Introduction
Leukemia is a malignant clonal disease of
hematopoietic stem cells (HSCs). During the process of maintaining
self-renewal and differentiation, HSCs accumulate a considerable
amount of DNA damage under the influence of endogenous or exogenous
factors. This damage, either irreparable or only very slowly
reparable, leads to genetic mutations, thus causing leukemogenesis
(1). At present, the major
treatment strategy for leukemia is pharmacotherapy. However, drug
resistance, which is exhibited in a considerable number of patients
with some mutational genes related to DNA damage repair (DDR)
response, has seriously limited its therapeutic effect (2). It is well known that there is a
complex but regulatable DDR mechanism in vivo that
selectively initiates specific repair mechanisms according to
different types of DNA damage. For example, DNA suffering from base
mismatch or deletion can initiate base excision repair (BER); DNA
single-strand breaks (SSBs) can be repaired by specific enzymes
in vivo such as apurinic/apyrimidinic endonuclease 1 (APE1),
polynucleotide kinase 3′-phosphatase (PNKP), and tyrosyl-DNA
phosphodiesterase 1 (TDP1); DNA double-strand breaks (DSBs) can
initiate the non-homologous end joining repair pathway (NHEJ) or
homologous recombination (HR) (3,4).
The single-strand annealing (SSA) repair pathway is
a bypass of HR. Unlike the classical pathway, SSA does not depend
on the presence of sister chromatids, so the mismatch rate
increases significantly (5). RAD52
(Radiation sensitive 52) is a key molecule in the regulation of the
SSA process after DNA damage end excision. Previous studies have
indicated that the MRN (MRE11/RAD50/NBS1) complex is linked to
these breaks, thereby initiating the strand exchange reaction of
DNA under the action of RAD52 (6,7). By
contrast, accumulating evidence has shown that BRCA1 and BRCA2 are
critical components of HR and are downregulated in acute myeloid
leukemia (AML) (8,9). Moreover, SSA bypass plays a key role
in DDR when HR is incomplete in cells with mutation or
downregulation of breast cancer type 1 susceptibility protein
(BRCA1)/breast cancer type 2 susceptibility protein (BRCA2)
(10). Furthermore, DNA
damage-related signaling pathways have been identified by several
reports indicating that the downstream cell cycle checkpoint
proteins are activated after DNA damage and thus affect DDR
(11,12).
The present study aimed to explore whether
RAD52-induced regulation of repair bypass occurs in AML cells and
to elucidate the underlying mechanism for this. To this end, we
applied an RAD52 aptamer to AML cells with downregulated BRCA1/2.
The effects of the RAD52 aptamer on cell proliferation, cell
apoptosis, and DNA damage repair were evaluated after drug
intervention. Moreover, expression of apoptosis-related proteins
and STAT3 signaling was also detected to explore the underlying
mechanism of RAD52 aptamer-induced regulation of AML cells.
Materials and methods
Peptide aptamers
Aptamers are short RNAs or DNAs made up by 20–80
nucleotides. Aptamers can fold into unique three-dimensional
conformations to specifically bind to targets (13). F79 synthetic peptide is a member of
the aptamers that contains a sequence of 13 amino acids surrounding
RAD52 (F79) (VINLANEMFGYNG-GGG-YARAAARQARA), and this was purchased
from Genemed Synthesis Inc.. F79 has a three-residue polyglycine
linker and protein transduction domain 4; these facilitate passage
across lipid bilayers and direct intracellular transduction of the
aptamers (14). Aptamers received
N-terminal tetramethyl-rhodamine and C-terminal amidation for
intracellular detection and reduction of proteolytic degradation
(15). F79 aptamers were purified
by high-performance liquid chromatography (HPLC) and characterized
by mass spectroscopy (MS).
Cell lines and reagents
Human acute myeloid leukemia (AML) cell lines
Kasumi-1 and KG1a were obtained from Guangzhou Institute of
Biomedicine and Health, Chinese Academy of Sciences, and no
mycoplasma infection was confirmed. The STAT3 inhibitor Stattic was
purchased from Cayman Chemical Co. (#14590) and was diluted in
DMSO. VP16 was purchased from Sigma-Aldrich/Merck KGaA (#E1383) and
diluted in DMSO.
Antibodies
CD34 antibody (#555821) and CD38 antibody (#555460)
for flow cytometry conjugated with biotin or specific dyes as
required were purchased from BD Biosciences; γ-H2AX antibody was
purchased from BioLegend (#613402). Western blot-related antibodies
were purchased from Cell Signaling Technologies [GAPDH (#2118);
BRCA1 (#9010); BRCA2 (#10741); DNA Damage Antibody Sampler Kit,
(#9947); H2AX (#2595); CHK2 (#2662); ATM (#2873); ATR (#2790); Bcl2
(#15071) BIM (#2819); STAT3 (#9139); p-STAT3 (#9145)].
Flow cytometry
Cells in logarithmic growth phase were collected at
a density of 1×106 per well, washed twice in 2% FBS
solution, blocked using Fc (Fc:2% FBS=1:100), and placed on ice for
10–15 min. Antibodies (diluted 1:200) were added, and the cells
were incubated for another 15 min in the dark. A blank tube and
single stain tubes were set as controls. Fluorescence intensity was
detected by BD FACSCalibur (BD Biosciences) and analyzed by FlowJo
software (version 7.6; FlowJo, LLC).
Collection of CD34-positive cells
Bone marrow samples were collected from healthy
donors, diluted in 2% FBS solution, superimposed on a Ficoll
lymphocyte separation solution (#1692254; MP Biomedicals), and
gently removed in a single nuclear layer after centrifugation.
Cells were resuspended, and red blood cells were lysed on ice for
8–10 min with 3–5 ml of FACS lysing solution (#349202; BD
Biosciences). Samples were resuspended in 5–10 ml of 2% FBS and
filtered through a 70-µm filter in a 15 ml centrifuge tube. Cells
were resuspended in 300 µl MACS buffer (PBS+0.5% BSA+2 mM EDTA),
blocked with FcR, and each sample received 100 µl of CD34 magnetic
beads (#130-046-702; Miltenyi Biotec, Inc.) in the dark. After
incubation for 30 min at 4°C, the cell suspension was instilled in
the column of the magnetic bead separator (#130-042-801; Miltenyi
Biotec). The fluid through the magnetic field was discarded, and
the remaining cells (CD34-positive) were collected after removal of
the magnetic field.
Healthy BM donor samples were collected from i) a
24-year old female, on 2019-8-25, ii) a 40-year-old male, on
2019-8-10, iii) a 38-year-old male, on 2020-9-15, iv) a 29-year-old
female, on 2019-9-8, v) a 33-year-old male, on 2019-9-10 and vi) a
28-year-old male, on 2019-9-28.
RT-qPCR
RNA was extracted using a Takara RNA extraction kit
(#9767; Shiga, Japan). After the purity and concentration were
determined, reverse transcription reaction was performed with the
Takara reverse transcription kit, followed by the Takara kit for
quantitative polymerase chain reaction. Thermocycling conditions
consisted of: Initial denaturation: 95°C for 30 sec, 39 cycles:
95°C 5 sec, 60°C 30 sec, 95°C for 1 sec, final extension: 65°C for
5 sec and 95°C for 5 sec. Data analysis was performed using the
2−ΔΔCq method (16) with
GAPDH as the baseline. Primer sequences were as follows: BRCA1
forward, 5′-GGAGGTCAGGAGTTCGAAACC-3′ and reverse,
5′-ACCGGCTAATTTCTGTATTTTTAGTAGAG-3′; BRCA2 forward,
5′-ACCTGTTAGTCCCATTTGTACATTTG-3′ and reverse,
5′-CACAACTCCTTGGTGGCTGAA-3′; GAPDH forward,
5′-ACCACAGTCCATGCCATCAC-3′ and reverse,
5′-TCCACCACCCTGTTGCTGTA-3′.
Drug trials
Cells (1×104) were seeded into 96-well
plates and then treated with 5 µM F79 and different concentrations
of VP16 (1, 2, 4, 8, 16 and 32 µg/ml) and incubated at 37°C in 5%
CO2 for 48 h. The inhibition rate (IR) was evaluated by
CCK-8 assay as described below. KG1a and Kasumi-1 cells were
treated with 0, 5 µM F79, IC50 VP16, 5 µM
F79+IC50 VP16 at 37°C in 5% CO2 for 48 h.
Flow cytometry and western blot analysis were conducted by using
the cells above. Stattic (0, 2.5, 5, 7.5 and 10 µM) was added to
the KG1a cells and incubation was carried at 37°C in 5%
CO2 for 24 h, and IR was evaluated by CCK-8 assay.
γ-H2AX expression level, cell apoptosis rate, cell cycle
distribution were measured using flow cytometry between cells
treated with 5 µM Stattic and the blank group for 24 h.
Cell proliferation
Different experimental groups were set with
triplicate wells each, and cell suspensions were plated in 96-well
plates at ~100 µl/well and incubated for 24 h. A 10 µl volume of
CCK-8 reagent (#CK04, Dojindo) was added to each well, plates were
incubated for 2 h, and the absorbance at 450 nm was measured. The
inhibition rate (IR) was calculated as follows, IR=100%-(Average
absorbance of the experimental group-Average absorbance of the
blank group)/(Average absorbance of the control group-Average
absorbance of the blank group). Data were analyzed by GraphPad
Prism software (GraphPad Software, Inc.).
Colony formation assay
Target cells were collected, and methylcellulose
(#HSC001; R&D Systems) was added to the cell suspension (1:2),
which was completely mixed and seeded in a 24-well plate at ~1
ml/well and cultured in an incubator for 14 days. Cell number
>100 was assumed for one colony. The number of colonies was
observed and recorded under a microscope (Zeiss Axio Vert. A1;
original magnification, ×40).
Comet assay
Samples were made according to the Trevigen
(#4250-050-K) instruction manual, and the slides were observed
under fluorescence microscopy (original magnification, ×200).
Western blot analysis
Cells were lysed for 30 min in an ice-cold buffer
containing RIPA lysate (~5×105 cells/ml), PMSF (1:100),
leupetin (1:1,000), and NaVO3 (5:1,000). The supernatant
was collected, the optical density (OD) value was detected, and the
samples were normalized. Samples were electrophoresed at room
temperature for 90 min and transferred to a PVDF membrane
(ISEQ00010, IPVH00010, Millipore, USA) on ice for 100 min. The
required internal reference protein and the target protein bands
were blocked at room temperature for 30 min-1 h after excision.
Membranes were placed in TBST buffer with the primary antibody
(1:2,000 diluted by 5% BSA) overnight at 4°C followed by the
incubation of the secondary antibody (1:5,000 diluted by 5% skim
milk, #9947) for 2 h and the addition of ECL solution, with washing
between each step. Band densities were observed with ImageJ
software (1.51; National Institutes of Health).
Image processing and statistical
analysis
Images were processed using Adobe Illustrator (CS6
13.0.0; Adobe Systems Inc.) and GraphPad Prism 6.01 (GraphPad
Software, Inc.). All data were analyzed using SPSS Statistics 19
(IBM Corp.) and are presented as mean ± SD. Each experiment was
repeated three times. Student's t-tests were performed using
statistical software GraphPad Prism v.6, and P-values are indicated
in the figure legends. Values are considered significant at
P<0.05, and the level of significance is noted in the figures as
follows: *P<0.05 or +P<0.05, **P<0.01 or
++P<0.01, ***P<0.001 or +++P<0.001
and ****P<0.0001 or ++++P<0.0001.
Results
Expression of BRCA1/BRCA2 is lower in
Kasumi-1 and KG1a cells than in primary hematopoietic stem
cells
To reflect the response of leukemia stem cells
(LSCs) after treatment with chemotherapeutic agents, cell lines
with a phenotype close to primary leukemia stem cells were selected
for this study. In previous experiments, we found that Kasumi-1 and
KG1a cell lines have the highest percentage of CD34-positive cells
among the various human acute myeloid leukemia cell lines.
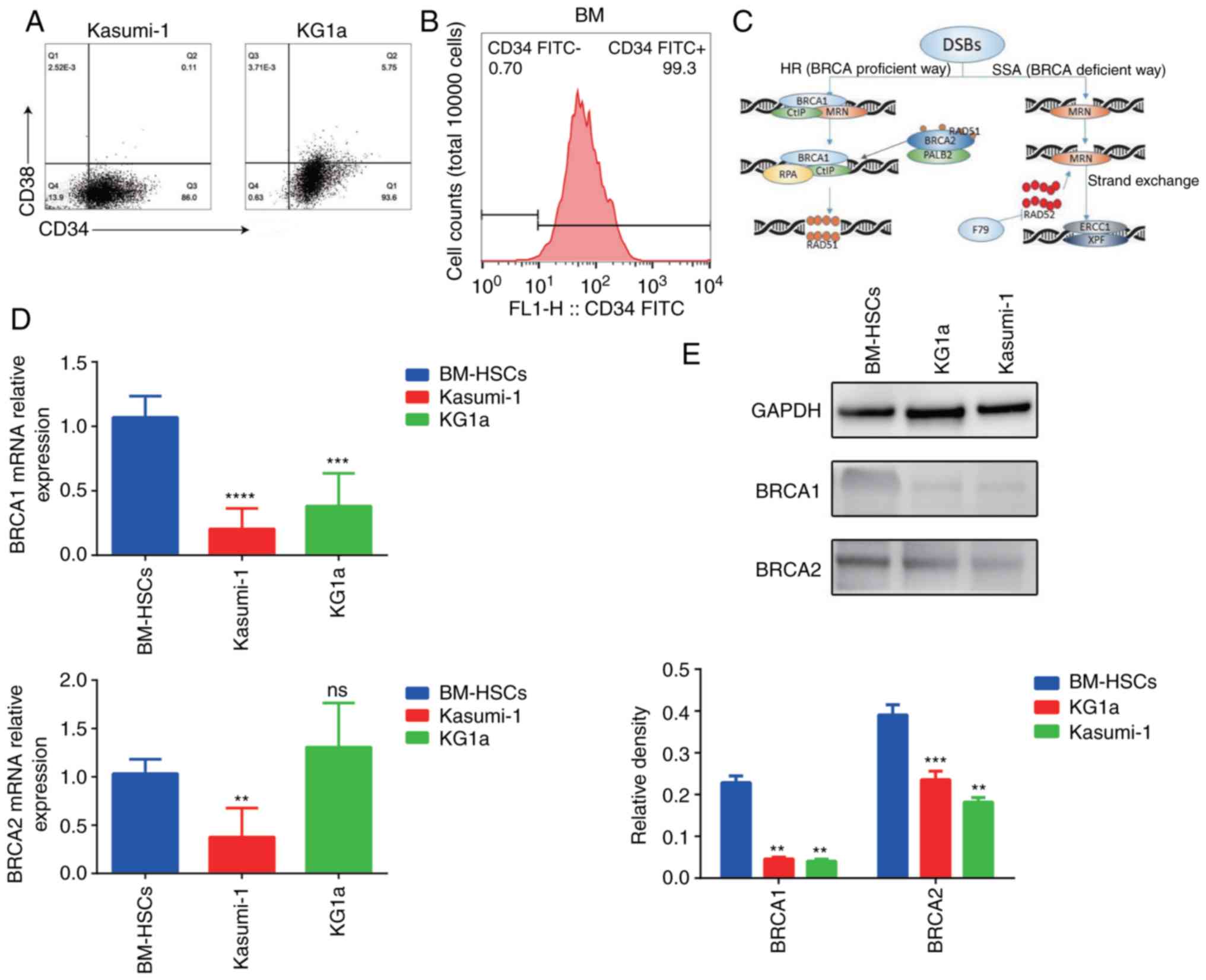
Therefore, we examined the ratio of
CD34+CD38− cells in these two cell lines
(Fig. 1A).
CD34+CD38− cells accounted for 86% of the
total Kasumi-1 cells and 93.6% of the total KG1a cells. In
addition, normal human bone marrow (BM) samples were collected for
sorting CD34+ mononuclear cells, and these were used as
the normal hematopoietic stem cell (HSC) control group in
subsequent experiments. As shown in Fig. 1B, the percentage of CD34+
cells reached 99% after sorting. In order to better understand the
role of BRCA1/2 in DDR, the diagram of related signaling pathways
is shown in Fig. 1C. The mRNA and
protein expression of BRCA1 and BRCA2 in normal CD34+
HSCs (from BM), Kasumi-1, and KG1a cells was detected by RT-qPCR
and western blot analysis (Fig. 1D and
E). Expression levels of BRCA1 in Kasumi-1 and KG1a cells were
significantly lower than that in the CD34+ HSCs. BRCA2
expression in Kasumi-1 cells was lower than that in the BM HSCs. No
statistical difference arose between the mRNA expression levels of
BRCA2 in KG1a and CD34+ HSCs, but the expression of
BRCA2 protein was significantly lower than that in the
CD34+ HSCs. Despite some exceptions, these results
indicated that expression of BRCA1/BRCA2 in Kasumi-1 and KG1a cells
was lower than that noted in the primary HSCs.
RAD52 aptamer inhibits the cell
proliferation of acute myeloid leukemia cells and promotes cell
apoptosis
To investigate the effects of the RAD52 aptamer on
cell proliferation and apoptosis, human myeloid leukemia cells
treated with F79 were subjected to CCK-8 assay to detect the IR of
cell proliferation by different concentrations of VP16 and were
compared with cancer cells without F79 treatment. As shown in
Fig. 2A, treatment with F79
achieved an obviously higher IR of cell proliferation in the
VP16-treated Kasumi-1 and KG1a cells. To further explore whether
F79 treatment affects cell survival, the cells were divided into a
control group (no treatment), F79 group (F79 treatment only), VP16
group (VP16 treatment only), and a combined group (F79+VP16
treatment). No significant difference arose in the apoptotic rate
between the F79 group and the control group in Kasumi-1 cells,
indicating the ignorable function of mere F79 treatment in cell
apoptosis (Fig. 2B). Notably, early
apoptosis, late apoptosis, and total apoptotic rate of the combined
group were statistically increased compared with those of the VP16
group (Fig. 2B). By contrast, in
KG1a cells, no significant difference was found between the F79
group and the control group, while the combined group had an
increased early apoptotic rate compared with that of the VP16 group
(Fig. 2B). Together, these results
clarified that F79 treatment promotes VP16-induced cell apoptosis
in human myeloid leukemia cells. In addition, cancer cells were
allowed to grow in methylcellulose medium for 14 days to form
colonies that were counted at the end of culture. We found that the
colony number formed by Kasumi-1 cells in the F79 group was
significantly decreased, while no difference between the two groups
could be observed in KG1a cells (Fig.
2C). Morphology demonstrated that the colonies formed in the
F79 group were generally smaller than those of the control group,
and the morphology was uneven (Fig.
2C), indicating that F79 inhibited the growth of the leukemia
cells.
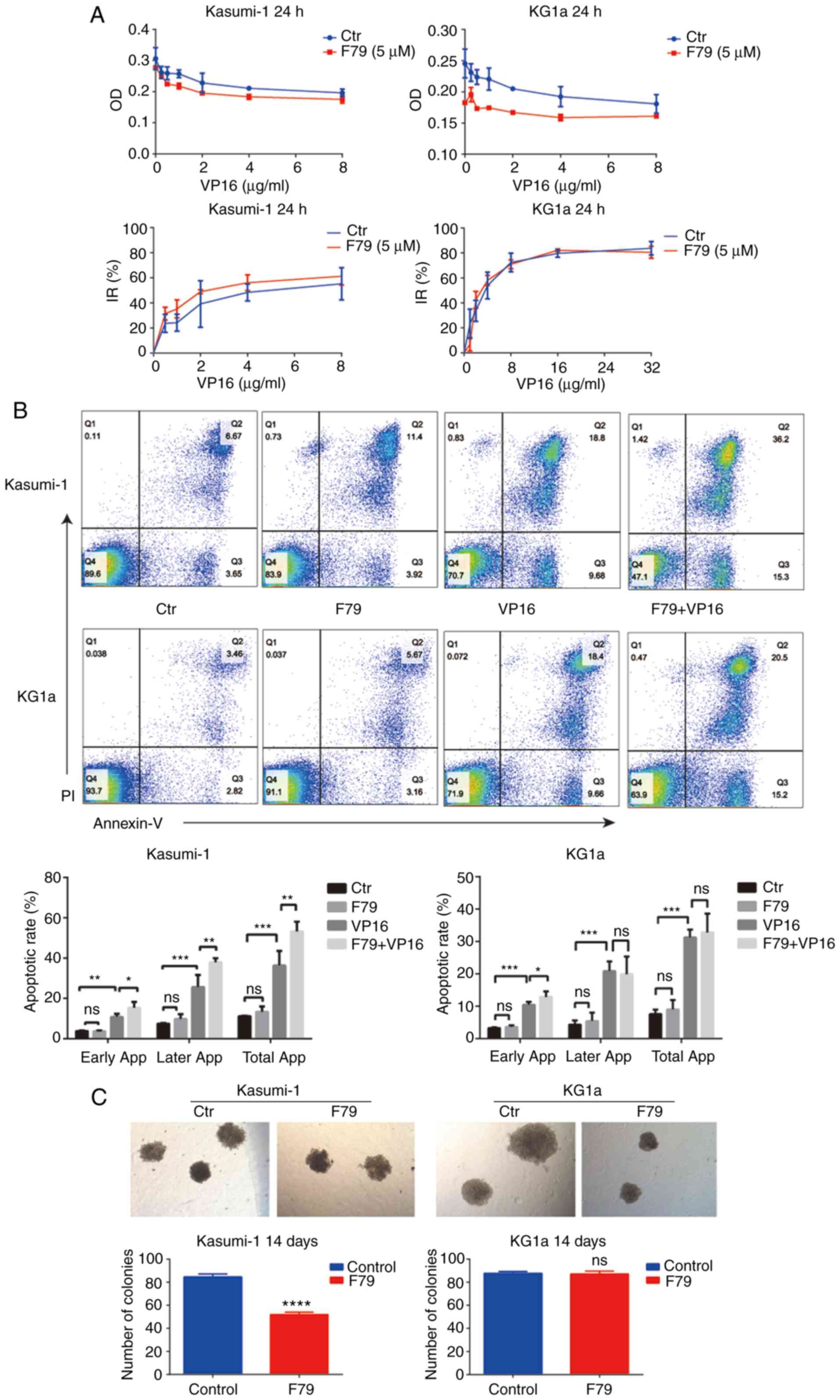 | Figure 2.RAD52 aptamer affects the
proliferation and apoptosis of leukemia cells. (A) CCK-8 assay was
used to detect the optical density (OD) values of the two groups of
cells treated with different concentrations of VP16. The OD values
of the experimental group were lower than those of the control
group. At the same time, the inhibition rate (IR) of cell
proliferation was higher in the F79 group than in the control group
(n=3). (B) Apoptotic rate of cells from each group was recorded
after 48 h of culture. Ctr, blank control group; F79 group, treated
with 5 µM F79; VP16 group, treated with IC50
concentration of VP16; F79+VP16 group, combination of the two
drugs. Data were plotted from triplicate plates. Unpaired Student's
t-test (two-tailed) was used for statistical analysis. Data are
shown as mean ± SD. *P<0.05, **P<0.01, ***P<0.001; ns,
P>0.05, n=3. (C) The number of cell colonies in each group was
counted after 14 days of culture. Cell number >100 was assumed
for one colony. Kasumi-1: Ctr: n=87, 82, 85; F79: n=50, 52, 54;
****P<0.0001. No colonies were formed in the VP16 group and the
combined group. KG1a: Ctr: n=89, 86, 88; F79: n=85, 86, 90; ns,
P>0.05. IC50, half maximal inhibitory concentration;
VP16, etoposide. |
Leukemia cells self-repair DNA
damage
Intervention of VP16 caused DNA double-strand breaks
in LSCs, thus inducing serious DNA damage. However, LSCs can repair
DNA damage by themselves. In this study, VP16 was utilized for the
pretreatment of Kasumi-1 and KG1a cells, and a comet assay and flow
cytometry were performed to detect the degree of DNA damage and
self-repairing capability after injury. As shown in Fig. 3A, the outcomes of the comet assay
exhibited that obvious comet tailing was observed in both Kasumi-1
and KG1a cells after treatment with VP16. In addition, DNA damage
repair behavior was observed after 2 h, and no more comet tail was
visualized after 4 or 6 h, indicating excellent DNA self-repairing
capability. Simultaneously, the mean fluorescence intensity (MFI)
of γ-H2AX detected by flow cytometry was significantly increased
after drug elution compared with the control group, which decreased
gradually in the next 4 h and remained unchanged after that
(Fig. 3B). The time variation was
roughly consistent with the results of the comet assay, confirming
that treatment with VP16 induced DNA damage in Kasumi-1 and KG1a
cells, and this was fixed by DNA self-repairing capability.
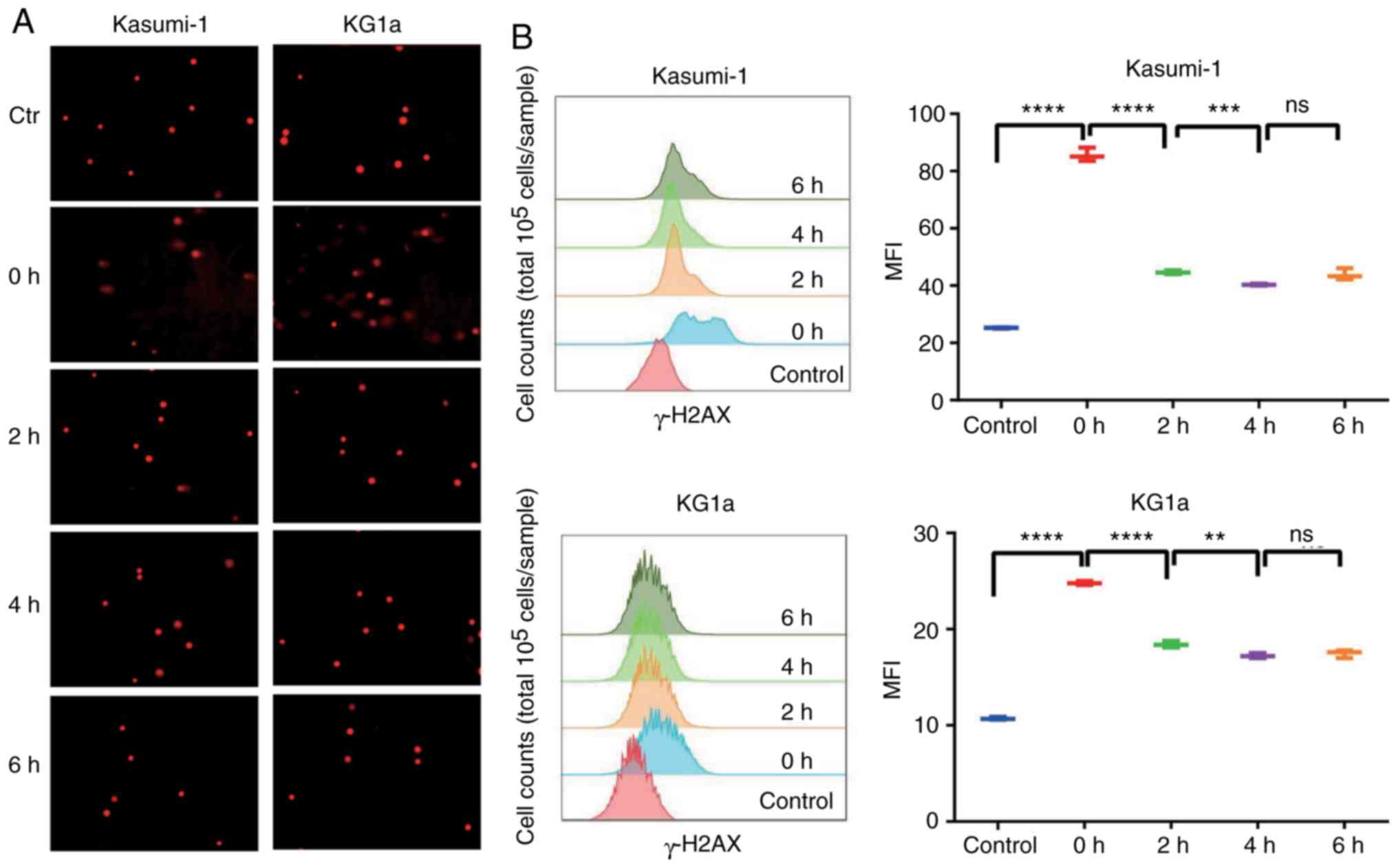 | Figure 3.Leukemia cells repair themselves when
damaged. (A) Cells were treated with VP16 for 2 h, and the drug was
then washed away. Expression of γ-H2AX was detected by comet assay
at 0, 2, 4, and 6 h. (B) After treatment of VP16 for 2 h, the
expression of γ-H2AX was detected by flow cytometry at 0, 2, 4, and
6 h. The results were expressed as mean fluorescence intensity
(MFI); n=3; 0 h, ****P<0.0001 (both cell lines); 2 h,
****P<0.0001 (both cell lines); 4 h, Kasumi-1 ***P<0.001,
KG1a **P<0.01. There was no significant difference (ns) at 6 h.
VP16, etoposide. |
RAD52 aptamer inhibits DNA damage
repair in LSCs
To investigate the effects of the RAD52 aptamer on
DNA self-repair capability of LSCs, the cancer cells pretreated
with VP16 were subjected to F79 treatment and flow cytometry for
detecting MFI of γ-H2AX and were compared with the cells without
F79 treatment. Firstly, cells were pretreated with VP16 and divided
into a control group and F79 group. After the drug was washed then
MFI was detected. As shown in Fig.
4A, although only minor differentiation was observed at 2 and 4
h, the MFI exhibited significant increase at 6 h in the F79 groups
of both Kasumi-1 and KG1a cells, indicating that F79 inhibited DNA
self-repair. In addition, untreated cells were divided directly
into four groups and MFI was detected, respectively. We further
verified that F79 increased the MFI of γ-H2AX in cells treated with
VP16 at both 2 and 24 h to a much larger extent than that without
VP16 treatment (Fig. 4B),
indicating the ability of F79 to inhibit the DNA self-repair
capability of LSCs with VP16-induced DNA damage.
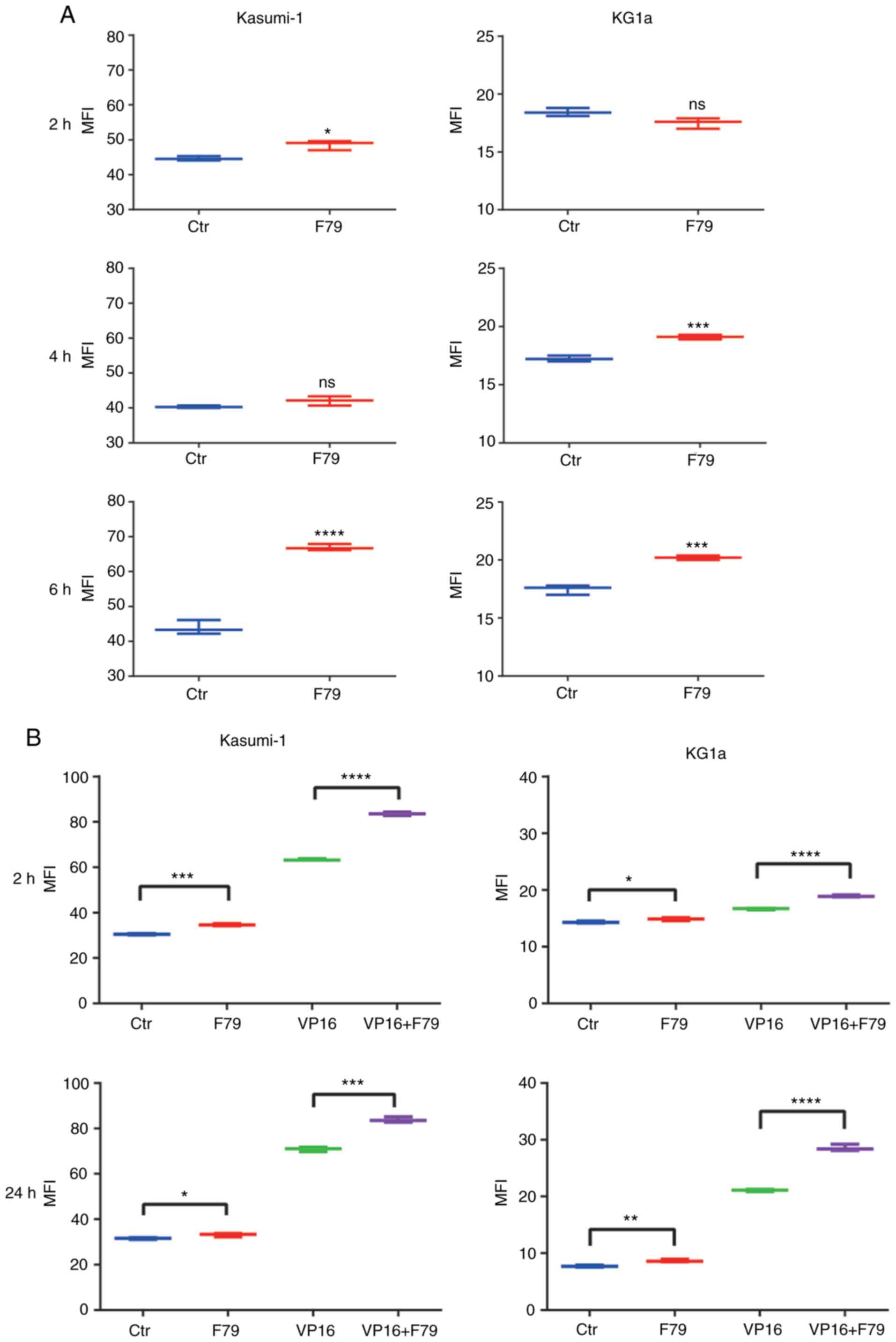 | Figure 4.Effect of RAD52 aptamer on DNA damage
repair in leukemia cells. (A) Cells were pretreated with VP16 and
divided into control group and F79 (5 µM) group. Flow cytometry was
used to detect the expression of γ-H2AX at indicated time points,
n=3. (B) Detection of mean fluorescence intensity (MFI) in
different groups at 2 and 24 h. Ctr, blank control group; F79
group, cells treated with 5 µM F79; VP16 group, cells treated with
IC50 concentration of VP16; F79+VP16 group, combined
treatment of the two drugs; n=3. *P<0.05, **P<0.01,
***P<0.001, ****P<0.0001; ns, P>0.05. IC50,
half maximal inhibitory concentration; VP16, etoposide. |
RAD52 aptamer persistently activates
cell cycle-associated checkpoint proteins after suffering DNA
damage
Herein, flow cytometry was performed to detect cell
cycle distribution of the control group compared with the VP16
group. The proportion of cells both in the S and G2/M phases was
significantly increased in the VP16 group compared with control
group, suggesting that most of the quiescent cells in the G0/G1
phase entered the cell cycle after VP16 treatment (Fig. 5A). Furthermore, we examined the
expression levels of phosphorylated H2AX (p-H2AX), phosphorylated
ataxia telangiectasia mutated (p-ATM), phosphorylated ataxia
telangiectasia and rad3 related (p-ATR), and phosphorylated
checkpoint kinase 2 (p-CHK2). F79 upregulated the expression of
p-H2AX (γ-H2AX), p-ATM, p-ATR, and p-CHK2 under the condition of
VP16 treatment (Fig. 5B).
Collectively, these results confirmed that F79 activated
cycle-related checkpoint proteins after the onset of DNA damage,
which continuously blocked the cells in the S/G2 phase of the cell
cycle, thereby hindering the damage repair process.
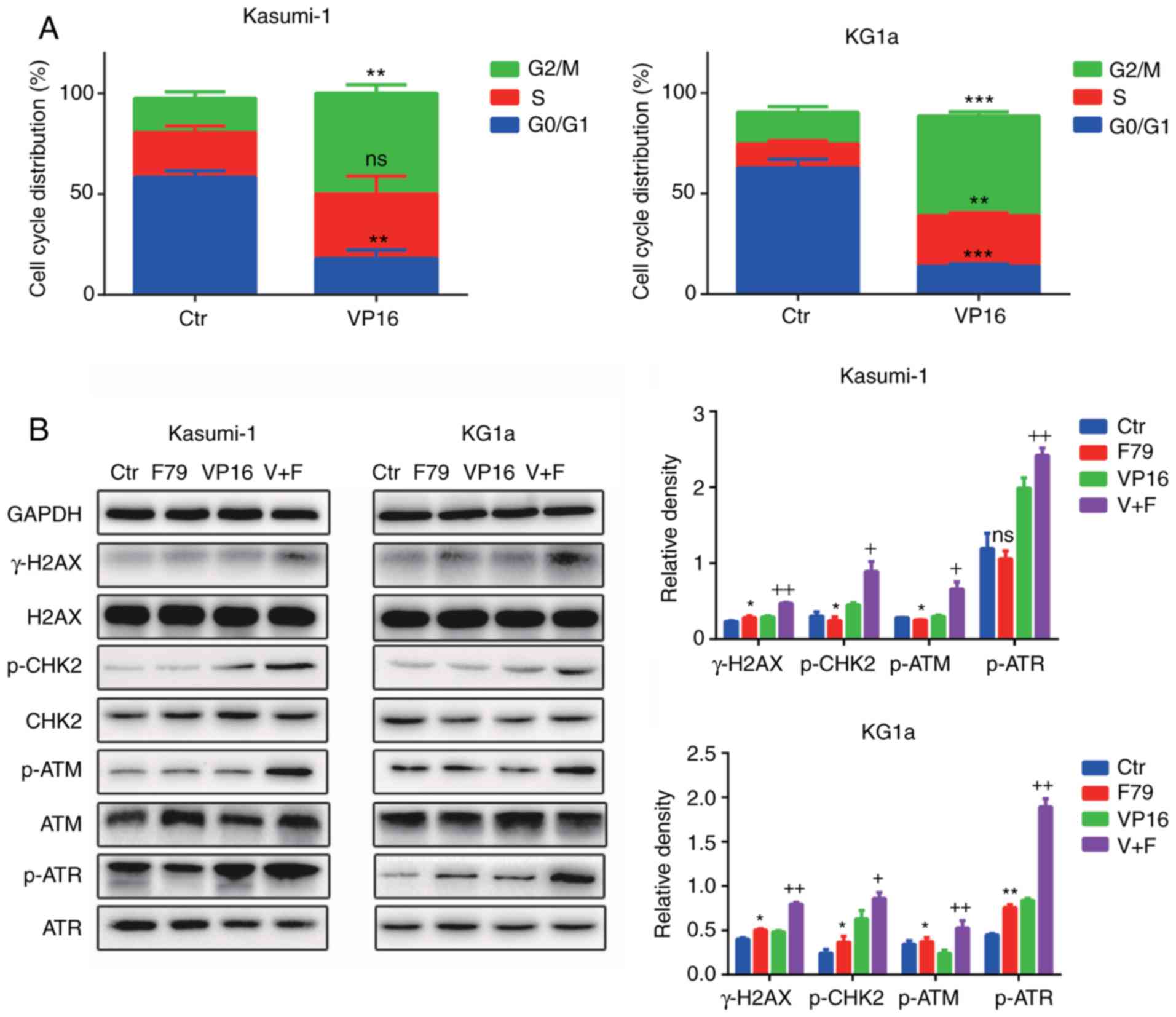 | Figure 5.The RAD52 aptamer activates
cell-cycle-associated checkpoint protein in damaged cells, allowing
cells to persist in the S/G2 phase. (A) Flow cytometry was used to
detect the cell cycle distribution before and after treatment with
VP16. **P<0.01, ***P<0.001 and ns, P>0.05, compared to the
Ctr, n=3. (B) The protein expression of p-H2AX (γ-H2AX), p-ATM,
p-ATR and p-CHK2 in cell lines was detected by western blot
analysis before and after treatment with VP16. Ctr, blank control
group; F79 group, cells treated with 5 µM F79; VP16 group, cells
treated with IC50 concentration of VP16; V+F group,
combined treatment of the two drugs. The results were compared by
relative band density (n=3). *P<0.05, F79 group compared with
the Ctr group; +P<0.05, ++P<0.01, the
combined group (V+F) compared with the VP16 group. p-ATM,
phosphorylated ataxia telangiectasia mutated; p-ATR, phosphorylated
ataxia telangiectasia and rad3 related; p-CHK2, phosphorylated
checkpoint kinase 2; VP16, etoposide. |
Stattic inhibits cell proliferation,
promotes cell apoptosis, and affects the process of DDR in AML
We detected the inhibition rate of KG1a cells
following treatment with the STAT3 inhibitor Stattic at 1, 2.5, 5,
and 10 µM concentration for 24 h. The results suggested that
Stattic had an inhibitory effect on KG1a cells in a
concentration-dependent manner (Fig.
6A). Flow cytometry was used to detect the apoptosis rate, MFI
of γ-H2AX, and the cell cycle distribution. Inhibition of STAT3 by
Stattic promoted cell apoptosis (Fig.
6B), which was statistically significant, and increased the MFI
of γ-H2AX in the cells (Fig. 6C).
In addition, the proportion of cells in the G2/M phase was
significantly increased in the Stattic-treated cells (Fig. 6D).
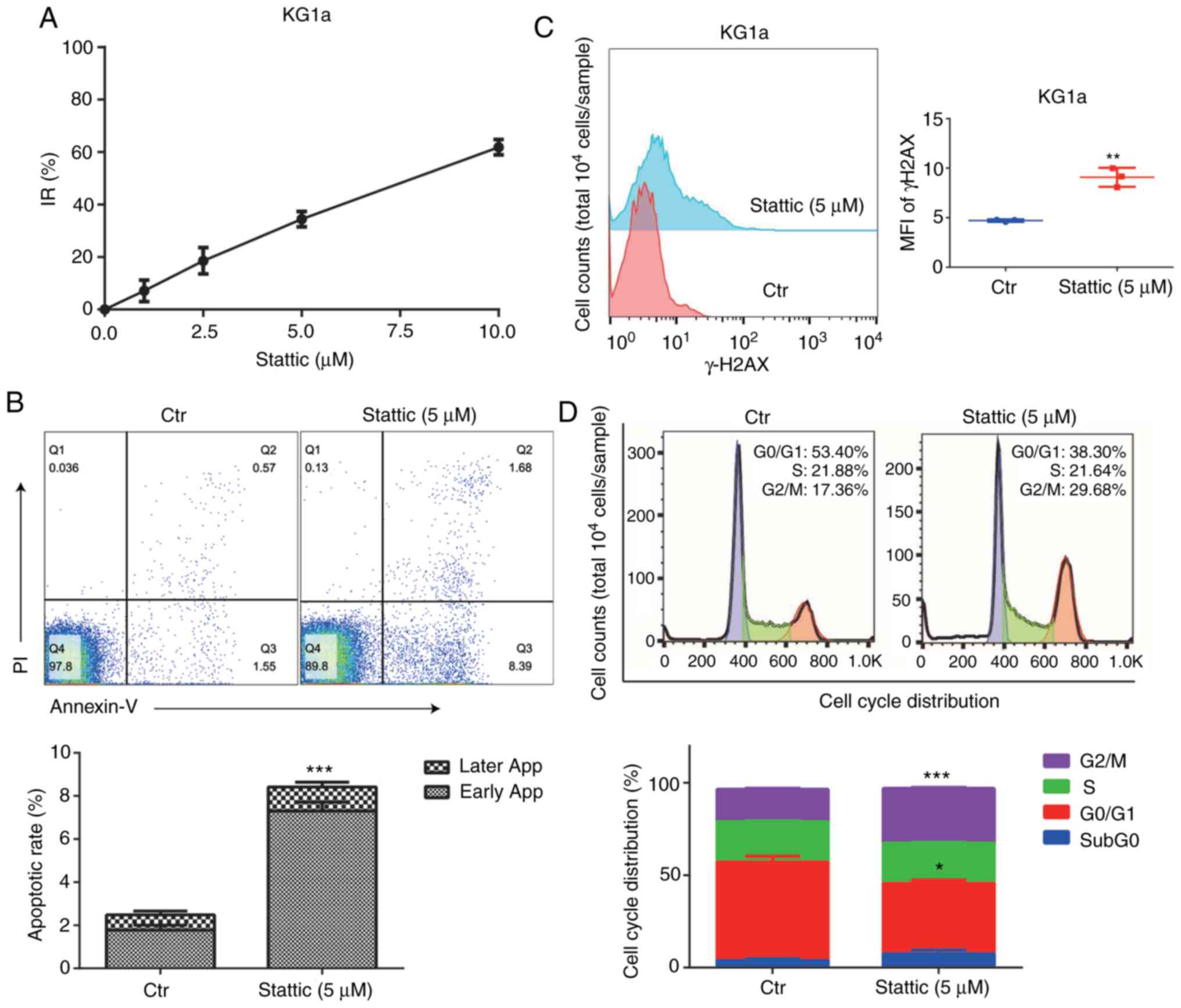 | Figure 6.Effect of STAT3 inhibitor Stattic on
cell survival and DNA damage repair in AML cells. (A) CCK-8 assay
was used to detect the optical density (OD) value of the cells
treated with different concentrations of Stattic for 24 h, and the
inhibition rate (IR) of cell proliferation was calculated. (B)
After treatment of Stattic (5 µM) for 24 h, the apoptotic rate was
detected by flow cytometry. ***P<0.001, compared with the Ctr
group, n=3. (C) Detection of mean fluorescence intensity (MFI) in
both groups: Ctr, blank group; Stattic group (5 µM). **P<0.01,
compared with the Ctr group, n=3. (D) Flow cytometry was used to
detect the cell cycle distribution before and after treatment with
Stattic. *P<0.05, ***P<0.001, compared with the Ctr group,
n=3. |
RAD52 aptamer affects the expression
of apoptotic signaling pathway proteins
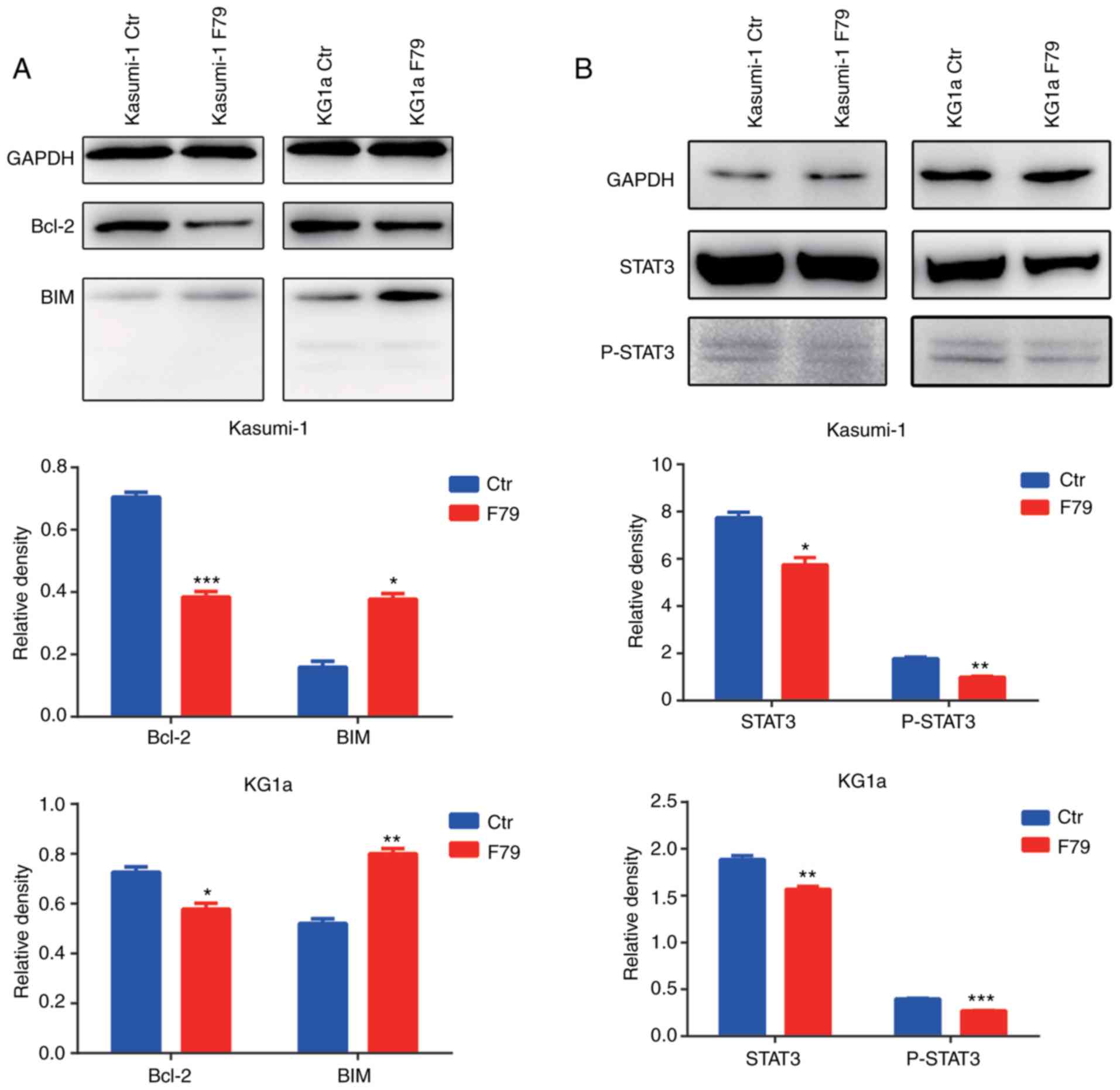
Our previous results showed that F79 increases the
apoptotic rate of cells, suggesting that the DNA damage repair
pathway may be involved in the activation of the apoptotic
signaling pathway. To test this hypothesis, western blot analysis
was performed to detect the expression of Bcl2 family members. We
found that expression of the anti-apoptotic protein Bcl2 was
significantly downregulated upon F79 treatment, while the
expression of the pro-apoptotic protein Bim was significantly
upregulated (Fig. 7A). Consistent
with these and the above experimental results, F79 is thus involved
in the activation of the Bcl2-related apoptotic pathway.
Continuous activation of STAT3 is commonly observed
in human myeloid leukemia cells. In this context, to explore
whether STAT3 participates in the SSA repair pathway, we utilized
western blot analysis to detect the protein expression of STAT3
protein and its activated, phosphorylated form (p-STAT3). The
results showed that F79 significantly downregulated the expression
of STAT3 and p-STAT3 after VP16-induced DNA damage (Fig. 7B), which indicated that SSA may
regulate cell proliferation and apoptosis by affecting the JAK-STAT
signaling pathway.
Discussion
In the present study, we verified the activity of
the single-strand annealing (SSA) repair bypass wherein RAD52
(Radiation sensitive 52) is involved in affecting the proliferation
and apoptosis of leukemia cells under BRCA1/BRCA2 downregulation
and detected the severity of DNA damage before and after VP16
(etoposide) intervention. We found that VP16-induced DNA damage was
repaired by the cells themselves in a short time, while treatment
of cells with RAD52 aptamer F79 prevented the cells from repairing
DNA damage and decreased this DNA self-repairing ability. Some
leukemia patients are resistant to chemotherapy drugs as leukemia
stem cells (LSCs) in vivo own a rapid DNA damage-repair
ability, preventing tumor cells from death (17). The homologous recombination (HR)
pathway plays a leading role in DNA damage repair (DDR) (18). However, the efficacy of HR is
greatly limited in acute myeloid leukemia cells with downregulated
BRCA1/BRCA2 expression. The SSA pathway leads to the lethality of
cells suffering from severe DNA damage after the HR pathway is
injured. Our research further emphasizes that synthetic lethality
can be achieved by blocking RAD52 in AML through different
experimental methods.
In addition, we found that cell-cycle checkpoint
proteins were upregulated during drug intervention. Meanwhile, the
expression of anti-apoptotic protein Bcl-2 was downregulated in
cells treated with F79, while the expression of pro-apoptotic
protein Bim was upregulated. When ataxia telangiectasia mutated
(ATM) and ataxia telangiectasia and rad3 related (ATR) are
recruited to double-strand breaks (DSBs), they phosphorylate
themselves and downstream signaling proteins such as p53 and
DDR-related cell cycle checkpoint proteins such as checkpoint
kinase (CHK)1, CHK2, and CDC25. Activation of checkpoint proteins
causes temporary cell-cycle arrest (2,19,20).
Once the damage is repaired, the cells can undergo a normal cycle,
while continuous cell cycle arrest activates the apoptotic pathway
proteins and promotes cell apoptosis. It has been reported that
when the cell cycle checkpoint is activated, p53 protein, which is
capable of regulating and promoting cell apoptosis by
downregulating the expression of Bcl-2, is also activated (3,21,22).
Our study indicates that AML cells after F79 intervention were in a
state of continuous cell cycle arrest and eventually led to their
death. We proposed that the SSA may activate upstream or downstream
proteins of the Bcl2-related apoptotic pathway, which in turn
affects cell survival and is likely to affect the expression of the
p53 protein. However, p53 has been found to be mutated in a variety
of tumor cells, and its mutation types are diverse, making it
difficult to be accurately targeted (23). Therefore, the presence of another
signal transduction pathway associated with DDR under the SSA
pathway has yet to be fully reported.
Clinically, we have seen an increase in acute
myeloid leukemia (AML) patients with STAT3 activation and found
that AML patients with STAT3 activation are prone to drug
resistance. Our study showed that inhibition of STAT3 played a role
in cell survival and DDR. Furthermore, STAT3 and its activated form
(p-STAT3) were downregulated in LSCs treated with RAD52 aptamer,
indicating that the SSA pathway may affect the expression of STAT3.
STAT3 is often seen activated in leukemia cells (24). However, the continuous activation of
STAT3 in myeloid leukemia cells may be related to drug resistance
and has become an urgent problem to be solved for killing leukemia
cells (25). In recent years, many
studies have found that STAT3 is involved in DDR, especially during
cell-cycle checkpoint activation, in the cells of other types of
malignant tumors (26–29). Reportedly, STAT3 prevents the
transmission of the ATR-CHK1 signaling pathway, thereby inhibiting
the progression of DDR in S phase (27). However, downregulation of STAT3
inhibits the activation of ATR-CHK1 and ATM-CHK2 signaling, which
in turn attenuates the activation of checkpoint proteins in the
S/G2 phase (26). Other studies
have shown that STAT3 is indeed involved in the regulation of DDR
and the downstream apoptosis pathway in cancer cells (30), but how STAT3 works in AML cells has
not been reported. Here, we preliminarily demonstrated that the
downregulation of STAT3 or p-STAT3 affects DDR, and RAD52 may
regulate the STAT3-related signaling pathway. However, the specific
mechanism remains unclear.
Our study had several limitations. The experiments
used to determine the anti-leukemia mechanism with F79 intervention
were limited. We simply focused on the inhibitory effect of DNA
repair by RAD52 and no further study was carried out on the
concentration-dependent changes after drug intervention. In
addition, we found that STAT3 and p-STAT3 were downregulated after
F79 treatment, while a similar phenomenon could be induced by the
STAT3 inhibitor. However, the evidence of amplifying STAT signaling
function in DDR and the relationship with the SSA pathway is
insufficient. Further studies are required to verify these
hypotheses.
In summary, we demonstrated that RAD52 has a
significant role in regulating cell survival and DDR in AML with
low expression of BRCA1/BRCA2. After F79 intervention, we found
leukemia cells to be in a poor condition of DNA repair, which may
be related to the expression of STAT3. Collectively, these data may
provide new insights into leukemia drug therapy.
Acknowledgements
Not applicable.
Funding
This work was supported by Basic Research Program
from the Guangdong Natural Science Foundation (S2013010016559 and
2014A030313138).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YX, YaL and YuL designed the study, conducted the
experiments and analyzed the data. YY, BL, ZF and LL collected the
human samples and were accountable for all aspects of the work in
ensuring that questions related to the accuracy or integrity of any
part of the work are appropriately investigated and resolved. JZ
supervised the study, wrote the manuscript and revised it
critically for important intellectual content. XZ put forward ideas
for research, reviewed the results and approved the final version
of the manuscript. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
All procedures performed in studies involving human
participants were in accordance with the ethical standards of the
National Science and Technology Ethics Committee and with the 1964
Helsinki Declaration and its later amendments or comparable ethical
standards. We obtained ethics approval from the Medical Ethics
Committee of the Third Affiliated Hospital of Sun Yat-sen
University, [(2020)-02-068-01]. All donors signed the informed
consent form.
Patient consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Moehrle BM and Geiger H: Aging of
hematopoietic stem cells: DNA damage and mutations? Exp Hematol.
44:895–901. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Esposito MT and So CW: DNA damage
accumulation and repair defects in acute myeloid leukemia:
Implications for pathogenesis, disease progression, and
chemotherapy resistance. Chromosoma. 123:545–561. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Biechonski S, Yassin M and Milyavsky M:
DNA-damage response in hematopoietic stem cells: An evolutionary
trade-off between blood regeneration and leukemia suppression.
Carcinogenesis. 38:367–377. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Iyama T and Wilson DM III: DNA repair
mechanisms in dividing and non-dividing cells. DNA Repair (Amst).
12:620–636. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bhargava R, Onyango DO and Stark JM:
Regulation of single-strand annealing and its role in genome
maintenance. Trends Genet. 32:566–575. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Symington LS: Role of RAD52 epistasis
group genes in homologous recombination and double-strand break
repair. Microbiol Mol Biol Rev. 66:630–670, table of contents.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rothenberg E, Grimme JM, Spies M and Ha T:
Human Rad52-mediated homology search and annealing occurs by
continuous interactions between overlapping nucleoprotein
complexes. Proc Natl Acad Sci USA. 105:20274–20279. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Faraoni I, Compagnone M, Lavorgna S,
Angelini DF, Cencioni MT, Piras E, Panetta P, Ottone T, Dolci S,
Venditti A, et al: BRCA1, PARP1 and γH2AX in acute myeloid
leukemia: Role as biomarkers of response to the PARP inhibitor
olaparib. Biochim Biophys Acta. 1852:462–472. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Scardocci A, Guidi F, D'Alo' F, Gumiero D,
Fabiani E, Diruscio A, Martini M, Larocca LM, Zollino M, Hohaus S,
et al: Reduced BRCA1 expression due to promoter hypermethylation in
therapy-related acute myeloid leukaemia. Br J Cancer. 95:1108–1113.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cramer-Morales K, Nieborowska-Skorska M,
Scheibner K, Padget M, Irvine DA, Sliwinski T, Haas K, Lee J, Geng
H, Roy D, et al: Personalized synthetic lethality induced by
targeting RAD52 in leukemias identified by gene mutation and
expression profile. Blood. 122:1293–1304. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Panier S and Boulton SJ: Double-strand
break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol.
15:7–18. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
van Attikum H and Gasser SM: Crosstalk
between histone modifications during the DNA damage response.
Trends Cell Biol. 19:207–217. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guan B and Zhang X: Aptamers as versatile
ligands for biomedical and pharmaceutical applications. Int J
Nanomedicine. 15:1059–1071. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Borghouts C, Kunz C and Groner B: Current
strategies for the development of peptide-based anti-cancer
therapeutics. J Pept Sci. 11:713–726. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Slupianek A, Dasgupta Y, Ren SY, Gurdek E,
Donlin M, Nieborowska-Skorska M, Fleury F and Skorski T: Targeting
RAD51 phosphotyrosine-315 to prevent unfaithful recombination
repair in BCR-ABL1 leukemia. Blood. 118:1062–1068. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ciccia A and Elledge SJ: The DNA damage
response: Making it safe to play with knives. Mol Cell. 40:179–204.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stadler J and Richly H: Regulation of DNA
repair mechanisms: How the chromatin environment regulates the DNA
damage response. Int J Mol Sci. 18:17152017. View Article : Google Scholar
|
|
19
|
Gaul L, Mandl-Weber S, Baumann P, Emmerich
B and Schmidmaier R: Bendamustine induces G2 cell cycle arrest and
apoptosis in myeloma cells: The role of ATM-Chk2-Cdc25A and
ATM-p53-p21-pathways. J Cancer Res Clin Oncol. 134:245–253. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xiao Z, Chen Z, Gunasekera AH, Sowin TJ,
Rosenberg SH, Fesik S and Zhang H: Chk1 mediates S and G2 arrests
through Cdc25A degradation in response to DNA-damaging agents. J
Biol Chem. 278:21767–21773. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rivlin N, Koifman G and Rotter V: p53
orchestrates between normal differentiation and cancer. Semin
Cancer Biol. 32:10–17. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vousden KH and Prives C: Blinded by the
light: The growing complexity of p53. Cell. 137:413–431. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bykov VJN, Eriksson SE, Bianchi J and
Wiman KG: Targeting mutant p53 for efficient cancer therapy. Nat
Rev Cancer. 18:89–102. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bruserud Ø, Nepstad I, Hauge M, Hatfield
KJ and Reikvam H: STAT3 as a possible therapeutic target in human
malignancies: Lessons from acute myeloid leukemia. Expert Rev
Hematol. 8:29–41. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Al Zaid Siddiquee K and Turkson J: STAT3
as a target for inducing apoptosis in solid and hematological
tumors. Cell Res. 18:254–267. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Barry SP, Townsend PA, Knight RA,
Scarabelli TM, Latchman DS and Stephanou A: STAT3 modulates the DNA
damage response pathway. Int J Exp Pathol. 91:506–514. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Koganti S, Hui-Yuen J, McAllister S,
Gardner B, Grasser F, Palendira U, Tangye SG, Freeman AF and
Bhaduri-McIntosh S: STAT3 interrupts ATR-Chk1 signaling to allow
oncovirus-mediated cell proliferation. Proc Natl Acad Sci USA.
111:4946–4951. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Deng WW, Hu Q, Liu ZR, Chen QH, Wang WX,
Zhang HG, Zhang Q, Huang YL and Zhang XK: KDM4B promotes DNA damage
response via STAT3 signaling and is a target of CREB in colorectal
cancer cells. Mol Cell Biochem. 449:81–90. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
De Groef S, Renmans D, Cai Y, Leuckx G,
Roels S, Staels W, Gradwohl G, Baeyens L, Heremans Y, Martens GA,
et al: STAT3 modulates β-cell cycling in injured mouse pancreas and
protects against DNA damage. Cell Death Dis. 7:e22722016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: A leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View Article : Google Scholar : PubMed/NCBI
|