Introduction
Cancer is a major public health problem worldwide
and is expected to be the leading cause of death and the single
most important barrier to increasing life expectancy in the 21st
century (1). It is now understood
that advanced age and exposure to ionizing radiation may be the
most important risk factors promoting carcinogenesis (2,3).
Substantial research progress has been made in recent years to
understand the basic mechanisms underlying cancer initiation and
progression and to identify potential targets for therapeutic
intervention. Thanks to the advance of next-generation sequencing
technology, extensive transcriptome and genome analyses of cancer
have identified multiple recurrently altered genes and pathways by
robustly assessing RNA sequencing (RNA-seq) and whole exome
sequencing data of tumor and matched normal tissue samples
(4,5). These pathways include the PI3K/AKT
signaling pathway, the Wnt signaling pathway and the DNA damage
response (DDR) pathway (6–8). Although errors in the DDR contribute
to genome instability, which may lead to tumor progression, they
also provide therapeutic opportunities (9,10). In
particular, the involvement of post-transcriptional regulation
within the DDR pathway is now becoming evident (11).
Post-transcriptional regulation is important for
modulation of gene expression and involves numerous different
aspects of biological processes, including RNA metabolism, pre-mRNA
splicing and translation (12). It
is widely recognized that alternative splicing (AS) occurs in
nearly all human genes and has a close connection to
cancer-associated pathways (13,14).
Compared to normal tissues, tumors present with abnormal splicing
patterns that have a pathogenic role in cancer progression
(15–18). For instance, AS of Cyclin D1
generates two isoforms, Cyclin D1a and D1b. Cyclin D1b is
upregulated in several types of cancer and is likely responsible
for anchorage-independent growth of tumor cells, which highlights
the role of AS in promoting sustained proliferation signals in
cancer (19–21).
A number of factors involved in AS regulation have
been well characterized, including serine/arginine-rich (SR)
RNA-binding proteins. SR proteins, also called SR splicing factors
(SRSF), contain one or two RNA-recognition motifs functioning
primarily in RNA binding and an SR domain enriched in arginine and
serine involved in protein-protein interactions (22,23).
There are 12 canonical members of the SR protein family that share
this classical domain structure (SRSF1-12). SR proteins have been
extensively characterized for their activities in regulating
constitutive and alternative pre-mRNA splicing (24). Abnormal expression of SR proteins
has been reported in various cancer types, including leukemia
(25,26), as well as breast (27), colon (28), skin (29), colorectal (30) and lung (28) cancer, and SRSF1, SRSF3 and SRSF6 are
always overexpressed in cancers (30–33).
SRSF6 is also known as SRP55 or SFRS6. In
Drosophila, its B52 homologue is able to influence cell
growth and the cell cycle, but not differentiation. Overexpression
of B52 promotes cell growth and upregulates Myc transcription
(34). SRSF6 also acts as a
proto-oncogene, which is frequently overexpressed in human skin
cancer, and its overexpression in transgenic mice produces
hyperplasia of sensitized skin and promotes aberrant AS (29). A study from 2016 demonstrated that
long intergenic non-protein coding RNA 1133 inhibits the
endothelial-mesenchymal transition and metastasis by directly
binding to SRSF6 as a target mimic, and thus, SRSF6 may serve as a
prognostic biomarker and effective therapeutic target for
colorectal cancer (35). Another
study revealed that SRSF6 is frequently upregulated in colorectal
cancer samples and is associated with poor prognosis, and confirmed
its function in promoting proliferation and metastasis (30). This evidence supports the robust
potential of SRSF6 protein to be useful biomarkers for diagnosis
and prognosis or effective therapeutic targets in cancer. However,
a comprehensive investigation of the transcriptional and
post-transcriptional regulation of SRSF6 in cancer remains to be
performed.
The present study extensively evaluated the
expression levels of SRSF6 in 16 cancer types available in The
Cancer Genome Atlas (TCGA) database, demonstrating an increase of
SRSF6 in cancers compared to normal tissues in most types of
cancer. To obtain further insight into how SRSF6 regulates gene
transcription and its involvement in cancer progression, an
SRSF6-overexpressing cell model was constructed. Overexpression of
SRSF6 was revealed to promote apoptosis and inhibit cell
proliferation. Using unbiased transcriptome analysis, the present
results indicated that overexpression of SRSF6 in cancer cells
induced AS of pre-mRNAs of hundreds of genes and also changed
transcript profiles. As an RNA binding protein and splicing factor,
SFRS6 has numerous predicted regulated targets, some of which are
enriched in ‘DNA repair’ and the ‘double-strand break repair via
the homologous recombination pathway’, as indicated by Gene
Ontology (GO) term analysis. Subsequently, 16 cervical tumor
samples, including 8 samples with high SRSF6 expression and 8
samples with low SRSF6 expression, were selected to further study
the potential impact of SRSF6 on AS regulation of the cancer
transcriptome. The SRSF6-regulated AS events (ASEs) that had been
detected in cancer cells were also validated in those cervical
cancer samples. Collectively, these results demonstrated that SRSF6
is able to regulate the transcriptome of genes involved in cancer
progression by mediating gene expression and AS. The present study
enhances the current understanding of the biological functions and
regulatory roles of SRSF6.
Materials and methods
Retrieval and analysis of TCGA
data
Analysis of TCGA data was performed with GEPIA
(36). RNA-seq expression data of
308 cervical tumor samples were downloaded from TCGA to determine
the expression levels of SRSF6. A total of 8 samples with high
SRSF6 expression and 8 samples with low SRSF6 expression were then
selected and their RNA-seq data were downloaded to analyze the
regulation of alternative splicing in cervical cancer.
SRSF6 overexpression
Primer pairs used for Hot Fusion were designed by CE
Design v1.04 (Vazyme Biotech Co., Ltd) with gene-specific sequences
and they included a portion of vector pIRES-hrGFP-1a sequences,
with a 17–30 bp overlap between primer pairs. The primer sequences
were as follows: Forward,
5′-AGCCCGGGCGGATCCGAATTCATGCCGCGCGTCTACATAGG-3′ and reverse,
5′-GTCATCCTTGTAGTCCTCGAGATCTCTGGAACTCGACCTGGACC-3′. The
SRSF6-overexpressing plasmid was constructed and HeLa cells were
transfected with SRSF6-overexpressing plasmid and empty plasmid as
described in a previous study by our group (37).
RT-qPCR
The housekeeping gene GAPDH was utilized as a
control gene to assess whether SRSF6 was overexpressed.
Complementary (c)DNA synthesis was performed by RT using the Kit
One-Step gDNA Removal and cDNA synthesis mix (cat. no. AT311-02;
Transgen Biotech) at 65°C for 5 min, 25°C for 10 min and 42°C for
30 min. qPCR was then performed using the Hieff™ qPCR
SYBR® Green Master Mix (Low Rox Plus; YEASEN) in a
Mycycler (Bio-Rad Laboratories) with the following thermocycling
conditions: 95°C for 5 min, followed by 40 cycles of 95°C for 10
sec and 60°C for 30 sec. Primer sequences are listed in Table SI. The concentration of each
transcript was then normalized to the level of GAPDH mRNA using the
2−ΔΔCq method (38).
Comparisons were performed by a paired Student's t-test using
GraphPad Prism software (v8.0; GraphPad Software, Inc.). RT-qPCR
was also performed in the present study for certain selected RASEs,
normalized to the reference gene GAPDH. The primers for detecting
pre-mRNA splicing are listed in Table
SI. To quantitatively analyze the two different splicing
isoforms of a specific ASE using a qPCR approach, two pairs of
primers were designed to specifically amplify each of these two
isoforms after the initial synthesis of the first-strand cDNA using
random primers (cat. no. AT311-02; Transgen). To achieve this
specificity, a primer complementary to the splice junction of the
constitutive exon and alternative exon was designed. The RNA
samples used for RT-qPCR were the same as those used for RNA-seq.
The RT step was the same as above. The PCR mixture was as described
above. The PCR conditions consisted of denaturation at 95°C for 10
min, followed by 40 cycles of denaturation at 95°C for 15 sec, and
annealing and extension at 60°C for 1 min each. PCR amplifications
were performed in triplicate for control and SRSF6 overexpression
(SRSF6-OE) samples and quantified using 2−ΔΔCq method
(38).
Western blot analysis
Proteins were extracted from samples by lysis with
wash buffer (1X PBS, 0.1% SDS, 0.5% nonidet-P-40 and 0.5% sodium
deoxycholate) and the protein concentration was determined using
the bicinchoninic acid (BCA) method (BCA detection Kit; cat. no.
P0012; Beyotime Institute of Biotechnology). Protein samples (20 µg
per lane) were loaded onto 10 or 12% SDS-PAGE gels depending on the
molecular weight which was determined using protein marker and then
transferred onto 0.45-mm polyvinylidene difluoride (PVDF) membranes
(cat. no. ISQE00010; EMD Millipore). The PVDF membranes were then
blocked with 5% skimmed milk (in a buffer containing 10 mM Tris, pH
8.0, 150 mM NaCl and 0.05% Tween-20) for 1 h at room temperature.
Subsequently, they were incubated with primary antibody at 4°C
overnight and then incubated with horseradish peroxidase-conjugated
secondary antibody for 1 h at room temperature. The membranes were
visualized through chemiluminescence reagent (cat. no. 32106;
Thermo Fisher Scientific, Inc.) and the is ChemiScope imaging
system from Clina. Protein bands were quantified using Image J
software (v 1.8.0; National Institutes of Health). The following
primary antibodies were used: anti-SRSF6 (1:1,000 dilution;
polyclonal antibody; cat. no. A0511; AB Clonal) and anti-actin
(1:1,000 dilution; polyclonal antibody; cat. no. AC001; ABClonal).
The following secondary antibody was used: Horseradish
peroxidase-conjugated goat anti-rabbit IgG (1:1,000 dilution; cat.
no. AS014; ABClonal).
Cell proliferation and apoptosis
assay
Cell proliferation was assessed using an MTT assay.
In total, 5×103 HeLa cells/well were cultured in 96-well
plates. The cells were transfected with the SRSF6-overexpressing
plasmid using Lipofectamine® 2000 according to the
manufacturer's protocol. After incubation at 37°C for 48 h, MTT
solution (0.025 ml at 5 mg/ml) was added to each well. The cells
were incubated for another 4 h and the supernatant was removed from
each well. The colored formazan crystals produced by MTT were
dissolved in DMSO (0.15 ml) and the optical density was measured
using an ultraviolet analyzer (Bio-Rad Laboratories, Inc.) at 490
nm.
For the flow cytometric analysis of cell apoptosis,
the transfected cells were incubated at 37°C for 48 h and the live
cells were then harvested and washed twice with ice-cold PBS.
Viable cells were double-stained with 7-amino actinomycin D and
FITC-conjugated Annexin V (Beijing 4A Biotech Co., Ltd.). The
percentage of apoptotic cells was calculated as the sum of the
right lower and upper quadrants. The number of stained cells was
quantified using a flow cytometer (CytoFLEX; Beckman Coulter,
Inc.). Cell cycle distribution was quantified using multi-cycle
software (FlowJo 10.5.3; FlowJo, LLC).
RNA extraction and high-throughput
sequencing
Total RNA was extracted using TRIzol reagent and was
further purified with two phenol-chloroform treatments. To remove
DNA, the purified RNA was then treated with RNase-free RQ1 DNase
(Promega Corp.) and its quality and quantity were determined by
measuring the absorbance at 260/280 nm (A260/A280) using a
Smartspec Plus (Bio-Rad Laboratories, Inc.). The integrity of RNA
was then verified by 1.5% agarose gel electrophoresis.
A total of 10 µg total RNA from each sample was used
to prepare a directional RNA-seq library. First, the polyadenylated
mRNAs were concentrated with oligo (dT)-conjugated magnetic beads
(Invitrogen; Thermo Fisher Scientific, Inc.). The concentrated
mRNAs were then iron-fragmented at 95°C, end-repaired and ligated
to a 5′ adaptor. RT was performed with RT primer harboring a 3′
adaptor sequence and randomized hexamer. The purified cDNAs were
amplified and stored at −80°C until they were used for sequencing.
Following the manufacturer's instructions, the libraries were
prepared for high-throughput sequencing. The Illumina HiSeq X Ten
system (Illumina, Inc.) was used to collect data from 150-bp
pair-end sequencing (BGI Inc.).
RNA-seq raw data clean and
alignment
Raw sequencing reads containing more than 2-N bases
were first discarded. Subsequently, the raw reads were trimmed of
adaptors and low-quality bases using a FASTX-Toolkit (v.0.0.13;
http://hannonlab.cshl.edu/fastx_toolkit/). In
addition, short reads of less than 16 nt were dropped to retain
clean reads, which were subsequently aligned to the GRch38 genome
by Tophat2 (39) with 4 mismatches.
Uniquely mapped reads were ultimately used to calculate read number
and paired-end fragments per kilobase of exon per million fragments
mapped (FPKM) for each gene.
Differentially expressed gene (DEG)
analysis
The expression levels of genes were evaluated using
FPKM. The software edgeR (40),
which is specifically used to analyze the differential expression
of genes, was applied to evaluate the FPKM value and screen the
RNA-seq data for DEGs. The results were analyzed based on the fold
change (FC≥2 or ≤0.5) and false discovery rate (FDR<0.05) to
determine whether a gene was differentially expressed.
Alternative splicing analysis
The ABLas pipeline as described previously (41) was used to define and quantify the
ASEs and regulated ASEs (RASEs) between the samples. In brief,
detection of seven types of canonical ASEs in each sample was based
on the splice junction reads. These ASEs were exon skipping (ES),
exon included [cassette exon (CE)], alternative 5′ splice site
(A5SS), alternative 3′ splice site (A3SS), mutual exclusive exon
skipping (MXE), MXE combined with an alternative polyadenylation
site and MXE combined with an alternative 5′ promoter.
Subsequently, the significant P-value was calculated using Fisher's
exact test, with the model reads of samples and alternative reads
as input data. The changed ratio of alternatively spliced reads and
constitutively spliced reads between compared samples, which was
defined as the RASE ratio, was calculated. A RASE ratio >0.2 and
P<0.05 were set as the threshold for RASE detection.
Functional enrichment analysis
Using the KOBAS 2.0 server (42), GO analyses and enriched Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathways were identified
to predict the functions of genes and calculate the distribution
frequency in each functional category. The enrichment of each
pathway (corrected P<0.05) was defined using hypergeometric
tests and the Benjamini-Hochberg FDR controlling procedure.
Reactome (http://reactome.org) pathway profiling
was also used for functional enrichment analysis of the sets of
selected genes.
Statistical analysis
One-way analysis of variance was used for comparison
of expression levels of SRSF6 among 16 TCGA cancer types with
GEPIA2, using disease state (Tumor or Normal) as the variable for
calculating differential expression. Student's t-test was used for
all other comparisons between the SRSF6-OE and control groups. For
each assay, the results are presented as the mean ± standard error
of the mean of three experiments. The data were analyzed using R
software (v3.5.3, http://www.r-project.org/). P<0.01 or FDR<0.05
was considered to indicate a statistically significant
difference.
Results
Differential expression of SRSF6 in 16
cancer types from TCGA
Inspired by previous discoveries on the
overexpression of SRSF6 in lung, breast, skin and colon cancer
samples (31), SRSF6 expression
data for 16 cancer types from TCGA which had at least 10 normal
samples were analyzed using GEPIA2 (36), a web-based tool that compares gene
expression between tumor and normal tissues from TCGA. SRSF6
expression in tumors had a higher median relative expression
compared with that in normal tissues in 13 of the 16 cancer types
(Fig. 1A). SRSF6 expression in
tumors was significantly upregulated (FDR<0.05) in four cancer
types and was significantly downregulated only in kidney
chromophobe (KICH). Furthermore, GEPIA was used to comprehensively
analyze the association of SRSF6 expression with survival rates in
various types of cancer. As presented in Fig. 1B, the hazard ratios were
significantly higher in KICH and liver hepatocellular carcinoma.
These results suggested that SRSF6 is a marker for cancers and may
have a role in tumorigenesis.
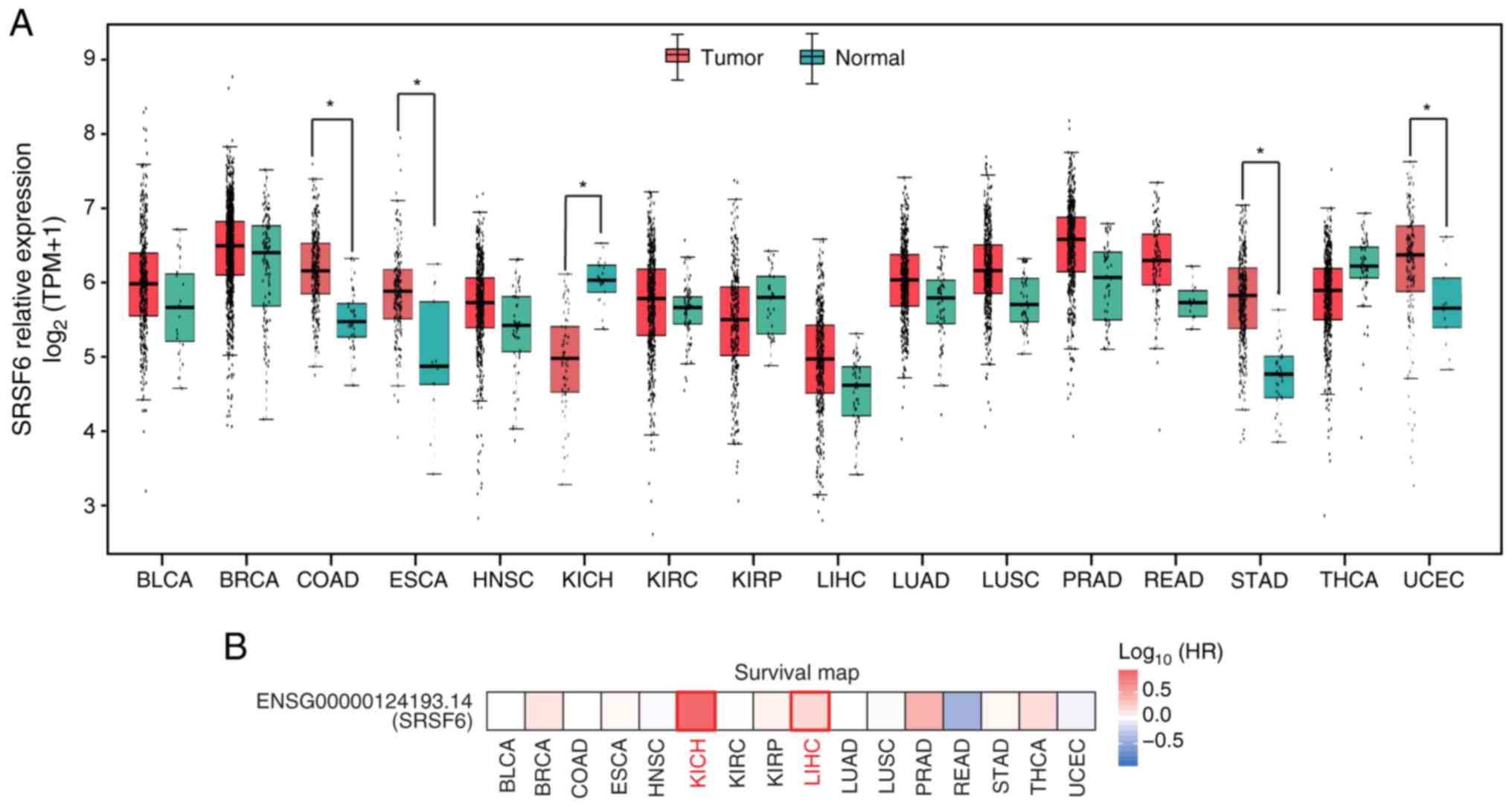 | Figure 1.Differential expression of SRSF6 in
tumor samples from The Cancer Genome Atlas. (A) Relative expression
(TPM) of SRSF6 in tumor samples (red) compared with normal samples
(green) from 16 cancer types. *P<0.05. (B) Association of SRSF6
expression with the survival rates in various types of cancer.
SRSF6, serine and arginine-rich splicing factor 6; HR, hazard
ratio; TPM, transcripts per million; BLCA, bladder urothelial
carcinoma; BRCA, breast invasive carcinoma; COAD, colon
adenocarcinoma; ESCA, esophageal carcinoma; HNSC, head and neck
squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney
renal clear-cell carcinoma; KIRP, kidney renal papillary cell
carcinoma; LIHC, liver hepatocellular carcinoma; LUAD, lung
adenocarcinoma; LUSC, lung squamous-cell carcinoma; PRAD, prostate
adenocarcinoma; READ, rectum adenocarcinoma; STAD, stomach
adenocarcinoma; THCA, thyroid carcinoma; UCEC, uterine corpus
endometrial carcinoma. |
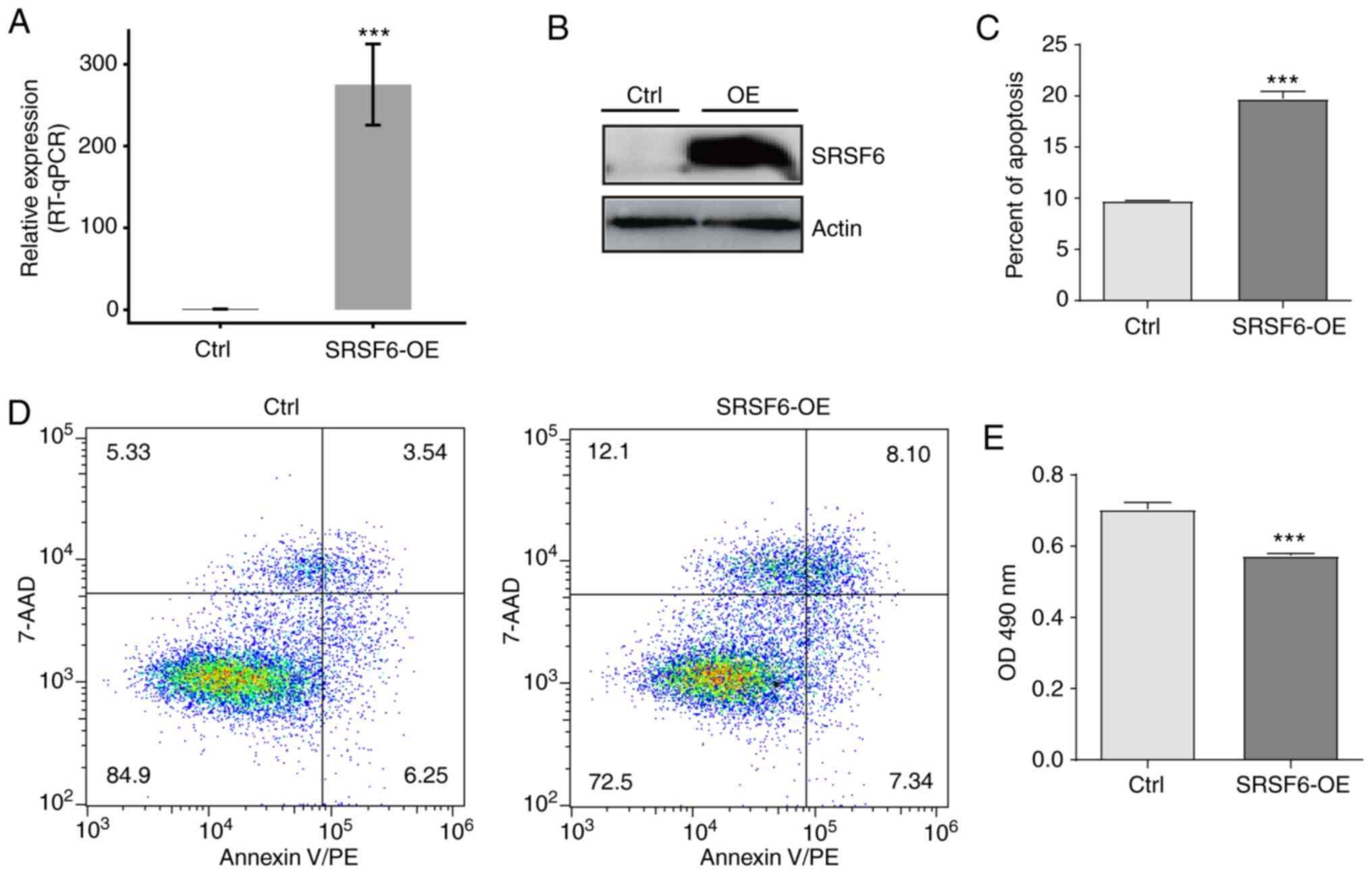
Overexpression of SRSF6 promotes HeLa
cell apoptosis and inhibits cell growth in vitro
To investigate the function of SRSF6 in cancer, an
SRSF6-overexpressing cell model was established by transfecting
HeLa cells with an SRSF6-overexpression plasmid. It was verified
that SRSF6 gene expression was significantly increased by RT-qPCR
(Fig. 2A) and western blot analysis
(Fig. 2B). To characterize the role
of SRSF6 in regulating cell apoptosis and proliferation of HeLa
cells, MTT assays and flow cytometric analyses were respectively
performed. The results suggested that cell apoptosis was
significantly increased in the SRSF6-OE group (P<0.01; Fig. 2C and D); however, cell proliferation
was significantly reduced in the SRSF6-OE group (P<0.01;
Fig. 2E). These results indicated
that SRSF6 may be involved in regulating cell apoptosis and cell
growth in HeLa cells.
SRSF6 overexpression induces
transcriptional differences
To further investigate SRSF6-mediated
transcriptional or post-transcriptional regulation, cDNA libraries
with SRSF6-OE cells and control cells were constructed for RNA-seq
on the Illumina HiSeq Xten platform. A total of two biological
replicates were used and a total of 82.4–85.7 Million (M)
150-nucleotide paired-end raw reads per sample were obtained. After
removing adaptors and low-quality reads, 75.2–82.1 M clean reads
were aligned to the human GRCH38 genome using TopHat2, of which
87.15–91.42% were aligned and 93.19–96.53% were uniquely aligned
(Table SII). The level of gene
expression was calculated in the units of FPKM. A total of 27,378
expressed genes were assessed by RNA-seq. Effective overexpression
of SRSF6 was further confirmed in parallel with the RNA-seq
analysis (Fig. 3A).
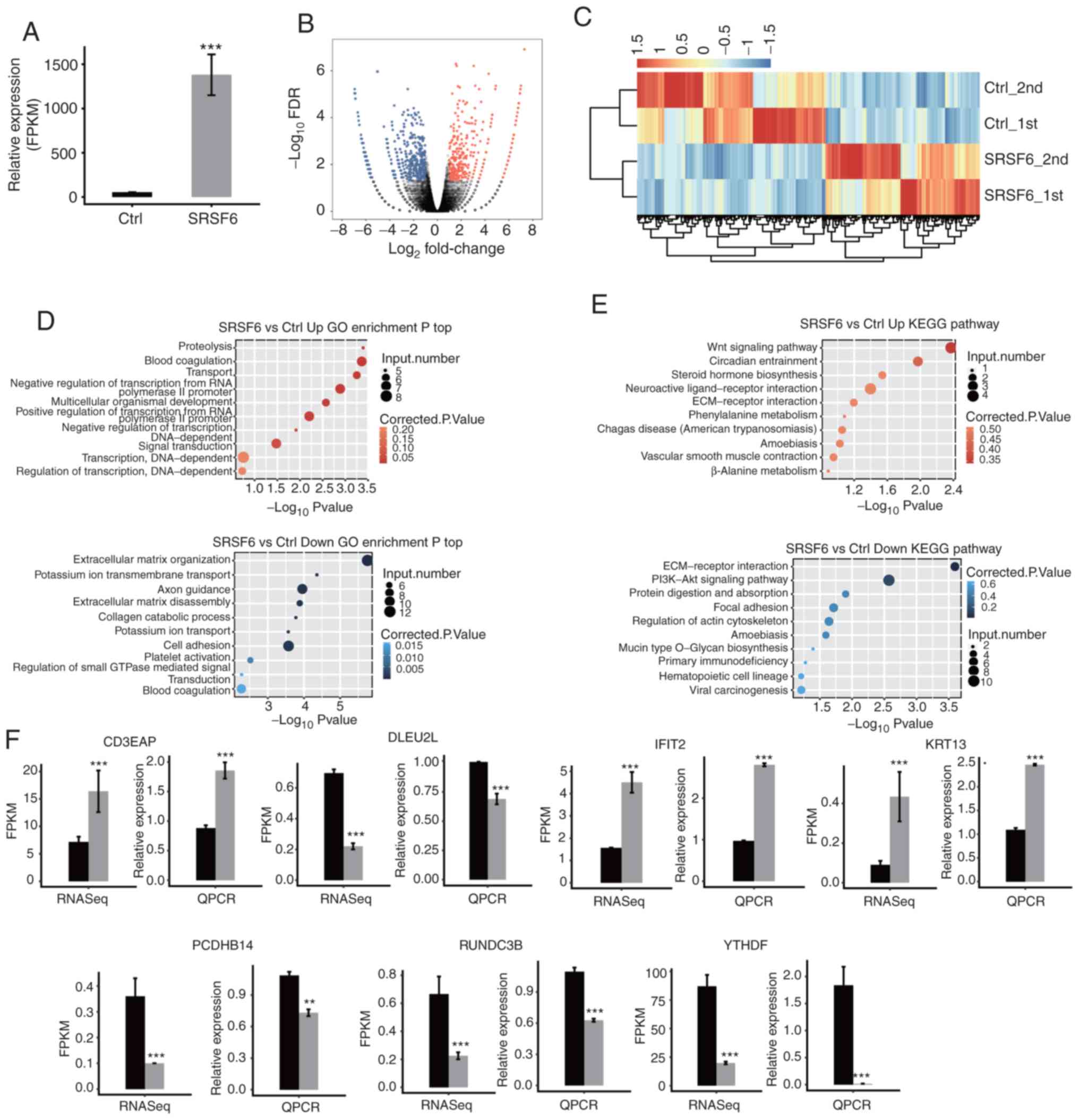 | Figure 3.RNA-seq analysis of gene expression
regulated by SRSF6 overexpression. (A) Following SRSF6
overexpression the mRNA expression level of SRSF6 was measured by
RNA-seq and FPKM values were calculated. (B) Volcano plot of the
genes regulated by SRSF6; upregulated genes (FC≥2; FDR<0.05) are
labeled in red and downregulated genes (FC≤-2; FDR<0.05) are
labeled in blue. (C) Heatmap of 837 DEGs between SRSF6
overexpression and control samples. Expression levels (FPKM) were
log2-transformed and then median-centered for each gene. (D and E)
The top 10 representative (D) GO biological process terms and (E)
KEGG pathways of upregulated and downregulated genes following
SRSF6 overexpression. (F) Reverse transcription-qPCR validation of
DEGs regulated by SRSF6 in cancer cells; black bars are for the
control group and grey bars for SRSF6 overexpression.
***P<0.001. Ctrl_1st and Ctrl_2nd, SRSF6_1st and SRSF6_2nd are
two biological replicates. RNA-seq, RNA sequencing; SRSF6, serine
and arginine-rich splicing factor 6; FPKM, fragments per kilobase
of transcript per million mapped reads; qPCR, quantitative PCR; FC,
fold change; FDR, false discovery rate; Ctrl, control; Up/Down,
up-/downregulated genes; KEGG, Kyoto Encyclopedia of Genes and
Genomes; ECM, extracellular matrix; GO, gene ontology; DEG,
differentially expressed gene; POLR1G, RNA polymerase I subunit G;
DLEU2L, deleted in lymphocytic leukemia 2 like; IFIT2, interferon
induced protein with tetratricopeptide repeats 2; KRT13, keratin
13; PCDHB14, protocadherin beta 14; RUNDC3B, RUN domain containing
3B; YTHDF1, YTH N6-methyladenosine RNA binding protein 1. |
The DEGs between the SRSF6-OE and control cells were
determined using an absolute FC ≥2 and a 5% FDR as criteria with
the edgeR package (42). A total of
422 upregulated and 515 downregulated DEGs were identified
(Table SIII). The DEGs associated
with SRSF6-OE are displayed in a volcano plot (Fig. 3B). The heatmap demonstrated
distinctly different transcription profiles between the SRSF6-OE
and control groups (Fig. 3C).
To further explore the potential biological roles of
these DEGs, GO and KEGG enrichment analyses were performed. The top
10 GO terms in the category biological process in SRSF6-OE cells,
including up- or downregulated of genes, are presented in Fig. 3D (details in Table SIV). The upregulated genes in the
SRSF6-OE cells were mainly enriched in proteolysis, blood
coagulation, transport and negative regulation of transcription
from RNA polymerase II promoter (Fig.
3D, upper panel). The downregulated genes were mostly
associated with extracellular matrix organization, potassium ion
transmembrane transport, axon guidance, extracellular matrix (ECM)
disassembly, cell adhesion and platelet activation (Fig. 3D, lower panel). According to the
KEGG analysis (details in Table
SV), the pathways of the upregulated gene sets were mainly
associated with the Wnt signaling pathway, circadian entrainment
and the ECM-receptor interaction signaling pathway (Fig. 3E, upper panel). Downregulated gene
sets were significantly enriched in ECM-receptor interaction,
PI3K/Akt signaling pathway, protein digestion and absorption, and
focal adhesion (Fig. 3E, lower
panel). These results indicated that SRSF6 is important in
regulating the cell cycle or ECM, which is directly associated with
cellular quiescence, proliferation and cancer.
To confirm the important regulatory function of
SRSF6 at the level of gene expression in the HeLa cell line, DEGs
that were important in tumorigenesis were validated by RT-qPCR
(Table SI). All of the seven
validated DEGs after SRSF6 overexpression, including YTH
N6-methyladenosine RNA binding protein 1, interferon induced
protein with tetratricopeptide repeats 2, RNA polymerase I subunit
G, deleted in lymphocytic leukemia 2 like, protocadherin beta 14,
RUN domain containing 3B and keratin 13, were confirmed (Fig. 3F).
SRSF6 significantly regulates AS of
genes involved in DDR pathways
To investigate the regulatory role of SRSF6 in AS,
the SRSF6-dependent ASEs were analyzed using the transcriptome
sequencing data of HeLa cells. A total of 40.52–41.97% of uniquely
mapped reads were spliced reads (Table
SVI). Furthermore, 240,773 out of 367,321 annotated exons, as
well as 158,256 annotated and 167,859 novel splice junctions were
detected from the RNA-seq data. The ABLas software tool (41,43)
was then used to explore ASEs. A total of 19,404 known ASEs were
detected, which were annotated in the reference genome and 61,995
novel ASEs were detected due to novel splice junctions (Table SVII).
By applying a stringent cut-off of P≤0.05 and an AS
ratio ≥0.2, 661 high-confidence RASEs were identified (Table SVIII). The RASEs included 76 CE, 92
ES, 109 A5SS, 91 A3SS, 13 cases of alternative last exon, 18 cases
of alternative first exon, 12 A3SS&ES, 11 A5SS&ES, 215 IR
and 24 MXE (Fig. 4A). The data
suggested that SRSF6 globally regulated ASEs in HeLa cells.
Excluding the changes in ASEs attributed to transcriptional
regulation, genes whose expression levels and AS were both
regulated by SRSF6 were also examined and eight genes were shared
between the DEGs and RASG: APC regulator of WNT signaling pathway
2, ATPase secretory pathway Ca2+ transporting 2,
TBC/LysM-associated domain containing 2, HOXB cluster antisense RNA
4, RP11-18H7.1, AC005253.2, leucine rich repeat containing 24 and
RP11-654A16.1 (Fig. 4B).
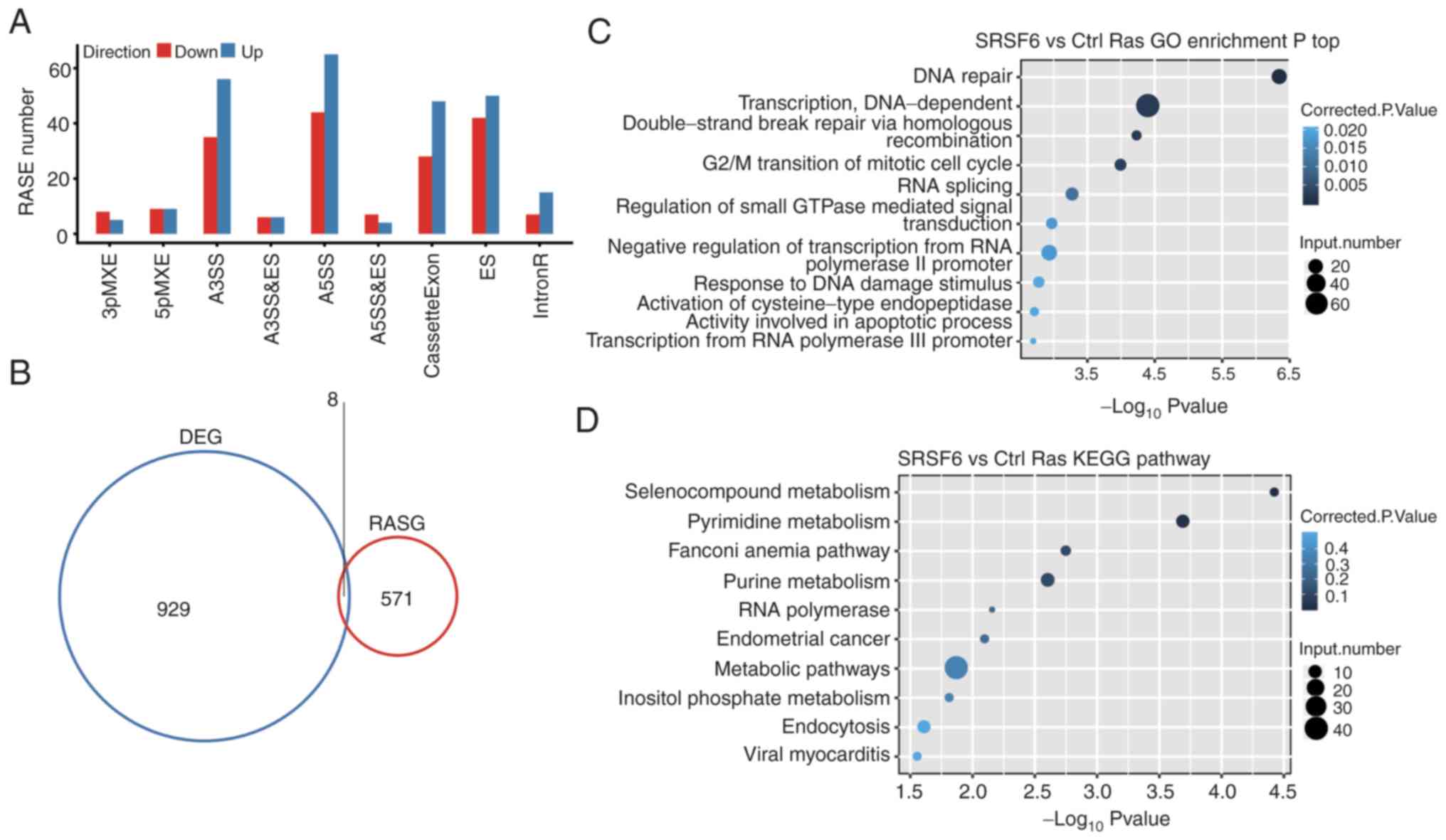 | Figure 4.AS analysis of cancer cells. (A)
Classification of differential AS types regulated by SRSF6
overexpression. (B) Overlap of DEGs and RASGs following SRSF6
overexpression. (C and D) The top 10 representative (C) GO
biological process terms and (D) KEGG pathways of RASGs. SRSF6,
serine and arginine-rich splicing factor 6; AS, alternative
splicing; DEG, differentially expressed gene; RASG, regulated AS
gene; GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and
Genomes; MXE, mutual exclusive exon skipping; 3pMXE, alternative
last exon; 5pMXE, alternative first exon; A3SS, alternative 3′
splice site; A5SS, alternative 5′ splice site; cassetteExon, exon
included; ES, exon skipping; IntronR, intron retention. |
The AS genes that were identified by GO analysis
were highly enriched in ‘DNA repair’, ‘double-strand break repair
via homologous recombination’, ‘DNA-dependent transcription’, ‘G2/M
transition of mitotic cell cycle’ and ‘response to DNA damage
stimulus pathways’ (GO biological process terms; Fig. 4C). The most enriched KEGG pathways
included those involved in ‘Selenocompound metabolism’, ‘Pyrimidine
metabolism’, ‘Fanconi anemia pathway’, ‘Purine metabolism’,
‘Endometrial cancer’, ‘metabolism’ and ‘inflammatory related
pathways’ (Fig. 4D and Table SIX). Collectively, these results
suggested that SRSF6 indeed regulated ASEs that were involved in
pathways associated with the DDR, which is strongly associated with
tumorigenesis (44).
Validation of SRSF6-regulated ASEs by
RT-qPCR
According to the present results, SRSF6 regulated AS
of genes primarily enriched in the DNA repair-related pathway.
Therefore, nine genes involved in this pathway were selected and
RT-qPCR assays were performed to validate the ASEs regulated by
SRSF6. PCR primer pairs (Table SI)
were designed to amplify the two different splicing isoforms (Model
and AS) in these ASEs for nine genes. Among the nine events tested,
the RT-qPCR results for six ASEs were consistent with the RNA-seq
results, including two Intron Retention (IR), one A5SS, 2 ES and 1
CE which were located on poly(ADP-ribose) polymerase family member
(PARP) 3, BRCA2 DNA repair associated (BRCA2), partner and
localizer of BRCA2, checkpoint kinase 1, PARP1 binding protein and
activating transcription factor 2 (Fig.
5). The consistent validation results demonstrated the
confidence in the SRSF6-regulated ASEs identified.
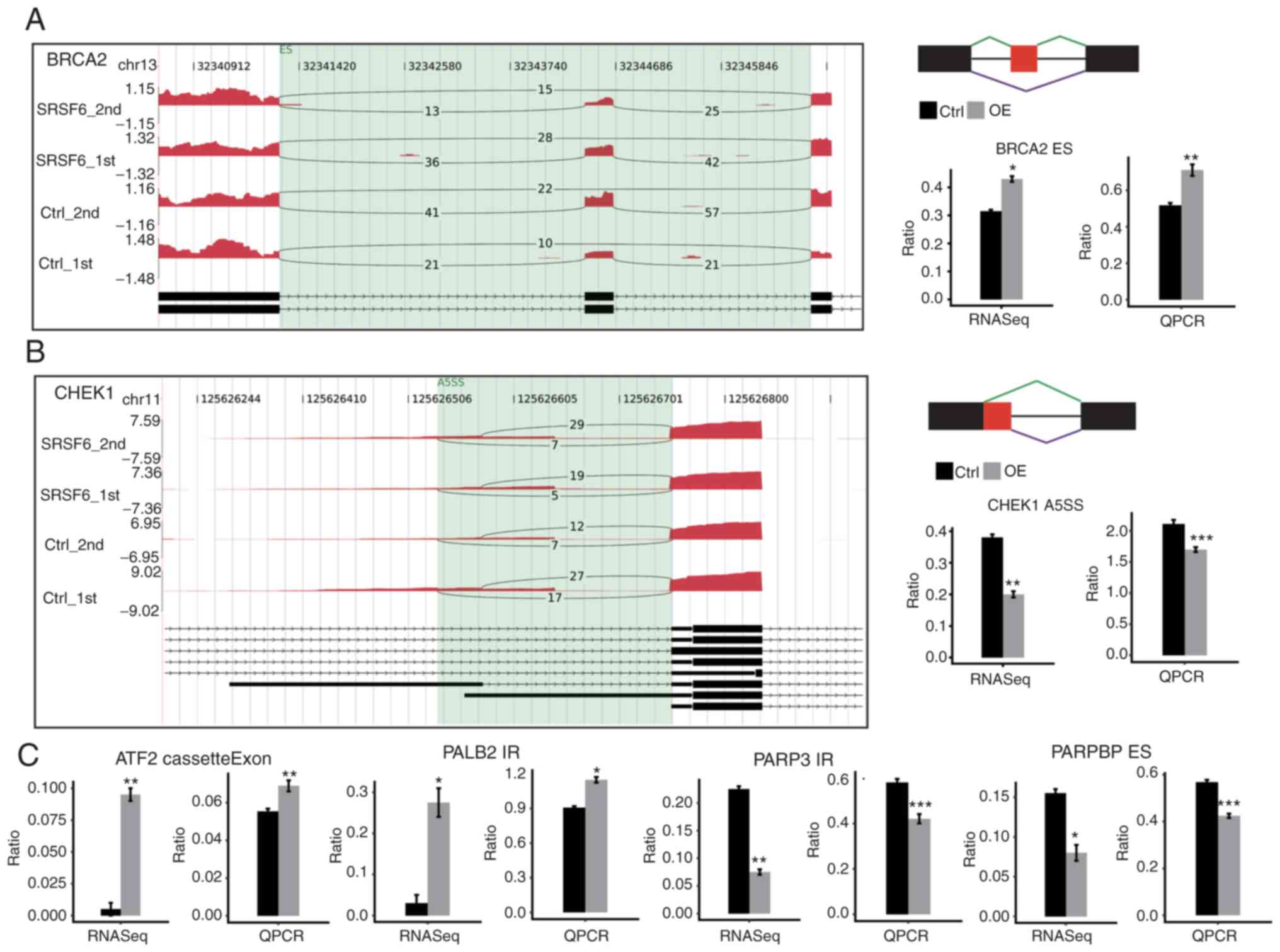 | Figure 5.Validation of ASEs in cancer cells.
(A and B) Genome visualization (left panel) indicates
SRSF6-regulated ASEs in SRSF6-overexpression and control cells. (A)
BRCA2 and (B) CHEK1. The number of junction reads was marked on the
line representing splice junctions composing ASEs. The structures
of the ASEs are depicted in the top-right panel. The altered ratio
of ASEs according to RNA-seq and reverse transcription-qPCR was
calculated and plotted (right panel, bottom). Ctrl_1st and
Ctrl_2nd, SRSF6_1st and SRSF6_2nd are two biological replicates.
(C) Validation results of the other four ASEs as in the right
panels in A and B. Black bars are for the control group and grey
bars for SRSF6 OE. *P<0.05, **P<0.01, ***P<0.001 vs. Ctrl.
SRSF6, serine and arginine-rich splicing factor 6; OE,
overexpression; Ctrl, control; chr, chromosome; qPCR, quantitative
PCR; RNA-seq, RNA sequencing; CHEK1, checkpoint kinase 1; ASE,
alternative splicing event; PARP3, poly(ADP-ribose) polymerase
family member 3; PALB2, partner and localizer of BRCA2; CHEK1,
checkpoint kinase 1; PARPBP, PARP1 binding protein; BRCA2, BRCA2
DNA repair associated; ATF2, activating transcription factor 2; ES,
exon skipping; cassetteExon, exon included; IR, intron retention;
A5SS, alternative 5′ splice site. |
Analysis of the SRSF6-regulated AS
network in cervical cancer samples
Since accumulating evidence has indicated a marked
association between altered DDR and cancer progression and poor
outcome (44,45), it was next queried whether
SRSF6-regulated ASEs identified in HeLa cells were also regulated
by SRSF6 in clinical cancer samples. A total of 16 cervical cancer
samples from TCGA were selected, 8 with high SRSF6 expression and 8
with low SRSF6 expression (Fig.
6A). A total of 170±99.4 M clean reads per sample were
downloaded from TCGA and 115.8±71.4 M reads per sample were
uniquely aligned to the human genome, of which 8.72–15.08% belonged
to junction reads (Table SX). By
using the ABLas software tool, 34,465 known ASEs and 52,623 novel
ASEs were detected (Table SXI).
The same stringent cut-off (P≤0.05 and T-value ≥0) was applied to
identify RASEs with high confidence and a total of 2,225
SRSF6-regulated ASEs were obtained in clinical cancer samples
(Table SXII), including 182 CE/237
ES, 484 A5SS, 394 A3SS and 647 IR (Fig.
6B). These results confirmed that SRSF6 is able to globally
regulate the AS process in cervical cancer.
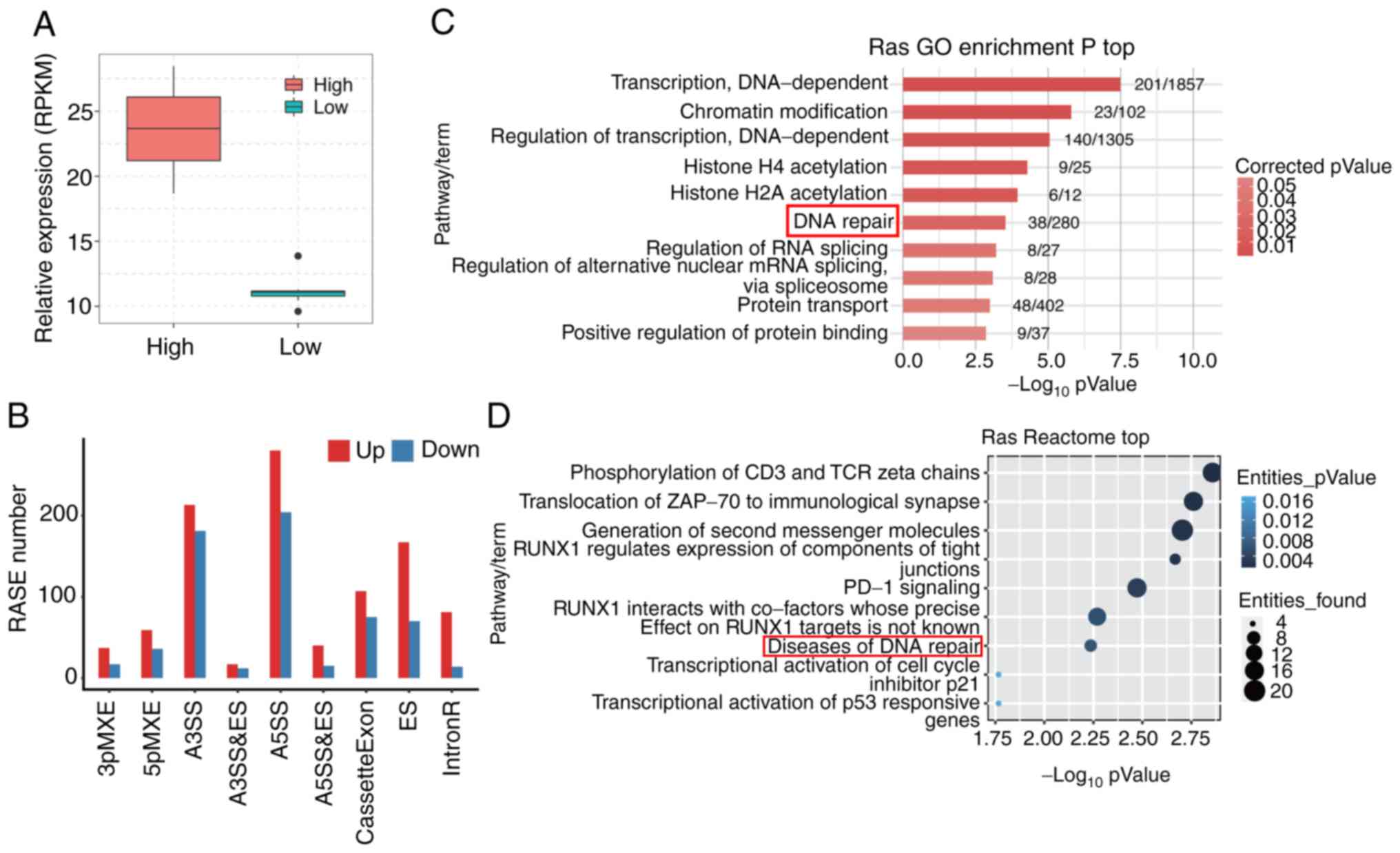 | Figure 6.Analysis of potential SRSF6-regulated
AS in cervical tumor samples. (A) A total of 16 tumor samples were
divided into two groups based on the expression level of SRSF6. Box
plots indicate the expression (RPKM) of SRSF6 in the two groups
with high or low expression of SRSF6. The horizontal lines indicate
the median, the boxes indicate the interquartile range and the
vertical lines indicate the standard deviation. (B) Classification
of regulated AS types in cervical tumors. (C) The top 10
representative GO biological process terms and (D) Reactome
pathways in which RASGs were enriched following SRSF6
overexpression. SRSF6, serine and arginine-rich splicing factor 6;
AS, alternative splicing; RASG, regulated AS gene; GO, gene
ontology; RUNX1, RUNX family transcription factor 1; PD-1,
programmed cell death 1; RPKM, reads per kilobase of transcript per
million reads mapped; 3pMXE, alternative last exon; 5pMXE,
alternative first exon; A3SS, alternative 3′ splice site; A5SS,
alternative 5′ splice site; cassetteExon, exon included; ES, exon
skipping; IntronR, intron retention. |
Further analysis of genes that had been
alternatively spliced revealed that these genes were highly
enriched in DNA-dependent regulation of transcription, chromatin
modification, histone acetylation, DNA repair and RNA splicing
pathways (GO biological process terms; Fig. 6C). Of note, biological pathways
enriched in SRSF6-regulated AS in HeLa cells were similar to those
in cervical cancer samples. These results highlighted again the
robust function of splicing factor SRSF6 in regulating ASEs
involved in different pathways of cancer, particularly the DNA
repair pathway, which was the most representative and meaningful
pathway. Reactome pathway analysis also revealed that
SRSF6-regulated AS genes were enriched in pathways of ‘Diseases of
DNA repair’ (Fig. 6D).
Furthermore, 9 significantly differential ASEs
involved in the genes of DDR-associated terms detected in cancer
cells were then compared with ASEs detected in 16 cervical cancer
samples from TCGA. Of the 9 tested ASEs, five ASEs were consistent.
The five validated splicing events were located in the following
genes: Minichromosome maintenance 8 homologous recombination repair
factor (MCM8), menin 1 (MEN1), mediator of DNA damage checkpoint 1
(MDC1), nei-like DNA glycosylase 1 (NEIL1) and SLX1 homolog A,
structure-specific endonuclease subunit (SLX1A) (Fig. 7). To summarize these results, SRSF6
appears to have a significant role in cancer progression by
regulating AS of important cancer-associated genes and pathways,
particularly DDR pathways.
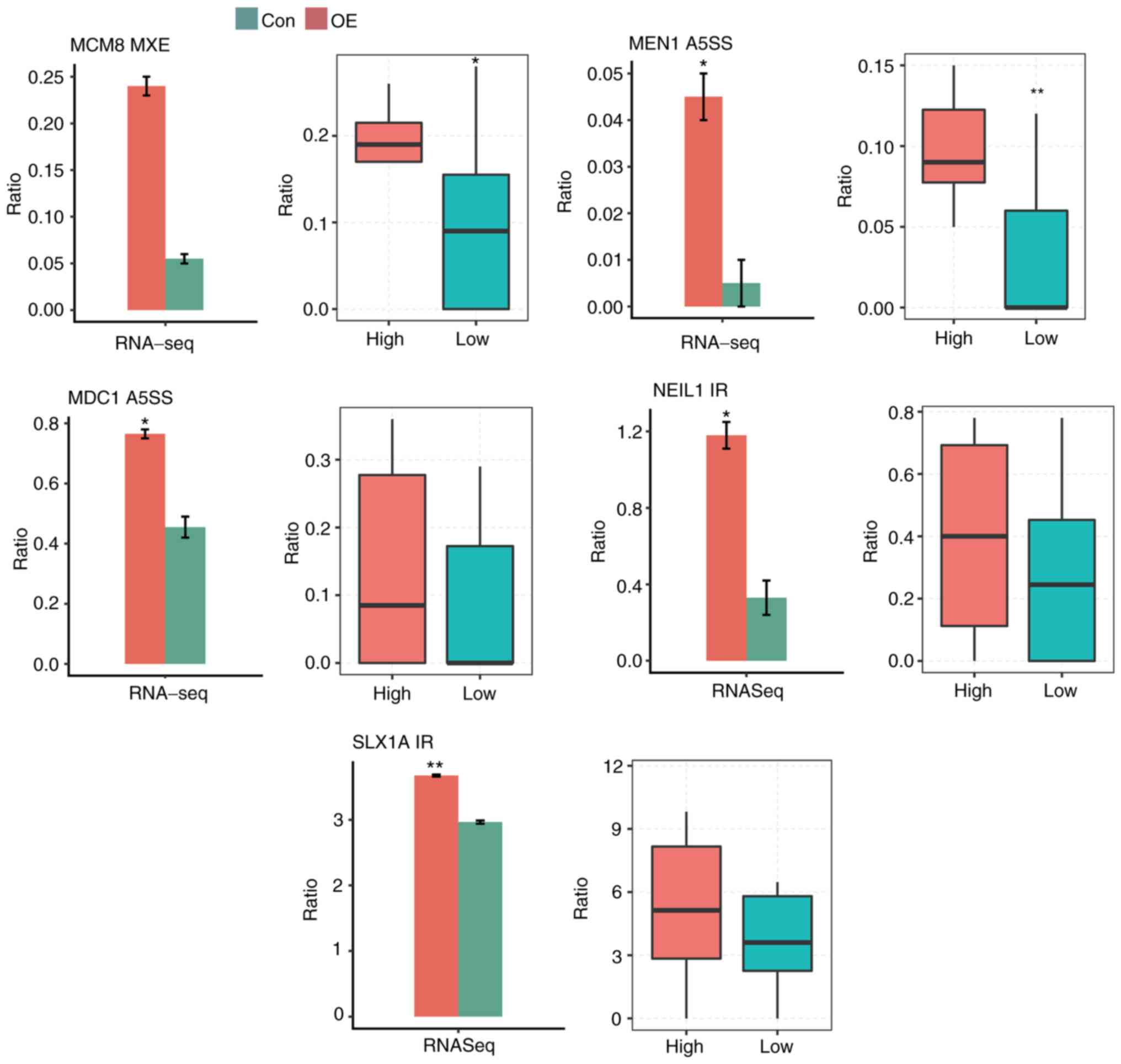 | Figure 7.Comparison of regulated ASEs in
cancer cells (left) and in tumor samples (right). Splicing ratio of
ASEs involved in DNA damage response-associated terms, which were
detected in cancer cell pathways, were compared. *P<0.05,
**P<0.01 vs. control. RNA-seq, RNA sequencing; Ctrl, control;
OE, overexpression; ASE, alternative splicing event; MCM8,
minichromosome maintenance 8 homologous recombination repair
factor; MEN1, menin 1; MDC1, mediator of DNA damage checkpoint 1;
NEIL1, nei-like DNA glycosylase 1; SLX1A, SLX1 homolog A,
structure-specific endonuclease subunit; IR, intron retention;
A5SS, alternative 5′ splice site; MXE, mutual exclusive exon
skipping. |
Discussion
In the present study, the role of SRSF6 in HeLa
cells was investigated and it was explored how SRSF6 regulates AS.
First, using GEPIA, it was revealed that 13 out of 16 cancer types
from TCGA exhibited upregulation of SRSF6, suggesting that SRSF6
may have an important role in tumor development. Based on this
premise, the HeLa cell line was used as a model to analyze the
consequences of overexpression of SRSF6. Overexpression of SRSF6
promoted cell apoptosis and inhibited cell proliferation. At the
cellular level, it was observed that overexpression of SRSF6 had a
broad effect on gene expression and functional clusters of DEGs
highly enriched in cancer-associated terms were obtained.
Furthermore, it was observed that overexpression of SRSF6
significantly regulated AS of genes involved in the DDR pathway.
These results revealed a role for SRSF6 in transcriptional and
post-transcriptional regulation during cancer progression. The data
of clinical samples were then analyzed and 16 cervical cancer
samples from TCGA were selected, including 8 with high SRSF6
expression and 8 displaying low SRSF6 expression, to further study
the potential impact of SRSF6 on AS regulation of the cancer
transcriptome. Significant differential ASEs in these two groups
were involved in different pathways in cervical carcinoma, with the
DNA repair pathway being the most representative.
Previous studies reported that SRSF6 was commonly
upregulated in lung (28), colon
(28), skin (29) and colorectal (30) cancers. SRSF6 overexpression
synergizes with MYC and its upregulation promotes the
transformation of lung epithelial cells and may trigger abnormal
proliferation (28). In the present
study, GEPIA was used to analyze data from TCGA and the results
suggested that 13 of 16 cancer types from TCGA exhibited higher
levels of SRSF6 in most types of tumor compared with matched normal
tissues, while 3 of 16 cancer types (KICH-kidney chromophobe,
KIRC-kidney renal clear-cell carcinoma and THCA-thyroid carcinoma)
had lower expression levels of SRSF6. Of note, in an in
vitro experiment of the present study, overexpression of SRSF6
in HeLa cells was indicated to promote cell apoptosis and inhibit
cell proliferation, which contradicts the results in colorectal and
lung cancer. Although oncogenes are more likely to be overexpressed
and tumor suppressor genes are more frequently disrupted, certain
genes have oncogenic and tumor-suppressor functions in different
tumor types or even within the natural evolution of the disease in
a single tumor (46). These
conflicting roles are a result of the complexity of biological
pathways, the heterogeneity of cancer cells and the higher network
degrees of the gene (46,47). In the present study, it was
speculated that SRSF6 may act as a tumor suppressor in certain
cancer types, including KICH, cervical squamous-cell carcinoma and
endocervical adenocarcinoma.
DDR is an essential function in the maintenance of
genome stability (48). When normal
repair procedures fail, irreversible DNA damage may occur and
uncontrolled cell division may lead to the formation of tumors or
cancers (49,50). Recent research on the genomic
landscape of prostate cancer has revealed that a significant number
of cases harbor DDR genetic aberrations, with BRCA2 as the most
common altered gene (6–8,51).
Molecular analysis of 333 primary prostate tumors identified
deleterious aberrations in DDR genes, including BRCA2, BRCA1,
cyclin-dependent kinase 12, ATM serine/threonine kinase, FA
complementation group D2 and RAD51 paralog C, in 19% of cases
(62/333) (7). In this study, an
in vitro model of SRSF6 overexpression was constructed and
RNA-seq analysis was performed, including DEG and AS analysis. A
certain variation among biological replicates of SRSF6-OE or Ctrl
samples was observed. This was most likely due to a lack of
sufficient repetition. Biological replicates are absolutely
essential for differential expression analysis. However, in
general, biological replicates tend to have more variability than
technical replicates and it is particularly difficult to determine
for cell lines. More biological repetitions provide a better
estimate of biological variation and a more accurate estimate of
average expression levels (52). A
total of 18 significant ASEs involved in DNA repair were filtered
out. Next, RNA-seq data in cancer cells were compared with RNA-seq
data in clinical samples and five DDR-associated genes with a
consistent response were identified: MCM8, MEN1, MDC1, NEIL1 and
SLX1. These five genes are generally linked to tumorigenesis and
progression, including cell proliferation, apoptosis and genome
stability (53–58). Their downstream targets are
regulated by SRSF6 and may influence cancer progression in cells or
in clinical samples.
Dysregulation of the cell cycle machinery causes
dysregulation of cell division, inducing cancer development. To
drive the cell cycle properly, expression levels of cell cycle
regulators are tightly regulated throughout the cell cycle. MCM8
and MCM9 are paralogues of the MCM2-7 eukaryotic DNA replication
helicase complex proteins. It is increasingly recognized that MCM8
and MCM9 are involved in HR repair as a heterohexameric MCM8-9
complex (59–61). Mutations of numerous helicases are
directly implicated in genetic diseases, including cancer and rapid
aging. MCM8/9 were recently added to the catalog of helicases and
mutations in MCM8/9, which correlate principally with primary
ovarian failure/insufficiency and infertility, indicating a meiotic
defect (56). MEN1 encodes menin, a
tumor suppressor associated with a syndrome known as multiple
endocrine neoplasia type I (57).
Menin is a scaffold protein that functions in histone modification
and epigenetic gene regulation. Menin specifically interacts with
FANCD2, a protein encoded by a gene involved in DNA repair that has
a critical role in repair of DNA damage (62). Interaction with NF-κB proteins and
modulation of NF-κB transactivation contribute to the function of
Menin as a tumor suppressor (63).
However, further investigations are required to confirm and
validate the association between SRSF6 and DDR, e.g. whether
SRSF6-OE is able to induce DNA damage by using the comet assay.
In conclusion, the present study highlighted the
functional importance of SRSF6 in mediating cancer progression by
regulating AS. It was demonstrated that both in cancer cells and
clinical tumor samples, SRSF6 regulates AS in genes enriched in
DDR-associated functions and pathways. To elucidate the
contribution of SRSF6 to cervical cancer, a series of analyses were
performed and six novel genes that may be useful for further cancer
research were isolated.
Supplementary Material
Supporting Data
Acknowledgements
The authors thank International Science Editing
(http://www.internationalscienceediting.com) for
editing this manuscript.
Funding
The present study was supported by grants from the
Preferential Projects supported by the Science and Technology
Department of Jilin province (grant no. 20191102012YY).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available under the gene expression omnibus series
accession no. GSE138636.
Authors' contributions
XY and BS initiated the study, conceived the
experiments and managed the project. XY, MW and BS wrote the
manuscript. HJ, SF, PZ, WT, MW and CC performed the experiments and
analyzed the data. All authors read and approved the final version
of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
DePinho RA: The age of cancer. Nature.
408:248–254. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Preston DL, Mattsson A, Holmberg E, Shore
R, Hildreth NG and Boice JD Jr: Radiation effects on breast cancer
risk: A pooled analysis of eight cohorts. Radiat Res. 158:220–235.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barbieri CE, Baca SC, Lawrence MS,
Demichelis JF, Blattner M, Theurillat JP, White TA, Stojanov P, Van
Allen E, Stransky N, et al: Exome sequencing identifies recurrent
SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet.
44:685–689. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kaczkowski B, Tanaka Y, Kawaji H, Sandelin
A, Andersson R, Itoh M, Lassmann T, Hayashizaki Y, Carninci P and
Forrest AR; FANTOM5 Consortium, : Transcriptome analysis of
recurrently deregulated genes across multiple cancers identifies
new pan-cancer biomarkers. Cancer Res. 76:216–226. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Robinson D, Van Allen EM, Wu YM, Schultz
N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC,
Attard G, et al: Integrative clinical genomics of advanced prostate
cancer. Cell. 161:1215–1228. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cancer Genome Atlas Research Network, .
The molecular taxonomy of primary prostate cancer. Cell.
163:1011–1025. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Grasso CS, Wu YM, Robinson DR, Cao X,
Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC,
et al: The mutational landscape of lethal castration-resistant
prostate cancer. Nature. 487:239–243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lord CJ and Ashworth A: The DNA damage
response and cancer therapy. Nature. 481:287–294. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shkreta L and Chabot B: The RNA splicing
response to DNA damage. Biomolecules. 5:2935–2977. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mata J, Marguerat S and Bähler J:
Post-transcriptional control of gene expression: A genome-wide
perspective. Trends Biochem Sci. 30:506–514. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Venables JP: Aberrant and alternative
splicing in cancer. Cancer Res. 64:7647–7654. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang ET, Sandberg R, Luo S, Khrebtukova I,
Zhang L, Mayr C, Kingsmore SF, Schroth GP and Burge CB: Alternative
isoform regulation in human tissue transcriptomes. Nature.
456:470–476. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
David CJ and Manley JL: Alternative
pre-mRNA splicing regulation in cancer: Pathways and programs
unhinged. Genes Dev. 24:2343–2364. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Srebrow A and Kornblihtt AR: The
connection between splicing and cancer. J Cell Sci. 119:2635–2641.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ward AJ and Cooper TA: The pathobiology of
splicing. J Pathol. 220:152–163. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Skotheim RI and Nees M: Alternative
splicing in cancer: Noise, functional, or systematic? Int J Biochem
Cell Biol. 39:1432–1449. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Howe D and Lynas C: The cyclin D1
alternative transcripts [a] and [b] are expressed in normal and
malignant lymphocytes and their relative levels are influenced by
the polymorphism at codon 241. Haematologica. 86:563–569.
2001.PubMed/NCBI
|
|
20
|
Knudsen KE, Diehl JA, Haiman CA and
Knudsen ES: Cyclin D1: Polymorphism, aberrant splicing and cancer
risk. Oncogene. 25:1620–1628. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Comstock CE, Augello MA, Benito RP, Karch
J, Tran TH, Utama FE, Tindall EA, Wang Y, Burd CJ, Groh EM, et al:
Cyclin D1 splice variants: Polymorphism, risk, and isoform-specific
regulation in prostate cancer. Clin Cancer Res. 15:5338–5349. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu JY and Maniatis T: Specific
interactions between proteins implicated in splice site selection
and regulated alternative splicing. Cell. 75:1061–1070. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kohtz JD, Jamison SF, Will CL, Zuo P,
Lührmann R, Garcia-Blanco MA and Manley JL: Protein-protein
interactions and 5′-splice-site recognition in mammalian mRNA
precursors. Nature. 368:119–124. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Long JC and Caceres JF: The SR protein
family of splicing factors: Master regulators of gene expression.
Biochem J. 417:15–27. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu J, Huang B, Xiao Y, Xiong HM, Li J,
Feng DQ, Chen XM, Zhang HB and Wang XZ: Aberrant expression of
splicing factors in newly diagnosed acute myeloid leukemia.
Onkologie. 35:335–340. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zou L, Zhang H, Du C, Liu X, Zhu S, Zhang
W, Li Z, Gao C, Zhao X, Mei M, et al: Correlation of SRSF1 and
PRMT1 expression with clinical status of pediatric acute
lymphoblastic leukemia. J Hematol Oncol. 5:422012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Anczuków O, Akerman M, Cléry A, Wu J, Shen
C, Shirole NH, Raimer A, Sun S, Jensen MA, Hua Y, et al:
SRSF1-Regulated alternative splicing in breast cancer. Mol Cell.
60:105–117. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cohen-Eliav M, Golan-Gerstl R, Siegfried
Z, Andersen CL, Thorsen K, Ørntoft TF, Mu D and Karni R: The
splicing factor SRSF6 is amplified and is an oncoprotein inlung and
colon cancers. J Pathol. 229:630–639. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jensen MA, Wilkinson JE and Krainer AR:
Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nat
Struct Mol Biol. 21:189–197. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wan L, Yu W, Shen E, Sun W, Liu Y, Kong J,
Wu Y, Han F, Zhang L, Yu T, et al: SRSF6-regulated alternative
splicing that promotes tumour progression offers a therapy target
for colorectal cancer. Gut. 68:118–129. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Karni R, de Stanchina E, Lowe SW, Sinha R,
Mu D and Krainer AR: The gene encoding the splicing factor SF2/ASF
is a proto-oncogene. Nat Struct Mol Biol. 14:185–193. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Anczukow O, Rosenberg AZ, Akerman M, Das
S, Zhan L, Karni R, Muthuswamy SK and Krainer AR: The splicing
factor SRSF1 regulates apoptosis and proliferation to promote
mammary epithelial cell transformation. Nat Struct Mol Biol.
19:220–228. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jia R, Li C, McCoy JP, Deng CX and Zheng
ZM: SRp20 is a proto-oncogene critical for cell proliferation and
tumor induction and maintenance. Int J Biol Sci. 6:806–826. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fernando C, Audibert A, Simon F, Tazi J
and Juge F: A role for the serine/arginine-rich (SR) protein
B52/SRSF6 in cell growth and myc expression in drosophila.
Genetics. 199:1201–1513. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kong J, Sun W, Li C, Wan L, Wang S, Wu Y,
Xu E, Zhang H and La M: Long non-coding RNA LINC01133 inhibits
epithelial-mesenchymal transition and metastasis in colorectal
cancer by interacting with SRSF6. Cancer Lett. 380:476–484. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tu Y, Wu X, Yu F, Dang J, Wei Y, Yu H,
Liao W, Zhang Y and Wang J: Tristetraprolin-RNA interaction map
reveals a novel TTP-RelB regulatory network for innate immunity
gene expression. Mol Immunol. 121:59–71. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim D, Pertea G, Trapnell C, Pimentel H,
Kelley R and Salzberg SL: TopHat2: Accurate alignment of
transcriptomes in the presence of insertions, deletions and gene
fusions. Genome Biol. 14:R362013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Robinson MD, McCarthy DJ and Smyth GK:
EdgeR: A bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xia H, Chen D, Wu Q, Wu G, Zhou Y, Zhang Y
and Zhang L: CELF1 preferentially binds to exon-intron boundary and
regulates alternative splicing in heLa cells. Biochim Biophys Acta
Gene Regul Mech. 1860:911–921. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong
S, Kong L, Gao G, Li CY and Wei L: KOBAS 2.0: A web server for
annotation and identification of enriched pathways and diseases.
Nucleic Acids Res. 39:W316–W322. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jin L, Li G, Yu D, Huang W, Cheng C, Liao
S, Wu Q and Zhang Y: Transcriptome analysis reveals the complexity
of alternative splicing regulation in the fungus verticillium
dahliae. BMC Genomics. 18:1302017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nombela P, Lozano R, Aytes A, Mateo J,
Olmos D and Castro E: BRCA2 and other DDR genes in prostate cancer.
Cancers (Basel). 12:3522019. View Article : Google Scholar
|
|
45
|
Schiewer MJ and Knudsen KE: DNA damage
response in prostate cancer. Cold Spring Harb Perspect Med.
2:a0304862019. View Article : Google Scholar
|
|
46
|
Shen L, Shi Q and Wang W: Double agents:
Genes with both oncogenic and tumor-suppressor functions.
Oncogenesis. 13:252018. View Article : Google Scholar
|
|
47
|
Barros-Filho MC, Guisier F, Rock LD,
Becker-Santos DD, Sage AP, Marshall EA and Lam WL: Tumour
suppressor genes with oncogenic roles in lung cancer. IntechOpen;
2019, https://www.intechopen.com/books/genes-and-cancer/tumour-suppressor-genes-with-oncogenic-roles-in-lung-cancerApril
16–2019 View Article : Google Scholar
|
|
48
|
Yoshida K and Miki Y: Role of BRCA1 and
BRCA2 as regulators of DNA repair, transcription, and cell cycle in
response to DNA damage. Cancer Sci. 95:866–871. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Helleday T, Petermann E, Lundin C, Hodgson
B and Sharma RA: DNA repair pathways as targets for cancer therapy.
Nat Rev Cancer. 8:193–204. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jeggo PA, Pearl LH and Carr AM: DNA
repair, genome stability and cancer: A historical perspective. Nat
Rev Cancer. 16:352016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kumar A, Coleman I, Morrissey C, Zhang X,
True LD, Gulati R, Etzioni R, Bolouri H, Montgomery B, White T, et
al: Substantial interindividual and limited intraindividual genomic
diversity among tumors from men with metastatic prostate cancer.
Nat Med. 22:369–378. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Klaus B: Statistical relevance-relevant
statistics, part I. EMBO J. 34:2727–2730. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ji X, Li J, Zhu L, Cai J, Zhang J, Qu Y,
Zhang H, Liu B, Zhao R and Zhu Z: CHD1L promotes tumor progression
and predicts survival in colorectal carcinoma. J Surg Res.
185:84–91. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Nihira NT and Yoshida K: Engagement of
DYRK2 in proper control for cell division. Cell Cycle. 14:802–807.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sailo BL, Banik K, Girisa S, Bordoloi D,
Fan L, Halim CE, Wang H, Kumar AP, Zheng D, Mao X, et al: FBXW7 in
cancer: What has been unraveled thus far? Cancers (Basel).
19:2462019. View Article : Google Scholar
|
|
56
|
Griffin WC and Trakselis MA: The MCM8/9
complex: A recent recruit to the roster of helicases involved in
genome maintenance. DNA Repair (Amst). 76:1–10. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kamilaris CD and Stratakis CA: Multiple
endocrine neoplasia type 1 (MEN1): An update and the significance
of early genetic and clinical diagnosis. Front Endocrinol
(Lausanne). 10:3392019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Peng J, Tang L, Cai M, Chen H, Wong J and
Zhang P: RECQL5 plays an essential role in maintaining genome
stability and viability of triple-negative breast cancer cells.
Cancer Med. 8:4743–4752. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lutzmann M, Grey C, Traver S, Ganier O,
Maya-Mendoza A, Ranisavljevic N, Bernex F, Nishiyama A, Montel N,
Gavois E, et al: MCM8- and MCM9-deficient mice reveal gametogenesis
defects and genome instability due to impaired homologous
recombination. Mol Cell. 47:523–534. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Nishimura K, Ishiai M, Horikawa K,
Fukagawa T, Takata M, Takisawa H and Kanemaki MT: Mcm8 and Mcm9
form a complex that functions in homologous recombination repair
induced by DNA interstrand crosslinks. Mol Cell. 47:511–522. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Park J, Long DT, Lee KY, Abbas T, Shibata
E, Negishi M, Luo Y, Schimenti JC, Gambus A, Walter JC and Dutta A:
The MCM8-MCM9 complex promotes RAD51 recruitment at DNA damage
sites to facilitate homologous recombination. Mol Cell Biol.
33:1632–1644. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jin S, Mao H, Schnepp RW, Sykes SM, Silva
AC, D'Andrea AD and Hua X: Menin associates with FANCD2, a protein
involved in repair of DNA damage. Cancer Res. 63:4204–4210.
2003.PubMed/NCBI
|
|
63
|
Heppner C, Bilimoria KY, Agarwal SK,
Kester M, Whitty LJ, Guru SC, Chandrasekharappa SC, Collins FS,
Spiegel AM, Marx SJ and Burns AL: The tumor suppressor protein
menin interacts with NF-kappaB proteins and inhibits
NF-kappaB-mediated transactivation. Oncogene. 20:4917–4925. 2001.
View Article : Google Scholar : PubMed/NCBI
|