Introduction
Renal cell carcinoma (RCC) is one of the most lethal
urological neoplasms, which contributed to ~5% of all malignant
carcinomas, worldwide (1).
Chemotherapy (such as sorafenib) is one of the primary treatment
strategies for RCC (2). However,
chemoresistance remains a major cause of cancer recurrence and
cancer-associated mortality (3).
Therefore, it is important to improve the understanding of RCC
pathogenesis and identify novel targets for the development of
diagnostic and therapeutic approaches.
Long non-coding RNAs (lncRNAs) are transcripts
>200 nucleotides in length, which are not protein coding
(4). Accumulating evidence
indicates that lncRNAs are involved in the physiological and
pathological processes in most types of cancer, such as
hepatocellular carcinoma, colorectal cancer and prostate cancer
(5,6). In previous reports, lncRNAs could
function as both oncogenes and tumor suppressors in RCC. For
example, lncRNA SARCC inhibited RCC progression by regulating
androgen receptor/miR-143 signals (7). On the other hand, lncRNA DUXAP8
promoted RCC tumorigenesis by downregulating miR-126 (8). Moreover, it has been reported that
lncRNAs have been associated with chemoresistance in cancer,
including RCC. For example, Xu et al (9) demonstrated that the knockdown of
lncRNA SRLR reduced chemoresistance to sorafenib in RCC cells.
PLK1S1 has been reported to be upregulated in tamoxifen-resistant
MCF7 cells, lung adenocarcinoma and bone metastasis (10–12).
However, the exact mechanisms of PLK1S1 in RCC remains unclear.
MicroRNAs (miRNAs/miRs) are another type of
endogenous non-coding RNAs with a length of 22–25 nucleotides,
which regulate gene expression at the post-transcriptional level
(13). miRNAs have been reported to
play vital roles in cell proliferation, apoptosis and metastasis in
human cancer (14). For example,
miR-296 inhibited cell invasion and migration in esophageal
squamous cell carcinoma by targeting STAT3 (15). Recently, miR-653 was found to
suppress non-small cell lung cancer and breast cancer tumorigenesis
(16,17). Nonetheless, whether miR-653 is
involved in RCC remains to be further clarified.
C-X-C chemokine receptors (CXCRs) are a family of
cellular G-protein coupled receptors (18). Among the CXCRs, the chemokine
receptor CXCR5, which is primarily expressed in B cells and CD4+ T
cells, has been associated with tumorigenesis and progression of
various types of cancer, such as breast, prostate and colon cancer
(19–21). Recently, Zheng et al
(22) reported that CXCR5 was
associated with clear cell renal cell carcinoma (ccRCC) progression
and predicted poor prognosis, which led to the hypothesis that
CXCR5 might be a vital modulator of RCC tumorigenesis. The findings
of the present study provide a further understanding into the
development of RCC, which may facilitate the development of a novel
therapeutic targets in the treatment of RCC.
Materials and methods
Clinical specimens
In total, 33 pairs of RCC tissues and adjacent
normal tissues were obtained from patients (19 males and 14
females), who had undergone surgical resection, with a median age
of 56 years (range, 31–78 years) between February 2016 and August
2018 at the Shanghai JiaoTong University School of Medicine.
Written informed consent was provided from all participators prior
to the start of the study. All tissues were instantly frozen in
liquid nitrogen, and then stored at −80°C for further analysis. The
clinicopathological data was obtained from the medical records at
the Shanghai Jiaotong University School of Medicine. All patients
were classified according to Union International Cancer Control and
the American Joint Committee on Cancer (23,24).
The study was approved by the Ethics Committee of Shanghai Jiaotong
University School of Medicine.
The Cancer Genome Atlas (TCGA)
analysis
The expression of PLK1S1 and TCGA kidney renal clear
cell carcinoma (TCGA-KIRC) clinical data were downloaded from the
TCGA data portal (https://tcga-data.nci.nih.gov/tcga/). Patients with
corresponding gene expression were included in the present study,
while those with missing overall survival data were excluded.
Cell culture
The human RCC cells (ACHN, Caki1, A498 and 786-O),
papillary renal cell carcinoma cells (Caki2) and the immortalized
renal proximal tubules epithelial cells (RPTEC) were purchased from
American Type Culture Collection. The cells were cultured in DMEM
(Gibco; Thermo Fisher Scientific, Inc.), and supplemented with 10%
fetal bovine serum (FBS) at 37°C in a humidified atmosphere
containing 5% CO2. To generate ACHN and 786-O resistant
cell lines (ACHN-R and 786-O-R, respectively), ACHN and 786-O cells
were incubated with increasing concentrations of sorafenib (1 to 20
µM) for >6 months.
Cell transfection
The short hairpin (sh)RNA specific to PLK1S1
(shPLK1S1; 5′-UCAGCUGCUGUCGUAUUCAUGAG-3′) and its negative control
(shNC; 5′-AAUUCUCCGAACGUGUCACGU-3′), miR-653 mimic
(5′-GUGUUGAAACAAUCUCUACUG-3′) and its negative control (NC mimic;
5′-GACAACUUACAAUCUCUACUG-3′), and the miR-653 inhibitor
(5′-AGCCUUGAUCGAGGUCGGGAU-3′) and its negative control (NC
inhibitor; 5′-CAGUAGAGAUUGUAAGUUGUC-3′), were synthesized by
Shanghai GenePharma Co., Ltd.. For the overexpression of CXCR5, the
CXCR5 cDNA was cloned into the pCDNA3.1 vector (Shanghai GenePharma
Co., Ltd). Transfection of the cells with vectors, shPLK1S1or shNC
and miR-653 mimic or NC mimic, miR-653 inhibitor or NC inhibitor,
and co-transfection with shPLK1S1 and miR-653 inhibitor (all at 10
nM) were conducted with Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.), according to the manufacturers
instructions. All functional experiments were performed 48 h
post-transfection.
Cell Counting Kit-8 (CCK-8) assay
To determine the IC50 value, ACHN and
786-O cells (1×104 cells/well) were seeded into 96-well
plates and then treated with varying concentrations of sorafenib
(2.5, 5, 10, 20, 40 and 80 µg/ml) for 48 h. For the detection of
cell viability, transfected ACHN and 786-O cells were treated with
sorafenib (2 µg/ml) for 48 h. Next, cell viability was determined
using the CCK-8 assay kit (Dojindo Molecular Technologies, Inc.).
Briefly, 10 µl CCK-8 solution was added to each well and incubated
for 3 h, and then the absorbance at 450 nm was measured using a
microplate reader (Thermo Fisher Scientific, Inc.). IC50
were determined as the concentration of the drug at which sorafenib
produced 50% growth inhibition, with higher IC50 values
indicated higher chemoresistance potential.
Colony formation assay
Transfected RCC cells were seeded in 6-well plates
at a density of 200 cells/well. Following culturing for two weeks,
PBS (Sigma-Aldrich; Merck KGaA) was used to rinse each well.
Subsequently, RCC cells were fixed in 4% paraformaldehyde
(Sigma-Aldrich; Merck KGaA), and then stained with 0.5% crystal
violet (Sigma-Aldrich; Merck KGaA) both at room temperature for 10
min. The number of colonies were then counted using a light
microscope (magnification, ×200). Groups of >50 cells were
considered a clone.
Matrigel assay
The invasion abilities of the RCC cells were
assessed using Transwell chambers (8.0-µm pore size; EMD Millipore)
pre-coated with Matrigel for 1 h (Corning Inc.) at room
temperature. Transfected cells (8×104 cells) were added
to the upper chamber containing 150 µl RPMI-1640 without FBS. In
addition, 550 µl RPMI-1640 medium was added to the lower chamber.
After 24 h, cells in the upper chamber were removed, and cells in
the lower membrane were fixed in 4% paraformaldehyde and stained
with 0.1% crystal violet (Beyotime Institute of Biotechnology) both
for 20 min at room temperature. Invaded cells were counted in 3
randomly selected visual fields using a light microscope
(magnification, ×200; Zeiss GmBH).
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL)
The TUNEL apoptosis kit (Roche Diagnostics GmbH) was
used to assess cell apoptosis. In brief, cells were washed with PBS
for 5 min, three times and fixed in 4% paraformaldehyde (cat. no.
AR1069; Wuhan Boster Biological Technology, Ltd.) at 4°C for 20
min. The cells were then incubated with the TUNEL enzyme for 60 min
at 3°C. Finally, the fluorescent reaction was counterstained with
DAPI (1:1,000 in PBS; Sigma-Aldrich; Merck KGaA) to stain the
nucleus for 10 min at room temperature. Antifade mounting medium
(cat. no. P0126; Beyotime Institute of Biotechnology) was used.
Images from 4 fields of view were used to obtain images with a
fluorescent microscope (magnification, ×20; Olympus
Corporation).
Western blot analysis
Proteins were extracted from transfected RCC cells
using RIPA buffer (Thermo Fisher Scientific, Inc.). Protein
concentration was measured using the bicinchoninic acid assay
(Beyotime Institute of Biotechnology). A total of 10 µg
protein/lane were separated using 10% SDS-PAGE (EMD Millipore), and
then transferred to PVDF membranes (Bio-Rad Laboratories, Inc.).
After blocking with 5% skimmed milk, membranes were probed with
primary antibodies against CXCR5 (1:1,000; cat. no. ab133706;
Abcam) and anti-GAPDH (1:1,000; cat. no. ab8245; Abcam) overnight
at 4°C. Subsequently, membranes were incubated with horseradish
peroxidase-conjugated secondary antibodies, goat anti-mouse IgG,
(cat. no. ab205719) and goat anti-rabbit IgG, (cat. no. ab205718)
(both 1,1000; both from Abcam) at room temperature for 2 h. The
protein bands were visualized using an enhanced chemiluminescence
kit (Bio-Rad Laboratories, Inc.). GAPDH served as the loading
control.
Reverse transcription-quantitative PCR
(RT-qPCR)
At 48-h post-transfection, total RNA was isolated
from tissues and cell lines using TRIzol® (Thermo Fisher
Scientific, Inc.). The RNA was reverse transcribed into cDNA using
a reverse transcriptase kit (Takara Bio, Inc.) or the
TaqMan® miRNA reverse transcription kit (Thermo Fisher
Scientific, Inc.) at 37°C for 15 min. RT-qPCR was performed on the
ABI 7900 Detection System (Applied Biosystems; Thermo Fisher
Scientific, Inc.) using the SYBR-Green PCR Master Mix kit (Takara
Bio, Inc). The following thermocycling conditions were used for the
qPCR: Initial denaturation at 95°C for 3 min; 40 cycles of 95°C for
5 sec and 60°C for 30 sec. The expression levels of genes were
calculated using the 2−ΔΔCq method (25). U6 and GAPDH were set as the internal
control. The sequences of the primers were as follows: PLK1S1
forward, 5′-CCCACATTCACACCGACAGA-3′ and reverse,
5′-ACTCTTGCCATGACGTGTGT-3′; miR-653 forward,
5′-ACCAGCTTCAAACAAGTTCACTG-3′ and reverse,
5′-GCTTCCATCTTATCATTCTTGCA-3′; CXCR5 forward,
5′-CCCTCATGGCCTCCTTCAAG-3′ and reverse, 5′-AGGGCAAGATGAAGACCAGC-3′;
GAPDH forward, 5′-GCACCGTCAAGGCTGAGAAC-3′; and reverse,
5′-GCCTTCTCCATGGTGGTGAA-3′; U6 forward,
5′-CTCGCTTCGGCAGCACATATACTA-3′ and reverse,
5′-ACGAATTTGCGTGTCATCCTTGCG-3′.
Luciferase reporter assay
StarBase (http://starbase.sysu.edu.cn) online tool was used to
predict the potential miRNAs that could bind to PLK1S1, while
TargetScan (http://www.targetscan.org) was used
to predict the potential downstream target of miR-653. Mutants
within the miR-653 binding site were created using the QuikChange
II Site Directed Mutagenesis kit (Agilent Technologies, Inc.). The
wild-type (WT) and mutant (Mut) PLK1S1 or CXCR5 was sub-cloned into
pmirGLO dual-luciferase vector (Shanghai GenePharma Co., Ltd.) to
construct PLK1S1-WT/Mut or CXCR5-WT/Mut vectors. Subsequently,
PLK1S1-WT/Mut or CXCR5-WT/Mut vectors were co-transfected with NC
mimic, miR-653 mimic and miR-653 inhibitor into 293T cells using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). Luciferase activity was evaluated using the
Dual-Luciferase Reporter Analysis system (Promega Corporation),
following incubation for 48 h. Firefly luciferase activity was
normalized to Renilla (Promega Corporation) luciferase gene
activity.
Xenograft experiment
A total of 10 male BALB/c-nu nude mice (age, 5 to
6-weeks-old; weight, 18–20 g) were randomly divided into two groups
and maintained under 26°C, 50% relative humidity, with a 12-h
light/dark cycle, with ad libitum access to food and water.
All in vivo experimental procedures were approved by the
Ethics Committee of Shanghai Jiaotong University School of
Medicine. The ACHN cells transfected with shNC or shPLK1S1 were
subcutaneously injected into the mice. Tumors were examined every 7
days. Mice were euthanized according to the following criteria: i)
the weight of the mouse was excessively reduced or increased; ii)
abnormal behavior; iii) tumor diameter was >1.5 cm; iv) the
tumor was ulcerated. No mouse died or had significant weight loss
during the experiment. On 28th day, the mice were sacrificed by
cervical dislocation following anesthesia with an intraperitoneal
injection of sodium pentobarbital (60 mg/kg). Animal death was
confirmed by cardiac and respiratory arrest, muscle relaxation and
lack of reflection. The tumors were photographed, and the tumor
weights were measured. Tumor volume was calculated using the
following formula: Volume = (length × width2)/2. The
maximum tumor volume was 709 mm3 and the maximum
diameter of tumor was 16 mm.
Statistical analysis
Statistical analysis was performed using SPSS v16.0
(SPSS, Inc.). The experiments were performed three times and the
data are presented as mean ± standard deviation. Comparisons among
multiple groups were performed using one-way analysis of variance,
followed by Tukey's post hoc test. Comparison between RCC and
adjacent normal tissue samples from patients with RCC was performed
using a paired Student's t-test, while comparison between the
experimental and control groups was performed using an unpaired
Student's t-test. Kaplan-Meier analysis and the log-rank test were
used to analyze survival curves. Cut-off values were determined
using the mean expression level of PLK1S1. P<0.05 was considered
to indicate a statistically significant difference.
Results
PLK1S1 is upregulated in RCC cells and
tissues and is associated with poor prognosis
The mRNA expression level of PLK1S1 was investigated
in RCC cells, and the results indicated that expression in the RCC
cells (Caki1, ACHN, A498 and 786-O) and the Caki2 papillary RCC
cell line was significantly upregulated compared with that in the
RPTEC cell line (Fig. 1A).
Furthermore, the PLK1S1 mRNA expression level was also
significantly increased in clinical tissues, (Fig. 1B). In addition, it was found that
PLK1S1 expression was associated with histological grade, tumor
stage, lymph node metastasis and distant metastasis, while there
was no association with age or sex (Table I).
 | Table I.Association between PLK1S1 mRNA
expression levels and clinicopathological features in patients with
renal cell carcinoma. |
Table I.
Association between PLK1S1 mRNA
expression levels and clinicopathological features in patients with
renal cell carcinoma.
|
|
| Expression of
PLK1S1 |
|
|---|
|
|
|
|
|
|---|
| Clinicopathological
features | Number | High, n (%) | Low, n (%) | P-value |
|---|
| Age, years |
|
|
| 0.374 |
|
≤60 | 15 | 7 (46.7) | 8 (53.3) |
|
|
>60 | 18 | 10 (55.6) | 8 (44.4) |
|
| Sex |
|
|
| 0.462 |
|
Male | 19 | 9 (47.4) | 10 (52.6) |
|
|
Female | 14 | 8 (57.1) | 6 (42.9) |
|
| Histological
grade |
|
|
| 0.011 |
|
Well | 21 | 9 (42.9) | 12 (57.1) |
|
|
Moderate and poor | 12 | 9 (75.0) | 3 (25.0) |
|
| Tumor stage |
|
|
| 0.028 |
| I and
II | 20 | 8 (40.0) | 12 (60.0) |
|
| III and
IV | 13 | 9 (69.2) | 4 (30.8) |
|
| Lymph node
metastasis |
|
|
| 0.011 |
|
Positive | 10 | 8 (80.0) | 2 (20.0) |
|
|
Negative | 23 | 11 (47.8) | 12 (52.2) |
|
| Distant
metastasis |
|
|
| 0.014 |
|
Positive | 11 | 9 (81.8) | 2 (18.2) |
|
|
Negative | 22 | 10 (45.5) | 12 (54.5) |
|
To investigate the mRNA expression levels further,
TCGA datasets were analyzed to determine the involvement of PLK1S1
in RCC. The data revealed that PLK1S1 mRNA expression levels in RCC
tissues was significantly increased compared with that in adjacent
normal tissues (Fig. 1C). In
addition, Kaplan-Meier analysis showed that patients with high
PLK1S1 mRNA expression level had a shorter overall survival time
compared with that in patients with low PLK1S1 expression (Fig. 1D). Notably, it was also found that
the PLK1S1 mRNA expression level was significantly increased in
patients with stage IV RCC (F, 3.18; adjusted P=0.0299) (Fig. 1E). Taken together, these data
indicated that PLK1S1 might be an oncogene for RCC progression.
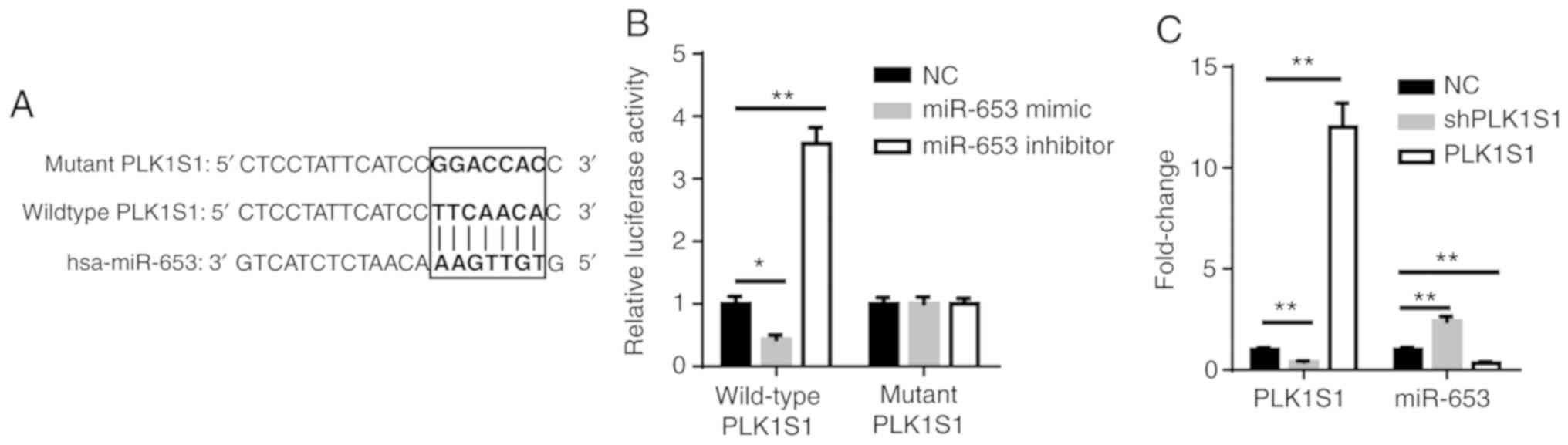
PLK1S1 interacts with miR-653
Using the StarBase bioinformatics analysis software,
PLK1S1 was found to bind to miR-653 via complementary base pairing
(Fig. 2A). The results from the
luciferase reporter assay indicated that miR-653 mimic
significantly reduced luciferase activity of pmirGLO-PLK1S1-WT
vectors, whereas the miR-653 inhibitor significantly increased
luciferase activity (Fig. 2B). In
addition, PLK1S1 was significantly downregulated in shPLK1S1
transfected ACHN cells and significantly upregulated in PLK1S1
overexpressed ACHN cells. The downregulation of PLK1S1
significantly increased the expression of miR-653 whereas the
upregulation of PLK1S1 significantly decreased the expression of
miR-653 (Fig. 2C). Taken together,
these results demonstrated that PLK1S1 inhibited miR-653 expression
by direct interaction.
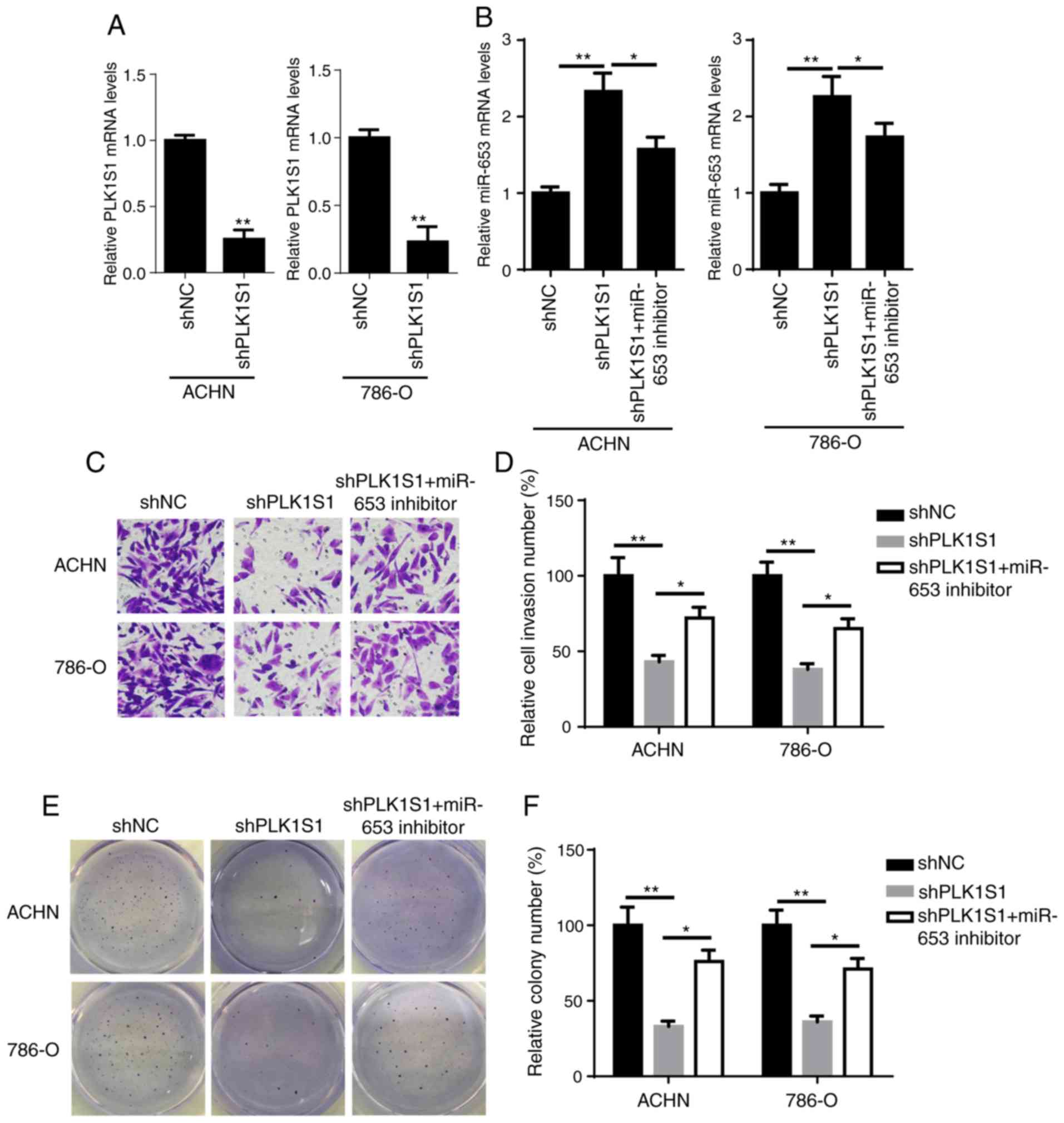
miR-653 inhibitor rescues PLK1S1
knockdown-attenuated tumorigenesis of RCC cells
To explore whether miR-653-mediated and
PLK1S1-regulated RCC tumorigenesis and progression, stable
PLK1S1-knockdown RCC cells (ACHN and 786-O) were generated
(Fig. 3A). Subsequently, the
miR-653 inhibitor was introduced into shPLK1S1-expressing ACHN and
786-O cells. RT-qPCR showed that the introduction of miR-653
inhibitor significantly reduced shPLK1S1-mediated miR-653
upregulation in RCC cells (Fig.
3B). Matrigel and colony formation assays revealed that the
miR-653 inhibitor abrogated the inhibitory effects of PLK1S1
knockdown on the invasion and proliferation of RCC cells (Fig. 3C-F). Based on these results, it was
confirmed that miR-653 was involved in PLK1S1-modulated
tumorigenesis of RCC cells.
PLK1S1 knockdown-mediated inhibitory
effect on sorafenib resistance is reduced by the miR-653 inhibitor
in sorafenib-resistant RCC cells
To investigate the potential role of PLK1S1 and
miR-653 in drug resistance to sorafenib in RCC cells,
sorafenib-resistant ACHN (ACHN-R) and 786-O cells (786-O-R) were
established. As shown in Fig. 4A and
B, the IC50 of sorafenib was significantly increased
in ACHN-R and 786-O-R cells, indicating that sorafenib-resistant
RCC cells were successfully generated. In addition, the miR-653
inhibitor partially reversed PLK1S1 knockdown-attenuated cell
viability in sorafenib-resistant RCC cells under sorafenib
treatment (Fig. 4C). Furthermore,
PLK1S1 knockdown significantly decreased the IC50 of
sorafenib in sorafenib-resistant RCC cells, while miR-653 inhibitor
partially reversed shPLK1S1-induced decrease in IC50
values (Fig. 4D and E). The TUNEL
assay revealed that the miR-653 inhibitor significantly decreased
cell apoptosis in shPLK1S1 sorafenib-resistant RCC cells (Fig. 4F and G). In summary, the data
suggested that PLK1S1 enhanced chemoresistance in RCC cells to
sorafenib by regulating miR-653.
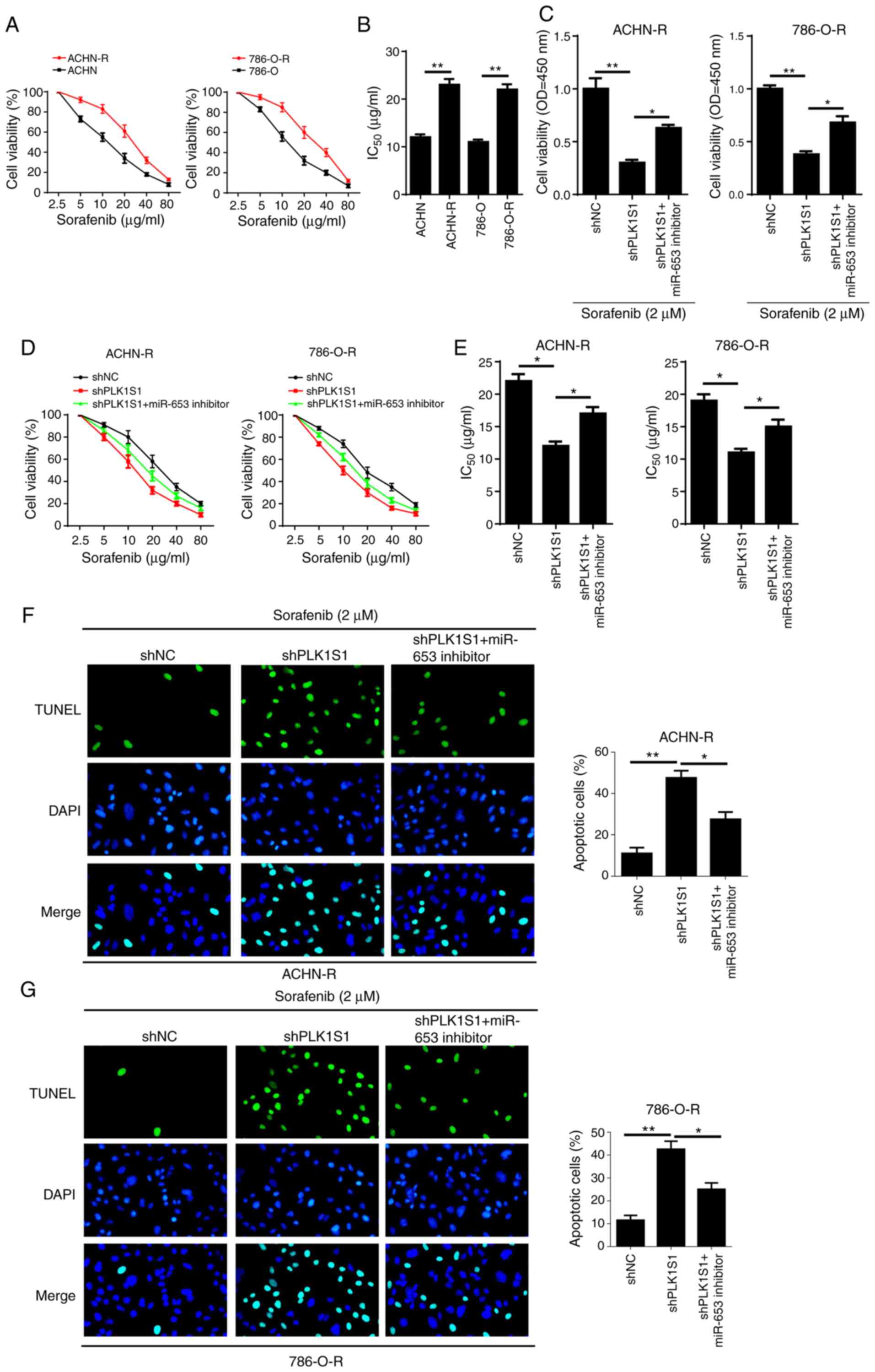 | Figure 4.PLK1S1 depletion-mediated inhibitory
effect on sorafenib resistance is eliminated by the miR-653
inhibitor in sorafenib-resistant renal cell carcinoma cell lines.
(A and B) The IC50 values of sorafenib in ACHN, ACHN-R,
786-O and 786-O-R cells was measured using the CCK-8 assay. CCK-8
was used to determine the (C) cell viability and (D and E)
IC50 values of sorafenib of ACHN-R and 786-O-R cells
transfected with shNC, shPLK1S1, shPLK1S1 plus miR-653 inhibitor
treated with or without 2 µM sorafenib. TUNEL assay was used to
investigate cell apoptosis in (F) ACHN-R and (G) 786-O-R cells
transfected with shNC, shPLK1S1, shPLK1S1 plus miR-653 inhibitor
treated with sorafenib. All data are represented as the mean ± SD.
*P<0.05; **P<0.01. miRNA, microRNA; sh, short hairpin; NC,
negative control; TUNEL, Terminal deoxynucleotidyl transferase dUTP
nick end labeling; CCK-8; Cell Counting Kit-8; R, resistant. |
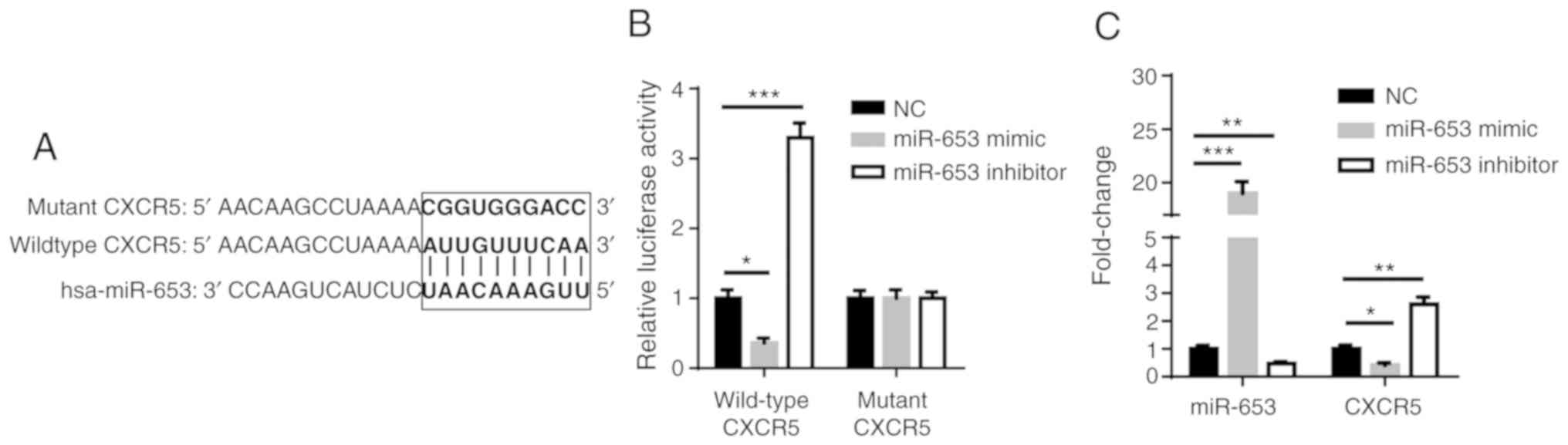
CXCR5 is a target of miR-653
Using TargetScan (http://www.targetscan.org), CXCR5 was predicted as a
downstream target of miR-653 (Fig.
5A). Furthermore, luciferase reporter assay demonstrated that
miR-653 mimic reduced luciferase activity of WT CXCR5, while the
opposite effect occurred in cells transfected with miR-653
inhibitor (Fig. 5B). In addition,
RT-qPCR revealed that miR-653 was significantly upregulated in
miR-653 mimic transfected ACHN cells and significantly
downregulated in miR-653 inhibitor transfected ACHN cells. The
upregulation of miR-653 significantly decreased the expression of
CXCR5, while the downregulation of miR-653 significantly increased
the expression of CXCR5 (Fig. 5C).
Thus, these data indicate that miR-653 directly targets CXCR5.
CXCR5 mediates
PLK1S1/miR-653-regulated RCC tumorigenesis
To further determine whether PLK1S1/miR-653 promoted
RCC progression through CXCR5, CXCR5 was introduced into shPLK1S1
and miR-653 mimic-expressing ACHN cells. Firstly, it was
demonstrated that overexpression of CXCR5 significantly reversed
the inhibitory effects of PLK1S1 knockdown and miR-653
overexpression on CXCR5 mRNA expression levels (Fig. 6A and B). Matrigel and colony
formation assays demonstrated that the overexpression of CXCR5
significantly reversed the shPLK1S1- and miR-653 mimic-attenuated
ACHN cell invasion and proliferation (Fig. 6C-H). Taken together, these data
revealed that the PLK1S1/miR-653/CXCR5 axis promoted the
development and progression of RCC.
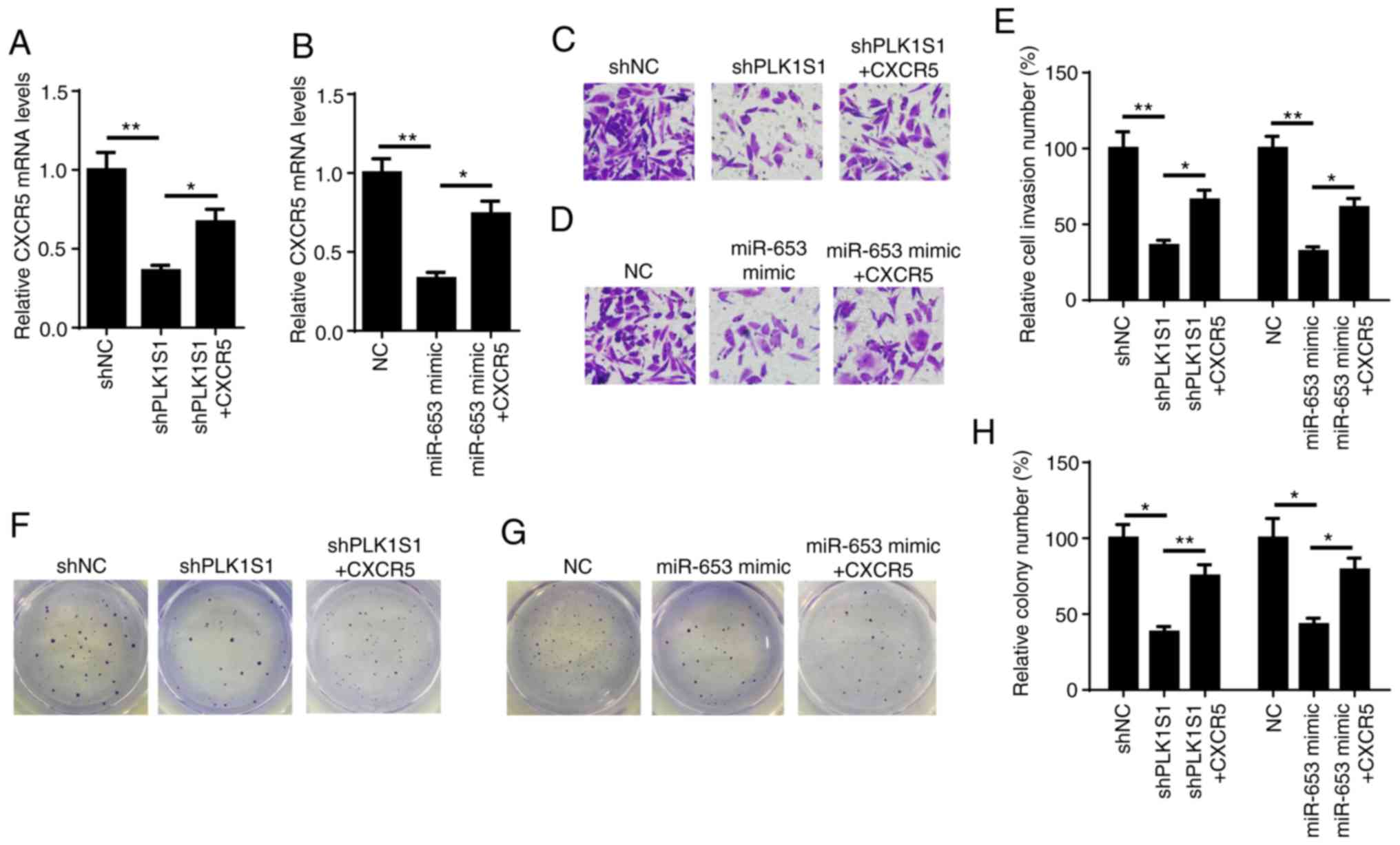 | Figure 6.CXCR5 mediates
PLK1S1/miR-653-regulated renal cell carcinoma tumorigenesis.
Reverse transcription-quantitative PCR was used to determine the
relative mRNA expression of CXCR5 in (A) ACHN cells transfected
with shNC, shPLK1S1, shPLK1S1 plus CXCR5 and (B) ACHN cells
transfected with NC mimic, miR-653 mimic, miR-653 mimic plus CXCR5.
Matrigel assay was used to investigate cell invasion in ACHN cells
transfected with (C) shNC, shPLK1S1, shPLK1S1 plus CXCR5 and (D) NC
mimic, miR-653 mimic, miR-653 mimic plus CXCR5 and the results were
subsequently (E) quantified. Colony formation assay was used to
determine the colony number of ACHN cells transfected with (F)
shNC, shPLK1S1, shPLK1S1 plus CXCR5 and (G) NC mimic, miR-653
mimic, miR-653 mimic plus CXCR5 and the results were subsequently
(H) quantified. All data are represented as the mean ± SD.
*P<0.05; **P<0.01. miRNA, microRNA; sh, short hairpin; NC,
negative control; CXCR5, C-X-C motif chemokine receptors 5. |
CXCR5 rescues PLK1S1 knockdown- or
miR-653 mimic-attenuated chemoresistance of RCC cells
Next, the role of CXCR5 in PLK1S1 and
miR-653-modulated drug resistance of RCC cells to sorafenib was
assessed. As shown in Fig. 7A,
CXCR5 increased the cell viability of sorafenib-resistant ACHN
cells expressing shPLK1S1 or miR-653 mimic in the presence of
sorafenib treatment. In addition, overexpression of CXCR5 reduced
the inhibitory effect of shPLK1S1 or miR-653 mimic on
IC50 values in sorafenib-resistant RCC cells (Fig. 7B and C). The TUNEL assay showed that
overexpression of CXCR5 significantly reversed shPLK1S1 or miR-653
mimic-mediated promotion of cell apoptosis in ACHN-R cells
following sorafenib treatment (Fig.
7D-G). Taken together, the data indicate that CXCR5 is a key
effector of PLK1S1 and miR-653-regulated sorafenib resistance in
RCC cells.
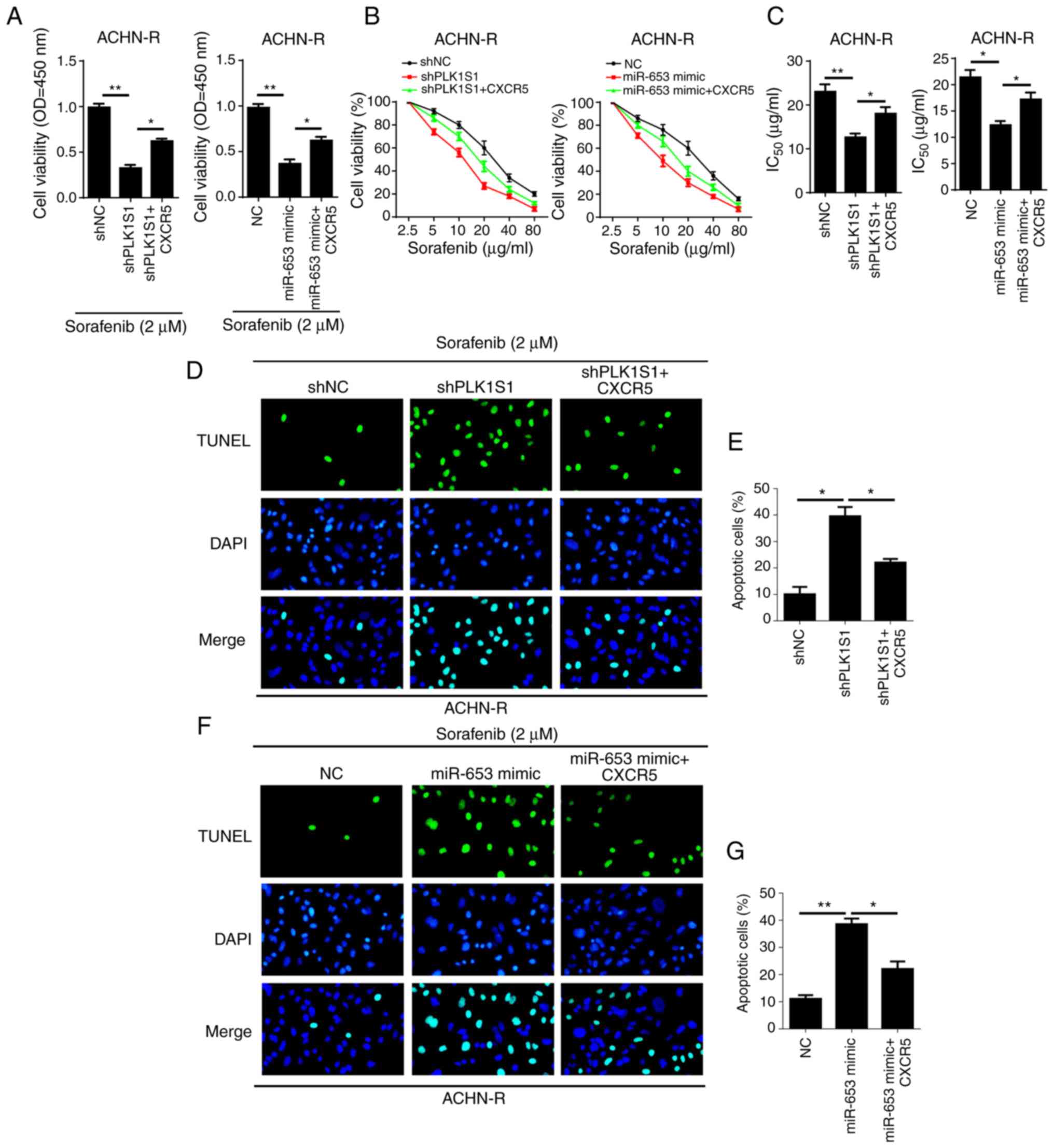 | Figure 7.CXCR5 rescues PLK1S1 knockdown- or
miR-653 mimic-attenuated chemoresistance of renal cell carcinoma
cell lines. Cell Counting Kit-8 assay was used to determine (A)
cell viability and (B and C) IC50 values in ACHN-R cells
transfected with shNC, shPLK1S1, shPLK1S1 plus CXCR5 and NC mimic,
miR-653 mimic, miR-653 mimic plus CXCR5 treated with and without 2
µM sorafenib, respectively. (D-G) TUNEL assay was used to
investigate cell apoptosis in ACHN-R cells transfected with (D)
shNC, shPLK1S1, shPLK1S1 plus CXCR5 and the results were
subsequently (E) quantified, and with (F) NC, miR-653 mimic,
miR-653 mimic plus CXCR5, treated with 2 µM sorafenib and the
results were (G) quantified. All data are represented as the mean ±
SD. *P<0.05; **P<0.01. miRNA, microRNA; sh, short hairpin;
NC, negative control; TUNEL, Terminal deoxynucleotidyl transferase
dUTP nick end labeling; R, resistant; CXCR5, C-X-C motif chemokine
receptors 5; OD, optical density. |
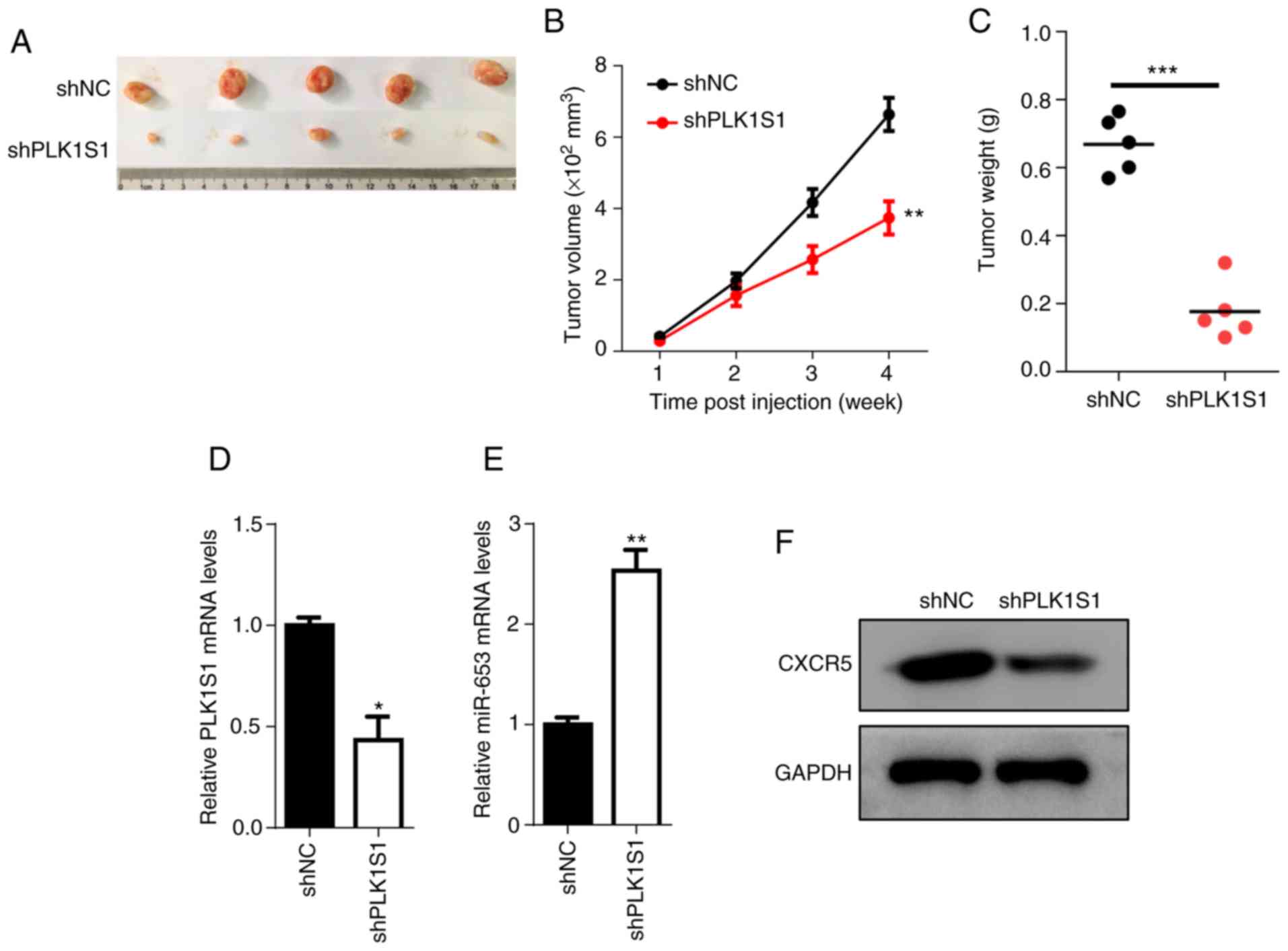
PLK1S1 knockdown prevents tumor growth
in vivo
To further determine whether PLK1S1 promotes RCC
growth in vivo, the xenograft experiment was performed. The
results demonstrated that PLK1S1 knockdown significantly reduced
the growth of the tumor compared with that in the shNC group
(Fig. 8A-C). RT-qPCR showed that
the mRNA expression level of PLK1S1 was significantly decreased in
the shPLK1S1 group compared with that in the shNC group, while the
expression level of miR-653 was significantly increased in the
shPLK1S1 group compared with that in the shNC group (Fig. 8D and E). Furthermore, western blot
analysis demonstrated that knockdown of PLK1S1 reduced the protein
expression level of CXCR5 (Fig.
8F). In summary, these data reveal that PLK1S1 promotes tumor
growth of RCC via the miR-653/CXCR5 axis.
Discussion
The present study revealed that PLK1S1 promoted RCC
tumorigenesis and enhanced sorafenib resistance of RCC through the
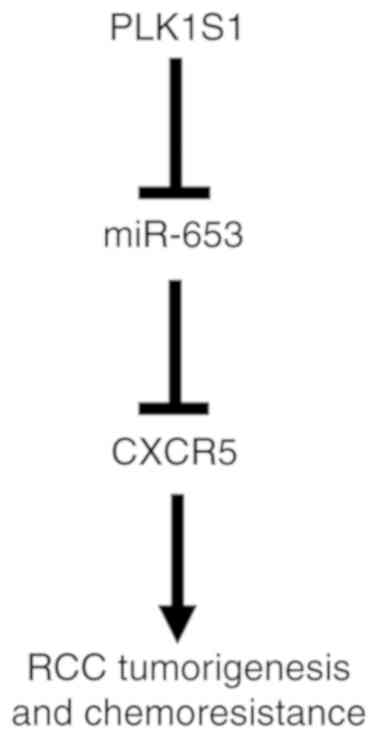
miR-653/CXCR5 axis (Fig. 9). The
present study not only discovered a novel regulatory mechanism in
RCC, but also identified a potential novel therapy for patients
with RCC.
Over the last decade, lncRNAs have attracted
increasing attention and demonstrated their pivotal roles in
various types of cancer, including RCC. For example, Dong et
al (26) reported that lncRNA
SNHG7 promoted proliferation and inhibited apoptosis of RCC cells
by inhibiting the protein expression level of CDKN1A. Yue et
al (27) revealed that the
knockdown of lncRNA DLEU1 inhibited RCC progression by regulating
the Akt and EMT signaling pathway. In the present study, it was
demonstrated that PLK1S1 was upregulated in RCC tissues and cells,
and that high expression of PLK1S1 was associated with an advanced
TNM stage and poor prognosis in patients with RCC.
The competing endogenous RNA (ceRNA) network
exhibits its regulatory function in human cancer, including RCC.
For example, Yang et al (28) found that the lncRNA TUG1 acted as a
ceRNA of miR-196a to accelerate the proliferation, migration and
invasion of RCC. Shi et al (29) demonstrated that the lncRNA ROR
sponged miR-206 to promote RCC progression through increasing the
protein expression level of VEGF. Furthermore, Xie et al
(17) demonstrated that
hsa_circ_0004771 facilitated the proliferation and inhibited
apoptosis in breast cancer by functioning as a ceRNA of miR-653 to
regulate the ZEB2 signaling pathway. In the present study, it was
confirmed that miR-653 had the ability to bind to PLK1S1. To the
best of our knowledge this is the first time that the sponging
effect of PLK1S1 on miR-653 in RCC has been discovered. In
addition, the inhibitory effects of shPLK1S1 on RCC progression and
chemoresistance of RCC cells to sorafenib were reduced by the
miR-653 inhibitor. Therefore, to the best of our knowledge it has
been demonstrated for the first time that PLK1S1 acted as a ceRNA
to sponge miR-653 to promote tumorigenesis and enhance the
chemoresistance of RCC.
CXCRs, comprising of CXCR 1 to 7, are not only
involved in the immune system but also in tumorigenesis and cancer
development (30). For example,
Saintigny et al (31) found
that CXCR2 was associated with a low 5-year survival rate and
promoted the invasion and metastasis of lung adenocarcinoma.
Furthermore, Sun et al (32)
reported that the knockdown of CXCR2 and CXCR3 suppressed the
migration, invasion, colony formation and sphere-forming abilities
of RCC cells. CXCR5 has been reported to be upregulated in ccRCC,
and CXCR5 knockdown reduced the promoting effect of CXCL13 on the
proliferation and migration of ccRCC cells (22). However, there are no previous
studies that investigated the involvement of CXCR5 in RCC. In the
present study, it was found that CXCR5 was a target of miR-653. In
addition, the overexpression of CXCR5 rescued PLK1S1 knockdown- or
miR-653 mimic-attenuated progression and chemoresistance of RCC
cells, suggesting that CXCR5 was crucial for
PLK1S1/miR-653-regulated progression of RCC.
In conclusion, the present study reported the
potential molecular mechanisms of PLK1S1 in the tumorigenesis and
chemoresistance of RCC. To the best of our knowledge it has been
demonstrated for the first time that PLK1S1 contributed to RCC
progression and enhanced the chemosensitivity of RCC cells via the
miR-653/CXCR5 pathway. The results provide a further understanding
in the treatment of RCC using a PLK1S1-targeted approach. However,
further investigation is still required. Firstly, other lncRNAs may
exist and serve as ceRNAs to regulate crucial gene expression in
RCC. Secondly, PLK1S1 can bind to a number of miRNAs, of which,
other miRNAs can also affect the development of RCC.
Acknowledgements
Not applicable.
Funding
This study was supported by the Science and
Technology Committee of Changning District of Shanghai (grant no.
CNKW2018Y03).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
WL, DL and ML designed the present study. DY and YZ
and SZ performed all the experiments. WL and ML analyzed the data
and prepared the figures. WL and DL drafted the initial manuscript.
ML reviewed and revised the manuscript. All authors approved the
final version of the manuscript.
Ethics approval and consent to
participate
All in vivo experimental procedures were
approved by the Ethics Committee of Shanghai Jiaotong University
School of Medicine.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Capitanio U, Bensalah K, Bex A, Boorjian
SA, Bray F, Coleman J, Gore JL, Sun M, Wood C and Russo P:
Epidemiology of renal cell carcinoma. Eur Urol. 75:74–84. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rini BI, Campbell SC and Escudier B: Renal
cell carcinoma. Lancet. 373:1119–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brugarolas J: Renal-cell
carcinoma-molecular pathways and therapies. N Engl J Med.
356:185–187. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schmitt AM and Chang HY: Long noncoding
RNAs in cancer pathways. Cancer Cell. 29:452–463. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huarte M: The emerging role of lncRNAs in
cancer. Nat Med. 21:1253–1261. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li M, Wang Y, Cheng L, Niu W, Zhao G, Raju
JK, Huo J, Wu B, Yin B, Song Y and Bu R: Long non-coding RNAs in
renal cell carcinoma: A systematic review and clinical
implications. Oncotarget. 8:48424–48435. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhai W, Sun Y, Guo C, Hu G, Wang M, Zheng
J, Lin W, Huang Q, Li G, Zheng J and Chang C: LncRNA-SARCC
suppresses renal cell carcinoma (RCC) progression via altering the
androgen receptor(AR)/miRNA-143-3p signals. Cell Death Differ.
24:1502–1517. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang T, Wang X, Yang X, Ji J, Wang Q, Yue
X and Dong Z: Long non-coding RNA DUXAP8 enhances renal cell
carcinoma progression via downregulating miR-126. Med Sci Monit.
24:7340–7347. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu Z, Yang F, Wei D, Liu B, Chen C, Bao Y,
Wu Z, Wu D, Tan H, Li J, et al: Long noncoding RNA-SRLR elicits
intrinsic sorafenib resistance via evoking IL-6/STAT3 axis in renal
cell carcinoma. Oncogene. 36:1965–1977. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xue X, Yang YA, Zhang A, Fong KW, Kim J,
Song B, Li S, Zhao JC and Yu J: LncRNA HOTAIR enhances ER signaling
and confers tamoxifen resistance in breast cancer. Oncogene.
35:2746–2755. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Peng F, Wang R, Zhang Y, Zhao Z, Zhou W,
Chang Z, Liang H, Zhao W, Qi L, Guo Z and Gu Y: Differential
expression analysis at the individual level reveals a lncRNA
prognostic signature for lung adenocarcinoma. Mol Cancer.
16:982017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dat Le T, Matsuo T, Yoshimaru T, Kakiuchi
S, Goto H, Hanibuchi M, Kuramoto T, Nishioka Y, Sone S and Katagiri
T: Identification of genes potentially involved in bone metastasis
by genome-wide gene expression profile analysis of non-small cell
lung cancer in mice. Int J Oncol. 40:1455–1469. 2012.PubMed/NCBI
|
|
13
|
Croce CM: Causes and consequences of
microRNA dysregulation in cancer. Nat Rev Genet. 10:704–714. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang RT, Xu M, Xu CX, Song ZG and Jin H:
Decreased expression of miR216a contributes to non-small-cell lung
cancer progression. Clin Cancer Res. 20:4705–4516. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang ZZ, Luo YR, Du J, Yu Y, Yang XZ, Cui
YJ and Jin XF: MiR-296-5p inhibits cell invasion and migration of
esophageal squamous cell carcinoma by downregulating STAT3
signaling. Eur Rev Med Pharmacol Sci. 23:5206–5214. 2019.PubMed/NCBI
|
|
16
|
Han W, Wang L, Zhang L, Wang Y and Li Y:
Circular RNA circ-RAD23B promotes cell growth and invasion by
miR-593-3p/CCND2 and miR-653-5p/TIAM1 pathways in non-small cell
lung cancer. Biochem Biophys Res Commun. 510:462–466. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xie R, Tang J, Zhu X and Jiang H:
Silencing of hsa_circ_0004771 inhibits proliferation and induces
apoptosis in breast cancer through activation of miR-653 by
targeting ZEB2 signaling pathway. Biosci Rep. 39:BSR201819192019.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin CY, Chen YM, Hsu HH, Shiu CT, Kuo HC
and Chen TY: Grouper (epinephelus coioides) CXCR4 is expressed in
response to pathogens infection and early stage of development. Dev
Comp Immunol. 36:112–120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao K, Yao Y, Luo X, Lin B, Huang Y, Zhou
Y, Li Z, Guo Q and Lu N: LYG-202 inhibits activation of endothelial
cells and angiogenesis through CXCL12/CXCR7 pathway in breast
cancer. Carcinogenesis. 39:588–600. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang J, Tang H, Huang J and An H:
Upregulation of CXCR7 is associated with poor prognosis of prostate
cancer. Med Sci Monit. 24:5185–5191. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bates RC, DeLeo MJ III and Mercurio AM:
The epithelial-mesenchymal transition of colon carcinoma involves
expression of IL-8 and CXCR-1-mediated chemotaxis. Exp Cell Res.
299:315–324. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zheng Z, Cai Y, Chen H, Chen Z, Zhu D,
Zhong Q and Xie W: CXCL13/CXCR5 axis predicts poor prognosis and
promotes progression through PI3K/AKT/mTOR pathway in clear cell
renal cell carcinoma. Front Oncol. 8:6822018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guinan P, Sobin LH, Algaba F, Badellino F,
Kameyama S, MacLennan G and Novick A: TNM staging of renal cell
carcinoma: Workgroup no. 3. Union international contre le cancer
(UICC) and the American joint committee on cancer (AJCC). Cancer.
80:992–993. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sobin LH and Fleming ID: TNM
classification of malignant tumors, fifth edition (1997). Union
internationale contre le cancer and the American joint committee on
cancer. Cancer. 80:1803–1804. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dong JS, Wu B and Jiang B: LncRNA SNHG7
promotes the proliferation and inhibits apoptosis of renal cell
cancer cells by downregulating CDKN1A. Eur Rev Med Pharmacol Sci.
23:10241–10247. 2019.PubMed/NCBI
|
|
27
|
Yue G, Chen C, Bai L, Wang G, Huang Y,
Wang Y, Cui H and Xiao Y: Knockdown of long noncoding RNA DLEU1
suppresses the progression of renal cell carcinoma by
downregulating the Akt pathway. Mol Med Rep. 20:4551–4557.
2019.PubMed/NCBI
|
|
28
|
Yang Y, Sun DM, Yu JF, Zhang M, Yi C, Yang
R, Dan BH and Li AJ: Long noncoding RNA TUG1 promotes renal cell
carcinoma cell proliferation, migration and invasion by
downregulating microRNA196a. Mol Med Rep. 18:5791–5798.
2018.PubMed/NCBI
|
|
29
|
Shi J, Zhang D, Zhong Z and Zhang W:
lncRNA ROR promotes the progression of renal cell carcinoma through
the miR-206/VEGF axis. Mol Med Rep. 20:3782–3792. 2019.PubMed/NCBI
|
|
30
|
Yu C and Zhang Y: Characterization of the
prognostic values of CXCR family in gastric cancer. Cytokine.
123:1547852019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Saintigny P, Massarelli E, Lin S, Ahn YH,
Chen Y, Goswami S, Erez B, O'Reilly MS, Liu D, Lee JJ, et al: CXCR2
expression in tumor cells is a poor prognostic factor and promotes
invasion and metastasis in lung adenocarcinoma. Cancer Res.
73:571–582. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sun KH, Sun GH, Wu YC, Ko BJ, Hsu HT and
Wu ST: TNF-α augments CXCR2 and CXCR3 to promote progression of
renal cell carcinoma. J Cell Mol Med. 20:2020–2028. 2016.
View Article : Google Scholar : PubMed/NCBI
|