Introduction
Chronic myeloid leukemia (CML), a hematological
malignancy, occurs as a result of the presence of the Bcr-Abl
fusion protein (1). The Bcr-Abl
fusion protein constitutively activates tyrosine kinase activity
and induces many downstream signaling pathways, such as
phosphoinositide 3 kinase (PI3K)/Akt, Janus kinase (JAK)/signal
transducer and activator of transcription (STAT) and
mitogen-activated protein kinase (MAPK)/extracellular
signal-regulated kinase (ERK) pathways, contributing to cell
survival and proliferation (2,3).
Imatinib, a tyrosine kinase inhibitor (TKI) that binds to the
ATP-binding site of ABL, has dramatically advanced the treatment of
CML by inducing long-term overall survival rates of > 90%
(4). However, approximately 35% of
patients develop pharmacological resistance to imatinib (5). To overcome this resistance,
second-generation (dasatinib, nilotinib, and bosutinib) and
third-generation (ponatinib) TKIs have been developed and have been
shown to be effective against refractory CML patients (6,7).
Although these drugs provide many benefits for patients with CML,
advanced patients show resistance even to these TKIs (8). Therefore, novel therapeutic strategies
are urgently needed for the treatment of TKI-resistant CML
patients.
The Aurora family of serine/threonine kinases is
essential for many cellular functions, including centrosome
function, spindle assembly, chromosome segregation, and cytokinesis
(9,10). They comprise Aurora A and Aurora B,
and elevated expression of Aurora A and Aurora B have been shown in
human cancers such as breast, ovarian, gastric, colon, and
pancreatic cancers (11,12). Furthermore, inhibition of Aurora A
and Aurora B has proven to be an attractive anticancer approach for
a variety of cancers including solid tumors and hematological
malignancies (13–15). Therefore, inhibiting Aurora kinase
is a potential target for the treatment of CML. JAK has long been
recognized to be involved in CML pathogenesis (16). JAKs are cytoplasmic protein tyrosine
kinases, and dysregulation of the JAK/STAT pathways is critically
involved in the survival and proliferation of CML cells (17). Therefore, blocking the JAK/STAT
pathway may also represent a novel therapeutic strategy for CML
patients.
AT9283 is a multi-targeted kinase inhibitor,
discovered using a fragment-based approach, with potent activity
against JAK, Aurora kinases, and Abl (18). Previous studies have shown that
AT9283 has therapeutic potential in leukemic cells,
myeloproliferative disorders, and colon cells (19). In our previous study, the K562/IR
cell line (imatinib-resistant CML cells) was established, and
K562/IR exhibited resistance to TKIs including imatinib, nilotinib,
dasatinib, bafetinib, and ponatinib (20). Here, we showed that AT9283
significantly decreased the cell viability of both TKI-sensitive
and TKI-resistant CML cells, including K562/IR. Furthermore, we
investigated the molecular mechanisms underlying the decrease in
the cell viability. Importantly, we found the critical role of
Aurora A and Aurora B in the antiproliferative effects of AT9283 in
both TKI-sensitive and TKI-resistant CML cells. Our studies suggest
that Aurora A and Aurora B are promising therapeutic targets in
TKI-sensitive and TKI-resistant CML, and AT9283 may have potential
clinical applications for CML treatment.
Materials and methods
Materials
AT9283 and AMG900 (Adooq Bioscience) were first
dissolved in dimethyl sulfoxide (DMSO) to a concentration of 50 mM
(stock solution) and stored at −20°C. These reagents were diluted
in phosphate-buffered saline (PBS) before use in the experiments
described below.
Cells and culture conditions
The human CML cell line K562 was obtained from the
Health Science Research Resources Bank (Osaka, Japan). K562/IR
cells were obtained from our laboratory. These cells were
maintained in RPMI-1640 medium (Sigma-Aldrich; Merck KGaA) with 10%
fetal bovine serum (FBS) (Gibco; Thermo Fisher Scientific, Inc.), 2
mM L-glutamine (Wako Pure Chemical Industries, Ltd.), 25 mM HEPES
(Wako Pure Chemical Industries, Ltd.), 100 µg/ml penicillin
(Gibco; Thermo Fisher Scientific, Inc.), and 100 U/ml streptomycin
(Gibco; Thermo Fisher Scientific, Inc.) at 37°C in 5%
CO2.
Trypan blue exclusion assay
CML cells were plated in 96-well plates at a
concentration of 2×104 cells/ml. Then, AT9283 (10, 30,
50, and 100 nM) or AMG900 (10, 50, 100, 300, and 500 nM) were added
to the well. After 3 days, CML cells were stained with trypan blue
and the number of stained cells was counted at a magnification of
×100 using an Olympus CK2 inverted microscope (Olympus Optical
Co.).
Cell cycle analysis
Assessment of the cell cycle analysis was performed
with the Muse™ cell cycle kit (Merck Millipore), according to the
manufacturer's protocol. CML cells were treated with 50 nM AT9283
or 300 nM AMG900 for 2 days. Then, Muse™ cell cycle reagent was
added and incubated at room temperature for 30 min in the dark.
After incubation, the cell cycle analysis was performed using a
Muse Cell Analyzer (Merck Millipore).
Annexin V assay
Measurement of cells undergoing apoptosis was
performed with the Muse™ Annexin-V and Dead Cell Assay kit (Merck
Millipore), according to the manufacturer's protocol. CML cells
were treated with 50 nM AT9283 or 300 nM AMG900 for 2 days. Then,
Muse™ Annexin V and dead cell reagent were added. After incubation
for 20 min at room temperature, apoptotic cells were applied to a
Muse Cell Analyzer (Merck Millipore).
Caspase-3/7 activity assay
Measurement of cells undergoing caspase-3/7
activation was performed using the Muse™ Caspase-3/7 kit (Merck
Millipore), according to the manufacturer's protocol. CML cells
were treated with 50 nM AT9283 or 300 nM AMG900 for 2 days. Then,
Muse™ Caspase-3/7 reagent was added and incubated at 37°C for 30
min. After incubation, Muse™ Caspase 3/7-AAD reagent was added and
incubated at room temperature for 5 min. The activation of
caspase-3/7 was applied to a Muse Cell Analyzer (Merck
Millipore).
Western blotting
CML cells were cultured with different
concentrations of AT9283 or AMG900 and lysed using lysis buffer as
previously described (21). The
protein concentration of the lysate was measured using the BCA
protein assay kit (Thermo Fisher Scientific, Inc.). The extracts
(40 µg) were separated by 10% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene
fluoride (PVDF) membranes (GE Healthcare). The membranes were
blocked in TBS plus 5% skim milk and incubated overnight at 4°C
with each of the following antibodies: Anti-phospho-Abl (cat. no.
2865; dilution 1:1,000), anti-Abl (cat. no. 2862; dilution
1:1,000), anti-phospho-STAT3 (cat. no. 9131; dilution 1:1,000),
anti-STAT3 (cat. no. 9132; dilution 1:1,000), anti-phospho-ERK1/2
(cat. no. 4370; dilution 1:2,000), anti-ERK1/2 (cat. no. 9102;
dilution 1:2,000), anti-phospho-Akt (cat. no. 9271; dilution
1:1,000), anti-Akt (cat. no. 9272; dilution 1:1,000),
anti-phospho-Aurora A (cat. no. 2914; dilution 1:1,000),
anti-Aurora A (cat. no. 4718; dilution 1:1,000),
anti-phospho-Aurora B (cat. no. 2914; dilution 1:1,000),
anti-Aurora B (cat. no. 3094; dilution 1:1,000),
anti-phospho-Histone H3 (cat. no. 3377; dilution 1:1,000),
anti-Histone H3 (cat. no. 4499; dilution 1:1,000; all from Cell
Signaling Technology, Inc.), anti-β-actin (product no. A2228;
dilution 1:3,000; Sigma-Aldrich; Merck KGaA), and anti-Lamin A/C
(cat. no. sc-20681; dilution 1:1,000; Santa Cruz Biotechnologies,
Inc.). Subsequently, the membranes were incubated with anti-rabbit
(cat. no. 7074; dilution 1:5,000; Cell Signaling Technology, Inc.)
or anti-mouse (cat. no. 7076; dilution 1:5,000; Cell Signaling
Technology, Inc.) secondary antibody. The proteins were visualized
using Luminata Forte Western HRP substrate (Merck Millipore)
according to the manufacturer's instructions. The quantities of
reactive proteins were determined based on densitometric
measurements using a CS analyzer (ATTO, Tokyo, Japan).
Statistical analysis
All results are expressed as the mean ± standard
deviation (SD) of three independent experiments. All analyses were
conducted using SPSS version 21.0 software (IBM Corp.), and
Shapiro-Wilk analysis and one-way analysis of variance (ANOVA)
followed by Dunnett's test. P-value <0.05 was considered as
indicative of a statistically significant result.
Results
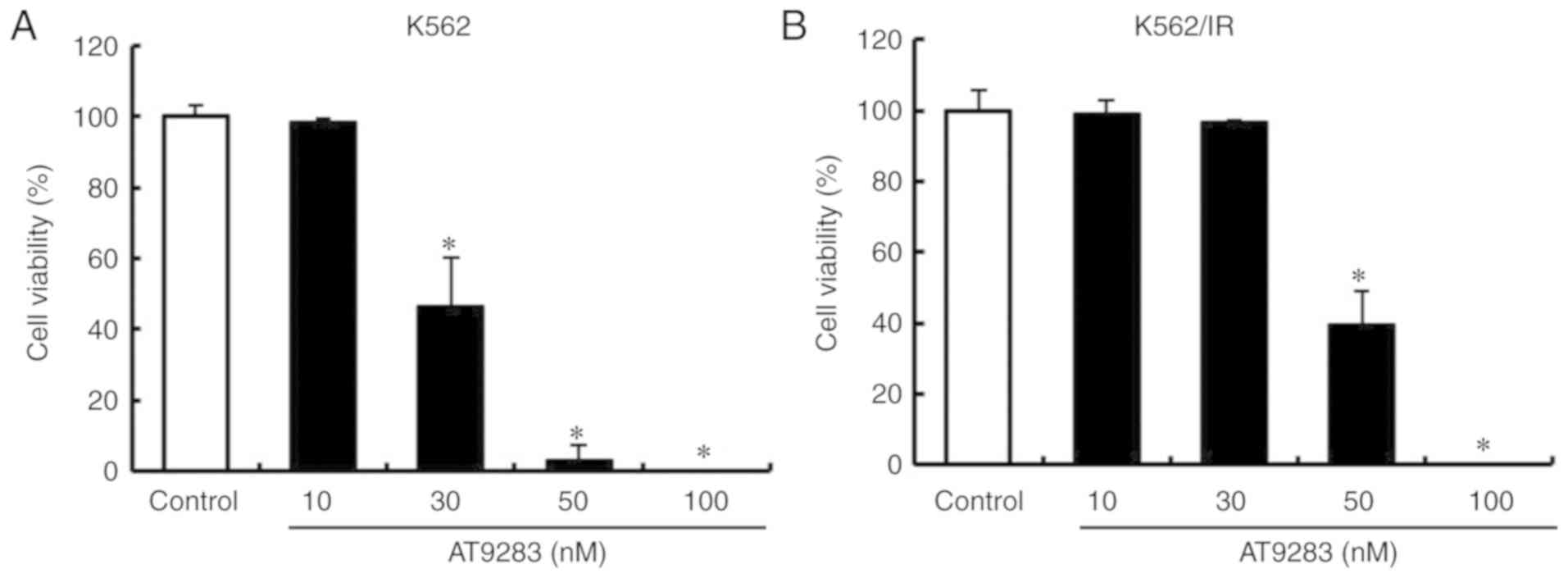
AT9283 decreases the cell viability of
TKI-sensitive and TKI-resistant CML cells
We first investigated the effect of AT9283 on the
cell viability using the trypan blue assay. We used the human CML
cell line K562 and TKI-resistance cell line K562/IR. In our
previous study, we showed that K562/IR exhibits resistance to TKIs
including imatinib, nilotinib, dasatinib, bafetinib, and ponatinib
(20). These cells were treated
with AT9283 (10, 30, 50, and 100 nM) for 3 days. We found that
AT9283 decreased the cell viability of both K562 (Fig. 1A) and K562/IR (Fig. 1B) cell lines in a
concentration-dependent manner. These results suggest that AT9283
decreases the cell viability of both TKI-sensitive and
TKI-resistant CML cells.
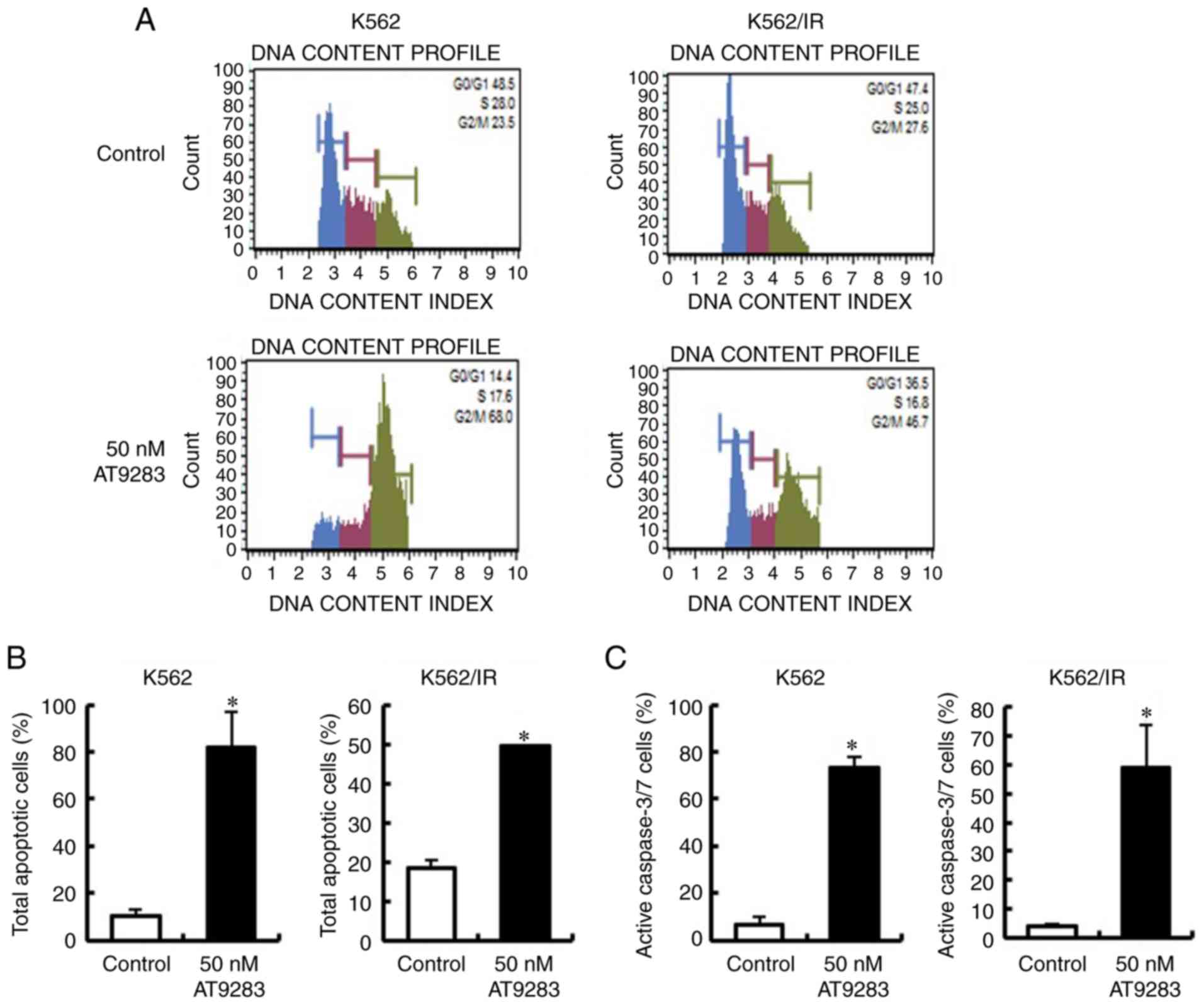
AT9283 increases the cell population
in the G2/M phase and induces apoptosis of TKI-sensitive and
TKI-resistant CML cells
AT9283 has been reported to arrest the G2/M phase in
tumor cells, ultimately leading to cell death by apoptosis
(18). Therefore, we confirmed the
effects of AT9283 on the cell cycle profile and the induction of
apoptosis of CML cells (K562 and K562/IR). CML cells were treated
with 50 nM AT9283 for 2 days, and the cell cycle profile was
determined using the Muse™ cell cycle kit. As expected,
AT9283-treated CML cells (K562 and K562/IR) showed an increase in
the G2/M phase (Fig. 2A). To
further examine whether AT9283 induced apoptosis in CML cells,
these cells were treated with 50 nM AT9283 for 2 days, and the
number of apoptotic cells was measured using the Muse Annexin V and
Dead Cell Assay kit. Our results showed that AT9283 treatment
increased the number of total apoptotic cells in both K562 cells
and K562/IR cell lines (Fig. 2B).
Apoptosis is induced by an interaction between various initiator
and effector caspases (22,23). Therefore, we confirmed the effects
of AT9283 on caspase-3/7 activity in CML cells (K562 and K562/IR).
CML cells were treated with 50 nM AT9283 for 2 days, and the
caspase-3/7 activity was determined using the Muse™ Caspase-3/7
kit. We showed that AT9283 increased caspase-3/7 activity in both
K562 cells and K562/IR cell lines (Fig.
2C). These results indicated that the decreased cell viability
upon treatment with AT9283 is attributed to an increase in the G2/M
phase and induction of apoptosis.
AT9283 suppresses Histone H3
phosphorylation via inhibition of Aurora A and Aurora B in
TKI-sensitive and TKI-resistant CML cells
AT9283 is a multi-targeted kinase inhibitor such as
Abl, Aurora kinases, and JAK (18).
To clarify the molecular mechanisms underlying the decrease in cell
viability upon treatment with AT9283, we investigated the
expression of phosphorylated Bcr-Abl, Aurora A, Aurora B,
downstream STAT3, ERK, Akt, and Histone H3 in CML cells with AT9283
(10, 30, and 50 nM) by western blotting. Interestingly, our results
showed that AT9283 significantly decreased the expression of Aurora
A, Aurora B, and Histone H3 phosphorylation in both K562 cells and
K562/IR cell lines, whereas we observed no changes in the levels of
Bcr-Abl, STAT3, ERK, and Akt phosphorylation (Fig. 3). These results suggest that the
decreased cell viability upon treatment with AT9283 may be involved
in the inhibition of Histone H3 by decreasing the expression of
phosphorylated Aurora A and Aurora B in both TKI-sensitive and
TKI-resistant CML cells.
 | Figure 3.AT9283 suppresses Histone H3
phosphorylation via the inhibition of Aurora A and Aurora B in
TKI-sensitive and TKI-resistant CML cells. (A) K562 and K562/IR
cells were treated with AT9283 (10, 30, and 50 nM) for 2 days. The
expression levels of phospho-Bcr-Abl, Bcr-Abl, phospho-STAT3,
STAT3, phospho-ERK1/2, ERK1/2, phospho-Akt, Akt, phospho-Aurora A,
Aurora A, phospho-Aurora B, Aurora B, phospho-Histone H3, and
Histone H3 were detected using western blotting. The expression
levels of β-actin and Lamin A/C were used as internal controls. (B)
Quantification of signals is presented as fold change relative to
phosphorylated protein vs. total protein. The results are
representative of three independent experiments. *P<0.05. TKI,
tyrosine kinase inhibitor; CML, chronic myeloid leukemia; STAT3,
signal transducer and activator of transcription 3; ERK,
extracellular signal-regulated kinase. |
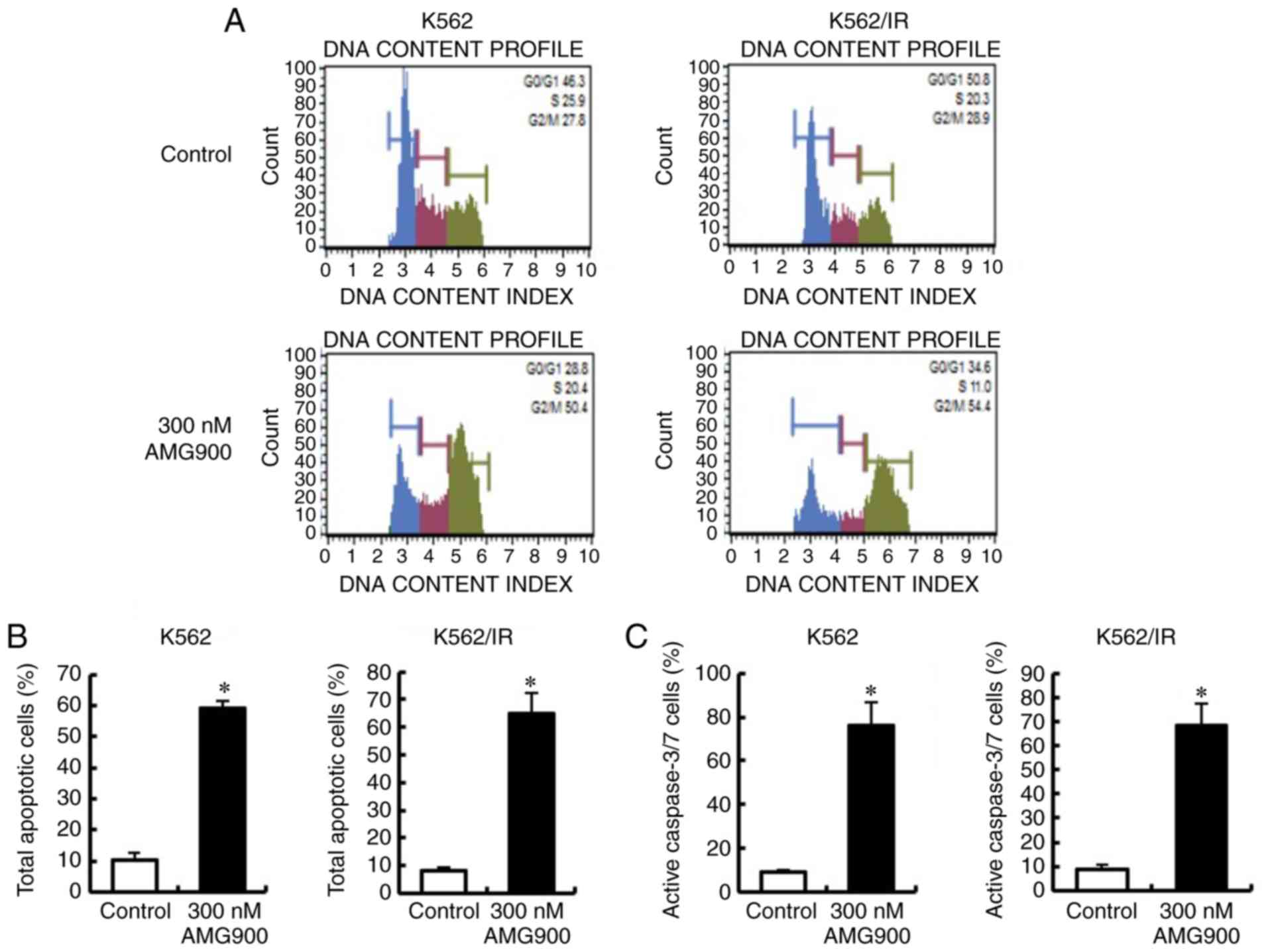
AMG900 increases G2/M phase and
induces apoptosis via inhibition of Aurora A and Aurora B of
TKI-sensitive and TKI-resistant CML cells
We further examined the role of Aurora A and Aurora
B in the anticancer activity of AT9283 by using a selective
AuroraA/B inhibitor AMG900. We showed that AMG900 decreased the
cell viability and the expression of Aurora A, Aurora B, and
Histone H3 phosphorylation in both K562 cells and K562/IR cell
lines (Fig. 4). We also observed
that AMG900 increased the G2/M phase population and significantly
induced apoptosis and caspase 3/7 activity (Fig. 5). These results support the critical
role of Aurora A and Aurora B in the antiproliferative effects of
AT9283 in both TKI-sensitive and TKI-resistant CML cells.
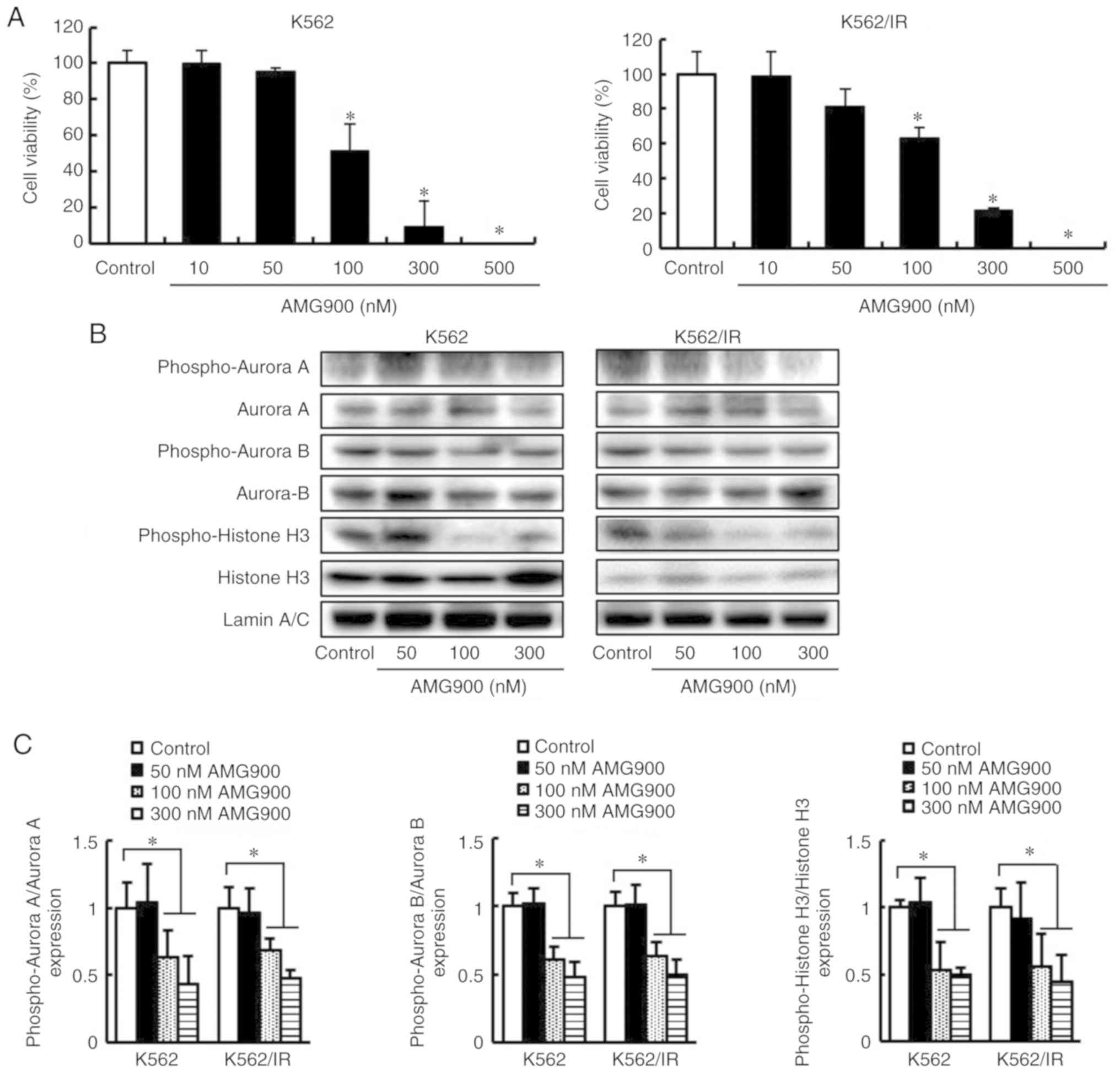 | Figure 4.AMG900 decreases the cell viability
via the inhibition of Aurora A and Aurora B of TKI-sensitive and
TKI-resistant CML cells. (A) K562 and K562/IR cells were treated
with AMG900 (10, 50, 100, 300, and 500 nM). Then, trypan blue
exclusion assay was performed on the cells after 3 days. The
results are expressed as mean ± SD of 3 experiments performed in
triplicate. *P<0.05 compared to the control. (B) K562 and
K562/IR cells were treated with AMG900 (50, 100, and 300 nM) for 2
days. The expression levels of phospho-Aurora A, Aurora A,
phospho-Aurora B, Aurora B, phospho-Histone H3, and Histone H3 were
detected using western blotting. The expression levels of Lamin A/C
were used as internal controls. (C) Quantification of signals is
presented as fold change relative to phosphorylated protein vs.
total protein. The results are representative of three independent
experiments. *P<0.05. TKI, tyrosine kinase inhibitor; CML,
chronic myeloid leukemia. |
Discussion
In the present study, we investigated the effects of
AT9283, a multi-targeted kinase inhibitor, in tyrosine kinase
inhibitor (TKI)-sensitive chronic myeloid leukemia (CML) cells
(K562) and TKI-resistant CML cells (K562/IR). We showed that AT9283
decreased the cell viability of both TKI-sensitive and
TKI-resistant CML cells. In addition, we found that AT9283
increased the G2/M phase population and cell death by apoptosis.
Preclinical studies have shown that AT9283 has potent cytotoxic and
cell growth inhibitory activity in vitro as well as
antitumor activity in vivo in a mouse xenograft model of
colon carcinoma (18,19). Furthermore, AT9283 has been reported
to increase the G2/M phase and induce apoptosis of multiple myeloma
and B-cell lymphoma cells in a time-dependent manner (19,24).
These results suggest that AT9283 may have potential clinical
applications for the treatment of TKI-sensitive and TKI-resistant
CML patients.
Differential sensitivity of AT9283 between K562 and
K562/IR was observed. In our previous study, we showed that
activation of MET in K562/IR cells was increased compared to K562
cells (20). MET is an oncogene
encoding tyrosine kinase receptor, and plays an important role in
embryogenesis, tumor growth, and metastasis (25). In addition, MET activation was
reported to be associated with poorer response to chemotherapy.
Therefore, differential sensitivity of AT9283 between K562 and
K562/IR cell lines may involve MET activation. Further research
will be carried out to investigate the differential sensitivity of
AT9283 between K562 and K562/IR cells in the future.
AT9283 is a multi-targeted kinase inhibitor against
Abl, Aurora kinases, and JAK (18).
However, the molecular mechanisms underlying the decrease in cell
viability of TKI-sensitive and TKI-resistant CML cells upon
treatment with AT9283 was not unclear. Interestingly, our results
showed that AT9283 decreased the expression of Aurora A, Aurora B,
and Histone H3 phosphorylation. In contrast, we observed no changes
in the levels of Bcr-Abl, STAT3, ERK, and Akt phosphorylation.
Furthermore, we showed that AMG900, a selective Aurora A and Aurora
B inhibitor, increased the G2/M phase cell population and induced
apoptosis via the inhibition of Aurora A and Aurora B of both
TKI-sensitive and TKI-resistant CML cells. Aurora A localizes to
centrosomes during the G2 phase and is required for the separation
and maturation of centrosomes through the recruitment of key
components of spindle assembly (26). Aurora B kinase is also a chromosomal
passenger protein that is essential for chromosome segregation
through the phosphorylation of mitotic histone H3 (27,28).
Recent studies have reported that a decrease in Aurora A or Aurora
B leads to G2/M arrest, spindle defects, and multi-nucleated cells,
which eventually undergo apoptosis (29,30).
In addition, Aurora B kinase RNAi was evidenced by the inhibition
of Histone H3 phosphorylation and endoreduplication, which leads to
apoptosis in colon cancer cells (31). These results support the critical
role of Aurora A and Aurora B in the antiproliferative effects of
AT9283 in TKI-sensitive and TKI-resistant CML cells.
Differential cell viability between AT9283 and
AMG900 in K562 and K562/IR was observed. In previous studies,
AT9283 inhibited Aurora kinase at low concentration compared with
AMG900 (18,32). Furthermore, our results showed that
30–50 nM of AT9283 inhibited the expression of Aurora A and Aurora
B phosphorylation, whereas 100–300 nM of AMG900 inhibited the
expression of Aurora A and Aurora B phosphorylation. Therefore,
AT9283 may inhibit Aurora A and Aurora B more strongly than
AMG900.
Different markers have been reported to predict the
efficacy of Aurora inhibitors such as DNA aneuploidy, mitotic
index, and percentage of aligned spindles in G2/M phase (33,34).
In normal cells, Aurora A and Aurora B are expressed in a cell
cycle-dependent manner, peaking during the G2/M phase (35). Several transcription factors, such
as E2F-1, E2F-4, DP-2 and FoxM1 have been reported to be implicated
in cell cycle-regulated transcription of Aurora A and Aurora B
(36–38). However, limited information is
available concerning regulation of Aurora A and Aurora B expression
in malignant tumors. Therefore, identifying the regulator of Aurora
A and Aurora B will be an important task to clarify possible
biomarkers of AT9283 in CML treatment.
Although the development of imatinib has led to an
extended lifespan for many CML patients, the development of
imatinib resistance represents a relevant clinical issue in the
treatment of CML (39). To overcome
this resistance, second-generation TKIs (nilotinib, dasatinib, and
bosutinib) have been developed and are effective against a range of
Bcr-Abl mutations (e.g., E255K, M351T), except for T315I (40,41).
In addition, third-generation TKI ponatinib has activity against
Bcr-Abl mutations, including T315I (42).
Although these drugs provide many benefits for
patients with CML, in 50% of imatinib-resistant CML patients, there
is no mutation in Bcr-Abl. Furthermore, the treatment of CML
patients with Bcr-Abl-independent imatinib resistance has been
disappointing. In the present study, we used K562/IR cells which
did not have any Bcr-Abl mutations (20). Interestingly, our results showed
that AT9283 decreased the cell viability of K562/IR cells in a
concentration-dependent manner. Furthermore, AT9283 inhibited the
expression of Aurora A, Aurora B, and downstream Histone H3
phosphorylation in K562/IR cells. Thus, our research provides new
insights that Aurora A and Aurora B are promising therapeutic
targets in Bcr-Abl-independent imatinib resistance, and AT9283 may
have potential clinical application for the treatment of
TKI-resistant CML patients with Bcr-Abl-independent imatinib
resistance.
In conclusion, AT9283 decreased the cell viability
of both TKI-sensitive and TKI-resistant CML cells. The decrease in
cell viability was attributed to an increase in the G2/M phase
population and induction of apoptosis via inhibition of Aurora A
and Aurora B. Our research suggests that Aurora A and Aurora B are
promising therapeutic targets in TKI-sensitive and TKI-resistant
CML, and AT9283 may have potential clinical applications for CML
treatment.
Acknowledgements
Not applicable.
Funding
This study was supported in part by a Grant-in-Aid
for Young Scientists from the Japan Society for the Promotion of
Science (JSPS).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
TT performed the analysis of the trypan blue
exclusion assay, cell cycle analysis, and western blotting and
drafted the manuscript. MT, SG, CN and YO carried out analysis of
the trypan blue exclusion assay, Annexin V assay, caspase-3/7
activity assay, and western blotting. MI and TS contributed to the
statistical analyses. SN designed the experiments and revised the
manuscript. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CML
|
chronic myeloid leukemia
|
|
TKI
|
tyrosine kinase inhibitor
|
|
K562/IR
|
imatinib-resistant K562 cells
|
|
JAK
|
Janus kinase
|
|
ERK1/2
|
extracellular signal-regulated kinase
1/2
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
References
|
1
|
Nie ZY, Yang L, Liu XJ, Yang Z, Yang GS,
Zhou J, Qin Y, Yu J, Jiang LL, Wen JK, et al: Morin inhibits
proliferation and induces apoptosis by modulating the
miR-188-5p/PTEN/AKT regulatory pathway in CML cells. Mol Cancer
Ther. 18:2296–2307. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mukaida N, Tanabe Y and Baba T: Chemokines
as a conductor of bone marrow microenvironment in chronic myeloid
leukemia. Int J Mol Sci. 18:18242017. View Article : Google Scholar
|
|
3
|
Chen SH, Chow JM, Hsieh YY, Lin CY, Hsu
KW, Hsieh WS, Chi WM, Shabangu BM and Lee CH: HDAC1,2 knock-out and
HDACi induced cell apoptosis in imatinib-resistant K562 cells. Int
J Mol Sci. 20:22712019. View Article : Google Scholar
|
|
4
|
Chorzalska A, Ahsan N, Rao RSP, Roder K,
Yu X, Morgan J, Tepper A, Hines S, Zhang P, Treaba DO, et al:
Overexpression of Tpl2 is linked to imatinib resistance and
activation of MEK-ERK and NF-κB pathways in a model of chronic
myeloid leukemia. Mol Oncol. 12:630–647. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bhamidipati PK, Kantarjian H, Cortes J,
Cornelison AM and Jabbour E: Management of imatinib-resistant
patients with chronic myeloid leukemia. Ther Adv Hematol.
4:103–117. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sarno F, Pepe G, Termolino P, Carafa V,
Massaro C, Merciai F, Campiglia P, Nebbioso A and Altucci L:
Trifolium Repens blocks proliferation in chronic myelogenous
leukemia via the BCR-ABL/STAT5 pathway. Cells. 9:3792020.
View Article : Google Scholar
|
|
7
|
Buffa P, Romano C, Pandini A, Massimino M,
Tirrò E, Di Raimondo F, Manzella L, Fraternali F and Vigneri PG:
BCR-ABL residues interacting with ponatinib are critical to
preserve the tumorigenic potential of the oncoprotein. FASEB J.
28:1221–1236. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cimino G, Pane F, Elia L, Finolezzi E,
Fazi P, Annino L, Meloni G, Mancini M, Tedeschi A, Di Raimondo F,
et al: The role of BCR/ABL isoforms in the presentation and outcome
of patients with Philadelphia-positive acute lymphoblastic
leukemia: A seven-year update of the GIMEMA 0496 trial.
Haematologica. 91:377–380. 2006.PubMed/NCBI
|
|
9
|
Lin YG, Immaneni A, Merritt WM, Mangala
LS, Kim SW, Shahzad MM, Tsang YT, Armaiz-Pena GN, Lu C, Kamat AA,
et al: Targeting aurora kinase with MK-0457 inhibits ovarian cancer
growth. Clin Cancer Res. 14:5437–5446. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Harrington EA, Bebbington D, Moore J,
Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C,
Hercend T, Diu-Hercend A, et al: VX-680, a potent and selective
small-molecule inhibitor of the Aurora kinases, suppresses tumor
growth in vivo. Nat Med. 10:262–267. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Matthews N, Visintin C, Hartzoulakis B,
Jarvis A and Selwood DL: Aurora A and B kinases as targets for
cancer: Will they be selective for tumors? Expert Rev Anticancer
Ther. 6:109–120. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lapenna S and Giordano A: Cell cycle
kinases as therapeutic targets for cancer. Nat Rev Drug Discov.
8:547–566. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rodrigues Alves AP, Machado-Neto JA,
Scheucher PS, Paiva HH, Simões BP, Rego EM and Traina F: Reversine
triggers mitotic catastrophe and apoptosis in K562 cells. Leuk Res.
48:26–31. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gavriilidis P, Giakoustidis A and
Giakoustidis D: Aurora kinases and potential medical applications
of aurora kinase inhibitors: A review. J Clin Med Res. 7:742–751.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ikezoe T, Yang J, Nishioka C, Tasaka T,
Taniguchi A, Kuwayama Y, Komatsu N, Bandobashi K, Togitani K,
Koeffler HP and Taguchi H: A novel treatment strategy targeting
Aurora kinases in acute myelogenous leukemia. Mol Cancer Ther.
6:1851–1857. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tabe Y, Jin L, Iwabuchi K, Wang RY,
Ichikawa N, Miida T, Cortes J, Andreeff M and Konopleva M: Role of
stromal microenvironment in nonpharmacological resistance of CML to
imatinib through Lyn/CXCR4 interactions in lipid rafts. Leukemia.
26:883–892. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang X, Tu H, Yang Y, Jiang X, Hu X, Luo
Q and Li J: Bone marrow-derived mesenchymal stromal cells promote
resistance to tyrosine kinase inhibitors in chronic myeloid
leukemia via the IL-7/JAK1/STAT5 pathway. J Biol Chem.
294:12167–12179. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Howard S, Berdini V, Boulstridge JA, Carr
MG, Cross DM, Curry J, Devine LA, Early TR, Fazal L, Gill AL, et
al: Fragment-based discovery of the pyrazol-4-yl urea (AT9283), a
multitargeted kinase inhibitor with potent aurora kinase activity.
J Med Chem. 52:379–388. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Qi W, Liu X, Cooke LS, Persky DO, Miller
TP, Squires M and Mahadevan D: AT9283, a novel aurora kinase
inhibitor, suppresses tumor growth in aggressive B-cell lymphomas.
Int J Cancer. 130:2997–3005. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tsubaki M, Takeda T, Kino T, Sakai K, Itoh
T, Imano M, Nakayama T, Nishio K, Satou T and Nishida S:
Contributions of MET activation to BCR-ABL1 tyrosine kinase
inhibitor resistance in chronic myeloid leukemia cells. Oncotarget.
8:38717–38730. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tsubaki M, Ogawa N, Takeda T, Sakamoto K,
Shimaoka H, Fujita A, Itoh T, Imano M, Satou T and Nishida S:
Dimethyl fumarate induces apoptosis of hematopoietic tumor cells
via inhibition of NF-κB nuclear translocation and down-regulation
of Bcl-xL and XIAP. Biomed Pharmacother. 68:999–1005. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tsubaki M, Fujiwara D, Takeda T, Kino T,
Tomonari Y, Itoh T, Imano M, Satou T, Sakaguchi K and Nishida S:
The sensitivity of head and neck carcinoma cells to statins is
related to the expression of their Ras expression status, and
statin-induced apoptosis is mediated via suppression of the Ras/ERK
and Ras/mTOR pathways. Clin Exp Pharmacol Physiol. 44:222–234.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takeda T, Tsubaki M, Tomonari Y, Kawashima
K, Itoh T, Imano M, Satou T and Nishida S: Bavachin induces the
apoptosis of multiple myeloma cell lines by inhibiting the
activation of nuclear factor kappa B and signal transducer and
activator of transcription 3. Biomed Pharmacother. 100:486–494.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Santo L, Hideshima T, Cirstea D, Bandi M,
Nelson EA, Gorgun G, Rodig S, Vallet S, Pozzi S, Patel K, et al:
Antimyeloma activity of a multitargeted kinase inhibitor, AT9283,
via potent Aurora kinase and STAT3 inhibition either alone or in
combination with lenalidomide. Clin Cancer Res. 17:3259–3271. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liang H and Wang M: MET oncogene in
non-small cell lung cancer: Mechanism of MET dysregulation and
agents targeting the HGF/c-Met axis. Onco Targets Ther.
13:2491–2510. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hirota T, Kunitoku N, Sasayama T, Marumoto
T, Zhang D, Nitta M, Hatakeyama K and Saya H: Aurora-A and an
interacting activator, the LIM protein Ajuba, are required for
mitotic commitment in human cells. Cell. 114:585–598. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tao Y, Zhang P, Girdler F, Frascogna V,
Castedo M, Bourhis J, Kroemer G and Deutsch E: Enhancement of
radiation response in p53-deficient cancer cells by the Aurora-B
kinase inhibitor AZD1152. Oncogene. 27:3244–3255. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Crosio C, Fimia GM, Loury R, Kimura M,
Okano Y, Zhou H, Sen S, Allis CD and Sassone-Corsi P: Mitotic
phosphorylation of histone H3: Spatio-temporal regulation by
mammalian Aurora kinases. Mol Cell Biol. 22:874–885. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Marumoto T, Honda S, Hara T, Nitta M,
Hirota T, Kohmura E and Saya H: Aurora-A kinase maintains the
fidelity of early and late mitotic events in HeLa cells. J Biol
Chem. 278:51786–51795. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Giet R and Glover DM: Drosophila aurora B
kinase is required for histone H3 phosphorylation and condensin
recruitment during chromosome condensation and to organize the
central spindle during cytokinesis. J Cell Biol. 152:669–682. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Curry J, Angove H, Fazal L, Lyons J, Reule
M, Thompson N and Wallis N: Aurora B kinase inhibition in mitosis:
Strategies for optimising the use of aurora kinase inhibitors such
as AT9283. Cell Cycle. 8:1921–1929. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Payton M, Bush TL, Chung G, Ziegler B,
Eden P, McElroy P, Ross S, Cee VJ, Deak HL, Hodous BL, et al:
Preclinical evaluation of AMG 900, a novel potent and highly
selective pan-aurora kinase inhibitor with activity in
taxane-resistant tumor cell lines. Cancer Res. 70:9846–9854. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chakravarty A, Shinde V, Tabernero J,
Cervantes A, Cohen RB, Dees EC, Burris H, Infante JR, Macarulla T,
Elez E, et al: Phase I assessment of new mechanism-based
pharmacodynamic biomarkers for MLN8054, a small-molecule inhibitor
of Aurora A kinase. Cancer Res. 71:675–685. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Malumbres M and Pérez de Castro I: Aurora
kinase A inhibitors: Promising agents in antitumoral therapy.
Expert Opin Ther Targets. 18:1377–1393. 2014.PubMed/NCBI
|
|
35
|
Zhang XH, Rao M, Loprieato JA, Hong JA,
Zhao M, Chen GZ, Humphries AE, Nguyen DM, Trepel JB, Yu X and
Schrump DS: Aurora A, Aurora B and survivin are novel targets of
transcriptional regulation by histone deacetylase inhibitors in
non-small cell lung cancer. Cancer Biol Ther. 7:1388–1397. 2009.
View Article : Google Scholar
|
|
36
|
Vader G and Lens SM: The Aurora kinase
family in cell division and cancer. Biochim Biophys Acta.
1786:60–72. 2008.PubMed/NCBI
|
|
37
|
Tanaka M, Ueda A, Kanamori H, Ideguchi H,
Yang J, Kitajima S and Ishigatsubo Y: Cell-cycle-dependent
regulation of human aurora A transcription is mediated by periodic
repression of E4TF1. J Biol Chem. 277:10719–10726. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kimura M, Uchida C, Takano Y, Kitagawa M
and Okano Y: Cell cycle-dependent regulation of the human aurora B
promoter. Biochem Biophys Res Commun. 316:930–936. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kantarjian HM, Talpaz M, Giles F, O'Brien
S and Cortes J: New insights into the pathophysiology of chronic
myeloid leukemia and imatinib resistance. Ann Intern Med.
145:913–923. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhou T, Commodore L, Huang WS, Wang Y,
Thomas M, Keats J, Xu Q, Rivera VM, Shakespeare WC, Clackson T, et
al: Structural mechanism of the Pan-BCR-ABL inhibitor ponatinib
(AP24534): Lessons for overcoming kinase inhibitor resistance. Chem
Biol Drug Des. 77:1–11. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Branford S, Melo JV and Hughes TP:
Selecting optimal second-line tyrosine kinase inhibitor therapy for
chronic myeloid leukemia patients after imatinib failure: Does the
BCR-ABL mutation status really matter? Blood. 114:5426–5435. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lan X, Zhao C, Chen X, Zhang P, Zang D, Wu
J, Chen J, Long H, Yang L, Huang H, et al: Platinum pyrithione
induces apoptosis in chronic myeloid leukemia cells resistant to
imatinib via DUB inhibition-dependent caspase activation and
Bcr-Abl downregulation. Cell Death Dis. 8:e29132017. View Article : Google Scholar : PubMed/NCBI
|