel drug-selectable fluorescent proteins: New building blocks for
cell marking and multi-gene analysis. Gene Ther. 17:511–520. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Feoktistova M, Geserick P and Leverkus M:
Crystal violet assay for determining viability of cultured cells.
Cold Spring Harb Protoc 2016. pdb.prot087379. 2016. View Article : Google Scholar
|
|
35
|
Niyazi M, Niyazi I and Belka C: Counting
colonies of clonogenic assays by using densitometric software.
Radiat Oncol. 2:42007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee E, Decker AM, Cackowski FC, Kana LA,
Yumoto K, Jung Y, Wang J, Buttitta L, Morgan TM and Taichman RS:
Growth Arrest-Specific 6 (GAS6) promotes prostate cancer survival
by g1 arrest/s phase delay and inhibition of apoptosis during
chemotherapy in bone marrow. J Cell Biochem. 117:2815–2824. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gustafsson A, Bostrom AK, Ljungberg B,
Axelson H and Dahlbäck B: Gas6 and the receptor tyrosine kinase Axl
in clear cell renal cell carcinoma. PLoS One. 4:e75752009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cummings CT, Deryckere D, Earp HS and
Graham DK: Molecular pathways: MERTK signaling in cancer. Clin
Cancer Res. 19:5275–5280. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Keating AK, Salzberg DB, Sather S, Liang
X, Nickoloff S, Anwar A, Deryckere D, Hill K, Joung D, Sawczyn KK,
et al: Lymphoblastic leukemia/lymphoma in mice overexpressing the
Mer (MerTK) receptor tyrosine kinase. Oncogene. 25:6092–6100. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sufit A, Lee-Sherick AB, DeRyckere D,
Rupji M, Dwivedi B, Varella-Garcia M, Pierce AM, Kowalski J, Wang
X, Frye SV, et al: MERTK inhibition induces polyploidy and promotes
cell death and cellular senescence in glioblastoma multiforme. PLoS
One. 11:e01651072016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wu J, Frady LN, Bash RE, Cohen SM,
Schorzman AN, Su YT, Irvin DM, Zamboni WC, Wang X, Frye SV, et al:
MerTK as a therapeutic target in glioblastoma. Neuro Oncol.
20:92–102. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Waizenegger JS, Ben-Batalla I, Weinhold N,
Meissner T, Wroblewski M, Janning M, Riecken K, Binder M,
Atanackovic D, Taipaleenmaeki H, et al: Role of Growth
arrest-specific gene 6-Mer axis in multiple myeloma. Leukemia.
29:696–704. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mandalà M, Falanga A and Roila F:
Management of venous thromboembolism (VTE) in cancer patients: ESMO
Clinical Practice Guidelines. Ann Oncol. 22 (Suppl 6):vi85–vi92.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hisada Y, Geddings JE, Ay C and Mackman N:
Venous thrombosis and cancer: From mouse models to clinical trials.
J Thromb Haemost. 13:1372–1382. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sheth RA, Niekamp A, Quencer KB, Shamoun
F, Knuttinen MG, Naidu S and Oklu R: Thrombosis in cancer patients:
Etiology, incidence, and management. Cardiovasc Diagn Ther. 7
(Suppl 3):S178–S185. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Iodice S, Gandini S, Löhr M, Lowenfels AB
and Maisonneuve P: Venous thromboembolic events and organ-specific
occult cancers: A review and meta-analysis. J Thromb Haemost.
6:781–788. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sohail MA and Saif MW: Role of
anticoagulation in the management of pancreatic cancer. JOP.
10:82–87. 2009.PubMed/NCBI
|
|
48
|
Walker FJ: Regulation of activated protein
C by a new protein. A possible function for bovine protein S. J
Biol Chem. 255:5521–5524. 1980.PubMed/NCBI
|
|
49
|
Walker FJ, Chavin SI and Fay PJ:
Inactivation of factor VIII by activated protein C and protein S.
Arch Biochem Biophys. 252:322–328. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ndonwi M and Broze G Jr: Protein S
enhances the tissue factor pathway inhibitor inhibition of factor
Xa but not its inhibition of factor VIIa-tissue factor. J Thromb
Haemost. 6:1044–1046. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chattopadhyay R, Sengupta T and Majumder
R: Inhibition of intrinsic Xase by protein S: A novel regulatory
role of protein S independent of activated protein C. Arterioscler
Thromb Vasc Biol. 32:2387–2393. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Plautz WE, Sekhar Pilli VS, Cooley BC,
Chattopadhyay R, Westmark PR, Getz T, Paul D, Bergmeier W, Sheehan
JP and Majumder R: Anticoagulant Protein S targets the factor IXa
Heparin-binding exosite to prevent thrombosis. Arterioscler Thromb
Vasc Biol. 38:816–828. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Saller F, Brisset AC, Tchaikovski SN,
Azevedo M, Chrast R, Fernández JA, Schapira M, Hackeng TM, Griffin
JH and Angelillo-Scherrer A: Generation and phenotypic analysis of
protein S-deficient mice. Blood. 114:2307–2314. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Burstyn-Cohen T, Lew ED, Traves PG,
Burrola PG, Hash JC and Lemke G: Genetic dissection of TAM
receptor-ligand interaction in retinal pigment epithelial cell
phagocytosis. Neuron. 76:1123–1132. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nakamura YS, Hakeda Y, Takakura N, Kameda
T, Hamaguchi I, Miyamoto T, Kakudo S, Nakano T, Kumegawa M and Suda
T: Tyro 3 receptor tyrosine kinase and its ligand, gas6, stimulate
the function of osteoclasts. Stem Cells. 16:229–238. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hafizi S and Dahlbäck B: Gas6 and Protein
S. Vitamin K-dependent ligands for the Axl receptor tyrosine kinase
subfamily. FEBS J. 273:5231–5244. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Linger RM, Keating AK, Earp HS and Graham
DK: TAM receptor tyrosine kinases: Biologic functions, signaling,
and potential therapeutic targeting in human cancer. Adv Cancer
Res. 100:35–83. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Graham DK, Salzberg DB, Kurtzberg J,
Sather S, Matsushima GK, Keating AK, Liang X, Lovell MA, Williams
SA, Dawson TL, et al: Ectopic expression of the proto-oncogene mer
in pediatric t-cell acute lymphoblastic leukemia. Clin Cancer Res.
12:2662–2669. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Shankar SL, O'Guin K, Kim M, Varnum B,
Lemke G, Brosnan CF and Shafit-Zagardo B: Gas6/Axl Signaling
activates the phosphatidylinositol 3-Kinase/Akt1 survival pathway
to protect oligodendrocytes from tumor necrosis factor
alpha-induced apoptosis. J Neurosci. 26:5638–5648. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Shankar SL, O'Guin K, Cammer M, McMorris
FA, Stitt TN, Basch RS, Varnum B and Shafit-Zagardo B: The growth
arrest-specific gene product Gas6 promotes the survival of human
oligodendrocytes via a phosphatidylinositol 3-kinase-dependent
pathway. J Neurosci. 23:4208–4218. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Moody G, Belmontes B, Masterman S, Wang W,
King C, Murawsky C, Tsuruda T, Liu S, Radinsky R and Beltran PJ:
Antibody-mediated neutralization of autocrine Gas6 inhibits the
growth of pancreatic ductal adenocarcinoma tumors in vivo. Int J
Cancer. 139:1340–1349. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Song X and Wang H, Logsdon CD, Rashid A,
Fleming JB, Abbruzzese JL, Gomez HF, Evans DB and Wang H:
Overexpression of receptor tyrosine kinase Axl promotes tumor cell
invasion and survival in pancreatic ductal adenocarcinoma. Cancer.
117:734–743. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Koorstra JB, Karikari C, Feldmann G, Bisht
S, Rojas PL, Offerhaus GJ, Alvarez H and Maitra A: The Axl receptor
tyrosine kinase confers an adverse prognostic influence in
pancreatic cancer and represents a new therapeutic target. Cancer
Biol Ther. 8:618–626. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Shen Y, Chen X, He J, Liao D and Zu X: Axl
inhibitors as novel cancer therapeutic agents. Life Sci.
198:99–111. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kim HA, Nam JY, Jung JY, Bae CB, An JM,
Jeon JY, Kim BS and Suh CH: Serum growth arrest-specific protein 6
levels are elevated in adult-onset Still's disease. Clin Rheumatol.
33:865–868. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Suh CH, Hilliard B, Li S, Merrill JT and
Cohen PL: TAM receptor ligands in lupus: Protein S but not Gas6
levels reflect disease activity in systemic lupus erythematosus.
Arthritis Res Ther. 12:R146. 2010. View
Article : Google Scholar : PubMed/NCBI
|