Introduction
Lung cancer is the leading cause of death from
cancer worldwide, with the highest incidence and mortality among
different types of cancers (1).
Lung adenocarcinoma (LAD) is the most common type of non-small cell
lung cancer (NSCLC), accounting for 40% of lung cancer (2). Although some targeted and
immunological drugs have been presented, cisplatin-based
combination chemotherapy plays a significant role in comprehensive
treatment programs of lung cancer (3). With the large-scale application of
cisplatin drugs, tumor cells become resistant to those drugs, and
the therapeutic effects of chemotherapy have markedly reduced.
(4). A previous study demonstrated
that long-term use of cisplatin drugs in patients with lung cancer
lead to a relapse rate of more than 60%, while the remission rate
of relapsed lung cancer chemotherapy drugs was less than 30%
(5). The remission rate of the
current chemotherapy regimen for lung cancer was only 30–40%, and
the five-year survival rate was lower than 15% in patients with
advanced LAD (6). Once the cancer
cells are resistant to cisplatin, they may be resistant to various
first-line chemotherapeutic drugs, such as doxorubicin,
vinblastine, fluorouracil, and mitomycin (7). Therefore, it is imperative to identify
specific molecular targets and biomarkers related to cisplatin
resistance in LAD to reverse resistance to cisplatin. Previous
findings have shown that cisplatin is a non-specific cell cycle
cytotoxic drug which plays a role mainly by inhibiting DNA
synthesis (8) and inducing
apoptosis (9) of tumor cells. The
resistance mechanism of cisplatin is extremely complex and involves
multiple genes, proteins and several pathways and is related to
some mechanisms, such as reduced drug uptake, increased drug
inactivation, and increased DNA damage repair (8–12).
Despite progress in the field of genomics and proteomics, the
resistance mechanism of cisplatin has remained elusive.
The NSCLC A549 cell line is sensitive to cisplatin.
The A549 cell is gradually induced by the cisplatin drug to produce
A549/DDP, which can maintain stable drug resistance to cisplatin.
In a previous study, we used high-throughput microarray technology
to compare LAD cisplatin-resistant A549/DDP cell and
cisplatin-sensitive A549 cell, and obtained differential mRNA
expression profiles of LAD cisplatin-resistant (13). Use of mRNA microarray and reverse
transcription-quantitative polymerase chain reaction RT-PCR
(RT-qPCR) demonstrated that protein kinase C delta binding protein
(PRKCDBP) is an mRNA molecule with the length of 1,039 bp which is
downregulated in A549/DDP. PRKCDBP is genetically and
epigenetically altered in several human malignancies and is a tumor
suppressor gene, and its low expression levels may be associated
with a high degree of methylation of PRKCDBP (14–17).
Thus, it was found that low expression of PRKCDBP could play a key
role in LAD resistance to cisplatin.
The role of PRKCDBP in LAD resistance to cisplatin
is not well understood. In the present study, we overexpressed
PRKCDBP in A549/DDP by lentiviral technology to determine the
underlying mechanism.
Materials and methods
Human LAD tissue specimen
Patients with LAD were recruited from the First
Affiliated Hospital of Wenzhou Medical University (Wenzhou, China)
from May 2010 to July 2015. The inclusion criterion was that
patients with primary LAD were in stages IIIB to IV. The first-line
chemotherapy was cisplatin (25 mg/m2) in the first 1 to
3 days, and combined with gemcitabine (1,000 mg/m2) or
paclitaxel (80 mg/m2) on the 1st and 8th days. A cycle
of chemotherapy was considered 21 days and each patient was treated
for 3 to 4 cycles. According to medical imaging examinations
[including computed tomography (CT), magnetic resonance imaging
(MRI)] and the results of serum lung tumor markers and response
evaluation criteria in solid tumor (RECIST) standards, the patients
were grouped into the ‘cisplatin sensitive group’ (complete
remission + partial remission) and ‘cisplatin insensitive group’
(progress). The specimens, which included 25 cisplatin-sensitive
and 32 cisplatin-insensitive specimens, were strictly identified by
the Department of Pathology (Wenzhou, China). The study was
approved by the Ethics Committee of the First Affiliated Hospital
of Wenzhou Medical University (no. 2014005). We analyzed PRKCDBP
expression level in the GEPIA website (http://gepia.cancer-pku.cn/index.html) and survival
analysis of PRKCDBP in Kaplan-Meier plotter website (http://kmplot.com/analysis/index.php?).
Cell culture
BEAS-2B, A549, and A549/DDP cells were cultured in
Roswell Park Memorial Institute (RPMI)-1640 medium containing 10%
fetal bovine serum (FBS), in which a single-cell suspension was
prepared by gentle pipetting in complete medium, and the cell
suspension was transferred to a cell culture flask with a pipette
and placed in an incubator at 37°C, with 5% CO2.
A549/DDP was added to 2 µg/ml cisplatin to maintain drug
resistance. When the cell growth status was satisfactory, the cell
passage was performed at a confluency of 70–90%.
RT-qPCR
Total RNA of tissues and cells was extracted using
TRIzol® reagent (Invitrogen) and reverse-transcribed
into cDNA using a PrimeScript RT Reagent kit (Takara), in
accordance with the manufacturer's instructions. The reverse
transcription reaction was carried out at 37°C for 15 min and the
inactivation reaction of reverse transcriptase at 85°C for 5 sec.
The levels of PRKCDBP, DNMT1, DNMT3a, tumor necrosis factor-α
(TNF-α) and β-actin were measured via Applied Biosystems 7500 Fast
Real-Time PCR System (Thermo Fisher Scientific). The primer
sequences used for PCR were: PRKCDBP, upstream:
5′-CAGGACACCGAGGAAGAT-3′, downstream: 5′-TCAGGCTACACTCTCCATT-3′.
DNMT1, upstream: 5′-AGGTGGAGAGTTATGACGAGGC-3′, downstream:
5′-TCAGGCTACACTCTCCATT-3′. DNMT3a, upstream:
5′-CGGCCATACGGTGGAGC-3′, downstream: 5′-TATCGTGGTCTTTGGAGGCG-3′.
TNF-α for upstream: 5′-TCCCCAGGGACCTCTCTCTA-3′, downstream:
5′-GAGGGTTTGCTACAACATGGG-3′. β-actin for upstream:
5′-CATGTACGTTGCTATCCAGGC-3′, downstream:
5′-CTCCTTAATGTCACGCACGAT-3′. Then, 20 µl PCR reaction volume was
determined with 6 µl double-distilled water, 10 µl SYBR-Green I
Premix mixture (2X, Takara), 1 µl PCR forward primer (10 mM), 1 µl
PCR reverse primer (10 mM) and 2 µl cDNA template. The qPCR
reaction program included a denaturation step of 10 min at 95°C,
followed by 40 cycles (for 5 sec at 95°C, and for 30 sec at 60°C),
and a final extension step for 5 min at 72°C. All samples were
normalized to β-actin. The relative gene expression data were
calculated using the median triplicate (ΔCq=Cq median target
gene-Cq median β-actin), and 2−ΔΔCq method (18).
Constructed lentivirus-mediated
overexpression and siRNA vector
The overexpression vector targeted PRKCDBP (OE) as
well as negative control vector (OE-NC)-transfected A549/DDP cells,
overexpression vector targeted TNF-α (TNF-α OE) as well as negative
control vector (TNF-α OE-NC)-transfected A549/DDP cells, and the
overexpressed vector targeted DNMT1 (DNMT1 OE) and negative control
vector (DNMT1 OE-NC) (Genechem)-transfected A549 cells. A549 cells
were transfected with siRNA vector targeting TNF-α (TNF-α siRNA)
and negative control siRNA (NC-siRNA) (TNF-α siRNA NC) (Genechem).
Transfection was performed by seeding 2×105 cells into a
six-well plate, and after 24 h the medium was aspirated and
incubated at 37°C for 8–12 h with transfection complex (including
vector and infection enhancer) according to the manufacturer's
protocol and the MOI value (MOI=10) was calculated. The A549/DDP
and A549 cells were infected with lentivirus for 72 h and treated
with 2 µg/ml puromycin, and the overexpression efficiency was
detected by RT-qPCR.
Decitabine processed A549/DDP
cells
As a specific DNA methyltransferase inhibitor,
decitabine can reverse the methylation process of DNA (19). After the cells were counted, 200,000
cells were seeded into 6-well plates. After 12 h, the solution was
changed and the drug was added: The final concentration of
decitabine in the experimental group was 5 µM, and the cells were
treated for 24 h.
Cell migration assays
Migration assays were performed with 8.0-µm pore
inserts (Millipore) in a 24-well plate. For the migration assay,
2×104 cells with culture medium (without serum) were
seeded into the upper compartment and RPMI-1640 medium containing
10% FBS to the lower chamber of the Transwell inserts. Migrated
cells were fixed with methanol and stained using 0.1 % (w/v)
crystal violet, then bleached with 33% acetic acid. Absorbance
value was measured at 570 nm on a microplate reader. Each
experiment was performed in triplicate.
Cell viability assay
Cell viability was evaluated by Cell Counting Kit-8
(CCK-8; Corning, Inc.) as per the manufacturer's instructions.
Briefly, 3,000 cells were re-suspended and seeded into a 96-well
plate supplemented in the presence of 10% FBS and cultured for a
week. The following day, the cells were incubated with CCK-8 for 1
h at 37°Cand the absorbance was measured at 450 nm using a
multifunctional microplate reader (Tecan).
Cisplatin sensitivity test
The cell inoculation density was 2,000 cells/well.
After 24 h of cell attachment 100 µl of complete medium (containing
cisplatin) was added to each well. The concentration of cisplatin
was 1, 2, 4, 10 and 25 µg/ml, and only the zeroing hole of the
medium and the control hole of the single-cell suspension was set
to zero. After 48 h of cultivation, the culture medium was replaced
with complete medium containing 10% CCK8. The cultivation was for
45 min. A microplate reader detected the absorbance at 450 nm
wavelength, cell viability %=(A plus-A blank)/(A0 plus drug-A
blank) ×100%, using the SPSS18.0 software profit regression model
to calculate the IC50 of the cell.
Western blot detection of PRKCDBP
The loading volume of each sample was 30 µg,
proteins from whole cell lysates were prepared in 1X sodium dodecyl
sulfate buffer, separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred to a PVDF (polyvinylidene difluoride) membrane
(Millipore). The membranes were blocked with 5% non-fat milk (RT, 1
h) and incubated with respective primary antibodies (PRKCDBP,
16250-1-AP, 1:1,000; actin, HRP-60008, 1:4,000; 4°C, overnight).
Membranes were then washed and incubated with horseradish
peroxidase-conjugated secondary antibody (anti-rabbit IgG Sc-2004,
1:3,000; 4°C, overnight). The size of PRKCDBP is 35–40KDa, and the
size of actin is 43 kDa. The signals were detected using ECL Plus
kit. Gray value was measured with image J.
Flow cytometry to detect apoptotic
rate
A549 cells were inoculated in 6-well plates at a
density of 1.0×106 cells/well. After various treatments
for 48 h, the cells were collected and digested with 0.25% trypsin
at 37°C for 3–4 min. The cells were gently pipetted and collected
by centrifugation at 800 × g for 5 min in room temperature. Then,
the cells were washed twice with cold phosphate-buffered saline
(PBS) and re-suspended in PBS prior to being stained with binding
buffer. Added 1 ul AnnexinV PE and mix well and reacted at room
temperature in the dark for 15 min. Then added 5 ul 7-AAD dye
solution, mixed well, and reacted at room temperature in the dark
for 15 min. Apoptosis was analyzed with a flow cytometer and the
percentage of apoptotic cells was determined.
Cell cycle assay
The cells were harvested by centrifugation at 85 × g
for 10 min in room temperature, fixed with 70% ethanol and
incubated at 4°C overnight. The cells were resuspended with 400 µl
PBS (containing 2 mg/ml RNA enzymes) and incubated at 37°C for 30
min, followed by the addition of 400 µl propidium iodide (0.1
mg/ml) for 10 min and detection of DNA content by a flow cytometry
analyzer (Cytomics FC 500; Beckman Coulter). The results were
analyzed using MultiCycle software.
Bisulfite (BSP) sequencing PCR
i) Sulfite treatment and purification (Qiagen) as
per the manufacturer's protocol, followed by 1 µg of DNA for
sulfite conversion, purification, and recovery. ii) Primer design
(the underlined sequence is the area to be tested, and the thick
black part base is the PCR primer position):
CCAACACAGTCTCTGCGCCCACTAAGATGCATGAAATAAAAATTTCCGTGACTCGCCCTTTGCAGTGGAGAACTGAAACAGGCACACCAGGGAATTGGAGCGGAGGAGGGTAACTCAAACTCAGAGTGAGAGGGTTTGCAGGGGGCCGATTTGGGGCCAACAGGCTTCCCAGCAGGCCCCCGGCGCGGGACAGCGGAAGGCGAAACGCTTTCAAGAGACCCCGCTGCCAACATCCCCACGCCCTCGCGCCCTCCCGCCGCCCCAGAAGGCCAACTCCGCCTGCCTGAGTCACAGCTGGAGCTGGGGAGGAGCCAGGGAAAGGAGGCCCCTGACCGTAGTGCGGCCAGCA.
PRKCDBP, upstream primer: 5′-TTTTTTGTAGTGGAGAATTGAAATAG-3′,
downstream primer: 5′-ATCAAAAACCTCCTTTCCCTAACT-3′. iii) PCR
thermocycling conditions were: PCR system (30 µl), including 3 µl
10X Taq buffer, 1 µl dNTP, 1 µl enzyme, 1 µl
H2O2, 1 µl upstream primer (10 µM), 1 µl
downstream primer (10 µM), 2 µl template. Reaction conditions were:
95°C 10 min, 40 cycles including 94°C for 30 sec, 55°C for 30 sec,
72°C for 40 sec. Finally, the extension was reacted at 72°C for 5
min. iv) T/A cloning and sequencing. The target fragment was
purified and XL10-Gold® ability, transformation,
resuscitation and vaccination were carried out according to the
manufacturer's protocol. Positive colonies were identified by
enzyme digestion to identify the plasmid used for sequencing.
Statistical analysis
Differences in variables among groups were tested
using one-way ANOVA with the least significant difference (LSD)
test as the post hoc test for the normal distribution or
Kruskal-Wallis test for the non-normal distribution. A comparison
between the two groups was performed by the LSD test or Student's
t-test or Mann-Whitney U test. Survival analysis is carried out
using Kaplan-Meier and a log-rank test. P<0.05 was considered
statistically significant.
Results
Expression level and survival analysis
of PRKCDBP in LAD and adjacent tissues
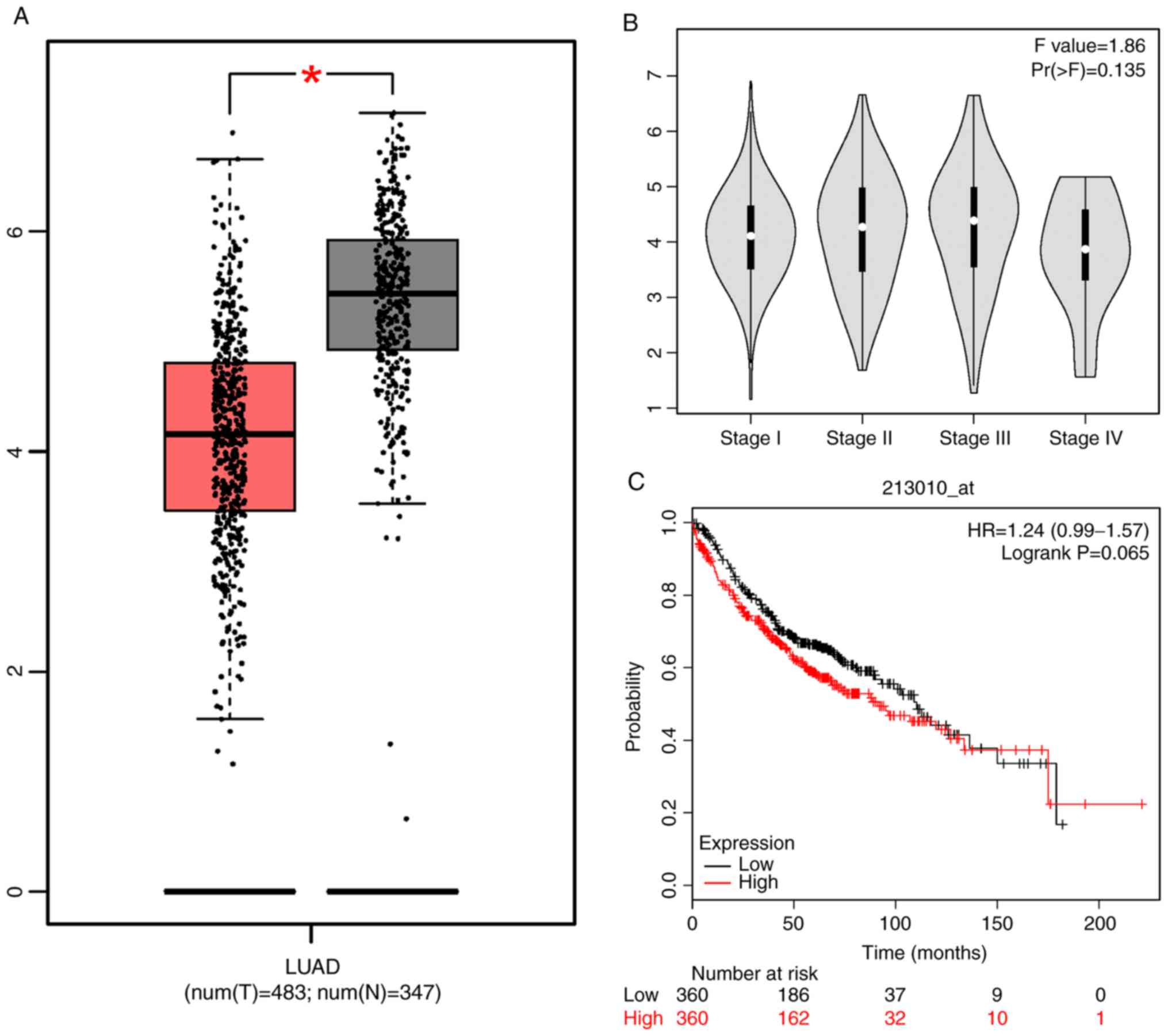
According to GEPIA website, Kaplan-Meier plotter
website, and Fig. 1, the expression
level of PRKCDBP in LAD tissues was markedly lower than that in
adjacent cancer tissues (http://gepia.cancer-pku.cn/detail.php?gene=PRKCDBP,
P<0.05; Fig. 1). The PRKCDBP
expression level was not significant in the histological
differentiation of lung cancer (F=1.86, P=0.135). It was found that
overall survival (OS) of high expression level of PRKCDBP was not
significantly different from low expression level of PRKCDBP
[hazard ratio (HR)=1.24, log-rank P=0.065], as indicated by the
Kaplan-Meier plotter (http://kmplot.com/analysis/index.php?p=service&start=1)
(Fig. 1A-C). DNMT1 mRNA level in
the cisplatin-insensitive group was markedly higher than that of
the cisplatin-sensitive group (t=7.233, P<0.0001, Fig. 2E) while PRKCDBP mRNA level in the
cisplatin-insensitive group was notably lower than that of
cisplatin-sensitive group (t=8.784, P<0.0001, Fig. 2F).
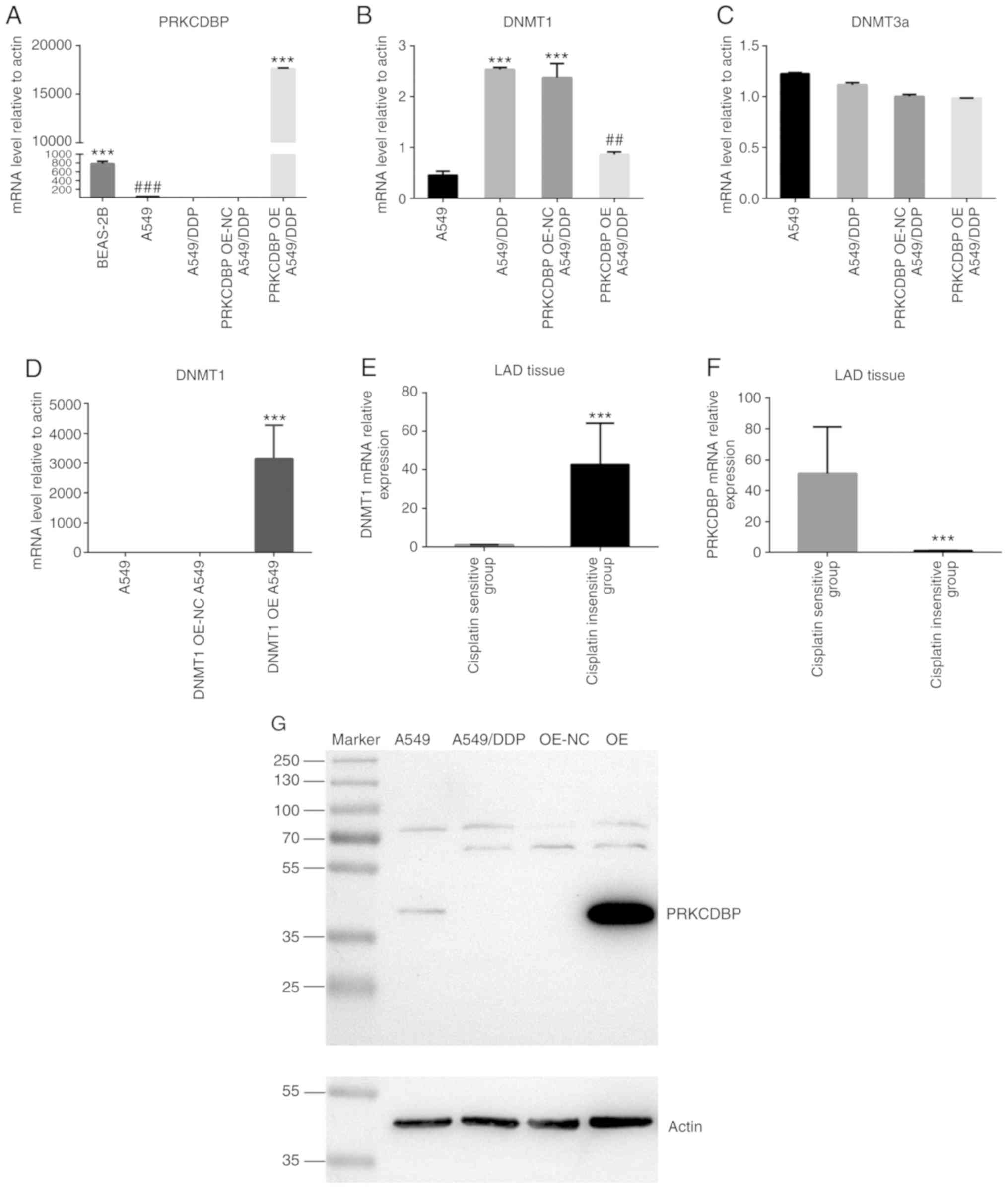 | Figure 2.The expression level of PRKCDBP,
DNMT1, DNMT3a from A549 and A549/DDP cells. (A) Compared to normal
human bronchial epithelial cell line BEAS-2B, PRKCDBP mRNA level of
A549 cells quickly decreased (t=12.97, P=0.0002). Compared to A549
cell, the expression levels of PRKCDBP mRNA level were signifantly
reduced (t=115.3, P<0.0001), After overexpression of PRKCDBP in
A549/DDP cells, the expression level of PRKCDBP mRNA was
signifantly increased compared to that of A549/DDP and A549/DDP NC
(F=28326, P<0.0001). (B) Compared to the A549/DDP NC group,
DNMT1 mRNA level of PRKCDBP OE 549/DDP was clearly decreased
(t=5.127, P=0.007). (C) DNMT3a mRNA level was not significantly
different in A549/DDP cells (t=4.525, P=0.110). (D) The expression
levels of DNMT1 mRNA of OE group were markedly higher than that of
A549 and A549 OE-NC (F=28326, P<0.0001). (E) DNMT1 mRNA level in
the cisplatin-insensitive group was markedly higher than that of
the cisplatin-sensitive group (t=7.233, P<0.0001). (F) PRKCDBP
mRNA level in the cisplatin-insensitive group was notably lower
than that of cisplatin-sensitive group (t=8.784, P<0.0001). (G)
Compared to A549 cells, the expression levels of PRKCDBP protein
level were signifantly reduced in A549/DDP (P<0.001), after
overexpression of PRKCDBP in A549/DDP cells, the expression levels
of PRKCDBP protein level were signifantly enhanced than that of
A549/DDP and PRKCDBP OE-NC A549/DDP (F=28326, P<0.0001).
##P<0.01, ###P<0.001, ***P<0.001.
PRKCDBP, protein kinase C delta binding protein. |
Expression levels of PRKCDBP, DNMT1,
DNMT3a in A549 and A549/DDP cells
Compared with BEAS-2B cells, the PRKCDBP mRNA level
in A549 cells was significantly decreased (t=12.97, P=0.0002).
Compared with A549 cells, the expression levels of PRKCDBP mRNA and
protein levels were markedly reduced (t=115.3, P<0.0001)
(Fig. 2A, D and G), while the DNMT1
mRNA level was markedly elevated in A549/DDP cell line (t=10.413,
P<0.001) (Fig. 2B). By contrast,
DNMT3a mRNA level was not significantly different in the A549/DDP
cell line (t=4.525, P=0.110) (Fig.
2C). After overexpression of PRKCDBP in A549/DDP cells, PRKCDBP
mRNA and protein levels were notably increased compared with
A549/DDP and A549/DDP NC (F=28326, P<0.0001) (Fig. 2A and E). Similarly, DNMT1 mRNA level
in DNMT1 OE A549 group was significantly higher than that of A549
and DNMT1 OE A549 NC (F=28326, P<0.0001) (Fig. 2D), which indicated successful
establishment of a lentiviral vector-mediated overexpression of
PRKCDBP in the A549/DDP group and overexpression of DNMT1 in the
A549 group. Compared to A549/DDP NC group, DNMT1 mRNA level was
markedly decreased (t=5.127, P=0.007) (Fig. 2B), while DNMT3a mRNA level did not
significantly change in PRKCDBP OE A549/DDP group (t=0.849,
P=0.444) (Fig. 2B). Due to the low
expression level of PRKCDBP in LAD tissues and A549 cells, DNMT1
mRNA level in the A549/DDP cell line was markedly elevated,
suggesting that PRKCDBP may be a hypermethylation state in A549/DDP
cell line by DNMT1.
PRKCDBP was not relative to cell
migration
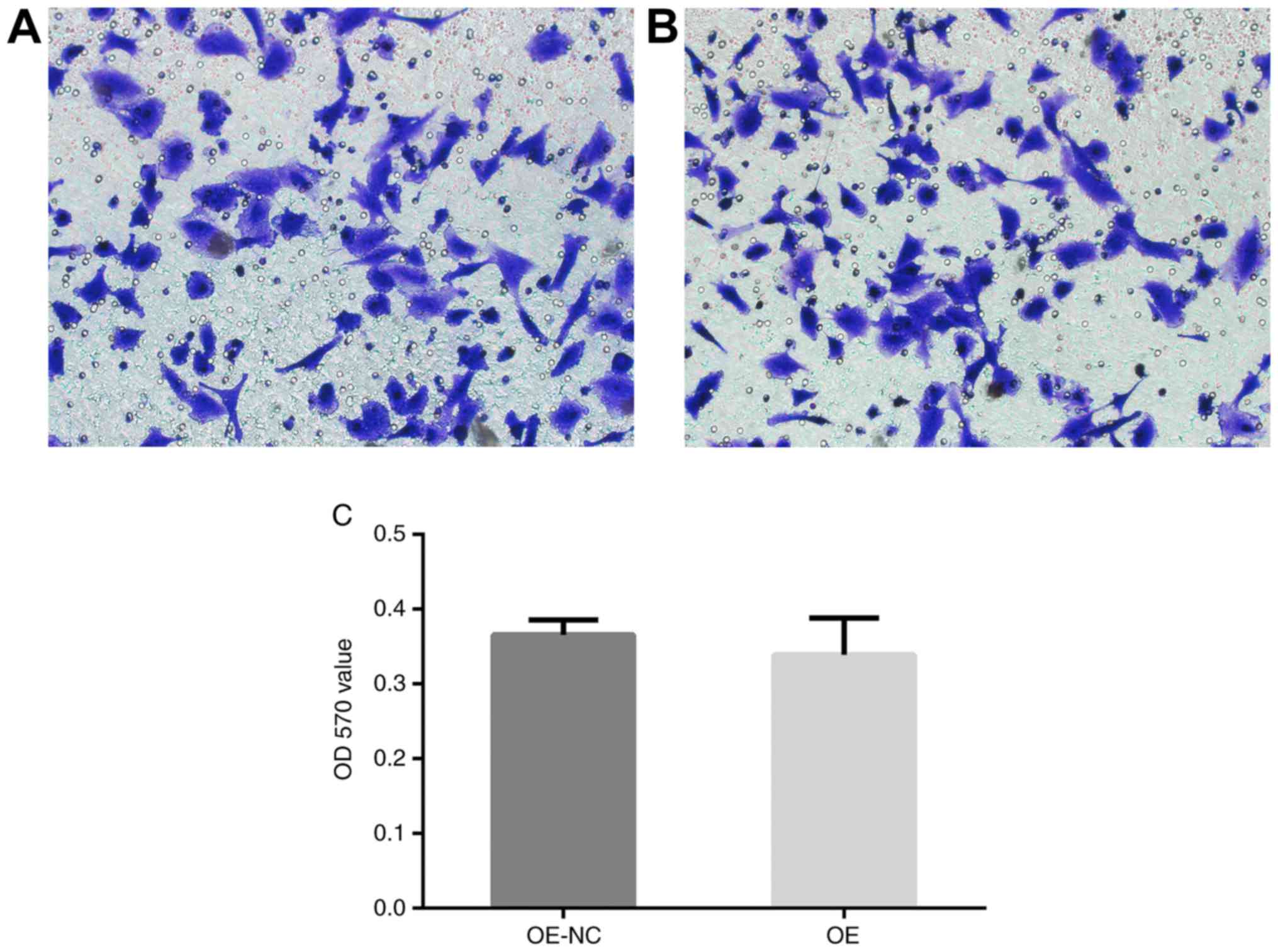
There was no significant difference in the OD570
value between PRKCDBP OE A549/DDP group and PRKCDBP OE A549/DDP NC
group (t=1.654, P=0.213) (Fig. 3).
This highlighted that overexpression of PRKCDBP did not influence
the migration ability of A549/DDP cell line.
Overexpression of PRKCDBP inhibited
cell proliferation and reduced IC50 in the A549/DDP cell line
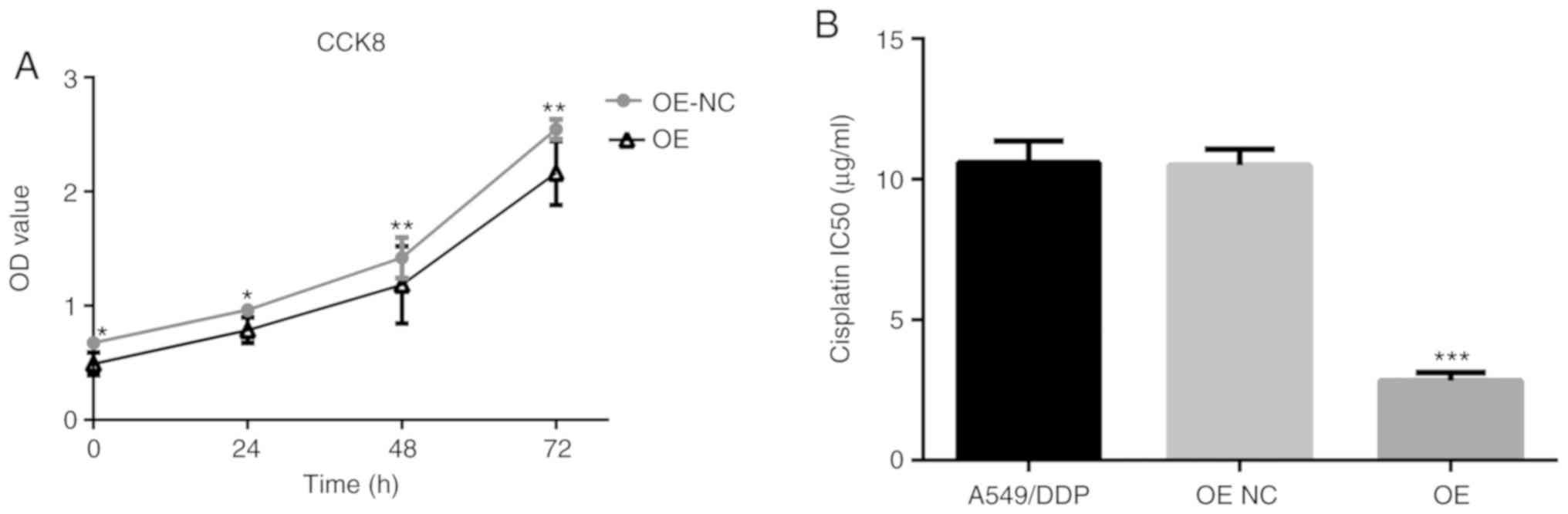
Compared with the PRKCDBP OE-NC A549/DDP group,
OD450 in the PRKCDBP OE A549/DDP group was markedly reduced at 0
(P<0.05), 24 (P<0.05), 48 (P<0.01), and 72 h (P<0.01)
(Fig. 4A). The IC50 of cisplatin in
the PRKCDBP OE A549/DDP group (5.98±1.20 µg/ml) was lower than that
in the A549/DDP (10.68±2.12 µg/ml, P<0.001) and PRKCDBP OE-NC
group (10.87±2.21 µg/ml, P<0.001) (Fig. 4B).
Overexpression of PRKCDBP promoted
cell apoptosis
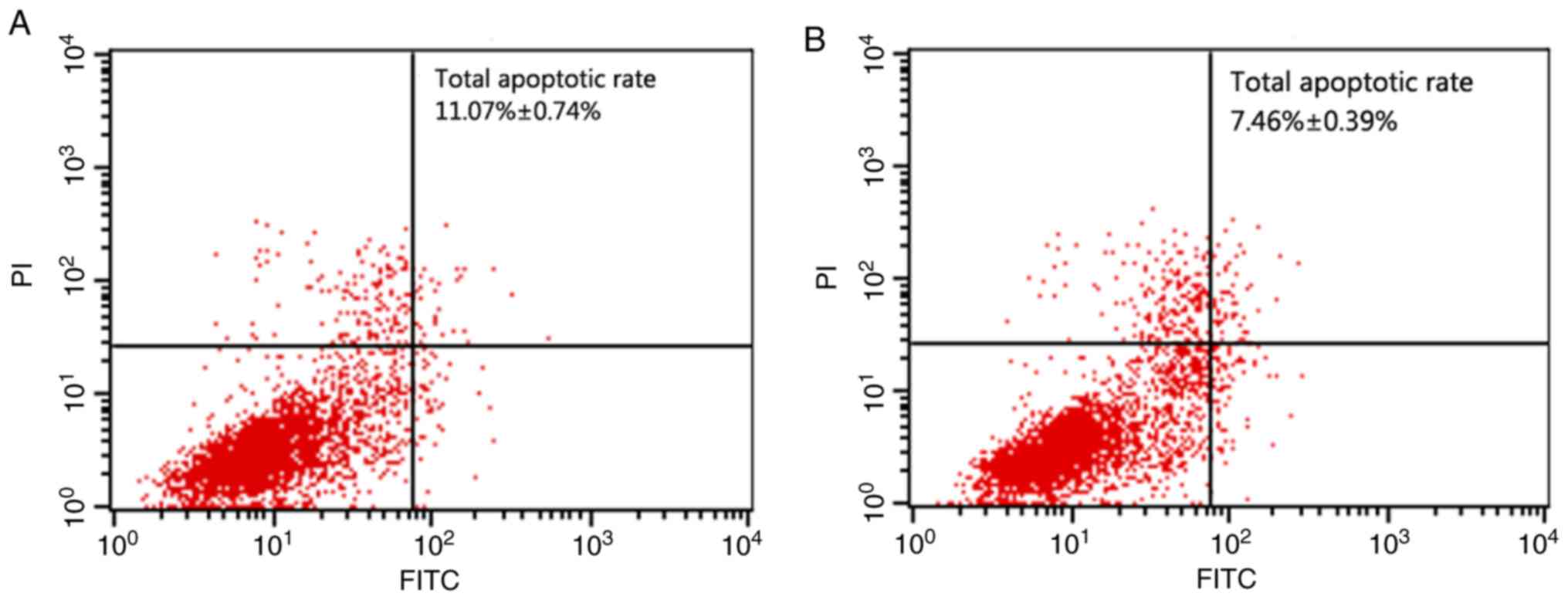
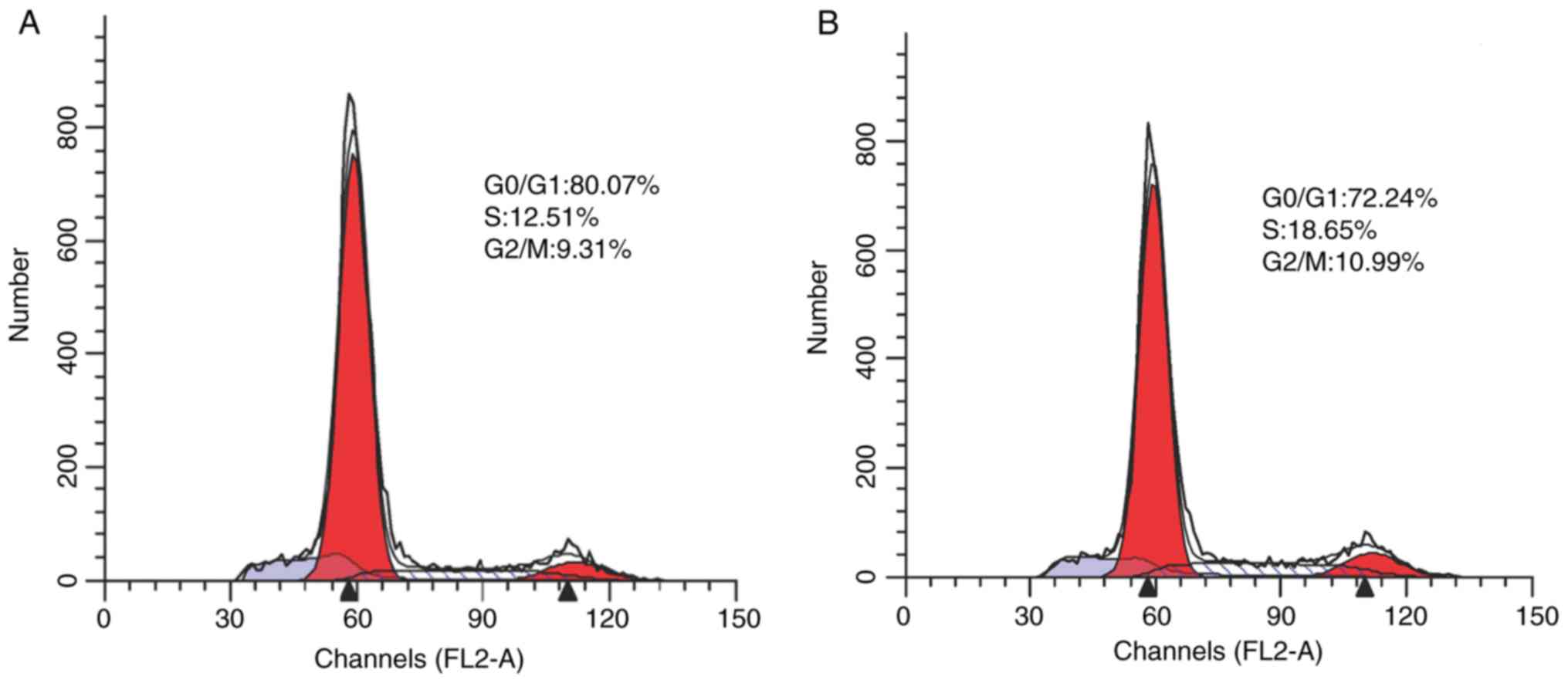
Flow cytometry showed an apoptotic rate of
11.07±0.74% in the PRKCDBP OE A549/DDP group was significantly
higher than that of 7.46±0.39% in the PRKCDBP OE-NC A549/DDP group
(t=8.652, P<0.001), which demonstrated that overexpression of
PRKCDBP promoted cell apoptosis (Fig.
5). Additionally, the cells in G0/G1 phase (80.07±2.4%) in the
PRKCDBP OE A549/DDP group were significantly increased (t=22.147,
P<0.001), compared with those in the PRKCDBP OE-NC A549/DDP
group (72.24±2.1%), while those in the S (12.51±0.21%) and G2/M
(9.31±0.11%) phases were markedly declined (18.65±0.13 and
10.99±0.12%) (t=−4.15, P<0.001; t=−8.02, P<0.001). The
results of the cell cycle assay further confirmed that
overexpression of PRKCDBP inhibited the proliferation ability of
A549/DDP cell line (Fig. 6).
Results of DNA methylation
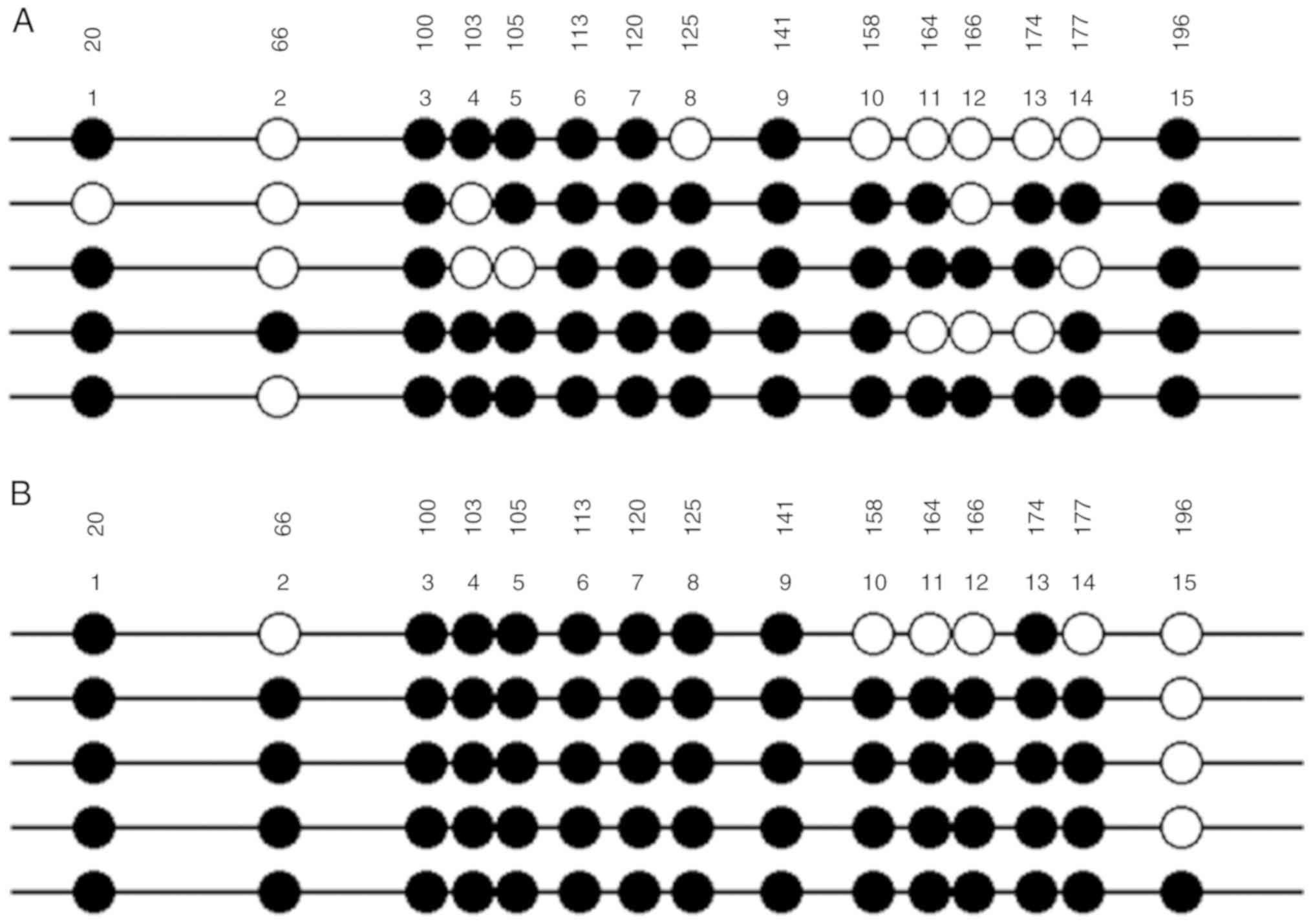
As shown in Fig. 7,
the degree of methylation of PRKCDBP in A549/DDP cell line (66/75)
was markedly higher than that in A549 cells (56/75) (Table I). Therefore, DNA methylation was
caused by DNA methyltransferase DNMT1, resulting in
hypermethylation of PRKCDBP promoter in A549/DDP cells and
reduction of PRKCDBP mRNA and protein levels. Further studies
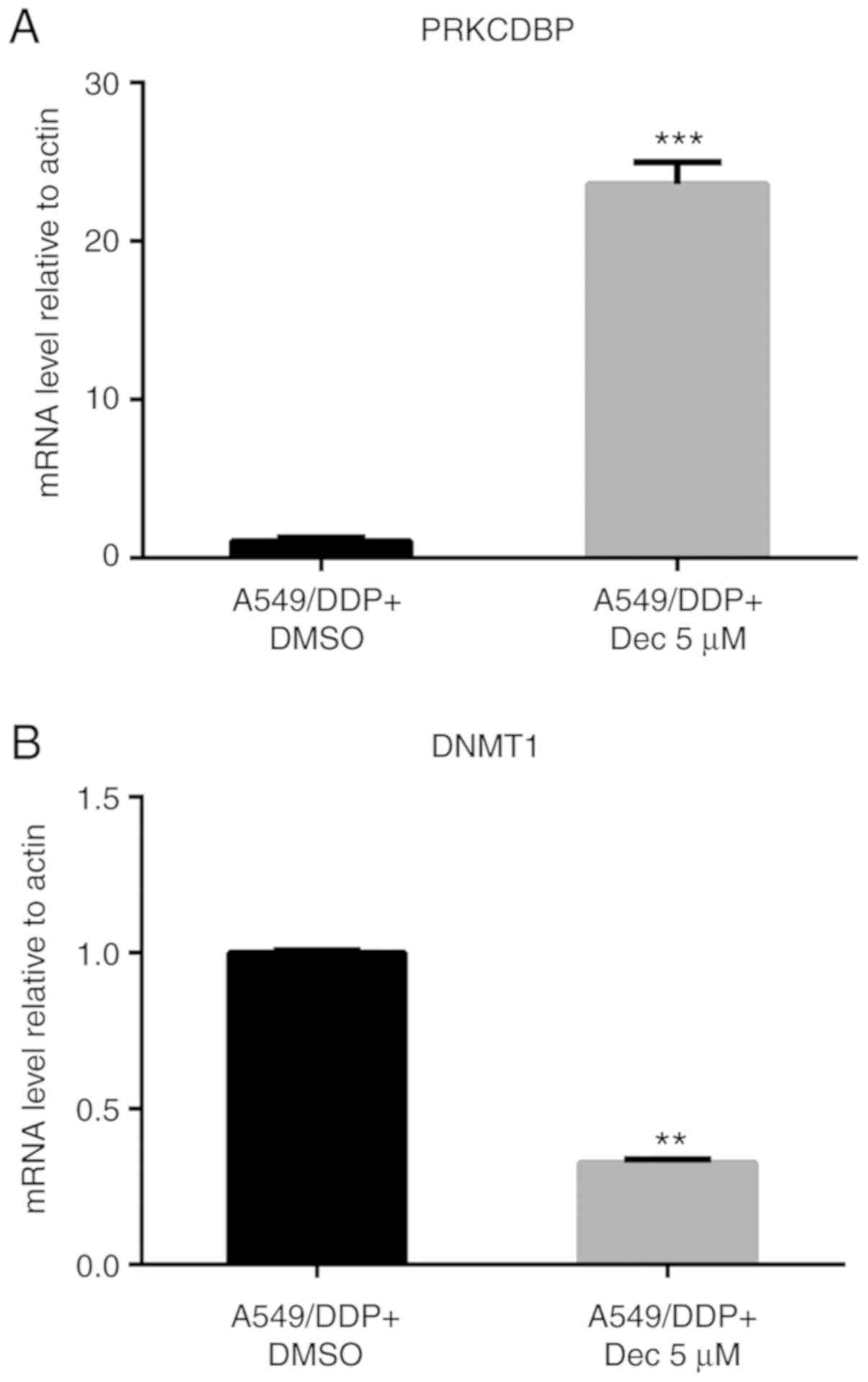
showed that PRKCDBP mRNA level was notably elevated with 5 µM
decitabine for 24 h (t=16.00, P<0.0001), while the DNMT1 mRNA
level was significantly decreased (t=48.422, P<0.0001) (Fig. 8). Therefore, decitabine is a
specific DNA methyltransferase inhibitor that can inhibit the DNMT1
mRNA level, thereby inhibiting the methylation of PRKCDBP, leading
to an increase in the PRKCDBP mRNA level.
 | Table I.PRKCDBP methylation status of each CpG
site among A549 and A549/DDP cell lines. |
Table I.
PRKCDBP methylation status of each CpG
site among A549 and A549/DDP cell lines.
| CpG position | 20 (%) | 66 (%) | 100 (%) | 103 (%) | 105 (%) | 113 (%) | 120 (%) | 125 (%) | 141 (%) | 158 (%) | 164 (%) | 166 (%) | 174 (%) | 177 (%) | 196 (%) | Total (%) |
|---|
| A549 Me-CpG | 4/580.0 | 1/520.0 | 5/5100.0 | 3/560.0 | 4/580.0 | 5/5100.0 | 5/5100.0 | 4/580.0 | 5/5100.0 | 4/580.0 | 3/560.0 | 2/540.0 | 3/560.0 | 3/560.0 | 5/5100.0 | 56/7574.7 |
| A549/DDP
Me-CpG | 5/5100.0 | 4/580.0 | 5/5100.0 | 5/5100.0 | 5/5100.0 | 5/5100.0 | 5/5100.0 | 5/5100.0 | 5/5100.0 | 4/580.0 | 4/580.0 | 4/580.0 | 5/5100.0 | 4/580.0 | 1/520.0 | 66/7588.0 |
TNF-α regulated PRKCDBP to influence
cisplatin resistance in LAD
We compared the PRKCDBP OE A549/DDP group and the
PRKCDBP NC A549/DDP group by mRNA expression profiling, and it was
found that TNF-α could regulate PRKCDBP. Our findings revealed that
the TNF-α mRNA level in A549/DDP cell line was lower than in A549
cells (t=41.52, P<0.0001) and BEAS-2B (t=42.50, P<0.0001)
(Fig. 9A). Lentiviral
vector-mediated overexpression of TNF-α A549/DDP and siRNA vector
transfection of A549 cell line (Fig.
9B) was established. After TNF-α was overexpressed, PRKCDBP
mRNA level was elevated (t=5.977, P=0.004, Fig. 9C), and IC50 was reduced (10.6±0.4
vs. 5.4±0.2 µg/ml, t=6.34, P=0.002, Fig. 9D) in A549/DDP. After TNF-α was
knocked down, PRKCDBP mRNA level was reduced (t=7.878, P=0.001,
Fig. 9C) and IC50 was increased
(1.4±0.1 vs. 3.7±0.2 µg/ml, t=10.45, P=0.007, Fig. 9D) in A549 cells. Thus, we speculated
that TNF-α could regulate PRKCDBP expression level to influence
cisplatin resistance in LAD.
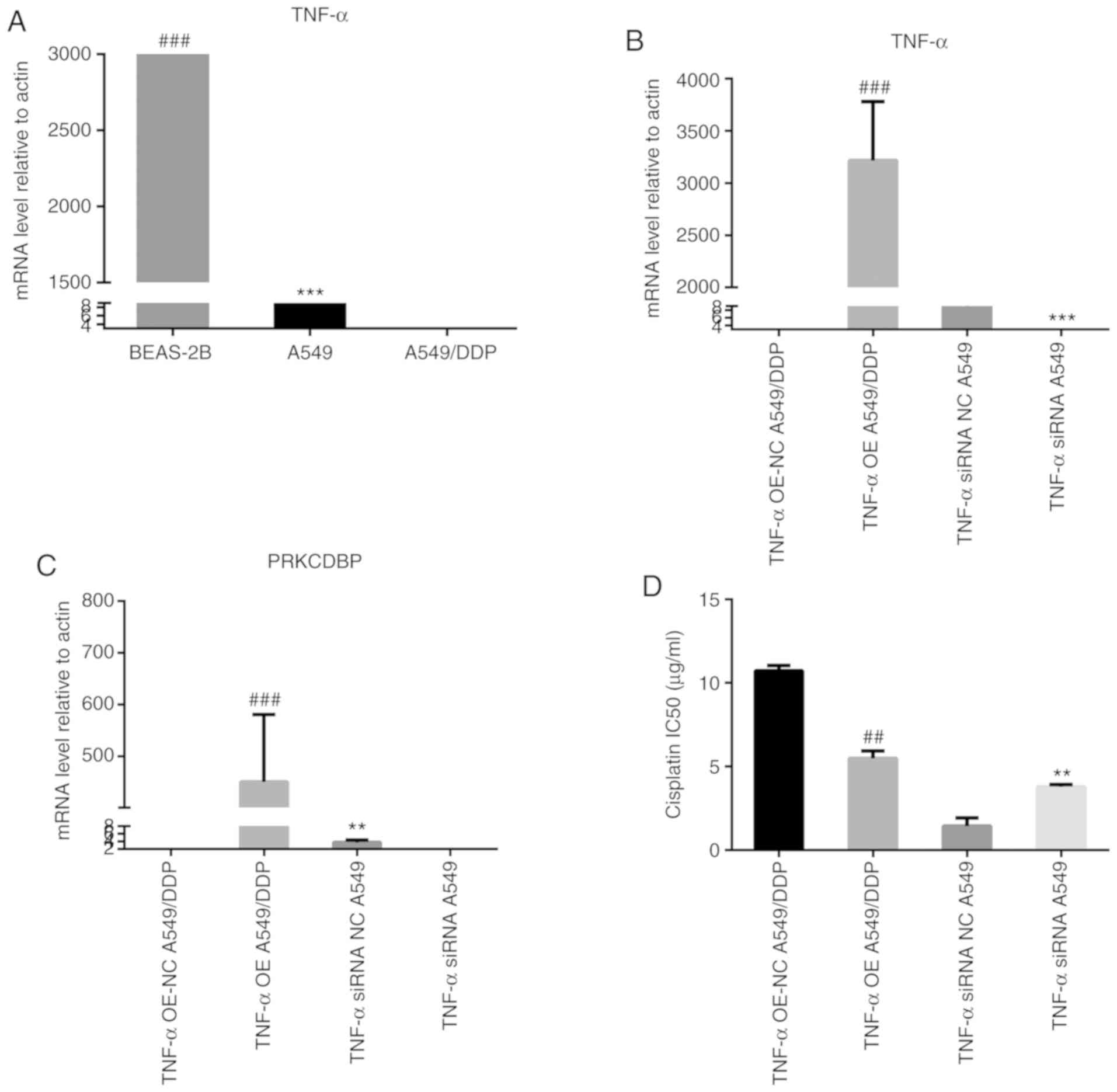 | Figure 9.TNF-α regulated PRKCDBP to influence
cisplatin resistance in LAD. (A) The TNF-α mRNA level in A549/DDP
was less than that in A549 cells (t=41.52, P<0.0001) and BEAS-2B
(t=42.50, P<0.0001). (B) Lentivirus-mediated TNF-α
overexpression and siRNA vector transfection cells was established.
(C) After TNF-α was overexpressed, PRKCDBP mRNA expression level
was increased (t=5.977, P=0.004) in A549/DDP. After TNF-α was
knocked down, the PRKCDBP mRNA expression level was reduced
(t=7.878, P=0.001) and in A549. (D) After TNF-α was overexpressed,
IC50 was decreased (IC=10.6±0.4 vs. 5.4±0.2 µg/ml, t=6.34, P=0.002)
in A549/DDP. After TNF-α was knocked down, IC50 was increased
(IC=1.4±0.1 vs. 3.7±0.2 µg/ml, t=10.45, P=0.007) in A549.
##P<0.01, ###P<0.001, **P<0.01,
***P<0.001. TNF, tumor necrosis factor; PRKCDBP, protein kinase
C delta binding protein. |
Overexpression of DNMT1 improved IC50
in A549 cells
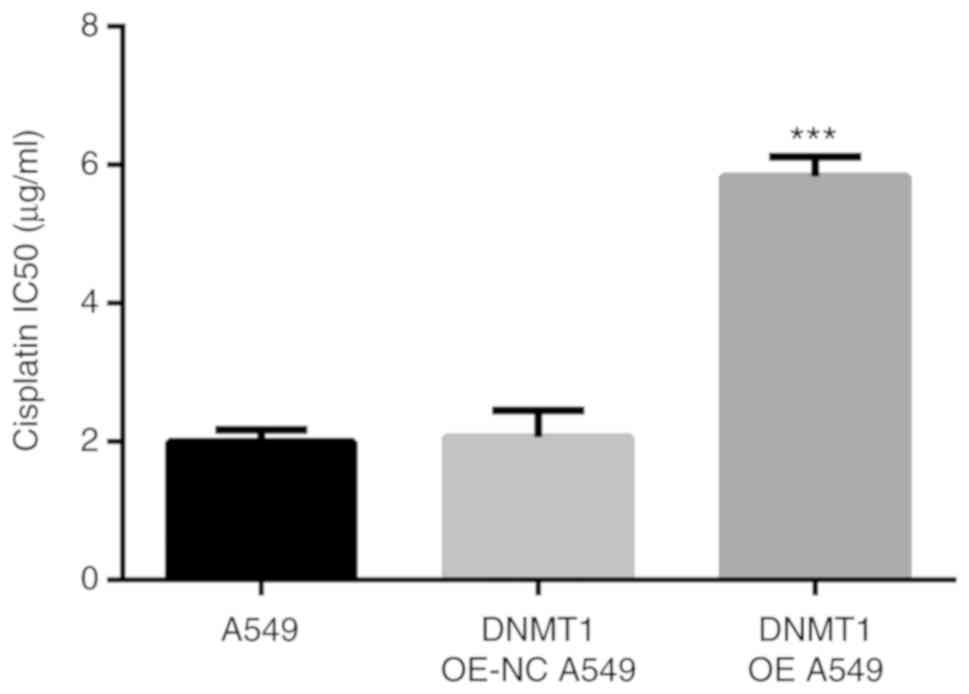
The IC50 of cisplatin in DNMT1 OE A549 group
(5.83±0.18 µg/ml) was markedly higher than that in A549 cells
(1.99±0.10 µg/ml, P<0.001) and DNMT1 OE-NC A549 group (2.06±0.22
µg/ml, P<0.001) (Fig. 10).
Therefore, overexpression of DNMT1 improved the IC50 of cisplatin
in A549 cells and increased cisplatin resistance.
Discussion
Cisplatin exerts anticancer effects via multiple
mechanisms, yet its most prominent (and best understood) mode of
action involves the generation of DNA lesions followed by
activation of the DNA damage response and the induction of
mitochondrial apoptosis. Despite a consistent rate of initial
responses, cisplatin treatment often results in the development of
chemoresistance, leading to therapeutic failure (8–12).
Cisplatin resistance arises through a multifactorial mechanism
involving reduced drug uptake, increased drug inactivation,
increased DNA damage repair, and inhibition of transmission of DNA
damage recognition signals to the apoptotic pathway, such as P53
pathway.
In the current study, our findings showed that
PRKCDBP level was markedly decreased in LAD tissues and A549/DDP
cell line compared to the adjacent cancer tissues and A549 cells,
while the DNMT1 mRNA level was significantly elevated. In addition,
the promoter of PRKCDBP was hypermethylated in A549/DDP. DNMT1 mRNA
level in cisplatin-insensitive group was markedly higher than that
in cisplatin-sensitive group while PRKCDBP mRNA expression level in
cisplatin-insensitive group was notably lower than that in
cisplatin-sensitive group. Compared with the PRKCDBP NC A549/DDP
group, the DNMT1 mRNA level was significantly reduced. Further
investigation showed that PRKCDBP mRNA level was significantly
elevated after treatment with 5 µM decitabine for 24 h, while DNMT1
mRNA level was markedly decreased. The above-mentioned findings
revealed that PRKCDBP is a tumor suppressor gene and the promoter
of PRKCDBP was hypermethylated in A549/DDP cell line by DNMT1.
After PRKCDBP was overexpressed, cell proliferation,
S phase and G2/M phase cells were notably reduced. By contrast,
apoptosis ability, G0/G1 phase cells and IC50 of cisplatin were
significantly elevated, although the migration ability did not
markedly change. We compared the PRKCDBP OE A549/DDP group and the
PRKCDBP NC A549/DDP group by mRNA expression profiling, and it was
found that TNF-α could regulate PRKCDBP. These results are
consistent with some literature reports (17,20,21).
TNF-α resulted in an increase of the PRKCDBP mRNA level, while
TNF-α siRNA led to a decrease thereof (P<0.001). These findings
demonstrated that low PRKCDBP could increase the proliferation
ability and IC50 of cisplatin. TNF-α appeared to regulate PRKCDBP
mRNA level to affect cisplatin resistance in LAD. Overexpression of
DNMT1 improved the IC50 of cisplatin in A549 cells and increased
cisplatin resistance.
In summary, results of the present study showed that
the promoter of PRKCDBP was hypermethylated in A549/DDP. Low
expression level of PRKCDBP could promote cisplatin resistance in
LAD by DNMT1 and TNF-α.
Acknowledgements
Not applicable.
Funding
This study was financially supported by the National
Natural Science Foundation of China (81672088), Zhejiang Provincial
Natural Science Foundation (LY19H200002), Zhejiang Provincial
Health Planning Commission (2018KY514), the Wenzhou Municipal
Science and Technology Bureau of China (Y20170208, Y20170718), and
Zhejiang University Student Science and Technology Innovation
Activity Plan (New Miao Talent Plan, 2020R413082).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JF, HZ and YW made substantial contributions to the
design of the study. JF and JC analyzed and interpreted the patient
data. JF, HZ, JC and YW performed the cell biological experiments.
All authors contributed to writing the manuscript. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Institutional Ethics
Review Committee of the First Affiliated Hospital of Wenzhou
Medical University. Patient consent was obtained.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Herbst RS, Morgensztern D and Boshoff C:
The biology and management of non-small cell lung cancer. Nature.
553:446–454. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pignon JP, Tribodet H, Scagliotti GV,
Douillard JY, Shepherd FA, Stephens RJ, Dunant A, Torri V, Rosell
R, Seymour L, et al: Lung adjuvant cisplatin evaluation: A pooled
analysis by the LACE Collaborative Group. J Clin Oncol.
26:3552–3559. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zarogoulidis K, Zarogoulidis P, Darwiche
K, Boutsikou E, Machairiotis N, Tsakiridis K, Katsikogiannis N,
Kougioumtzi I, Karapantzos I, Huang H and Spyratos D: Treatment of
non-small cell lung cancer (NSCLC). J Thorac Dis. 5 (Suppl
4):S389–S396. 2013.PubMed/NCBI
|
|
5
|
Bunn PA Jr and Kelly K: New combinations
in the treatment of lung cancer: A time for optimism. Chest. 117 (4
Suppl 1):138S–143S. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Spiro SG and Silvestri GA: One hundred
years of lung cancer. Am J Respir Crit Care Med. 172:523–529. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Choi MK and Kim DD: Platinum transporters
and drug resistance. Arch Pharm Res. 29:1067–1073. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Martin LP, Hamilton TC and Schilder RJ:
Platinum resistance: The role of DNA repair pathways. Clin Cancer
Res. 14:1291–1295. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wangpaichitr M, Wu C, You M, Kuo MT, Feun
L, Lampidis T and Savaraj N: Inhibition of mTOR restores cisplatin
sensitivity through down-regulation of growth and anti-apoptotic
proteins. Eur J Pharmacol. 591:124–127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Seve P and Dumontet C: Chemoresistance in
non-small cell lung cancer. Curr Med Chem Anticancer Agents.
5:73–88. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ohmichi M, Hayakawa J, Tasaka K, Kurachi H
and Murata Y: Mechanisms of platinum drug resistance. Trends
Pharmacol Sci. 26:113–116. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu C, Wangpaichitr M, Feun L, Kuo MT,
Robles C, Lampidis T and Savaraj N: Overcoming cisplatin resistance
by mTOR inhibitor in lung cancer. Mol Cancer. 4:252005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hu L, Chen J, Zhang F, Wang J, Pan J, Chen
J and Wang Y: Aberrant long noncoding RNAs expression profiles
affect cisplatin resistance in lung adenocarcinoma. Biomed Res Int.
2017:74981512017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li Y, Melnikov AA, Levenson V, Guerra E,
Simeone P, Alberti S and Deng Y: A seven-gene CpG-island
methylation panel predicts breast cancer progression. BMC cancer.
15:4172015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fukasawa M, Kimura M, Morita S, Matsubara
K, Yamanaka S, Endo C, Sakurada A, Sato M, Kondo T, Horii A, et al:
Microarray analysis of promoter methylation in lung cancers. J Hum
Genet. 51:368–374. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Moutinho C, Martinez-Cardus A, Santos C,
Navarro-Pérez V, Martínez-Balibrea E, Musulen E, Carmona FJ,
Sartore-Bianchi A, Cassingena A, Siena S, et al: Epigenetic
inactivation of the BRCA1 interactor SRBC and resistance to
oxaliplatin in colorectal cancer. J Natl Cancer Inst.
106:djt3222014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee JH, Kang MJ, Han HY, Lee MG, Jeong SI,
Ryu BK, Ha TK, Her NG, Han J, Park SJ, et al: Epigenetic alteration
of PRKCDBP in colorectal cancers and its implication in tumor cell
resistance to TNFα-induced apoptosis. Clin Cancer Res.
17:7551–7562. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yilmaz A, Menevse S, Konac E and Alp E:
The DNA methyl transferase inhibitor, 5′-aza-2-deoxycitidine,
enhances the apoptotic effect of Mevastatin in human leukemia HL-60
cells. Hum Exp Toxicol. 33:414–423. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim JW, Lee CK, Kim HJ, Shim JJ, Jang JY,
Dong SH, Kim BH, Chang YW and Chi SG: Polymorphisms in PRKCDBP, a
transcriptional target of TNF-α, Are associated with inflammatory
bowel disease in Korean. Intest Res. 13:242–249. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim JW, Kim HJ, Lee CK, Shim JJ, Jang JY,
Dong SH, Kim BH, Chang YW and Chi SG: Elevation of PRKCDBP, a novel
transcriptional target of TNF-α, and its downregulation by
infliximab in patients with ulcerative colitis. Dig Dis Sci.
59:2947–2957. 2014. View Article : Google Scholar : PubMed/NCBI
|