Introduction
Lung cancer is the most common type of cancer, in
terms of both occurrence and mortality. Non-small cell lung cancer
(NSCLC), which includes adenocarcinoma, squamous cell carcinoma and
large cell carcinoma, accounts for ~85% of all lung cancer cases
and is associated with a poor prognosis (1). The principle chemotherapeutic agent
for patients with NSCLC is platinum-based compounds, especially
cisplatin. However, the overall 5-year survival rate of NSCLC
treatment with platinum-based regimens remains low at 16% (2). Platinum-based regimens also appear to
have a higher toxicity compared with non-platinum-based regimens
(3). Therefore, it would be
beneficial to identify novel anticancer agents that can improve the
efficacy and reduce the toxicity of platinum-based chemotherapy for
NSCLC treatment.
The protein kinase B (AKT)/nuclear factor-κB (NF-κB)
signaling pathway is vital for cell growth, survival and apoptosis
(4). The serine/threonine-protein
kinase AKT is activated via specific phosphorylation at Thr-308 or
Ser-473 by phosphatidylinositol-3-kinase (PI3K) (5), and the activated AKT can subsequently
regulate the activation of NF-κB to control the expression of cell
survival regulators (6,7). The AKT/NF-κB signaling pathway is
constitutively activated in several types of cancer, including
NSCLC (6). Furthermore, the high
AKT/NF-κB activation has also been demonstrated as a key mechanism
of acquired cisplatin resistance by increasing the threshold for
cell death induction (8,9). Exposure to cisplatin activates
AKT/NF-κB signaling, which consequently induces the expression of
NF-κB target genes, including anti-apoptotic genes and genes
involved in cell survival, such as Bcl-xL and survivin. These
events lead to the inhibition of cisplatin-induced apoptosis and
subsequently the development of cisplatin resistance (10,11).
Currently, there is an increasing worldwide interest in plant
phytochemicals from a health perspective, due to epidemiological
and clinical studies reporting the benefits of plant consumption in
lowering the risk of cancer (12).
The inactivation of AKT/NF-κB signaling by phytochemicals, such as
genistein (13,14), baicalein (15) and tunicamycin (16), has been revealed to contribute to a
reduction in cancer cell viability and tumor progression and
enhance the efficacy of therapeutic cisplatin in NSCLC in
vitro and in vivo. Targeting the AKT/NF-κB pathway using
plant chemicals is a promising approach for enhancing cisplatin
sensitivity in NSCLC.
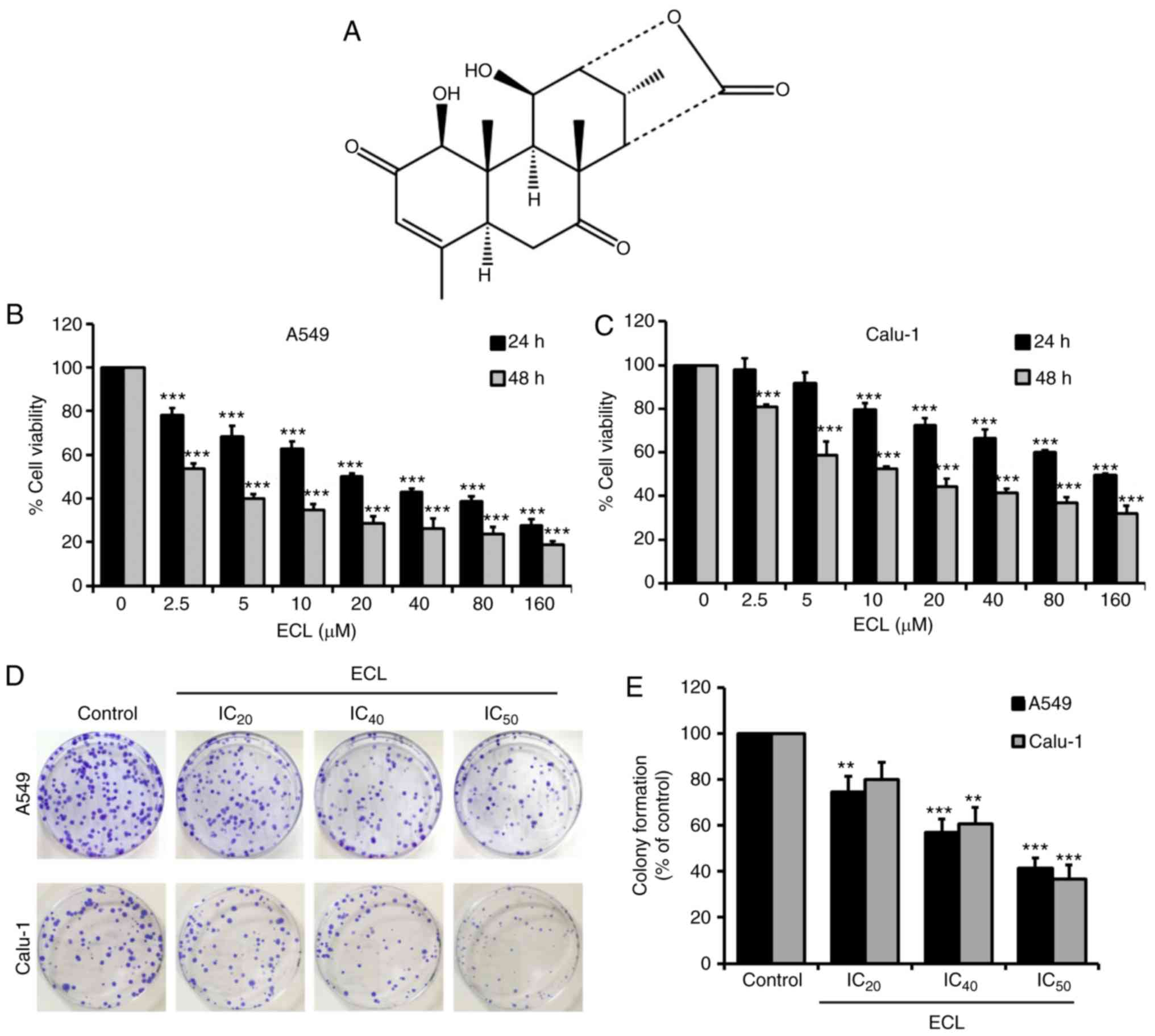
The present study focused on eurycomalactone (ECL;
Fig. 1A), an active natural C-19
quassinoid compound isolated from Eurycoma longifolia Jack,
a popular herbal medicine used in Southeast Asian countries
(17). Its root and rhizome extract
have been traditionally used to treat various conditions and
diseases, including sexual dysfunction, malaria, diabetes, anxiety,
aches, fever, constipation and cancer (18). The in vitro preliminary
screening for the anticancer potential of several quassinoids,
identified the main bioactive compounds derived from E.
longifolia. Notably, among the isolated quassinoids, ECL has
displayed the most potent anticancer effect against various cancer
cell lines, including human NSCLC A549 cell line (19,20).
However, the mechanisms underlying the strong cytotoxicity of ECL
against human NSCLC cells have not been investigated. Our previous
study reported that ECL efficiently sensitized X-ray-induced
apoptosis of NSCLC A549 and COR-L23 cells by inducing cell cycle
arrest at the G2/M phase and inhibiting the repair of
radiation-induced DNA double-strand breaks (21). Notably, ECL was also reported to
inhibit the NF-κB activity in TNF-α-activated 293/NF-κB-luc cells,
a stable cell line containing an NF-κB driven luciferase reporter
gene (22). Another study suggested
that ECL may act as a protein synthesis inhibitor, which suppressed
the expression of the NF-κB-dependent target genes ICAM-1, VCAM-1
and E-selectin in TNFα-activated human endothelial cells (23). It was therefore hypothesized that
ECL may exert an anticancer effect by suppressing the AKT/NF-κB
signaling pathway in human NSCLC cells, leading to the induction of
apoptosis and the enhancement of cisplatin-induced
cytotoxicity.
Materials and methods
Chemicals, reagents and
antibodies
ECL, with a purity of 93.6%, was purchased from
Biopurify Phytochemicals Ltd. Cisplatin was purchased from Merck
KGaA. Culture media Roswell Park Memorial Institute (RPMI)-1640,
Dulbecco's modified Eagle's medium (DMEM), penicillin/streptomycin
antibiotics and trypsin-ethylenediaminetetraacetic acid
(trypsin-EDTA) were purchased from Thermo Fisher Scientific, Inc.
Fetal bovine serum (FBS) was obtained from GE Healthcare Life
Sciences. MTT was obtained from AppliChem GmbH. Muse®
Cell Cycle kit and Muse™ Annexin V & Dead Cell kit were
purchased from EMD Millipore. Mammalian Protein Extraction buffer
was purchased from GE Healthcare Life Sciences. Antibodies specific
to AKT (product no. 9272) and phosphorylated (p)-AKT (S473; product
no. 9271), NF-κB p65 (product no. 6956), p-NF-κB-p65 (S536; product
no. 3033), caspase-3 (product no. 9662), cleaved caspase-3 (Asp175;
product no. 9661), poly (ADP-ribose) polymerase (PARP; product no.
9542) and survivin (product no. 2808) were purchased from Cell
Signaling Technology, Inc. Antibodies against Bcl-xL (product code
ab32370 and β-actin (cat. no. A2066) were obtained from Abcam and
Merck KGaA, respectively. Horseradish peroxidase-conjugated goat
anti-rabbit (cat. no. 1706515) or goat anti-mouse (cat. no.
1706516) immunoglobulin G were purchased from Bio-Rad Laboratories,
Inc. Cisplatin was dissolved in normal saline (1 mg/ml) and
maintained at room temperature. ECL was dissolved in dimethyl
sulfoxide (DMSO) and stored at −20°C until use. The final
concentration of DMSO was <0.5% (v/v), which was also present in
the corresponding controls at the same concentrations.
Cell lines and cell culture
The human adenocarcinoma NSCLC A549 cell line was
obtained from the American Type Culture Collection. The human lung
squamous cell carcinoma Calu-1 cell line was purchased from the CLS
Cell Lines Service. The A549 and Calu-1 cells were grown and
maintained in DMEM and RPMI-1640 media, respectively. These culture
media were supplemented with 10% (v/v) FBS, 100 U/ml penicillin and
100 µg/ml streptomycin. All cell lines were incubated at 37°C in a
humidified atmosphere containing 5% CO2. All cell lines
were mycoplasma-free. The continuous cell lines were routinely
checked every other month by PCR using a service from the Center
for Veterinary Diagnosis, Faculty of Veterinary Science, Mahidol
University Salaya Campus, Nakorn Pathom (Thailand).
Cell viability assay
The A549 (3,500 cells/well) and Calu-1 (6,000
cells/well) cells were seeded on a 96-well plate for 24 h and then
exposed to 0, 2.5, 5, 10, 20, 40, 80 and 160 µM cisplatin or ECL
for 24 and 48 h. The number of viable cells was determined by MTT
assay, as previously described (24). The optical density (OD) of the
formazan dye was measured at 570 nm using a microplate reader
(Bio-Rad Laboratories, Inc.). The percentage of cell viability was
calculated using the formula: Cell
viability=(ODtreated/ODcontrol) ×100%. The
ECL concentrations required for 20, 40 and 50% inhibition of cell
viability (IC20, IC40 and IC50,
respectively) in A549 and Calu-1 cells at 24 h were then determined
from the viability curves and selected for further experiments.
Colony formation assay
The antiproliferative effects of ECL on human NSCLC
A549 and Calu-1 cell lines were confirmed using a colony formation
assay. Briefly, the A549 (2×105 cells/well) or Calu-1
(3×105 cells/well) cells, were seeded on a 6-well plate
and incubated for 24 h. The cells were then treated with ECL at an
IC20, IC40 and IC50 concentration
for 24. Next, the cells were collected by trypsinization with 0.25%
trypsin-EDTA (Gibco; Thermo Fisher Scientific, Inc.) and replated
in 10-cm culture dishes at a low density of 400 cells/dish to allow
colony formation for 2 weeks, with a change of the fresh growth
media every 3 days. The colonies were washed with cold
phosphate-buffered saline (PBS), fixed with 100% methanol for 10
min, and stained with 0.5% crystal violet for 1 h at room
temperature. The colonies containing >50 cells were counted
using an inverted microscope at a magnification of ×40.
Cell cycle analysis by flow
cytometry
The A549 (2×105 cells/well) or Calu-1
(3×105 cells/well) cells, were seeded on a 6-well plate
and incubated for 24 h. Following ECL treatment at the indicated
concentrations and incubation times, cells were collected, fixed
gently in 70% ethanol and stored at −20°C overnight. The fixed
cells were then stained using a Muse® Cell Cycle kit for
30 min at room temperature in the dark. The cell cycle phase
distributions were determined by Muse® Cell Analyzer and
Muse® Cell Cycle software module (version 1.0.0.0.; EMD
Millipore). The cell cycle phases subG1, G1, S and G2/M were
analyzed using GuavaSoft 2.7 software (EMD Millipore).
Combination index (CI) analysis for
combined treatment with ECL and cisplatin
The IC20 and IC40 of ECL were
selected to further study cisplatin co-treatment, to compare the
effects of sub-cytotoxic and highly toxic concentrations of ECL on
cisplatin sensitization. A549 or Calu-1 cells were treated with
0–160 µM cisplatin either alone or in combination with ECL at the
IC20 (2 or 10 µM, respectively) or the IC40
(12 or 80 µM, respectively) for 24 h. Cell viability was then
determined by MTT assay, as previously described. The CI, which
reflects the nature of drug interactions in combination
chemotherapy, was calculated using the Chou-Talalay Method
(25) based on the following
equation:
CI=[(D)1/(Dx)1]+[(D)2/(Dx)2],
where (D)1 and (D)2 represent the
concentrations of compounds 1 (Cisplatin) or 2 (ECL) in the
co-treatment that attains a 50% inhibition, and (Dx)1
and (Dx)2 represent the concentrations of compounds 1 or
2 in the co-treatment that attains a 50% inhibition when present
alone (26). A CI of <0.9,
0.9–1.1 and >1.1 indicated synergistic, additive and
antagonistic effects, respectively.
Cell apoptosis assay by flow
cytometry
The A549 (2×105 cells/well) or Calu-1
(3×105 cells/well) cells were seeded on a 6-well plate
and incubated for 24 h. The cells were then treated with ECL alone,
cisplatin alone or ECL (IC20 or IC40) plus
cisplatin for 24 h. Apoptosis was quantified by staining with
Muse® Annexin V & Dead Cell kit (EMD Millipore) for
20 min at room temperature in the dark, according to the
manufacturer's instructions. The quantitative analysis of cell
living, early and late apoptosis, and cell death, were obtained by
the Muse® Cell Analyzer (EMD Millipore) using the
Muse® Annexin V & Dead Cell software module (version
1.0.0.0) with a minimum of 2,000 events per sample.
Western blotting
Protein expression levels of p(S473)-AKT,
AKT, p(S536)-NF-κB p65, NF-κB p65, caspase-3, PARP,
Bcl-xL and survivin were measured by immunoblotting, as described
in our previous study (27).
Briefly, cells collected from each treatment group were lysed in a
Mammalian Protein Extraction buffer. Protein concentration was
determined using the Bradford assay. Protein (30 µg) from each
sample was separated by 12% SDS-PAGE and electro-transferred to
PVDF membranes. Primary antibodies (1:1,000 dilution) were added
and incubated at 4°C overnight; after which the appropriate
HRP-conjugated secondary antibody (1:5,000 dilution) was added and
incubated for 2 h at room temperature. Chemiluminescence signals
were detected on the X-ray film. As an internal control, the
β-actin primary antibody was also probed. The normalized mean
density of the immunological cross-reactive band was quantified by
ImageJ software (version 1.51j8; National Institutes of
Health).
Statistical analysis
Data are expressed as the mean ± standard deviation
of at least three independent experiments. Significant differences
among groups were analyzed and compared by one-way analysis of
variance and Tukey's post hoc test using SPSS 22.0 software (IBM
Corp.). P<0.05 was considered to indicate a statistically
significant difference.
Results
ECL inhibits the cell viability and
colony-forming capacity of NSCLC cells
To determine the effect of ECL on NSCLC cell
viability in vitro, human lung cancer A549 (adenocarcinoma)
and Calu-1 (squamous cell carcinoma) cells were incubated with
various concentrations of ECL (0–160 µM) for 24 or 48 h, and cell
viability was then examined by MTT assay. As revealed in Fig. 1B and C, ECL treatment significantly
reduced the cell viability of both NSCLC cell lines in a
concentration- and time-dependent manner. The ECL concentrations
required for a 50% inhibition of cell viability (IC50)
of A549 cells at 24 and 48 h (20.81±1.86 and 3.15±0.36 µM,
respectively) were markedly lower compared with those required for
the same inhibition of cell viability in Calu-1 cells (151.87±4.75
and 12.95±0.85 µM, respectively), indicating that ECL has a higher
toxicity in A549 than Calu-1 cells. Table I summarizes the ECL concentrations
at IC20, IC40 and IC50 after 24
and 48 h of incubation. The IC20, IC40 and
IC50 concentrations of ECL at 24 h (2, 12 and 20 µM for
A549 cells, and 10, 80 and 150 µM for Calu-1 cells) were used for
subsequent experiments.
 | Table I.Cytotoxic effect of ECL against two
different types of NSCLC cells (A549 and Calu-1). |
Table I.
Cytotoxic effect of ECL against two
different types of NSCLC cells (A549 and Calu-1).
|
|
| 24 h | 48 h |
|---|
|
|
|
|
|
|---|
|
|
| ECL (µM) | ECL (µM) |
|---|
|
|
|
|
|
|---|
| NSCLC cell
lines | Subtypes |
IC20 |
IC40 |
IC50 |
IC20 |
IC40 |
IC50 |
|---|
| A549 | Adenocarcinoma | 2.29±0.44 | 12.02±1.44 | 20.81±1.86 | 1.04±0.19 | 2.12±0.16 | 3.15±0.36 |
| Calu-1 | Squamous cell
carcinoma | 10.14±1.67 | 80.77±6.84 | 156.30±5.95 | 2.60±0.12 | 5.09±0.65 | 12.95±0.85 |
Subsequently, the anti-proliferative effect of ECL
on the A549 and Calu-1 cells was confirmed by the inhibition of
colony formation, as revealed in Fig.
1D and E. The indicated cells were treated with ECL at the
IC20, IC40, and IC50
concentrations for 24 h before the cells were replated (300
cells/plate) and allowed to form colonies. The numbers of the
colonies formed proportionally reflect the IC20,
IC40 and IC50 concentrations of ECL.
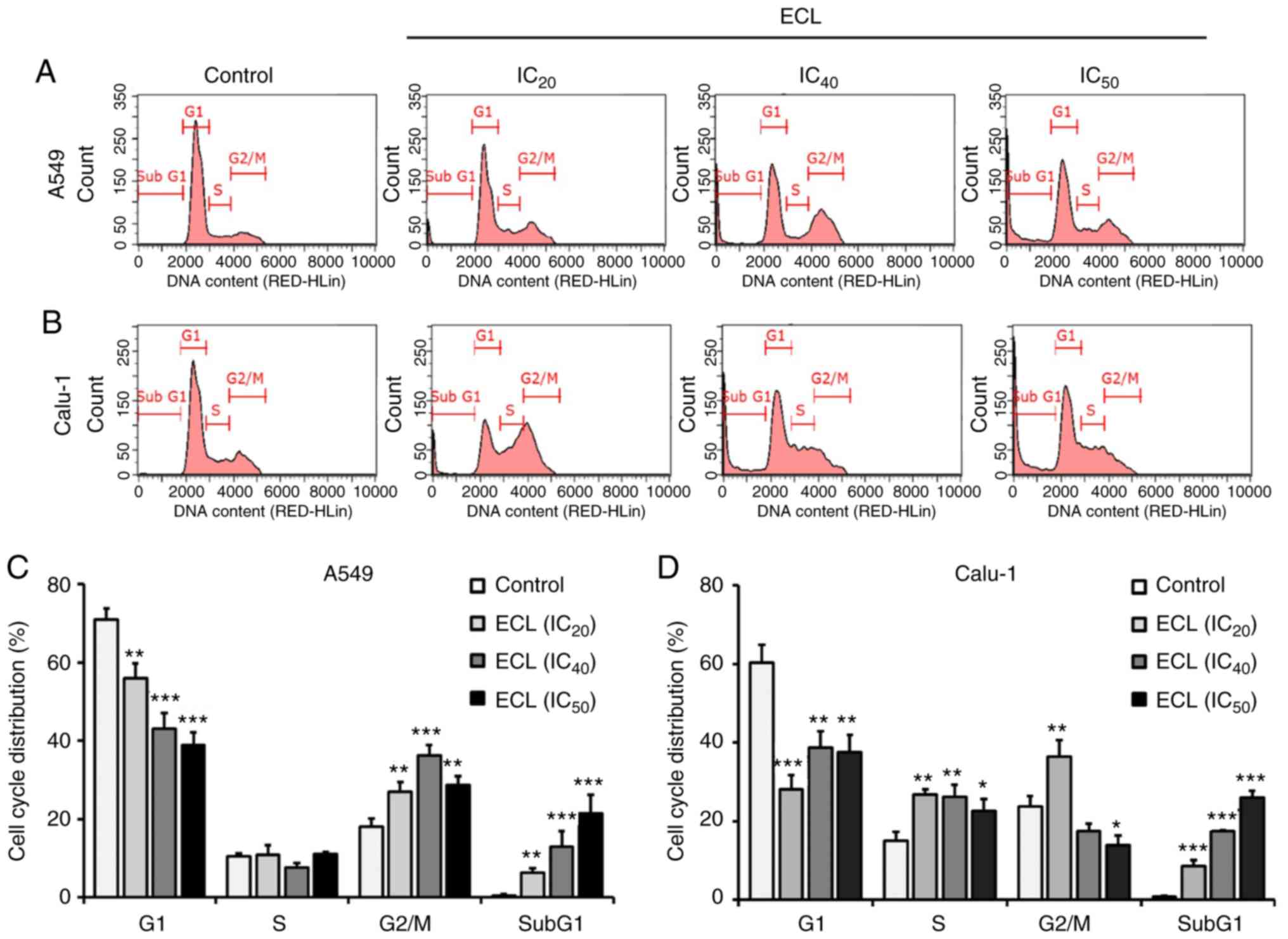
ECL induces cell cycle arrest in NSCLC
cells
To investigate the mechanism through which ECL
inhibits cell growth, cell cycle distribution was analyzed by flow
cytometry following 24 h of ECL treatment with the IC20,
IC40 and IC50 concentrations. As revealed in
Fig. 2A and C, ECL caused cell
cycle arrest at the G2/M phase in A549 cells. In Calu-1 cells, ECL
caused the S phase arrest when treated with IC20,
IC40 or IC50 concentrations (Fig. 2B and D) and induced both S and G2/M
phase arrest when treated with the IC20 concentration.
Such arrest in both NSCLC cells was associated with a concomitant
decrease in the percentage of cells at the G1 phase. Moreover, the
accumulation of a sub-G1 population, which comprised apoptotic
cells containing only fractional DNA content, was observed in a
dose-dependent manner in the A549 and Calu-1 cells. These results
indicated that ECL could induce NSCLC cell death following the
induction of cell cycle arrest.
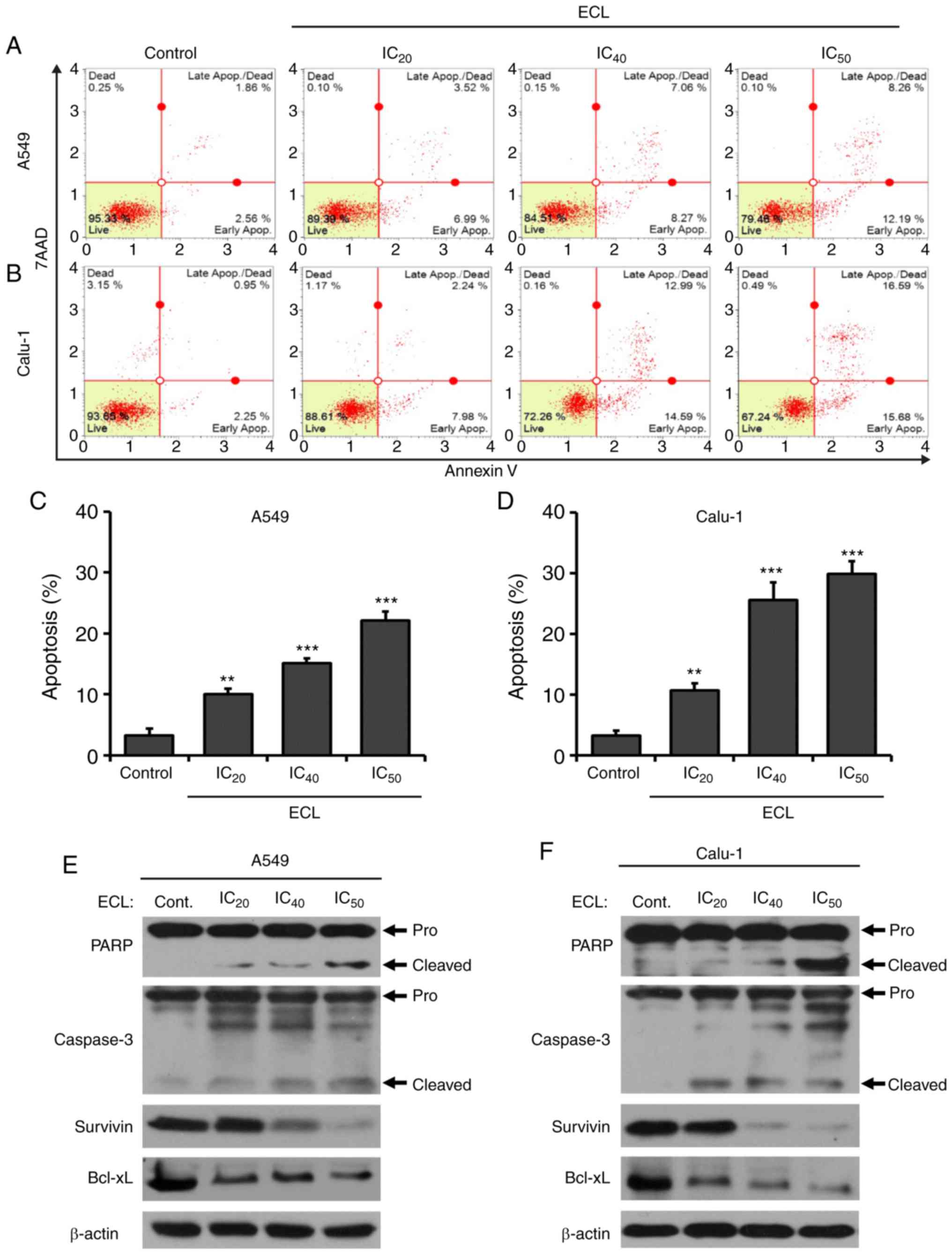
ECL promotes NSCLC cell apoptosis
The effect of ECL on the NSCLC cell apoptosis was
confirmed using Annexin V-FITC/7-AAD double-staining, followed by
flow cytometry (Fig. 3A and B).
Following the 24-h ECL treatment, the apoptotic rates of A549
(Fig. 3C) and Calu-1 (Fig. 3D) cells in the ECL-treated group
were significantly increased in a dose-dependent manner, when
compared with the control group. Next, the expression of apoptosis
regulators, including the pro-apoptotic caspase-3 and PARP
proteins, and the anti-apoptotic Bcl-xL and survivin proteins, was
determined by immunoblotting in the A549 and Calu-1 cells (Fig. 3E and F, respectively). The
expression levels of active caspase-3 and active PARP (cleaved
form) were both markedly induced, while that of Bcl-xL and survivin
were significantly decreased following treatment with various
concentrations of ECL for 24 h, when compared with the control
group. These results confirmed that ECL could trigger apoptotic
cell death in NSCLC cells.
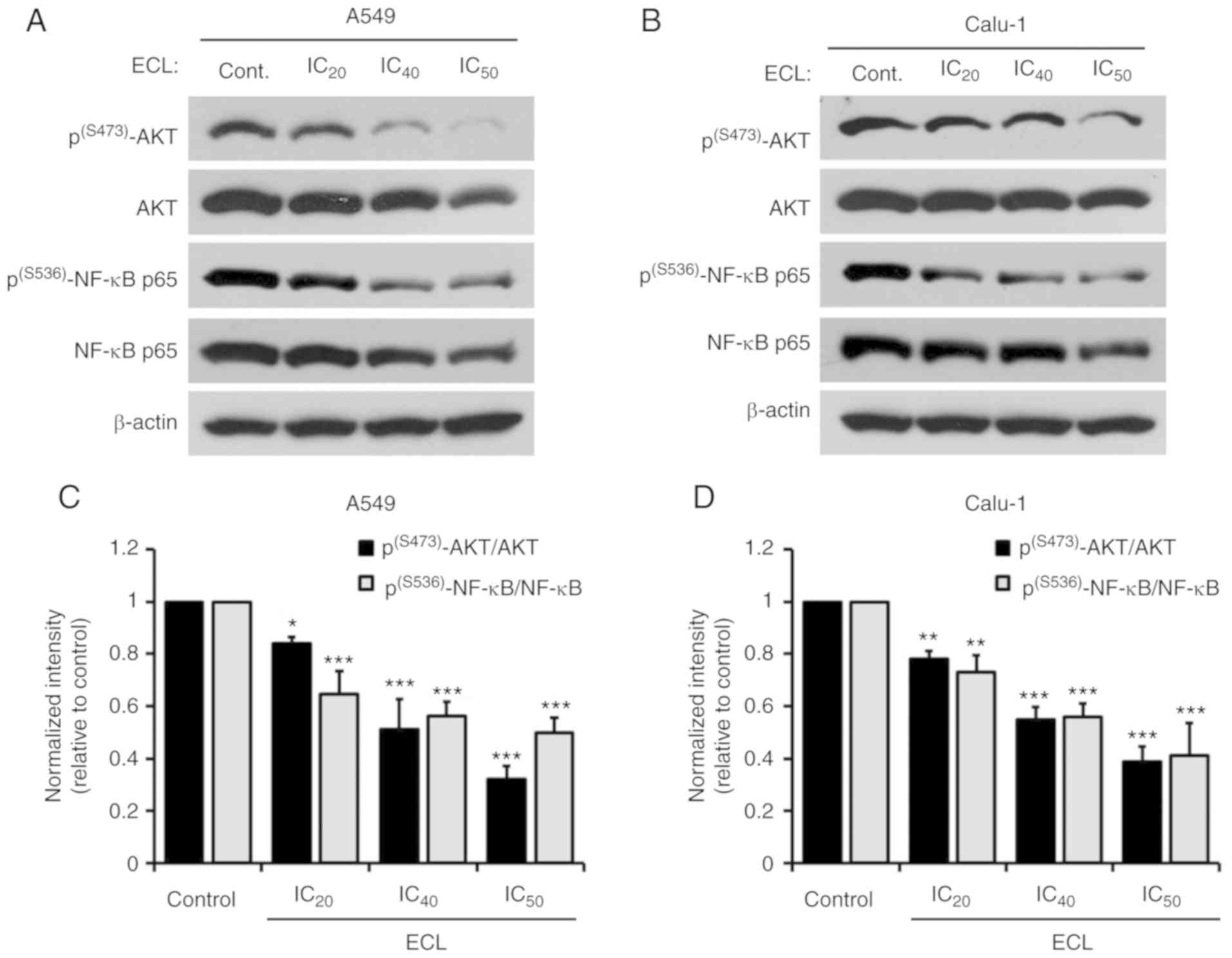
ECL suppresses AKT/NF-κB activation in
NSCLC cells
The inhibitory effect of ECL on the viability of
NSCLC cells by triggering apoptotic cell death prompted us to
investigate the AKT/NF-κB signaling pathway, which participates in
not only multiple steps of lung cancer progression, but also the
resistance to chemotherapy (28,29).
Phosphorylation at serine 473 (S473) in the C-terminal hydrophobic
motif of AKT is involved in AKT activation (30), and phosphorylation at the serine 536
(S536) position of the NF-κB p65 subunit is required for the
activation and nuclear translocation of NF-κB (31). ECL significantly inhibited the
expression levels of p(S473)-AKT, total AKT,
p(S536)-NF-κB p65 and total NF-κB p65 in both A549
(Fig. 4A) and Calu-1 (Fig. 4B) cells. In fact, ECL downregulated
the expression of p(S473)-AKT and total AKT in both A549
and Calu-1 cells when normalized to β-actin (Fig. S1). Likewise, the significant
depletion of both p(S536)-NF-κB p65 and total NF-κB p65
levels when normalized to β-actin was observed in the NSCLC cells
treated with ECL (Fig. S2).
Notably, the ratio of p(S473)-AKT/AKT and
p(S536)-NF-κB p65/NF-κB p65 were also significantly
decreased in the A549 (Fig. 4C) and
Calu-1 (Fig. 4D) cells, supporting
the involvement of AKT/NF-κB inactivation in ECL-induced apoptosis
in the NSCLC cells.
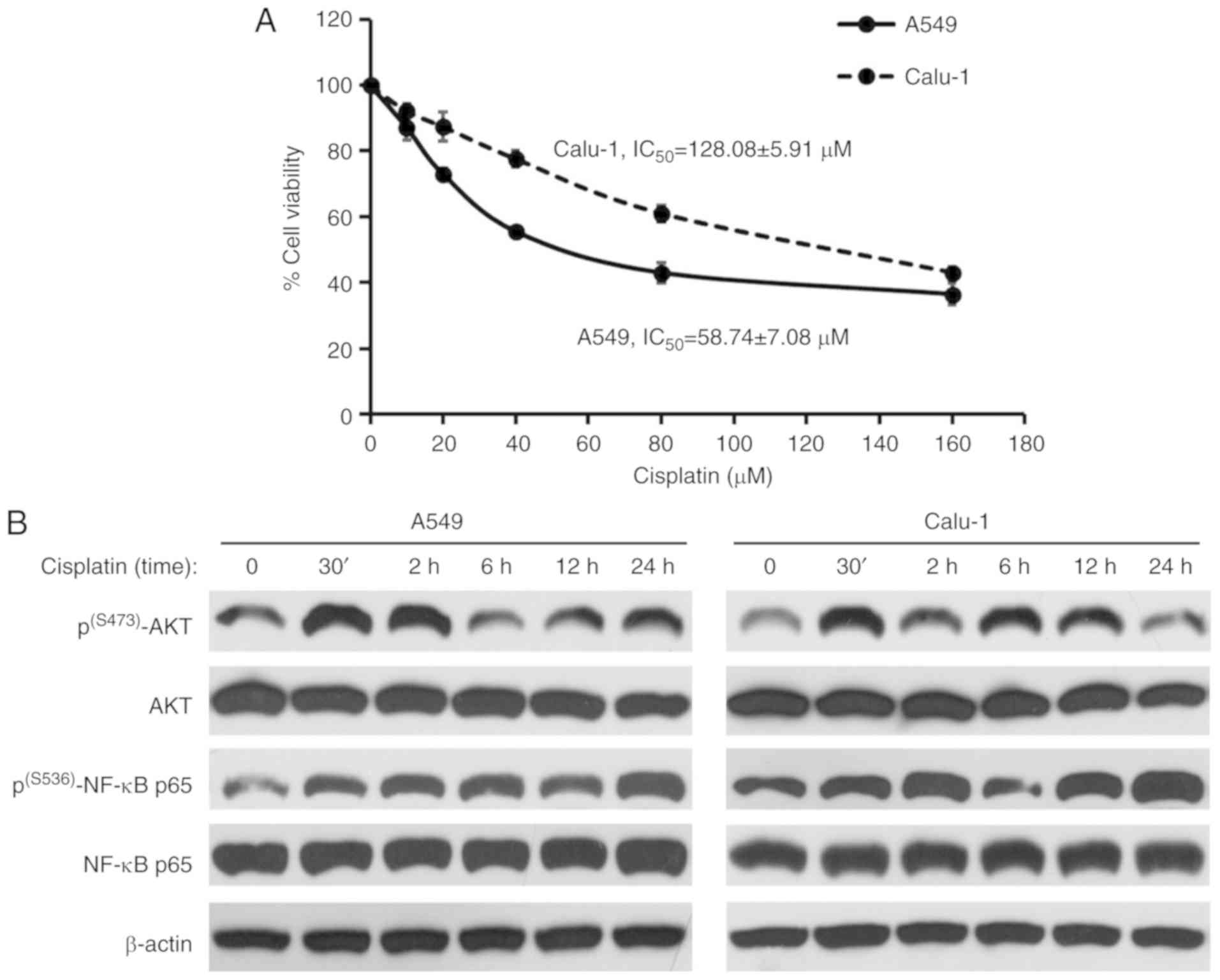
Cisplatin treatment induces the
phosphorylation of AKT and NF-κB at the activation sites
To observe their cisplatin sensitivity, the A549 and
Calu-1 cells were treated with cisplatin at various concentrations
for 24 h, and cell viability was determined by MTT assay. As
revealed in Fig. 5A, the A549 cells
displayed a 2-fold greater sensitivity to cisplatin than the Calu-1
cells, when comparing their IC50 values. Next, it was
determined whether cisplatin treatment (at IC20) could
stimulate AKT/NF-κB signaling in the NSCLC cells using
immunoblotting of p(S473)-AKT, total AKT,
p(S536)-NF-κB p65 and total NF-κB p65. The results in
Fig. 5B revealed that the levels of
p(S473)-AKT and p(S536)-NF-κB p65 were
markedly increased in the A549 and Calu-1 cells following cisplatin
treatment. The highest levels of p(S473)-AKT and
p(S536)-NF-κB p65 were detected at 30 min and 24 h,
respectively, following cisplatin treatment in both NSCLC cell
lines. Therefore, the activation of AKT and NF-κB was responsive to
cisplatin treatment in the NSCLC cells.
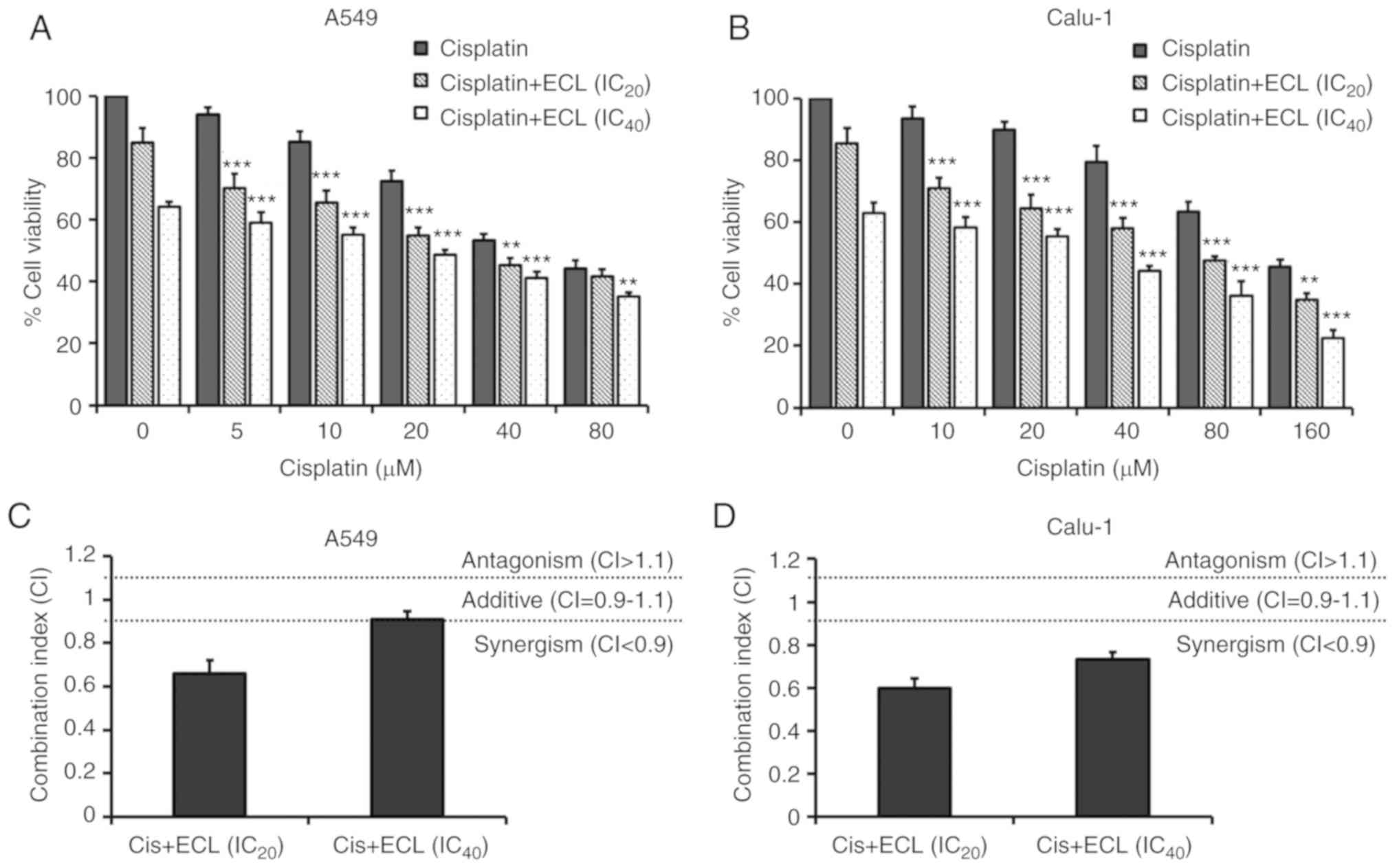
ECL significantly enhances cisplatin
sensitivity in NSCLC cells
Next, it was examined whether the combination of ECL
and cisplatin exerts a lethal enhancement in NSCLC cells. Following
co-treatment with the IC20 or IC40
concentrations of ECL and various concentrations of cisplatin for
24 h, MTT assays were performed. As demonstrated in Fig. 6A and B, the co-treatment
significantly reduced the viability of both NSCLC cells when
compared with cisplatin alone. Table
II summarizes the IC50 values of cisplatin in the
co-treatment with ECL (IC20 or IC40); the CI
values were calculated to define the drug interaction of cisplatin
and ECL, when administered in combination, as synergistic, additive
or antagonistic effects. Co-treatment with ECL led to the positive
dose (IC50) reduction of cisplatin in both NSCLC cell
lines. The CI values of cisplatin combined with ECL at the
IC20 and IC40 concentrations in the A549
cells were 0.66±0.06, indicating synergistic effects, and
0.91±0.04, indicating additive effects (Fig. 6C). Moreover, the CI values of
cisplatin plus ECL at the IC20 and IC40
concentrations in the Calu-1 cells were 0.60±0.04 and 0.73±0.03,
respectively, indicating their synergistic effects (Fig. 6D).
| Table II.IC50 values of cisplatin
in NSCLC cells when administered alone or in combination with ECL,
the CI and drug interactions of ECL and cisplatin combination. |
Table II.
IC50 values of cisplatin
in NSCLC cells when administered alone or in combination with ECL,
the CI and drug interactions of ECL and cisplatin combination.
| NSCLC cell
lines | Treatment | IC50
(µM) | Combination index
(CI) | Drug
interactions |
|---|
| A549 | Cisplatin | 53.58±3.05 | – | – |
|
| Cisplatin +
ECL(IC20) | 30.16±2.62 | 0.66±0.06 | Synergism |
|
| Cisplatin +
ECL(IC40) | 17.91±2.11 | 0.91±0.04 | Additive |
| Calu-1 | Cisplatin | 135.49±7.83 | – | – |
|
| Cisplatin +
ECL(IC20) | 72.49±2.05 | 0.60±0.04 | Synergism |
|
| Cisplatin +
ECL(IC40) | 29.53±3.71 | 0.73±0.03 | Synergism |
Combination of cisplatin with ECL
enhances apoptosis induction in NSCLC cells
Since the effect of ECL on potentiating cisplatin
sensitivity was found in the A549 and Calu-1 cells, the mechanisms
of ECL on the enhancement of cisplatin cytotoxicity were further
investigated by examining the induction of apoptosis. The flow
cytometric results presented in Fig. 7A
and B revealed that co-treatment of cisplatin at the
IC20 value with ECL at the IC20 or
IC40 concentrations significantly induced a higher
number of A549 cells to undergo apoptosis compared with either
cisplatin or ECL alone. Similar results were obtained in Calu-1
cells (Fig. 7C and D). The effect
on apoptosis was confirmed by immunoblotting of pro-apoptotic and
anti-apoptotic proteins. As revealed in Fig. 7E and F, caspase-3 cleavage was
induced by either ECL or cisplatin as a single agent, whereas the
co-treatment resulted in a higher increase of cleaved caspase-3,
with a corresponding decrease in the pro-form of caspase-3. By
contrast, the co-treatment of cisplatin with ECL markedly decreased
the expression of anti-apoptotic Bcl-xL and survivin proteins, when
compared with cisplatin alone. These findings demonstrated that the
combination of cisplatin and ECL treatment further induced
apoptosis in the NSCLC cells more effectively than cisplatin
alone.
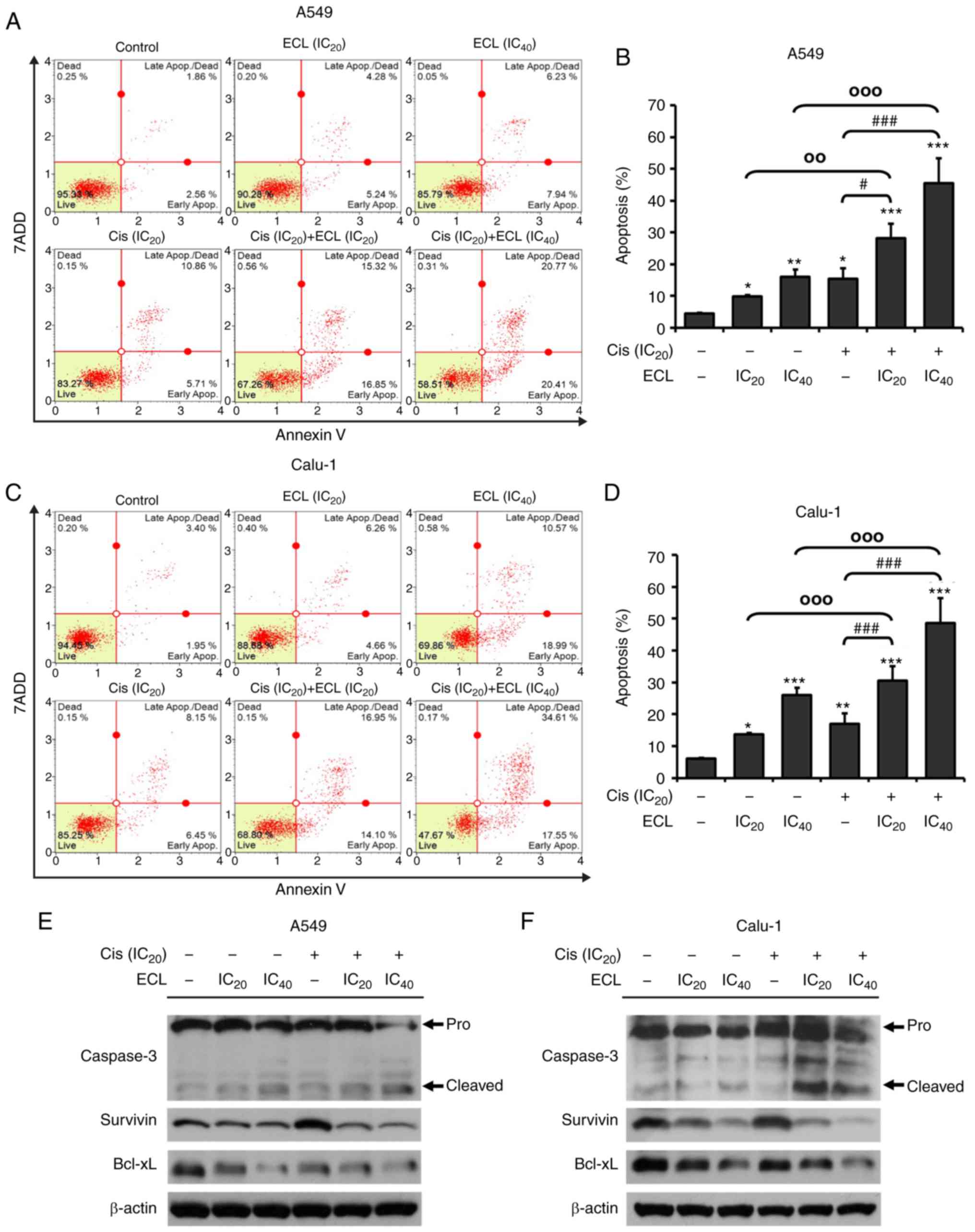 | Figure 7.Effect of cisplatin and ECL alone or
in the combination on apoptosis induction of NSCLC cells. (A) The
representative dot plots display the apoptosis and (B) the
quantitative analysis of total apoptotic cell percentage in the
A549 cells treated with ECL at IC20 or IC40
alone, cisplatin at IC20 alone, or in the combination
for 24 h. (C) The representative dot plots and (D) the quantitative
analysis of total apoptotic cells percentage in the Calu-1 cells
treated with the indicated agents. Cell apoptosis was analyzed by
Annexin V-FITC/7AAD double-staining with flow cytometry.
*P<0.05, **P<0.01, ***P<0.001 vs. the control
(non-treatment group); #P<0.05,
###P<0.001 vs. cisplatin alone;
°°P<0.01, °°°P<0.001 vs. ECL alone at
each concentration. Expression of caspase-3, survivin, and Bcl-xL
proteins was detected in (E) A549 and (F) Calu-1 cells by western
blot analysis following 24 h of treatment with cisplatin alone or
the IC20 or IC40 concentrations of ECL alone,
or both agents. β-actin was used as an internal control. ECL,
eurycomalactone; NSCLC, non-small cell lung cancer. |
ECL inhibits cisplatin-induced AKT and
NF-κB phosphorylation in NSCLC cells
To understand the cisplatin sensitization mechanisms
of ECL, our attention turned to the cisplatin resistance-related
AKT/NF-κB signaling pathway, as ECL alone could suppress AKT/NF-κB
activation in the NSCLC cells, as demonstrated in the previous
results (Fig. 4). The expression of
p(S473)-AKT, total AKT, p(S536)-NF-κB p65 and
total NF-κB p65 was therefore detected by western blotting in the
A549 (Fig. 8A) and Calu-1 (Fig. 8B) cells following treatment with ECL
or cisplatin alone, or in combination. ECL alone could
significantly suppress the ratio of p(S473)-AKT/AKT
(Fig. 8C and D) and
p(S536)-NF-κB p65/NF-κB p65 (Fig. 8E and F) in the A549 and Calu-1
cells, when compared to the non-treatment control. On the other
hand, cisplatin alone significantly increased the ratio of
p(S473)-AKT/AKT and p(S536)-NF-κB p65/NF-κB
p65 in both tested NSCLC cells, indicating that cisplatin could
induce AKT and NF-κB activation. Notably, the co-treatment of ECL
with cisplatin significantly decreased the ratio of
p(S473)-AKT/AKT and p(S536)-NF-κB p65/NF-κB
p65, when compared to cisplatin alone. In combination, ECL
sensitized the cisplatin-induced cytotoxicity in NSCLC cells at
least partially by inhibiting the activation of the AKT/NF-κB
signaling pathway.
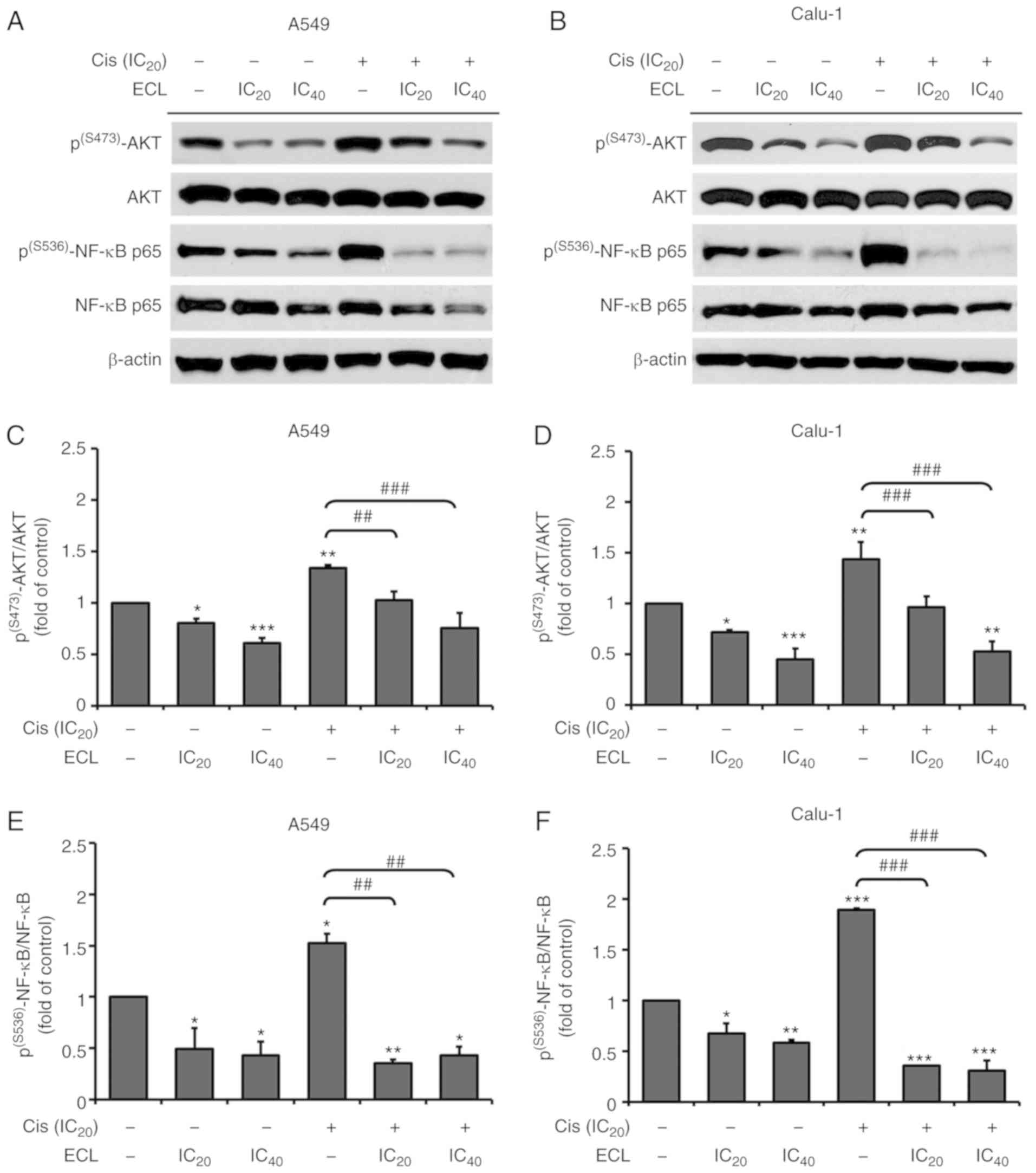 | Figure 8.Inhibitory effect of ECL on
cisplatin-induced AKT/NF-κB signaling activation in human NSCLC
cells. The A549 and Calu-1 cells were treated with cisplatin at the
IC20 or ECL at the IC20 or IC40
concentrations alone, or in combination for 24 h. Representative
immunoblotting images of (A) A549 and (B) Calu-1 cells stained for
p(S473)-AKT, AKT, p(S536)-NF-κB p65 and NF-κB
p65. β-actin was included as an internal control. Bar graphs
revealed the average relative expression of (C and D)
p(S473)-AKT/AKT ratio and (E and F)
p(S536)-NF-κB p65/NF-κB p65 ratio in the A549 and Calu-1
cells, respectively. *P<0.05, **P<0.01, ***P<0.001 vs. the
control (non-treatment group); ##P<0.01;
###P<0.001 vs. cisplatin alone. ECL, eurycomalactone;
AKT, protein kinase B; NF-κB, nuclear factor-κB; NSCLC, non-small
cell lung cancer. |
Discussion
As a leading cause of cancer-related mortality
worldwide, lung cancer has the greatest annual burden among all
types of cancer (32). The
development of novel agents for the treatment of lung cancer is
urgently required. Plant-derived compounds with diverse
bioactivities have attracted increasing attention for their
pharmaceutical potential in cancer treatment, by being used either
alone or as part of combination therapy, to potentiate the effect
of chemotherapeutic drugs (12).
The potential for plant extracts to act as anticancer therapeutic
agents is due to their abilities to promote apoptosis and inhibit
tumor growth and metastasis with few side effects (33).
In the screening of cytotoxicity, ECL, a natural
active quassinoid from E. longifolia Jack, demonstrated a
strong cytotoxicity activity toward various human cancer cell types
including human breast cancer MCF-7 and human NSCLC A549 cell lines
(19,20). Herein, we attempted to elucidate the
anticancer effect of ECL on the survival, proliferation, apoptosis
and cisplatin sensitization in NSCLC A549 and Calu-1 cells, as well
as the related cell signaling mechanism. Another quassinoid
compound of the E. longifolia Jack family, eurycomanone, has
been reported to have an anticancer mechanism, through which it
decreased the activity of prohibitin in lung cancer cells (34) and the expression of p53 in
hepatocellular carcinoma cells (35). Both proteins regulate the cell
cycle, proliferation and apoptosis. Moreover, eurycomanone was
revealed to act on leukemia cells by inhibiting NF-κB signaling
through the inhibition of inhibitor of κB (IκB)α phosphorylation
and upstream mitogen-activated protein kinase signaling (36). Notably, the action of ECL as an
NF-κB inhibitor has been established using an NF-κB-driven
luciferase reporter gene assay in TNF-α-activated 293/NF-κB-luc
cells (22). These findings
prompted us to hypothesize that the anticancer activity of ECL is
likely a result of the inhibition of NF-κB, as well as its upstream
signal transduction pathway, the AKT signaling pathway. The present
study is the first to the best of our knowledge, to reveal the
anticancer mechanism of ECL in the NSCLC A549 and Calu-1 cells via
the induction of cell cycle arrest and cell apoptosis. Moreover,
ECL was found to cause the upregulation of pro-apoptotic (cleaved)
caspase-3 and cleaved PARP, as well as the downregulation of the
expression of anti-apoptotic Bcl-xL and survivin proteins. As
anticipated, ECL could also inhibit AKT and NF-κB signaling in
NSCLC cells.
The activation of the AKT pathway is frequently
dysregulated in several types of cancer, including lung cancer, and
is an important factor in the growth, survival and chemotherapeutic
resistance of cancer cells (37).
Increased AKT activation in human cancers can result from
constitutive phosphorylation of AKT protein at the Ser473 site, due
to aberrant PI3K activation (30).
One of the important downstream signaling targets of AKT is NF-κB.
AKT controls the activity of NF-κB via the phosphorylation of IκB
kinase (IKK) and subsequent degradation of the IκB, which results
in the release and translocation of NF-κB into the nucleus
(38). NF-κB is a transcription
factor that regulates the expression of numerous genes that are
critical for the survival or inhibition of apoptotic cell death
(39). Moreover, AKT/NF-κB is one
of the most important signaling pathways that promote lung
carcinogenesis and regulate the inactivation of apoptosis in lung
cancer (40,41). Therefore, AKT/NF-κB
signaling-induced apoptosis is a suitable target for anticancer
therapy.
Suppression of AKT activity by specific synthetic
inhibitors of PI3K such as wortmannin and LY294002 has been
revealed to exert antitumor activity against several human solid
tumor models (42). Notably,
several natural products, such as epigallocatechin gallate (EGCG)
and safflower polysaccharide (SPS) inhibited AKT and induced
apoptosis in NSCLC cells (43,44).
EGCG significantly reduced the level of phosphorylated-AKT, but the
same treatment did not affect the levels of total AKT expression
(43). Conversely, SPS inhibited
both the expression of total and phosphorylated AKT (44). The present data demonstrated the
inhibition of AKT by ECL. A significant decrease of
p(S473)-AKT/AKT ratio by ECL confirmed that ECL could
suppress the AKT activation by inhibiting the phosphorylation of
AKT at Serine473 (S473), which is the regulatory site responsible
for its activity (30). Moreover,
the p(S536)-NF-κB p65/NF-κB p65 ratio was significantly
reduced by ECL suggesting the inactivation of NF-κB by ECL by
suppressing the phosphorylation at the serine 536 (S536) position
of the NF-κB p65 subunit, which is required for the activation and
nuclear translocation of NF-κB (31). Since the suppression of AKT
activation by an inhibitor of PI3K (LY294002) inhibited the
phosphorylation of S536-NF-κB p65, NF-κB activation via
phosphorylated S536 NF-κB p65 is largely relied on AKT (45). Various natural agents such as
piperlongumine (46) and oenothein
B (4) have also been revealed to
inhibit lung tumor growth and/or induce NSCLC cell apoptosis by
blocking the NF-κB activation. The suppression of NF-κB activation
by ECL is likely modulated through the inhibition of its upstream
signaling, AKT kinase.
The AKT/NF-κB signaling pathway is influential in
the regulation of cell survival, due to the activation of
anti-apoptotic downstream effectors. The pro-apoptotic potential of
some anticancer agents is highly correlated with the inactivation
of the AKT/NF-κB pathway (10,47).
The NF-κB-regulated gene products include anti-apoptotic and cell
survival genes, such as Bcl-xL and surviving (48). Since ECL induces apoptosis in the
NSCLC cells, this effect is associated with the inhibition of
anti-apoptotic proteins, Bcl-xL and survivin, whose expression is
controlled by NF-κB. Subsequently, the high activation of
pro-apoptotic caspase-3 and PARP occurred following treatment with
ECL. The downregulation of anti-apoptotic proteins in conjunction
with the upregulation of pro-apoptotic proteins in both A549 and
Calu-1 cells treated with ECL likely serve to shift the balance
from pro-survival to proapoptotic signaling. Therefore, ECL may
induce the NSCLC cell apoptosis by inhibiting the AKT/NF-κB
activation.
An alternative mode of action of ECL has been
proposed as an inhibitor of protein synthesis. A study in
TNF-α-activated human endothelial cells revealed that ECL, rather
than inhibiting the early NF-κB signaling, post-transcriptionally
downregulated the expression of the NF-κB-dependent target genes,
including ICAM-1, VCAM-1 and E-selectin. Notably, ECL alone could
not inhibit the expression of survivin in endothelial cells
(23). Not only the activation of
AKT and NF-κB, but also the expression of survivin, were found to
be higher in cancer cells than in normal cells (6,49).
Thus, ECL may effectively exert different modes of actions
depending on the differences in cell types and differentiation.
Cisplatin is one of the most widely used
chemotherapeutic drugs in the treatment of lung cancer (50). It is a DNA-damaging agent, which can
covalently bind to DNA leading to different types of DNA lesions.
Subsequently, cisplatin-DNA adducts cause various cellular
responses, such as replication arrest, transcription inhibition,
cell-cycle arrest, DNA repair, and apoptotic cell death (51). However, the efficacy of cisplatin in
NSCLC treatment is limited due to either intrinsic or acquired
cisplatin resistance. Several mechanisms have been proposed to
account for the resistance of NSCLC tumor cells to cisplatin,
including the disruption of apoptotic cell death pathways (52). In addition, a high AKT/NF-κB
activity in the NSCLC cells has been revealed to be highly
associated with the apoptosis inhibition-related cisplatin
resistance (53,54). Notably, cisplatin treatment can
activate the AKT/NF-κB signaling pathway in NSCLC cells, resulting
in an anti-apoptotic effect that may also counteract the cisplatin
cytotoxicity and lead to the development of cisplatin resistance
(11). Moreover, IKK
phosphorylation of NF-κB at serine 536 could contribute to acquired
cisplatin resistance (55).
Therefore, inhibition of NF-κB signaling favorably serves as a
critical target for enhancing the efficacy of cisplatin in NSCLC
treatment. The inhibition of the AKT/NF-κB pathway by plant-derived
compounds, such as baicalein (15)
and tunicamycin (16) could enhance
the cisplatin sensitivity of NSCLC cells. Significantly, the
results of the present study demonstrated for the first time that
ECL is in fact capable of sensitizing the NSCLC cells to
cisplatin-mediated apoptosis, which is evidenced by the activation
of caspase-3 as well as the downregulation of anti-apoptotic Bcl-xL
and survivin proteins. Furthermore, cisplatin mediated the
activation of AKT and NF-κB phosphorylation in both NSCLC cell
lines, while ECL blocked the AKT activation and almost completely
inhibited the NF-κB activation induced by cisplatin. Likewise, two
of the anti-apoptosis-related targets of NF-κB, survivin and
Bcl-xL, were further downregulated by combination treatment with
cisplatin and ECL, as compared to treatment with cisplatin alone.
Since cisplatin is still wildly used as a chemotherapeutic drug,
the long-term treatment of cisplatin causes cancer cells to develop
chemoresistance by various mechanisms, including the activation of
AKT/NF-κB signaling (11).
Therefore, ECL not only enhanced chemosensitivity, but may also
further reduce the chemoresistance of NSCLC cells to cisplatin.
In conclusion, the present in vitro study has
provided evidence of an underlying anticancer mechanism of ECL in
NSCLC cells, through which ECL inactivates the AKT/NF-κB-signaling
pathway, leading to the induction of cancer cell apoptosis and
chemosensitization to cisplatin. The insight gained from the
present study indicated the potential application of ECL for
potentiating cisplatin chemosensitivity for the effective treatment
of NSCLC. Additional in vivo studies are required to confirm
the result of ECL in combination with cisplatin, before entering
this combination into a clinical trial that may offer a novel
treatment option for patients with NSCLC.
Supplementary Material
Supporting Data
Acknowledgements
The authors wish to acknowledge the Faculty of
Medicine, Chiang Mai University, Chiang Mai, Thailand for providing
research facilitates.
Funding
The present study was supported by the Royal Golden
Jubilee PhD (RGJ-PHD) Programme, Thailand Science Research and
Innovation (TSRI) (grant no. PHD/0088/2558), the Faculty of
Medicine Research Fund (grant no. 090-2560), Faculty of Medicine,
Chiang Mai University, and a grant from the Faculty of Dentistry,
Mahidol University, Thailand.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ND, KC, PP and AW contributed to the conception,
design and follow-up of the study. ND, KC and JK contributed to the
acquisition, analysis, and interpretation of data. ND performed the
statistical analysis and wrote the manuscript. AI and WT
participated in the interpretation of data and provided critical
revisions to the scientific content of the manuscript. PP and AW
reviewed and edited the manuscript, and AW supervised the project.
All the authors approved the final version of the manuscript and
agree to be accountable for all aspects of this work in ensuring
that questions related to the accuracy or integrity of any part of
this work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liloglou T, Bediaga NG, Brown BR, Field JK
and Davies MP: Epigenetic biomarkers in lung cancer. Cancer Lett.
342:200–212. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
D'Addario G, Pintilie M, Leighl NB, Feld
R, Cerny T and Shepherd FA: Platinum-based versus
non-platinum-based chemotherapy in advanced non-small-cell lung
cancer: A meta-analysis of the published literature. J Clin Oncol.
23:2926–2936. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pei X, Xiao J, Wei G, Zhang Y, Lin F,
Xiong Z, Lu L, Wang X, Pang G, Jiang Y and Jiang L: Oenothein B
inhibits human non-small cell lung cancer A549cell proliferation by
ROS-mediated PI3K/Akt/NF-κB signaling pathway. Chem Biol Interact.
298:112–120. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hafsi S, Pezzino FM, Candido S, Ligresti
G, Spandidos DA, Soua Z, McCubrey JA, Travali S and Libra M: Gene
alterations in the PI3K/PTEN/AKT pathway as a mechanism of
drug-resistance (review). Int J Oncol. 40:639–644. 2012.PubMed/NCBI
|
|
6
|
Hakan Kucuksayan H, Sakir Akgun S and Akca
H: Pl3K/Akt/NF-κB signalling pathway on NSCLC invasion. Med Chem.
6:42016.
|
|
7
|
Naugler WE and Karin M: NF-kappaB and
cancer-identifying targets and mechanisms. Curr Opin Genet Dev.
18:19–26. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen W, Liu X, Yuan S and Qiao T: HSPA12B
overexpression induces cisplatin resistance in non-small-cell lung
cancer by regulating the PI3K/Akt/NF-κB signaling pathway. Oncol
Lett. 15:3883–3889. 2018.PubMed/NCBI
|
|
9
|
Zou W, Ma X, Hua W, Chen B and Cai G:
Caveolin-1 mediates chemoresistance in cisplatin-resistant ovarian
cancer cells by targeting apoptosis through the Notch-1/Akt/NF-κB
pathway. Oncol Rep. 34:3256–3263. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Godwin P, Baird AM, Heavey S, Barr MP,
O'Byrne KJ and Gately K: Targeting nuclear factor-kappa B to
overcome resistance to chemotherapy. Front Oncol. 3:1202013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Albanell J, Chattopadhyay S, Tapia M,
Rovira A, Belda-Iniesta C, Manguán-García JDC, Machado R, Gonzalez
Barón M and Perona R: JNK/MKP-1 and PI3K/NF-κB: Critical pathways
controlling cellular response towards cisplatin in non-small cell
lung cancer (NSCLC). J Clin Oncol. 23:2033. 2005. View Article : Google Scholar
|
|
12
|
Nobili S, Lippi D, Witort E, Donnini M,
Bausi L, Mini E and Capaccioli S: Natural compounds for cancer
treatment and prevention. Pharmacol Res. 59:365–378. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li Y, Ahmed F, Ali S, Philip PA, Kucuk O
and Sarkar FH: Inactivation of nuclear factor kappaB by soy
isoflavone genistein contributes to increased apoptosis induced by
chemotherapeutic agents in human cancer cells. Cancer Res.
65:6934–6942. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu D, Yan L, Wang L, Tai W, Wang W and
Yang C: Genistein enhances the effect of cisplatin on the
inhibition of non-small cell lung cancer A549 cell growth in
vitro and in vivo. Oncol Lett. 8:2806–2810. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu M, Qi B, Wu X, Xu J and Liu X:
Baicalein increases cisplatin sensitivity of A549 lung
adenocarcinoma cells via PI3K/Akt/NF-κB pathway. Biomed
Pharmacother. 90:677–685. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ahmmed B, Khan MN, Nisar MA, Kampo S,
Zheng Q, Li Y and Yan Q: Tunicamycin enhances the suppressive
effects of cisplatin on lung cancer growth through PTX3
glycosylation via AKT/NF-kappaB signaling pathway. Int J Oncol.
54:431–442. 2019.PubMed/NCBI
|
|
17
|
Miyake K, Tezuka Y, Awale S, Li F and
Kadota S: Quassinoids from Eurycoma longifolia. J Nat Prod.
72:2135–2140. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rehman SU, Choe K and Yoo HH: Review on a
traditional herbal medicine, Eurycoma longifolia Jack (tongkat
ali): Its traditional uses, chemistry, evidence-based pharmacology
and toxicology. Molecules. 21:3312016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kuo PC, Damu AG, Lee KH and Wu TS:
Cytotoxic and antimalarial constituents from the roots of Eurycoma
longifolia. Bioorg Med Chem. 12:537–544. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Miyake K, Li F, Tezuka Y, Awale S and
Kadota S: Cytotoxic activity of quassinoids from Eurycoma
longifolia. Nat Prod Commun. 5:1009–1012. 2010.PubMed/NCBI
|
|
21
|
Dukaew N, Konishi T, Chairatvit K,
Autsavapromporn N, Soonthornchareonnon N and Wongnoppavich A:
Enhancement of radiosensitivity by eurycomalactone in human NSCLC
cells through G2/M cell cycle arrest and delayed DNA double-strand
break repair. Oncol Res. 28:161–175. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tran TV, Malainer C, Schwaiger S, Atanasov
AG, Heiss EH, Dirsch VM and Stuppner H: NF-κB inhibitors from
Eurycoma longifolia. J Nat Prod. 77:483–488. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Malainer C, Schachner D, Sangiovanni E,
Atanasov AG, Schwaiger S, Stuppner H, Heiss EH and Dirsch VM:
Eurycomalactone inhibits expression of endothelial adhesion
molecules at a post-transcriptional level. J Nat Prod.
80:3186–3193. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Phannasorn W, Khanaree C, Wongnoppavich A
and Chewonarin T: The effect of purple rice (Oryza sativa L.
indica) extract on the inflammatory response in a colon cancer cell
line and dextran sulfate-induced tumor promotion in the rat colon.
Mol Cell Toxicol. 13:433–442. 2017. View Article : Google Scholar
|
|
25
|
Peters GJ, van der Wilt CL, van Moorsel
CJ, Kroep JR, Bergman AM and Ackland SP: Basis for effective
combination cancer chemotherapy with antimetabolites. Pharmacol
Ther. 87:227–253. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wongnoppavich A, Dukaew N, Choonate S and
Chairatvit K: Upregulation of maspin expression in human cervical
carcinoma cells by transforming growth factor beta1 through the
convergence of Smad and non-Smad signaling pathways. Oncol Lett.
13:3646–3652. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kaltschmidt C, Banz-Jansen C, Benhidjeb T,
Beshay M, Förster C, Greiner J, Hamelmann E, Jorch N, Mertzlufft F,
Pfitzenmaier J, et al: A role for NF-κB in organ specific cancer
and cancer stem cells. Cancers (Basel). 11:6552019. View Article : Google Scholar
|
|
29
|
Huang WC and Hung MC: Induction of Akt
activity by chemotherapy confers acquired resistance. J Formos Med
Assoc. 108:180–194. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liao Y and Hung MC: Physiological
regulation of Akt activity and stability. Am J Transl Res. 2:19–42.
2010.PubMed/NCBI
|
|
31
|
Hu J, Nakano H, Sakurai H and Colburn NH:
Insufficient p65 phosphorylation at S536 specifically contributes
to the lack of NF-kappaB activation and transformation in resistant
JB6 cells. Carcinogenesis. 25:1991–2003. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Barta JA, Powell CA and Wisnivesky JP:
Global epidemiology of lung cancer. Ann Glob Health. 85:82019.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ijaz S, Akhtar N, Khan MS, Hameed A, Irfan
M, Arshad MA, Ali S and Asrar M: Plant derived anticancer agents: A
green approach towards skin cancers. Biomed Pharmacother.
103:1643–1651. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wong PF, Cheong WF, Shu MH, Teh CH, Chan
KL and AbuBakar S: Eurycomanone suppresses expression of lung
cancer cell tumor markers, prohibitin, annexin 1 and endoplasmic
reticulum protein 28. Phytomedicine. 19:138–144. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zakaria Y, Rahmat A, Pihie AH, Abdullah NR
and Houghton PJ: Eurycomanone induce apoptosis in HepG2 cells via
up- regulation of p53. Cancer Cell Int. 9:162009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hajjouli S, Chateauvieux S, Teiten MH,
Orlikova B, Schumacher M, Dicato M, Choo CY and Diederich M:
Eurycomanone and eurycomanol from Eurycoma longifolia Jack as
regulators of signaling pathways involved in proliferation, cell
death and inflammation. Molecules. 19:14649–14666. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tokunaga E, Oki E, Egashira A, Sadanaga N,
Morita M, Kakeji Y and Maehara Y: Deregulation of the Akt pathway
in human cancer. Curr Cancer Drug Targets. 8:27–36. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bai D, Ueno L and Vogt PK: Akt-mediated
regulation of NFkappaB and the essentialness of NFkappaB for the
oncogenicity of PI3K and Akt. Int J Cancer. 125:2863–2870. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Oeckinghaus A and Ghosh S: The NF-kappaB
family of transcription factors and its regulation. Cold Spring
Harb Perspect Biol. 1:a0000342009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen W, Li Z, Bai L and Lin Y: NF-kappaB
in lung cancer, a carcinogenesis mediator and a prevention and
therapy target. Front Biosci (Landmark Ed). 16:1172–1185. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Umemura S, Mimaki S, Makinoshima H, Tada
S, Ishii G, Ohmatsu H, Niho S, Yoh K, Matsumoto S, Takahashi A, et
al: Therapeutic priority of the PI3K/AKT/mTOR pathway in small cell
lung cancers as revealed by a comprehensive genomic analysis. J
Thorac Oncol. 9:1324–1331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Garcia-Echeverria C and Sellers WR: Drug
discovery approaches targeting the PI3K/Akt pathway in cancer.
Oncogene. 27:5511–5526. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gu JJ, Qiao KS, Sun P, Chen P and Li Q:
Study of EGCG induced apoptosis in lung cancer cells by inhibiting
PI3K/Akt signaling pathway. Eur Rev Med Pharmacol Sci.
22:4557–4563. 2018.PubMed/NCBI
|
|
44
|
Li JY, Yu J, Du XS, Zhang HM, Wang B, Guo
H, Bai J, Wang JH, Liu A and Wang YL: Safflower polysaccharide
induces NSCLC cell apoptosis by inhibition of the Akt pathway.
Oncol Rep. 36:147–154. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kwon HJ, Choi GE, Ryu S, Kwon SJ, Kim SC,
Booth C, Nichols KE and Kim HS: Stepwise phosphorylation of p65
promotes NF-κB activation and NK cell responses during target cell
recognition. Nat Commun. 7:116862016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zheng J, Son DJ, Gu SM, Woo JR, Ham YW,
Lee HP, Kim WJ, Jung JK and Hong JT: Piperlongumine inhibits lung
tumor growth via inhibition of nuclear factor kappa B signaling
pathway. Sci Rep. 6:263572016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Khan KH, Yap TA, Yan L and Cunningham D:
Targeting the PI3K-AKT-mTOR signaling network in cancer. Chin J
Cancer. 32:253–265. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shukla S, Shankar E, Fu P, MacLennan GT
and Gupta S: Suppression of NF-κB and NF-κB-regulated gene
expression by apigenin through IκBα and IKK pathway in TRAMP mice.
PLoS One. 10:e01387102015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cheung CH, Huang CC, Tsai FY, Lee JY,
Cheng SM, Chang YC, Huang YC, Chen SH and Chang JY:
Survivin-biology and potential as a therapeutic target in oncology.
Onco Targets Ther. 6:1453–1462. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Rinaldi M, Cauchi C and Gridelli C: First
line chemotherapy in advanced or metastatic NSCLC. Ann Oncol. 17
(Suppl 5):v64–v67. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Köberle B, Tomicic MT, Usanova S and Kaina
B: Cisplatin resistance: Preclinical findings and clinical
implications. Biochim Biophys Acta. 1806:172–182. 2010.PubMed/NCBI
|
|
53
|
Baby J, Pickering BF, Vashisht Gopal YN
and Van Dyke MW: Constitutive and inducible nuclear factor-kappaB
in immortalized normal human bronchial epithelial and non-small
cell lung cancer cell lines. Cancer Lett. 255:85–94. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chen B, Shen Z, Wu D, Xie X, Xu X, Lv L,
Dai H, Chen J and Gan X: Glutathione peroxidase 1 promotes NSCLC
resistance to cisplatin via ROS-induced activation of PI3K/AKT
pathway. Biomed Res Int. 2019:76405472019.PubMed/NCBI
|
|
55
|
Li Z, Yang Z, Lapidus RG, Liu X, Cullen KJ
and Dan HC: IKK phosphorylation of NF-κB at serine 536 contributes
to acquired cisplatin resistance in head and neck squamous cell
cancer. Am J Cancer Res. 5:3098–3110. 2015.PubMed/NCBI
|