Introduction
Although tyrosine kinase inhibitors (TKIs) have
revolutionized the treatment of chronic myeloid leukemia (CML),
some CML patients experience TKI resistance (1,2), TKI
intolerance (3) or economic plight
(4). Thus, there is still a need to
identify new approaches for CML treatment.
Despite the vast improvements in therapeutic
strategies, DNA damage-based chemotherapy agents currently remain
the preferred choice to treat most leukemias. DNA damage can induce
cell cycle arrest and activate repair pathways, which are the main
mechanisms of resistance to these preferred chemotherapy drugs.
Therefore, combining DNA damage response (DDR) pathway inhibitors
with DNA damage-based chemotherapeutics is a potential new strategy
for treating leukemia, including CML.
In general, DNA damage induces cell cycle arrest by
activating cell cycle checkpoints, which gives cells enough time to
repair the damage. Cancer cells mostly rely on the G2/M
checkpoint to repair damaged DNA due to inactivating mutations or
loss of p53 (5,6). Thus, we were inspired to target
important components of the G2/M checkpoint to treat
cancer, especially p53-mutant cancer. Two major signaling pathways
are involved in the DDR: ATM/checkpoint kinase 2 (CHK2) and
ATR/CHK1 (7–9). Research has shown that in malignant
cells, the components involved in the ATR/CHK1 pathway are often
upregulated, while ATM/CHK2 components are often deficient
(10). Hence, the ATR/CHK1
signaling pathway, which is also involved in the G2/M
checkpoint, is a potential target for the treatment of cancer.
CHK1 is a Ser/Thr protein kinase that was first
discovered in fission yeast in 1993 (11). In many different cancers, resistance
to chemotherapy and radiotherapy is associated with CHK1
overexpression. CHK1 is phosphorylated by ATR in response to DNA
damage and subsequently phosphorylates its downstream substrates to
induce cell cycle arrest (12–14).
Subsequent research confirmed that both the G2/M
checkpoint and CHK1 play critical roles in the S phase and mitotic
checkpoints (15,16). The CHK1 pathway can directly
facilitate homologous recombination (HR) by regulating the
expression, activity or localization of related proteins (17,18).
Therefore, CHK1 is an attractive candidate for anticancer
therapy.
To date, several CHK1 inhibitors have been developed
and administered either alone or in combination with other
chemotherapeutic agents in preclinical or clinical trials (19–25).
Among them, CCT245737
((R)-5-((4-((morpholin-2-ylmethyl)amino)-5-(trifluoromethyl)pyridin-2-yl)amino)pyrazine-2-carbonitrile)
is a selective CHK1 inhibitor that has oral bioavailability
(26,27). Previous research has shown that
CCT245737 can enhance the anticancer effects of gemcitabine and
carboplatin in non-small cell lung cancer (NSCLC) and B-cell
lymphoma mouse models (26).
However, no study has tested the potential of this inhibitor to
increase the efficacy of chemotherapy or elucidated the mechanism
by which this inhibitor enhances chemotherapeutic effects in
CML.
In the present study, CHK1 was inhibited by RNA
interference (RNAi) or CCT245737 in K562 cells to determine its
role in the chemosensitivity of CML cells. We confirmed that CHK1
inhibition significantly and specifically enhanced the
chemosensitivity of K562 cells to etoposide (VP16) by eliminating
VP16-induced G2/M arrest and suppressing HR efficiency.
Moreover, we also preliminarily investigated the therapeutic effect
of CCT245737 in imatinib-treated K562 cells and showed that this
approach could be applicable to TKI treatment of CML in the
future.
Materials and methods
Cell lines
K562 (human CML) and 293T/17 cells were purchased
from the Cell Bank, Chinese Academy of Sciences (Shanghai, China);
the KCL22 (human CML) cell line was obtained from the German
Collection of Cell Cultures (Braunschweig, Germany); and the CAM191
(human normal lymphocyte) cells were purchased from the Cell Bank,
Chinese Academy of Sciences (Kunming, China). K562 and KCL22 cells
were grown in IMDM (Gibco; Thermo Fisher Scientific, Inc.), CAM191
cells were grown in RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc.), and 293T/17 cells were grown in DMEM (Gibco;
Thermo Fisher Scientific, Inc.). All media were supplemented with
100 U/ml penicillin, 0.1 mg/ml streptomycin and 10% fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc.). The cells were
incubated at 37°C in a humidified atmosphere containing 5%
CO2 and were used for experiments in the logarithmic
growth phase.
Reagents
VP16, imatinib and the CHK1 inhibitor CCT245737 were
obtained from Selleckchem (USA) and dissolved in DMSO
(Sigma-Aldrich; Merck KGaA) for the in vitro studies. In the
shRNA-mediated CHK1 knockdown study, cells were seeded at
105/ml and then treated with 5 µM VP16 for 4.5 h to
induce DNA double-strand breaks (DSBs). After incubation, the cells
were washed with PBS and maintained in IMDM for 48 h to allow the
repair of VP16-induced DSBs before further analysis (28,29).
In the imatinib experiments, K562 cells were seeded at
105/ml and then treated with 1 µM imatinib for 24 h.
After incubation, live cells were acquired by washing with PBS and
incubated for another 24 h, and then, the cells were collected for
subsequent experiments. In most CCT245737-induced CHK1 inhibition
studies, in order to induce sustained DNA damage in cells and
detect the role of CCT245737 on CHK1 activation more efficiently
and to promote this pharmacological combination approach in the
clinic more conveniently in the future, cells were treated with
VP16 (5 µM) and CCT245737 (500 nM) either alone or in combination
at the density of 105/ml for 24 h as in previous
research (22). Before further
analysis, cells were washed with PBS. Primary antibodies against
pSer296 CHK1 (cat. no. 2349), pSer317 CHK1 (cat. no. 2344), pSer345
CHK1 (cat. no. 2341), total CHK1 (cat. no. 2360), total H2AX (cat.
no. 7631), pSer139 H2AX (cat. no. 9718), cleaved-PARP (cat. no.
5625), total H3 (cat. no. 4499), pSer10 H3 (cat. no. 53348),
pSer216 CDC25c (cat. no. 4901), total CDK1 (cat. no. 9116),
pTyr15CDK1 (cat. no. 4539), Rad51 (cat. no. 8875), Rad50 (cat. no.
3427), BRCA1 (cat. no. 9010) and GAPDH (cat. no. 2118) were
purchased from Cell Signaling Technology, and antibodies against
total CDC25c (product code 32444), MRE11 (product code 208020),
Ku70 (product code 92450), Ku80 (product code 80592) and Lig4
(product code 193353) were purchased from Abcam.
RNAi-mediated gene knockdown
The shRNA sequence GCAACAGTATTTCGGTATAAT was used to
knock down CHK1 (shCHK1-KD), whereas the sequence
GCGCGCTTTGTAGGATTCG, which is unrelated to any sequence in humans,
served as a negative control (shRNA-NC). The pLVX-shRNA vector
system containing either a puromycin resistance cassette or ZsGreen
(Clontech Laboratories) was used to carry these sequences. In
addition to shRNA-NC, the empty pLVX-shRNA vector was also used as
a control (shRNA-Vector). To produce lentiviral particles, all of
these plasmids (with two packaging plasmids at a ratio of 4:3:2)
were cotransfected into 293T/17 cells via the calcium phosphate
precipitation method. Viral infection was performed with 10 µg/ml
polybrene, and GFP expression in cells was observed by a
fluorescence microscope after 48–72 h. After positive cells were
sorted, qPCR and western blotting were performed to detect the
knockdown efficiency. Cells with significant target gene knockdown
were used for subsequent experiments.
RNA extraction and qPCR
Total RNA was extracted with a MiniBEST Universal
RNA Extraction Kit (Takara), and cDNA was produced by the
PrimeScript RT Master Mix (Takara). The TB Green Premix Ex Taq™ II
(Tli RNaseH Plus) Kit (Takara) was used to detect relative mRNA
expression by LightCycler 96 system (Roche) with 40 cycles of PCR
thermocycling. The primer sequences were as follows: β-actin
forward, 5′-GGATTCCTATGTGGGCGACGA−3′ and reverse,
5′-GCGTACAGGGATAGCACAGC-3′; CHK1 forward,
5′-CCAGATGCTCAGAGATTCTTCCA-3′ and reverse,
5′-TGTTCAACAAACGCTCACGATTA-3′. The mRNA expression of the target
gene relative to β-actin expression was calculated by the
2−ΔΔCq method (30).
Protein extraction and western
blotting
Cells were harvested and lysed in RIPA lysis buffer
(Thermo Fisher Scientific, Inc.) containing protease and
phosphatase inhibitors (Epizyme Scientific, China). Lysates were
centrifuged to obtain protein extracts for western blotting. The
BCA method (Beyotime Institute of Biotechnology) was used to detect
the protein concentration. SDS-PAGE gels (10% gel) (Epizyme, China)
were used for protein electrophoresis (20–40 µg per sample), and
the separated proteins were transferred to PVDF membranes
(Millipore). Next, the membranes were blocked in 5% nonfat milk for
2 h at room temperature and washed 3 times with TBS-T. Then, the
membranes were incubated with specific primary antibodies at the
dilution of 1:1,000, at 4°C overnight. On the next day, the
membranes were washed and then incubated for 2 h at room
temperature with HRP-conjugated anti-rabbit or anti-mouse secondary
antibodies at the dilution of 1:1,000 (cat. nos. 7074 and 7076;
both from Cell Signaling Technology). Protein bands were detected
after incubation with ECL solution (Thermo Fisher Scientific, Inc.)
for 2 min in the dark. GAPDH served as a loading control and the
densitometry of band intensities were detected by Amersham Imager
600 (GE Healthcare).
Cell cycle distribution analysis
After the cells were washed twice and collected in
100 µl prechilled PBS, they were fixed with 95% ethanol overnight
at 4°C. The next day, the cells were collected and washed once with
prechilled PBS before they were treated with FxCycle PI/RNase
Staining Solution (Invitrogen; Thermo Fisher Scientific, Inc.). The
cell cycle distribution was determined by a Flow cytometer (BD
Biosciences) and analyzed by ModFit LT 3.2 (Verity Software
House).
Alkaline comet assay
The cells were washed twice with prechilled PBS and
diluted to 1.5×105/ml. The comet assay was performed
using CometAssay HT kits (Trevigen). Aliquots of 50 µl of cells
(7,500 cells total) were mixed with 500 µl of LM-Agarose and
rapidly pipetted over the entire sample area of comet slides, which
were then placed at 4°C for 30–45 min in the dark. After the
agarose solidified, the slides were immersed in ice-cold lysis
solution for 2 h at 4°C in the dark. Excess buffer was drained
after lysis, and the slides were immersed in freshly prepared
alkaline unwinding solution for 30 min at room temperature in the
dark. The slides were placed in an electrophoresis slide tray,
covered with slide tray overlay and electrophoresed at 21 V for 25
min. After electrophoresis, the slides were washed twice with
deionized water, immersed in 70% ethanol for 10 min in the dark,
and placed in a drying oven for 30 min to remove excess buffer.
SYBR Gold was pipetted onto each sample, and the slides were
incubated for 30 min at room temperature in the dark. Images were
collected by an Olympus microscope and charge-coupled device (CCD)
camera at a final magnification of ×200. The tail moment was
analyzed using the Comet Assay Software Project 4 (CASP 4;
Perspective Instruments, Ltd.) as an indicator of DNA damage.
Colony formation assay
K562 cells were seeded evenly at 200 cells/well in
24-well plates. A two-layer soft agarose assay was performed with
0.6% agarose as the bottom layer and 0.3% agarose as the top layer.
After the cells were seeded, 100 µl of culture medium was added to
each well every 2 days. After 10 days, colony number and size were
analyzed, and representative images were captured by microscope
(Nikon) and charge-coupled device (CCD) camera.
CCK-8 assay
Cells were seeded in 96-well plates and treated as
indicated. Then, 10 µl of CCK-8 reagent (Dojindo) was added to each
well, and the plates were incubated for 2 h at 37°C in the dark.
The absorbance at 450 nm was detected by a SpectraMax microplate
reader (Molecular Devices). The combination index (CI) between
CCT245737 and VP16 was calculated through Chou-Talalay method by
CompuSyn software (CompuSyn Inc., USA).
Statistical analysis
All statistical analyses were performed using
GraphPad Prism 6.0 (GraphPad Software, Inc.). Data are presented as
the mean ± standard deviation. All quantitative experiments were
conducted with a minimum of three independent experiments. An
unpaired two-tailed t test or one-way ANOVA followed by Turkey,
Dunnett-T3 or Dunnett post-doc test was used as appropriate to
determine statistical significance (*P<0.05, **P<0.01, and
***P<0.001). Statistical analyses were performed with IBM SPSS
Statistics 19.0 (SPSS Inc., USA) and GraphPad Prism 7 (GraphPad
Software, USA).
Results
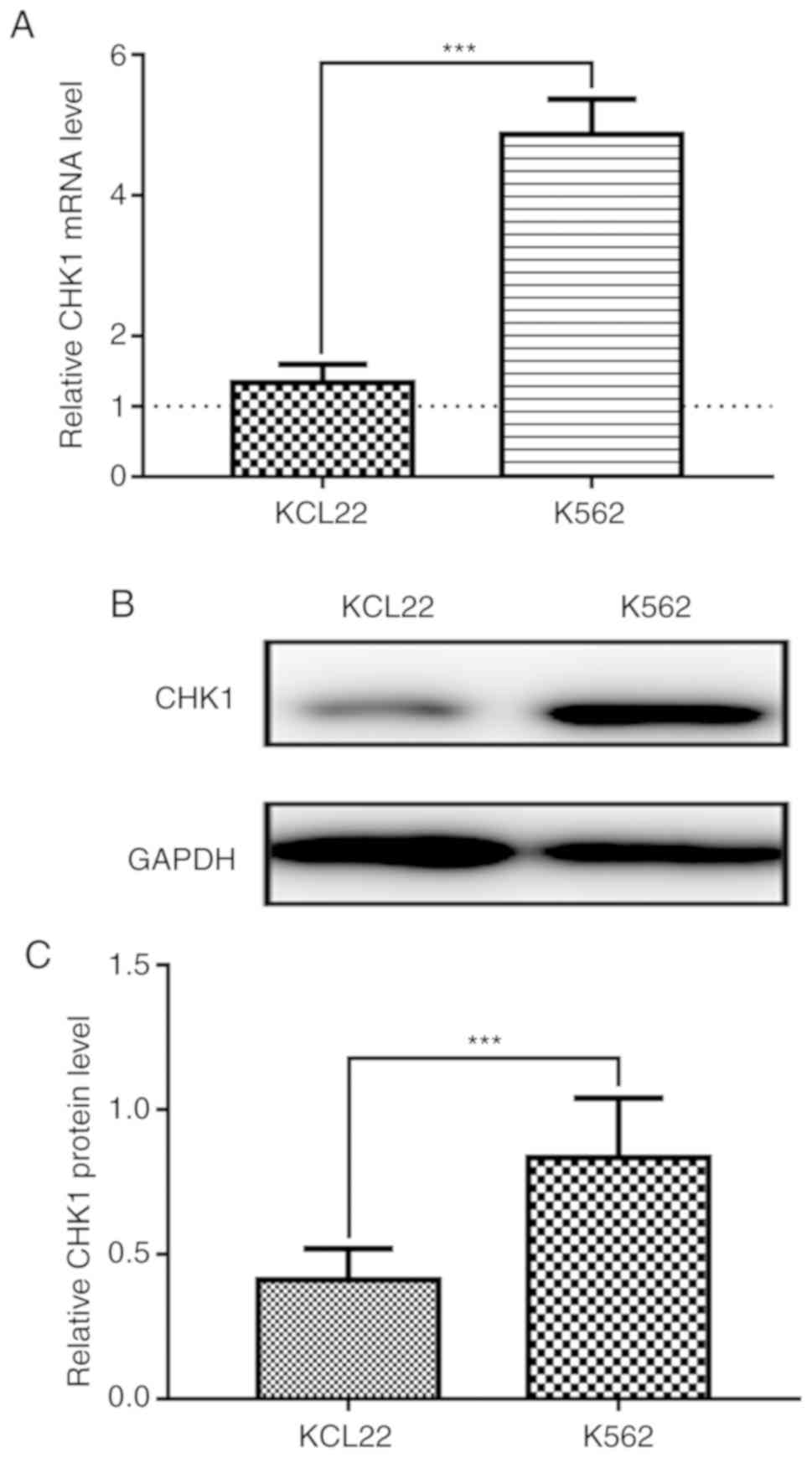
CHK1 displays higher expression in
K562 cells
The KCL22 and K562 cell lines are the two most
common CML cell lines used in experiments. To determine CHK1 mRNA
levels in these two cell lines, we performed qPCR with CAM-191
cells as a control as they have the lowest CHK1 mRNA expression of
the three cell lines tested (the dotted line). As shown in Fig. 1A, CHK1 mRNA expression was
significantly higher in K562 cells than in KCL22 cells.
Consistently, western blot analysis demonstrated that CHK1 protein
expression was also significantly higher in K562 cells than in
KCL22 cells (Fig. 1B and C). Thus,
K562 cells were selected for the subsequent experiments.
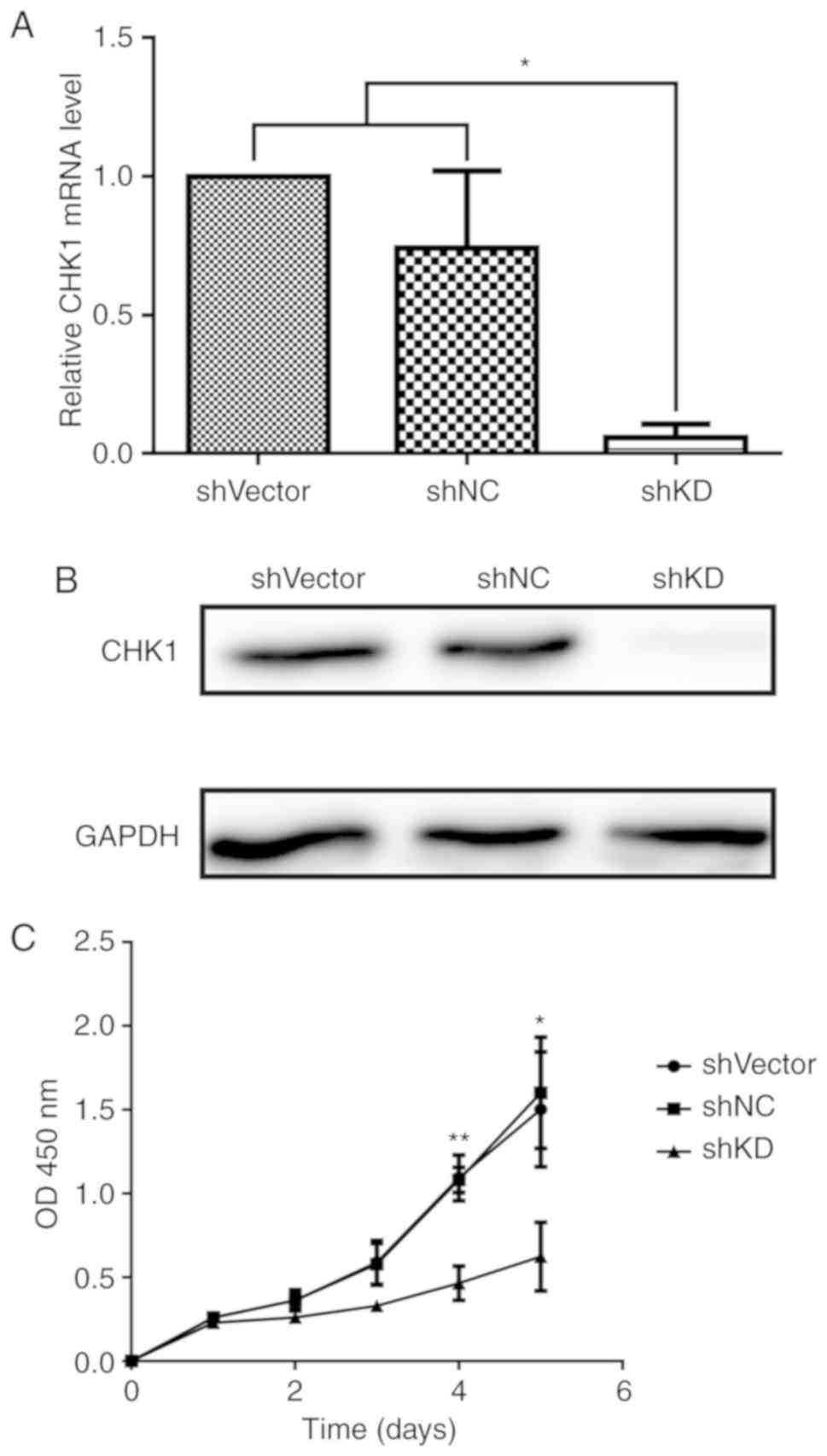
shRNA-mediated CHK1 knockdown
efficiently suppresses cell proliferation
To determine the role of CHK1 in K562 cell
proliferation, we transduced K562 cells with lentiviral vectors
containing shRNA specifically targeting CHK1 (shCHK1-KD), a
negative control sequence (shRNA-NC) or an empty pLVX-shRNA vector
(shRNA-Vector). shCHK1-KD significantly reduced CHK1 expression at
both the mRNA and protein levels (Fig.
2A and B). Compared with shRNA-NC and shRNA-Vector, shCHK1-KD
significantly suppressed K562 cell proliferation (Fig. 2C). Thus, CHK1 is a potential target
for the treatment of CML.
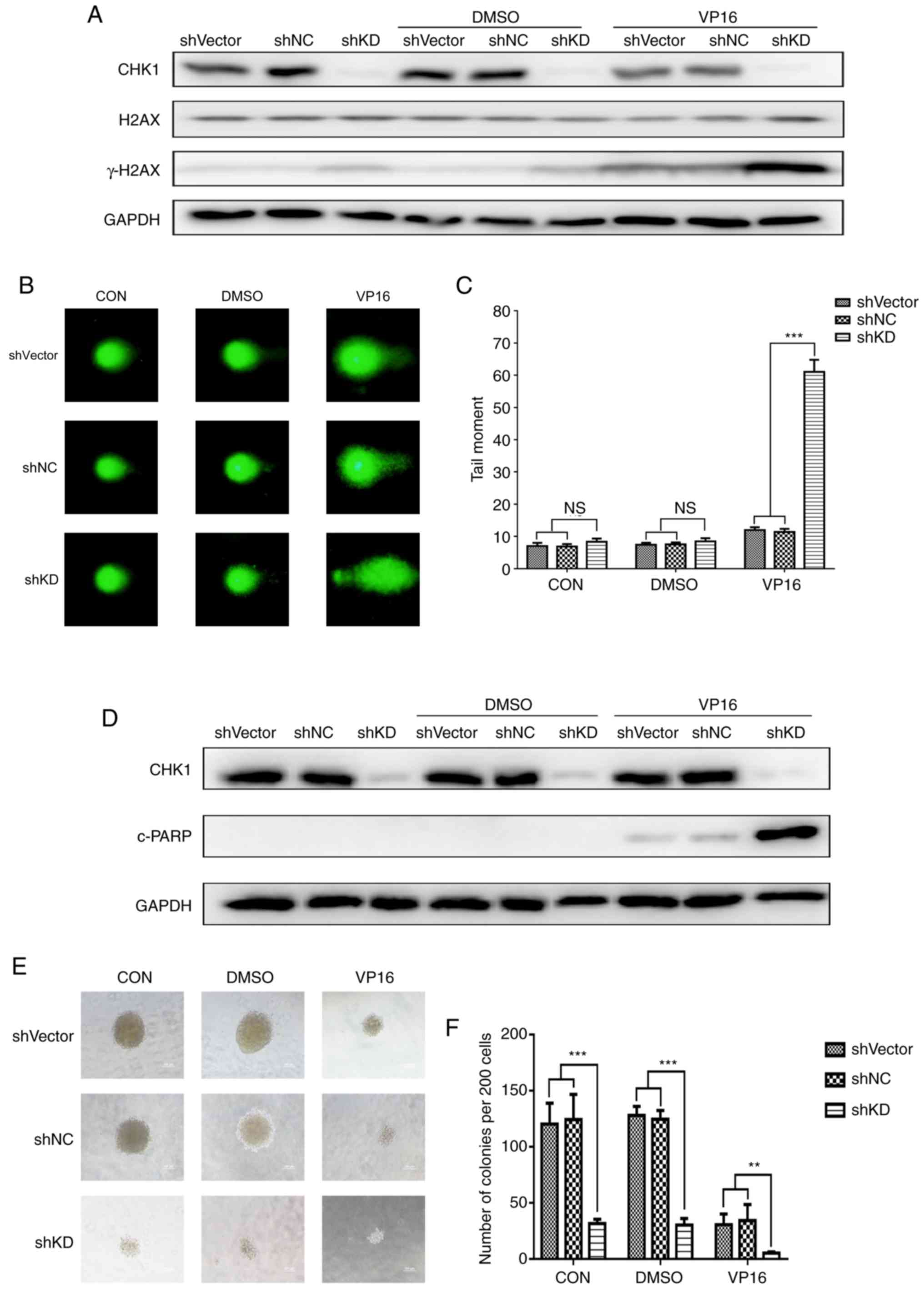
CHK1 knockdown enhances the
cytotoxicity of VP16 in K562 cells
To investigate whether silencing CHK1 increases the
cytotoxicity of VP16 in K562 cells, K562 cells were transduced with
shCHK1-KD, shRNA-NC or shRNA-Vector and then treated with 5 µM
VP16; we then analyzed DSBs, apoptosis and colony formation. First,
we investigated the level of phosphorylated H2AX (γH2AX), a protein
marker of DSBs. The results demonstrated that CHK1 knockdown
increased γH2AX levels compared to the two controls, especially
when combined with VP16 treatment (Fig.
3A). Subsequently, we performed comet assays and analyzed
different parameters using CASP. The Olive Tail Moment (OTM) was
used to describe the level of DNA damage in cells. Similar to the
change in γH2AX accumulation, the comet assay showed that CHK1
knockdown slightly, but not obviously, increased OTM values under
both normal growth and DMSO treatment conditions. However, after
VP16 treatment, the CHK1 knockdown group exhibited a significant
increase in DNA damage (Fig. 3B and
C). These results revealed that CHK1 knockdown can increase
VP16-induced DNA damage in K562 cells. Next, as shown in Fig. 3D, CHK1 knockdown did not
significantly increase the expression of cleaved poly(ADP-ribose)
polymerase (c-PARP), an apoptosis marker, under either normal
growth or DMSO treatment conditions. However, after incubation with
VP16, cells with CHK1 knockdown showed distinctly increased levels
of c-PARP, which indicates elevated levels of apoptosis.
Furthermore, we performed colony formation assays to determine the
role of CHK1 in cell proliferation. Silencing of CHK1 was
associated with a significant decrease in colony formation ability
under all conditions, regardless of the presence of VP16 (Fig. 3E and F). These results suggest that
downregulation of CHK1 enhances the cytotoxic effect of VP16 on
K562 cells.
shRNA-mediated CHK1 knockdown reduces
G2/M arrest and HR repair in K562 cells
Accurate and timely DNA damage repair is vital for
cells to survive chemotherapy-induced DNA damage. As we showed that
CHK1 knockdown sensitized K562 cells to VP16, we aimed to ascertain
whether CHK1 knockdown impairs the DNA damage repair pathway in
K562 cells. First, we analyzed the cell cycle distribution in the
shRNA-NC and shCHK1-KD groups and found that CHK1 knockdown
eliminated G2/M arrest under both conditions, especially
following VP16 treatment (Fig. 4A and
B). Consistent with the cell cycle distribution results, VP16
treatment increased the levels of the G2/M arrest
markers pS216 CDC25c and pY15 CDK1, and shRNA-mediated CHK1
knockdown suppressed the induction of pS216 CDC25c and pY15 CDK1 by
VP16 (Fig. 4C). Moreover, CHK1
knockdown reversed the VP16-induced decrease in the levels of the
mitotic marker pS10 H3 (Fig. 4D).
These results indicate that shRNA-mediated CHK1 knockdown mitigates
chemotherapy-induced G2/M arrest and forces cells to
enter mitosis with unrepaired DNA damage. HR and non-homologous end
joining (NHEJ) are two major pathways for repairing damaged DNA.
Although a previous study showed that CHK1 is required for the HR
repair pathway (17), the detailed
mechanism is still unclear. Here, we examined the expression of
proteins related to NHEJ and HR to determine which pathway is
affected by CHK1 silencing. CHK1 knockdown had no impact on the
expression of the NHEJ-related proteins Ku70/80 and ligase 4 (Lig4)
(Fig. 4E and F). In contrast, the
HR pathway was impaired based on the suppression of VP16-induced
BRCA1 elevation, which represents the location capacity of Rad51 to
DNA damage loci. However, shRNA-mediated CHK1 ablation had no
direct influence on the VP16-mediated induction of the HR-related
proteins MRE11, Rad50 and Rad51 (Fig.
4G and H). Taken together, the results showed that CHK1
knockdown abolishes G2/M arrest and reduces the
efficiency of HR repair; thus, knockdown cells are forced to enter
mitosis with unrepaired DNA damage.
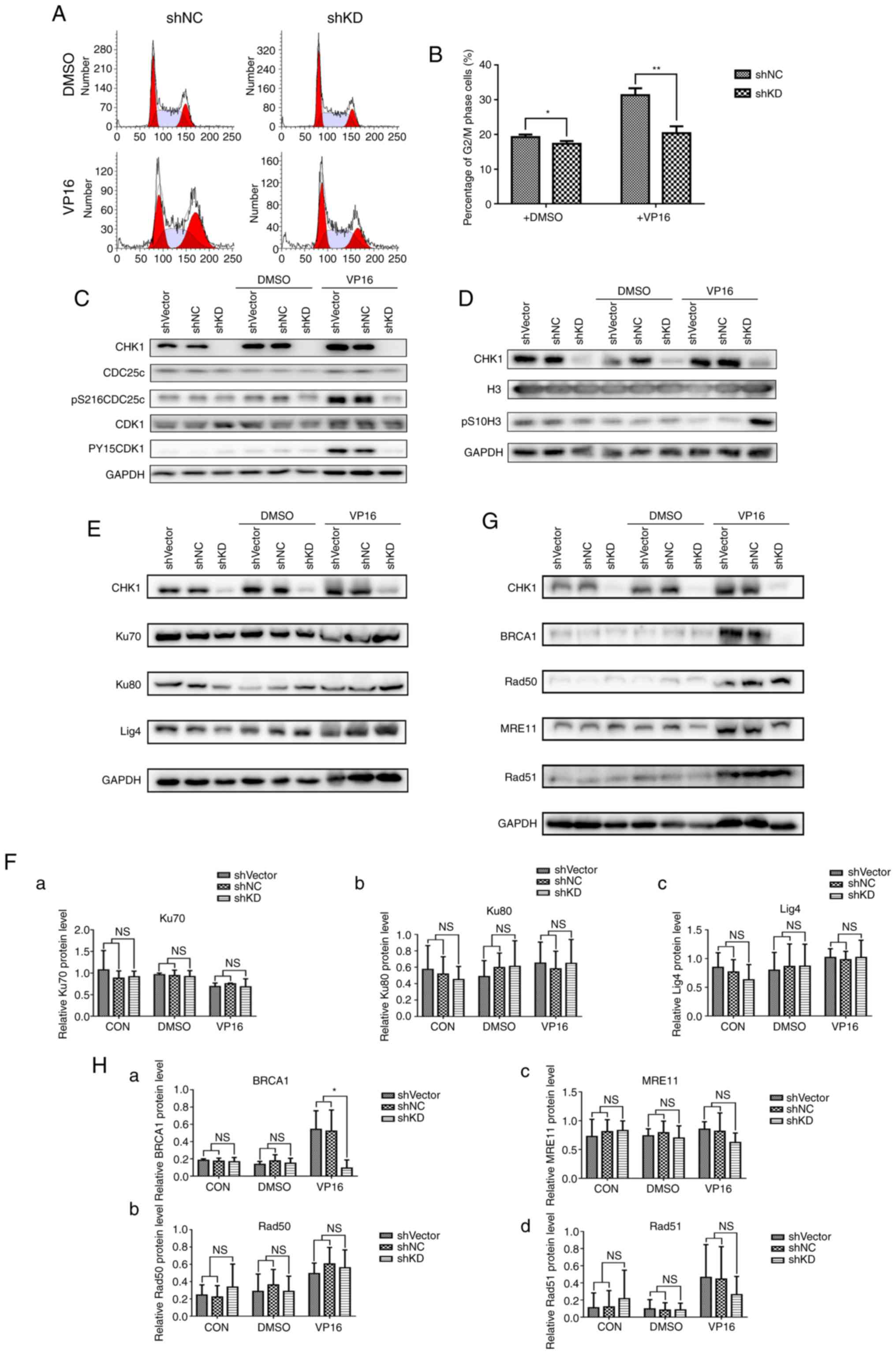 | Figure 4.shRNA-mediated CHK1 knockdown reduces
G2/M arrest and HR repair in K562 cells. (A) The cell
cycle distribution of K562 cells transduced with shNC or shCHK1
(shKD) was detected following treatment with DMSO (control) or
VP16. (B) Statistical analysis of the G2/M phase
percentages (%) shown in A (*P<0.05, **P<0.01). (C) Western
blot analysis of the G2/M arrest markers pS216CDC25c,
pY15CDK1 and total CDC25c and CDK1 in K562 cells infected with
lentivirus carrying shVector, shNC or shCHK1 (shKD) under normal
conditions and in the presence of DMSO or VP16. (D) Western blot
analysis of the mitotic marker pS10-H3 and total H3 in K562 cells
infected with lentivirus carrying shVector, shNC or shCHK1 (shKD)
under normal conditions and in the presence of DMSO or VP16. (E)
Western blot analysis of the NHEJ pathway-related proteins Ku70,
Ku80 and ligase 4 (Lig4) in shVector-, shNC-, or shCHK1-transduced
(shKD) K562 cells under normal conditions and after treatment with
DMSO or VP16. (F) Relative quantification of Ku70 (a), Ku80 (b) and
Lig4 (c) expression shown in E by densitometric analysis; target
band intensity was normalized to that of GAPDH. (G) Western blot
analysis of the HR pathway-related proteins BRCA1, Rad50, MRE11 and
Rad51 in shVector-, shNC-, or shCHK1-transduced (shKD) K562 cells
under normal conditions and after treatment with DMSO or VP16. (H)
Relative quantification of BRCA1 (a), Rad50 (b), MRE11 (c) and
Rad51 (d) expression shown in G by densitometric analysis; band
intensity was normalized to that of GAPDH (*P<0.05). NS, not
significant. CHK1, checkpoint kinase 1; HR, homologous
recombination; VP16, etoposide. |
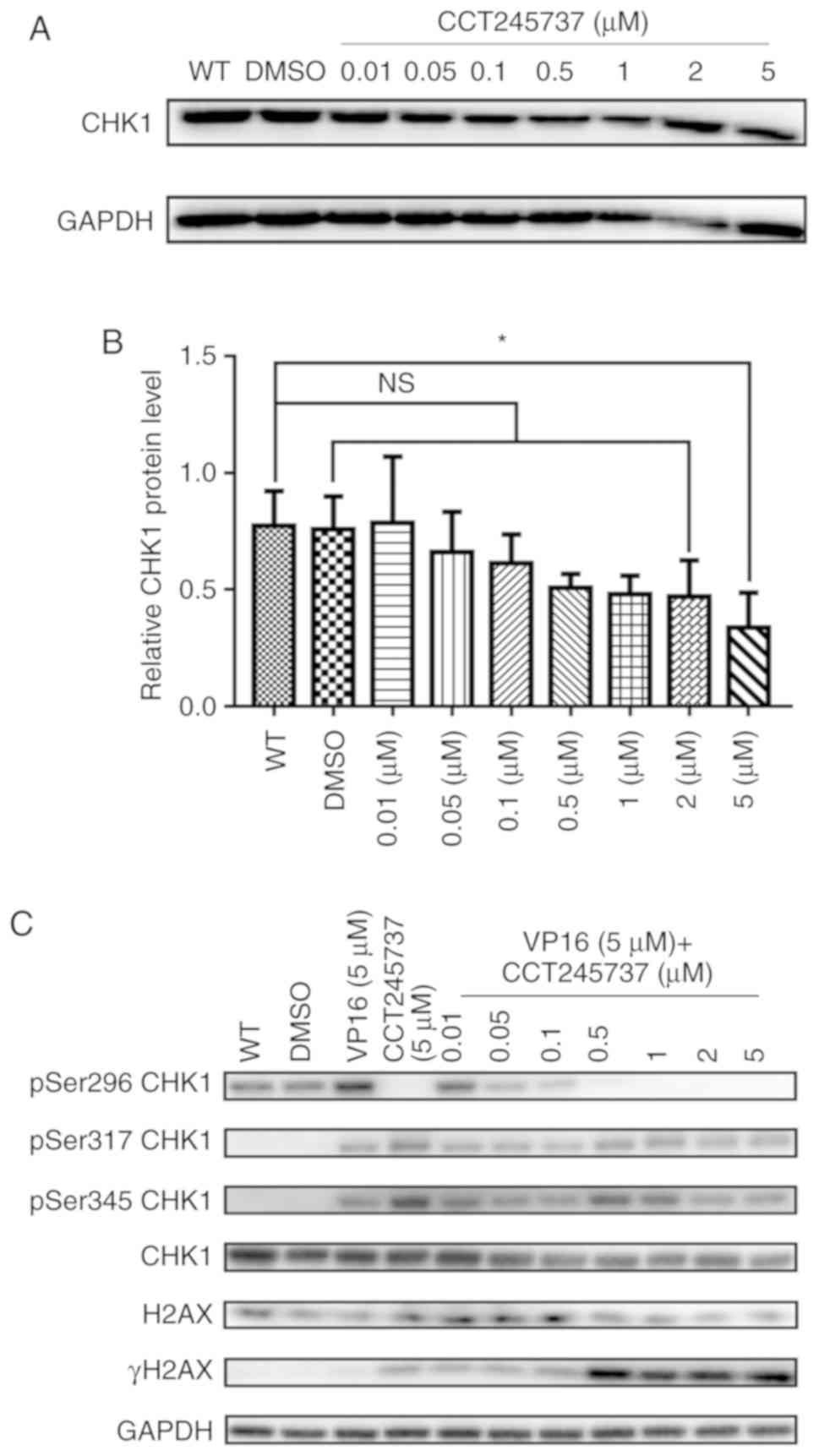
CCT245737 efficiently inhibits CHK1
phosphorylation and promotes the accumulation of DNA damage
CCT245737 is a potent CHK1 inhibitor with good oral
bioavailability. As shown in a previous study (26,27),
CCT245737 is a selective CHK1 inhibitor with a half maximal
inhibitory concentration (IC50) value of 1.4±0.3 nM and
>1,000-fold selectivity over CHK2 and CDK1.
To confirm the ability of CCT245737 in inhibiting
CHK1, we first analyzed total CHK1 protein expression in cells
treated with different concentrations of CCT245737. As shown in
Fig. 5A and B, increasing
concentrations of CCT245737 did not significantly decrease total
CHK1 protein levels, and CHK1 was inhibited at only a high
concentration of CCT245737 (5 µM). Then, we treated K562 cells with
VP16 and increasing concentrations of CCT245737 alone or in
combination to investigate whether CCT245737 influences
phosphorylated CHK1 levels. VP16 obviously induced CHK1
autophosphorylation at Ser296 and phosphorylation at Ser317 and
Ser345 (Fig. 5C). In the presence
of CCT245737, VP16-induced Ser296 autophosphorylation was
dramatically suppressed at 0.05 µM and completely eliminated at 0.5
µM. In contrast, the influence of CCT245737 on other CHK1
phosphorylation sites (Ser317 and Ser345) and total CHK1 levels was
minimal. Meanwhile, the inhibition of CHK1 autophosphorylation at
Ser296 coincided with the accumulation of γH2AX. Therefore, these
results suggest that CCT245737 can efficiently inhibit the
activation of CHK1 by suppressing VP16-induced CHK1
autophosphorylation at Ser296 and promoting the accumulation of DNA
damage in VP16-treated K562 cells.
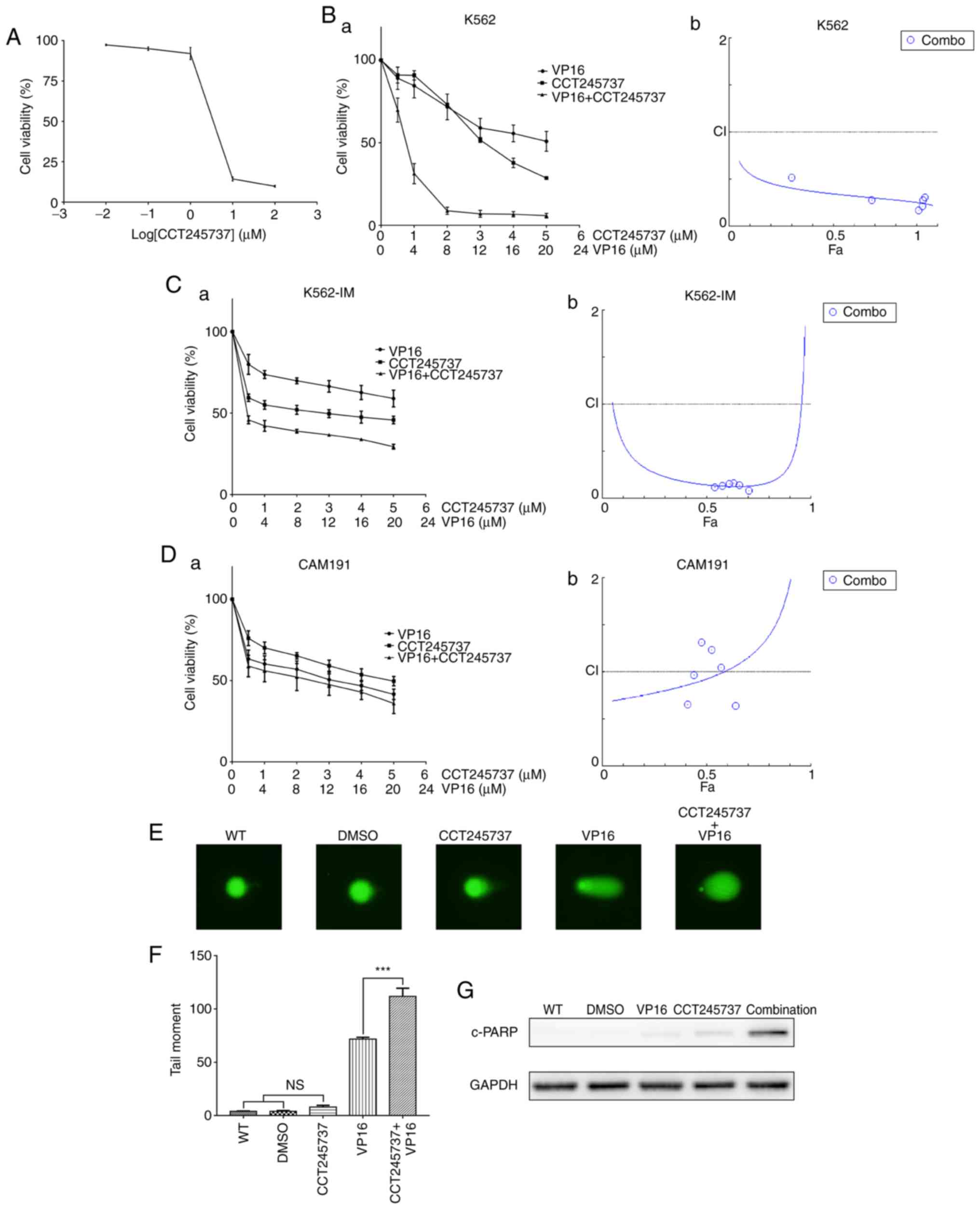
CCT245737 sensitizes K562 cells to
VP16
To determine the cytotoxicity of CCT245737, K562
cells were treated with increasing concentrations of CCT245737 for
24 h, and cell viability was measured by CCK-8 assays. Our results
showed that K562 cell viability was significantly reduced by
CCT245737 in a dose-dependent manner (Fig. 6A). Furthermore, we demonstrated that
CCT245737 and VP16 had a synergistic effect on the viability of
both K562 and imatinib-treated K562 cells (K562-IM), with a CI of
0.35 and 0.15 at a 50% effective dose (ED50) as
calculated by the Chou-Talalay analysis (Fig. 6B and C) (31). To clarify that this drug combination
is specific for cancer cells, we performed the same experiment with
a normal human lymphocyte cell line, CAM191. CCT245737 showed weak
synergism with VP16 in CAM191 cells compared with vehicle- and
imatinib-treated K562 cells, with a CI of 0.93 at the
ED50 (Fig. 6D).
Moreover, consistent with the γH2AX accumulation shown in Fig. 4C, comet assays demonstrated that
CCT245737 obviously increased the level of VP16-induced DNA damage
(Fig. 6E and F). Furthermore, the
increased c-PARP levels in the combination treatment group
suggested the upregulation of apoptosis (Fig. 6G). These data revealed that
CCT245737 may have a considerable and specific synergistic
anticancer effect with VP16 in K562 cells treated with or without
imatinib.
CCT245737 abrogates G2/M
arrest and reduces HR repair capacity in K562 cells
To elucidate the mechanism of the anticancer synergy
between CCT245737 and VP16 in leukemia cells, we first analyzed the
cell cycle distribution. CCT245737 abolished G2/M arrest
in K562 cells under normal conditions and after acute VP16 exposure
(5 µM for 1.5 h) (Fig. 7A and B).
The western blot results showed that CCT245737 efficiently reduced
the VP16-induced increases in the G2/M arrest markers
pS216 CDC25c and pY15 CDK1 (Fig.
7C). Moreover, the VP16-induced decrease in the mitotic marker
pS10 H3 was rescued by CCT245737 (Fig.
7D). Next, the expression of critical proteins related to the
NHEJ and HR pathways was investigated to elucidate the potential
mechanism by which CCT245737 enhances chemosensitivity. Consistent
with the results of shRNA-induced CHK1 silencing, CCT245737 had no
obvious impact on NHEJ-related protein expression (Fig. 7E and F). However, in contrast to the
results of the shCHK1-KD experiments, the induction of both Rad51
and BRCA1 by VP16 was markedly suppressed by CCT245737 (Fig. 7G and H). This result indicates that
CCT245737 does not only reduce the local availability of Rad51 but
also directly suppresses Rad51 expression after DSBs. Due to the
results above, BRCA1 and Rad51 were chosen to be the most important
proteins involved in CHK1-related DNA damage repair pathway in K562
wild-type (K562-WT) cells. Thus, we prioritized the Rad51 and BRCA1
expression in imatinib-treated K562 (K562-IM) cells over other
assays to certify that CHK1 inhibition by CCT245737 could also
suppress HR efficiency in K562-IM cells through downregulating
these two proteins expression and similar results were obtained in
K562-IM cells. Compared with K562-WT cells, K562 cells pretreated
with imatinib showed a nonsignificant VP16-induced increase in
Rad51 and BRCA1 expression, perhaps because this pretreatment could
induce abundant Rad51 and BRCA1 expression in these cells.
CCT245737 also obviously inhibited Rad51 and BRCA1 expression in
imatinib-treated K562 cells, indicating that CCT245737 can also
effectively suppress HR efficiency in TKI-treated CML cells
(Fig. 7I and J). In conclusion,
these results suggest that CCT245737 can abolish the VP16-induced
activation of the G2/M checkpoint, efficiently inhibit
the HR repair pathway, and promote the progression of cells with
unrepaired DNA damage into mitosis.
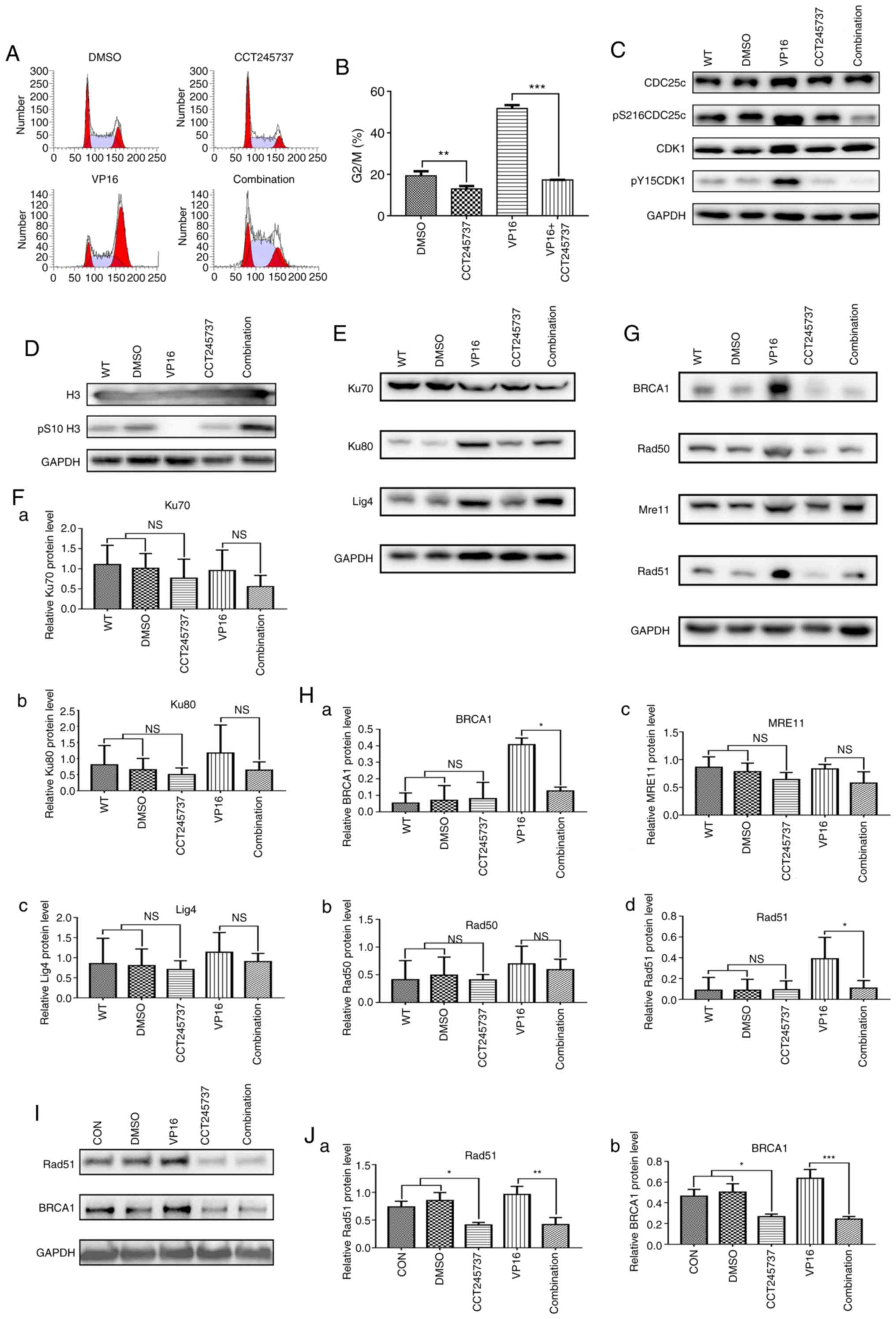 | Figure 7.CCT245737 abrogates G2/M
arrest and reduces HR repair capacity in K562 cells. (A) Effects of
CCT245737 (500 nM, 24 h) on cell cycle arrest in K562 cells treated
with acute VP16 (5 µM, 1.5 h). (B) Statistical analysis of the
G2/M phase percentage (%) described in A (**P<0.01,
***P<0.001). (C) Western blot analysis of the G2/M
arrest markers pS216CDC25c, pY15CDK1 and total CDC25c and CDK1 in
K562 cells treated with CCT245737 (500 nM) and/or VP16 (5 µM) for
24 h. (D) Western blot analysis of the mitosis marker pS10-H3 and
total H3 in K562 cells treated with CCT245737 (500 nM) and/or VP16
(5 µM) for 24 h. (E) Western blot analysis of the NHEJ
pathway-related proteins Ku70, Ku80 and ligase 4 (Lig4) in K562
cells treated with CCT245737 (500 nM) and/or VP16 (5 µM) for 24 h.
(F) Relative quantification of Ku70 (a), Ku80 (b) and Lig4 (c)
expression shown in E by densitometric analysis of band intensities
normalized to those of GAPDH. (G) Western blot analysis of the HR
pathway-related proteins BRCA1, Rad50, MRE11 and Rad51 in K562
cells treated with CCT245737 (500 nM) and/or VP16 (5 µM) for 24 h.
(H) Relative quantification of BRCA1 (a), Rad50 (b), MRE11 (c) and
Rad51 (d) expression shown in G by densitometric analysis of band
intensities normalized to those of GAPDH (*P<0.05). (I) Western
blot analysis of BRCA1 and Rad51 protein expression in
imatinib-treated K562 cells after exposure to CCT245737 (500 nM)
and/or VP16 (5 µM) for 24 h. (J) Relative quantification of Rad51
(a) and BRCA1 (b) expression shown in I by densitometric analysis
of band intensities normalized to those of GAPDH (*P<0.05,
**P<0.01, ***P<0.001). NS, not significant; HR, homologous
recombination; CHK1, checkpoint kinase 1; VP16, etoposide. |
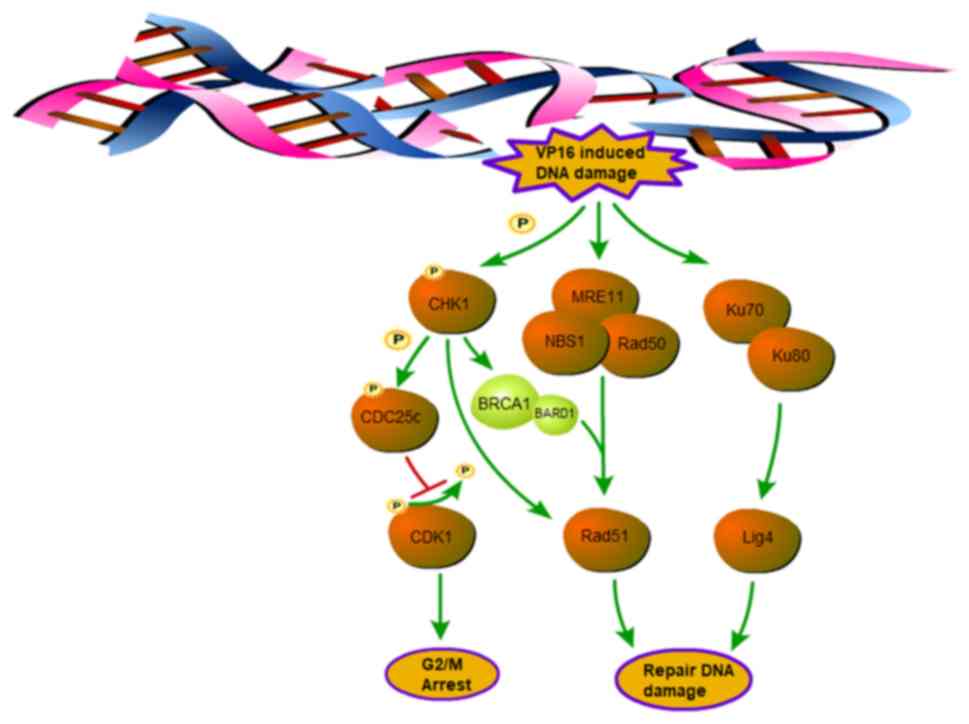
Discussion
Although the development of various chemotherapeutic
drugs has greatly improved cancer therapy, there remain patients
who are resistant to certain therapies. Due to the critical role of
the DNA damage response (DDR) in chemotherapy resistance, blocking
this pathway has emerged as a new approach to address this problem.
Checkpoint kinase 1 (CHK1), a well-known cell cycle checkpoint
protein, is associated with the DDR pathway, especially in
p53-mutant cells, which account for a large subset of cancer cells
(6,32). CHK1 is involved in not only cell
cycle regulation but also the homologous recombination (HR) pathway
through direct or indirect regulation of Rad51 (Fig. 8). Thus, CHK1 is potentially an ideal
candidate for the treatment of chemotherapy-resistant cancer.
CCT245737, a selective CHK1 inhibitor with greater
than 1,000-fold selectivity over CHK2 and CDK1, has an oral
bioavailability nearly 100% (26).
The present study demonstrated that CCT245737 suppressed CHK1
activation by abolishing autophosphorylation at Ser296 but had a
minimal impact on Ser317 and Ser345 phosphorylation or total CHK1
levels. As pSer317 CHK1 and pSer345 CHK1 levels are influenced by
the levels of DNA damage and PP2A phosphatase activity, we utilized
pSer296 CHK1 as a specific biomarker of CHK1 activity, consistent
with previous studies (19,33). Therefore, CCT245737 could be
considered a candidate CHK1 inhibitor to enhance the efficacy of
chemotherapy.
In the present study, we first downregulated CHK1
expression in CML cells with shRNA and found that CHK1 silencing
significantly increased DNA damage, promoted apoptosis, and reduced
the proliferation and colony formation of cells. Subsequently,
CCT245737 was shown to have a favorable and specific synergistic
effect with VP16 in CML cells. To uncover the potential mechanism,
we performed related experiments to investigate the influence of
CHK1 inhibition on the cell cycle and the DNA damage repair
pathway.
VP16, a DNA topoisomerase II (TOP2) inhibitor that
can induce and stabilize DNA double-strand breaks (DSBs) by
disrupting the TOP2 catalytic cycle at the DNA ligation step, is
commonly included in anticancer therapy regimens (34). A previous study showed that
VP16-mediated DSBs induce G2/M arrest by activating the
G2/M cell cycle checkpoint to repair DNA damage
(35). In our study, we revealed
that both RNAi-mediated CHK1 silencing and CCT245737 efficiently
abolished the VP16-induced activation of the G2/M cell
cycle checkpoint, eliminating the time necessary for DNA repair by
overruling the G2/M arrest. Hence, cells may enter
mitosis with accumulated unrepaired damage.
Non-homologous end joining (NHEJ) and HR are the two
major mechanisms for repairing DSBs (36). NHEJ can occur during almost the
whole cell cycle, while HR only occurs in S/G2 phase
after DNA replication. In NHEJ repair, DSB ends are resected by
various endonucleases or exonucleases to generate a microhomology
sequence (<4 nucleotides) (37).
Then, Ku70 and Ku80 heterodimers (Ku70/80) recognize and bind DSBs
to recruit nucleases, ligases, and polymerases, such as DNA ligase
IV, to repair the breaks. We discovered that neither shRNA-mediated
CHK1 knockdown nor CCT245737 significantly impacted the expression
of Ku70/80 or DNA ligase IV, which indicated that the NHEJ repair
pathway might not be influenced by CHK1 inhibition.
In contrast to NHEJ, which evokes error-prone
repair, HR repair is a more precise method of repairing DNA damage.
Once DSBs occur, the Mre11-Nbs1-Rad50 (MRN) complex converts the
DSB ends into single-strand DNA (ssDNA). Subsequently, a key
component of the HR pathway, Rad51, is recruited to replication
protein A (RPA)-coated single-strand breaks (SSBs) and induces
D-loop formation to repair the breaks. BRCA1 is involved in various
biological processes, including mRNA splicing (38), DNA damage signaling (39) and HR repair (40,41).
BRCA1 can form a stable complex with BRCA1-associated ring domain
protein 1 (BARD1) (42), and one
study showed that it coimmunoprecipitates with Rad51 (43). A more recent report indicated that
the BRCA1-BARD1 complex can directly interact with human Rad51,
enhance the affinity of Rad51 for DNA damage loci and facilitate
D-loop formation (44). In our
study, shRNA-mediated Chk1 silencing markedly inhibited BRCA1
expression but not MRE11, Rad50 and Rad51 expression after VP16
treatment. Thus, shRNA-mediated CHK1 downregulation may suppress HR
repair efficiency by reducing BRCA1-induced Rad51 localization and
D-loop formation rather than by directly repressing Rad51
expression. However, in contrast to shRNA-mediated CHK1 inhibition,
which only suppressed BRCA1 induction by VP16, CCT245737
considerably decreased the VP16-induced increases in both Rad51 and
BRCA1 levels. A similar result was also found in imatinib-treated
K562 cells. These results demonstrate that CCT245737 might block
the HR pathway by both directly suppressing Rad51 expression and
indirectly inhibiting the BRCA1-mediated localization of Rad51 at
DNA damage and D-loop formation. These results are consistent with
those of a recent study, which found that prexasertib, another
selective CHK1 inhibitor, suppressed Rad51 and BRCA1 expression in
triple-negative breast cancer cells by promoting ubiquitin-mediated
proteasome degradation, but RNAi-induced CHK1 downregulation only
affected the focus-forming capacity of Rad51 (45). However, the detailed mechanism by
which CCT245737 inhibits Rad51 and BRCA1 expression in CML cells
and whether these effects depend on the timing of drug
administration remain to be determined in subsequent experiments.
Based on the CCK-8 and western blot results in K562-IM cells, we
speculated that imatinib (IM) may force K562 cells into a stress
state which helped cells to be more resistant to other
pharmacological stimulation. Therefore, a more in-depth comparison
of the pharmacological combination of CCT245737 and VP16 between
K562-WT and K562-IM cells in terms of DNA damage repair would also
be investigated in our future research.
In addition to the critical role of CHK1 in the cell
cycle and DNA damage repair pathway, a recent study revealed that a
CHK1 inhibitor can also trigger Bcr-Abl protein degradation in CML
(46). This is another potential
mechanism by which targeting CHK1 could functionally treat CML.
In conclusion, CHK1 is a promising target for the
development of CML therapies. CHK1 inhibition can efficiently
impair DSB-mediated cell cycle arrest and the HR repair pathway.
Therefore, the lack of time and capacity to repair DNA damage after
CHK1 inhibition will induce greater accumulation of DNA damage and
increase apoptosis, resulting in reductions in proliferation and
colony formation. Furthermore, CCT245737 is potentially an ideal
candidate CHK1 inhibitor that demonstrates dramatic and specific
synergism with VP16 in both wild-type and IM-treated K562 cells.
These results provide convincing evidence to promote this strategy
in future clinical regimens for CML or even TKI-resistant CML.
Acknowledgements
Not applicable.
Funding
This present work was supported by the Ministry of
Science and Technology of China (grant no. 2016YFE0107200), the
National Major Scientific and Technological Special Project for
‘Significant New Drugs Development’ (2018ZX09201002-005), the
National Natural Science Foundation of China (31471029, 31671055,
81461138037) and Xu Jun's expert work station (2017IC025).
Availability of data and materials
The datasets used and/or analyzed in the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZF, the first author of this article, designed the
research, collected the samples, performed the experiments,
analyzed the data and wrote the manuscript. HL, JZ, FW, WZ, JW, SL,
QL, YX, GW, AL and JX participated in sample collection, data
collection and analysis. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing
interests.
References
|
1
|
Bhamidipati PK, Kantarjian H, Cortes J,
Cornelison AM and Jabbour E: Management of imatinib-resistant
patients with chronic myeloid leukemia. Ther Adv Hematol.
4:103–117. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kaleem B, Shahab S, Ahmed N and Shamsi TS:
Chronic myeloid leukemia-prognostic value of mutations. Asian Pac J
Cancer Prev. 16:7415–7423. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Valent P, Hadzijusufovic E, Schernthaner
GH, Wolf D, Rea D and le Coutre P: Vascular safety issues in CML
patients treated with BCR/ABL1 kinase inhibitors. Blood.
125:901–906. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Experts in Chronic Myeloid Leukemia, . The
price of drugs for chronic myeloid leukemia (CML) is a reflection
of the unsustainable prices of cancer drugs: From the perspective
of a large group of CML experts. Blood. 121:4439–4442. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dillon MT, Good JS and Harrington KJ:
Selective targeting of the G2/M cell cycle checkpoint to
improve the therapeutic index of radiotherapy. Clin Oncol (R Coll
Radiol). 26:257–265. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Del Nagro CJ, Choi J, Xiao Y, Rangell L,
Mohan S, Pandita A, Zha J, Jackson PK and O'Brien T: Chk1
inhibition in p53-deficient cell lines drives rapid chromosome
fragmentation followed by caspase-independent cell death. Cell
Cycle. 13:303–314. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cimprich KA and Cortez D: ATR: An
essential regulator of genome integrity. Nat Rev Mol Cell Biol.
9:616–627. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yazinski SA and Zou L: Functions,
regulation, and therapeutic implications of the ATR checkpoint
pathway. Annu Rev Genet. 50:155–173. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jazayeri A, Falck J, Lukas C, Bartek J,
Smith GC, Lukas J and Jackson SP: ATM- and cell cycledependent
regulation of ATR in response to DNA double-strand breaks. Nat Cell
Biol. 8:37–45. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sarmento LM, Póvoa V, Nascimento R, Real
G, Antunes I, Martins LR, Moita C, Alves PM, Abecasis M, Moita LF,
et al: CHK1 overexpression in T-cell acute lymphoblastic leukemia
is essential for proliferation and survival by preventing excessive
replication stress. Oncogene. 34:2978–2990. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Walworth N, Davey S and Beach D: Fission
yeast chk1 protein kinase links the rad checkpoint pathway to cdc2.
Nature. 363:368–371. 1993. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bartek J and Lukas J: Chk1 and Chk2
kinases in checkpoint control and cancer. Cancer Cell. 3:421–429.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang Y and Hunter T: Roles of Chk1 in
cell biology and cancer therapy. Int J Cancer. 134:1013–1023. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Patil M, Pabla N and Dong Z: Checkpoint
kinase 1 in DNA damage response and cell cycle regulation. Cell Mol
Life Sci. 70:4009–4021. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maya-Mendoza A, Petermann E, Gillespie DA,
Caldecott KW and Jackson DA: Chk1 regulates the density of active
replication origins during the vertebrate S phase. EMBO J.
26:2719–2731. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tang J, Erikson RL and Liu X: Checkpoint
kinase 1 (Chk1) is required for mitotic progression through
negative regulation of polo-like kinase 1 (Plk1). Proc Natl Acad
Sci USA. 103:11964–11969. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sorensen CS, Hansen LT, Dziegielewski J,
Syljuåsen RG, Lundin C, Bartek J and Helleday T: The cell-cycle
checkpoint kinase Chk1 is required for mammalian homologous
recombination repair. Nat Cell Biol. 7:195–201. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang X, Kennedy RD, Ray K, Stuckert P,
Ellenberger T and D'Andrea AD: Chk1-mediated phosphorylation of
FANCE is required for the Fanconi anemia/BRCA pathway. Mol Cell
Biol. 27:3098–3108. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Walton MI, Eve PD, Hayes A, Valenti MR, De
Haven Brandon AK, Box G, Hallsworth A, Smith EL, Boxall KJ,
Lainchbury M, et al: CCT244747 is a novel potent and selective CHK1
inhibitor with oral efficacy alone and in combination with
genotoxic anticancer drugs. Clin Cancer Res. 18:5650–5661. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Calvo E, Braiteh F, Von Hoff D, McWilliams
R, Becerra C, Galsky MD, Jameson G, Lin J, McKane S, Wickremsinhe
ER, et al: Phase I study of CHK1 inhibitor LY2603618 in combination
with gemcitabine in patients with solid tumors. Oncology.
91:251–260. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Laquente B, Lopez-Martin J, Richards D,
Illerhaus G, Chang DZ, Kim G, Stella P, Richel D, Szcylik C,
Cascinu S, et al: A phase II study to evaluate LY2603618 in
combination with gemcitabine in pancreatic cancer patients. BMC
Cancer. 17:1372017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yin Y, Shen Q, Zhang P, Tao R, Chang W, Li
R, Xie G, Liu W, Zhang L, Kapoor P, et al: Chk1 inhibition
potentiates the therapeutic efficacy of PARP inhibitor BMN673 in
gastric cancer. Am J Cancer Res. 7:473–483. 2017.PubMed/NCBI
|
|
23
|
Italiano A, Infante JR, Shapiro GI, Moore
KN, LoRusso PM, Hamilton E, Cousin S, Toulmonde M, Postel-Vinay S,
Tolaney S, et al: Phase I study of the checkpoint kinase 1
inhibitor GDC-0575 in combination with gemcitabine in patients with
refractory solid tumors. Ann Oncol. 29:1304–1311. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Restelli V, Lupi M, Vagni M, Chila R,
Bertoni F, Damia G and Carrassa L: Combining ibrutinib with Chk1
inhibitors synergistically targets mantle cell lymphoma cell lines.
Target Oncol. 13:235–245. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Booth L, Roberts J, Poklepovic A and Dent
P: The CHK1 inhibitor SRA737 synergizes with PARP1 inhibitors to
kill carcinoma cells. Cancer Biol Ther. 19:786–796. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Walton MI, Eve PD, Hayes A, Henley AT,
Valenti MR, De Haven Brandon AK, Box G, Boxall KJ, Tall M, Swales
K, et al: The clinical development candidate CCT245737 is an orally
active CHK1 inhibitor with preclinical activity in RAS mutant NSCLC
and Emicro-MYC driven B-cell lymphoma. Oncotarget. 7:2329–2342.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Osborne JD, Matthews TP, McHardy T, Proisy
N, Cheung KM, Lainchbury M, Brown N, Walton MI, Eve PD, Boxall KJ,
et al: Multiparameter lead optimization to give an oral checkpoint
kinase 1 (CHK1) inhibitor clinical candidate:
(R)-5-((4-((Morpholin-2-ylmethyl)amino)-5-(trifluoromethyl)pyridin-2-yl)amino)pyr
azine-2-carbonitrile (CCT245737). J Med Chem. 59:5221–5237. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang M, Tian X, Fan Z, Yu W, Li Z, Zhou J,
Zhang W and Liang A: Targeting RAD51 enhances chemosensitivity of
adult T-cell leukemia-lymphoma cells by reducing DNA double-strand
break repair. Oncol Rep. 42:2426–2434. 2019.PubMed/NCBI
|
|
29
|
Li L, Ye S, Yang M, Yu W, Fan Z, Zhang H,
Hu J, Liang A and Zhang W: SIRT1 downregulation enhances
chemosensitivity and survival of adult T-cell leukemia-lymphoma
cells by reducing DNA double-strand repair. Oncol Rep.
34:2935–2942. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tirrò E, Massimino M, Romano C, Pennisi
MS, Stella S, Vitale SR, Fidilio A, Manzella L, Parrinello NL,
Stagno F, et al: Chk1 inhibition restores inotuzumab ozogamicin
citotoxicity in CD22-positive cells expressing mutant p53. Front
Oncol. 9:572019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Parsels LA, Qian Y, Tanska DM, Gross M,
Zhao L, Hassan MC, Arumugarajah S, Parsels JD, Hylander-Gans L,
Simeone DM, et al: Assessment of chk1 phosphorylation as a
pharmacodynamic biomarker of chk1 inhibition. Clin Cancer Res.
17:3706–3715. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Morimoto S, Tsuda M, Bunch H, Sasanuma H,
Austin C and Takeda S: Type II DNA topoisomerases cause spontaneous
double-strand breaks in genomic DNA. Genes (Basel). 10:8682019.
View Article : Google Scholar
|
|
35
|
Schonn I, Hennesen J and Dartsch DC:
Cellular responses to etoposide: Cell death despite cell cycle
arrest and repair of DNA damage. Apoptosis. 15:162–172. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shibata A and Jeggo PA: DNA double-strand
break repair in a cellular context. Clin Oncol (R Coll Radiol).
26:243–249. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chang HHY, Pannunzio NR, Adachi N and
Lieber MR: Non-homologous DNA end joining and alternative pathways
to double-strand break repair. Nat Rev Mol Cell Biol. 18:495–506.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Savage KI, Gorski JJ, Barros EM, Irwin GW,
Manti L, Powell AJ, Pellagatti A, Lukashchuk N, McCance DJ,
McCluggage WG, et al: Identification of a BRCA1-mRNA splicing
complex required for efficient DNA repair and maintenance of
genomic stability. Mol Cell. 54:445–459. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Roy R, Chun J and Powell SN: BRCA1 and
BRCA2: Different roles in a common pathway of genome protection.
Nat Rev Cancer. 12:68–78. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Moynahan ME, Chiu JW, Koller BH and Jasin
M: Brca1 controls homology-directed DNA repair. Mol Cell.
4:511–518. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Caestecker KW and Van de Walle GR: The
role of BRCA1 in DNA double-strand repair: Past and present. Exp
Cell Res. 319:575–587. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wu LC, Wang ZW, Tsan JT, Spillman MA,
Phung A, Xu XL, Yang MC, Hwang LY, Bowcock AM and Baer R:
Identification of a RING protein that can interact in vivo with the
BRCA1 gene product. Nat Genet. 14:430–440. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Scully R, Chen J, Plug A, Xiao Y, Weaver
D, Feunteun J, Ashley T and Livingston DM: Association of BRCA1
with Rad51 in mitotic and meiotic cells. Cell. 88:265–275. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhao W, Steinfeld JB, Liang F, Chen X,
Maranon DG, Jian Ma C, Kwon Y, Rao T, Wang W, Sheng C, et al:
BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature.
550:360–365. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mani C, Jonnalagadda S, Lingareddy J,
Awasthi S, Gmeiner WH and Palle K: Prexasertib treatment induces
homologous recombination deficiency and synergizes with olaparib in
triple-negative breast cancer cells. Breast Cancer Res. 21:1042019.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lei H, Jin J, Liu M, Li X, Luo H, Yang L,
Xu H and Wu Y: Chk1 inhibitors overcome imatinib resistance in
chronic myeloid leukemia cells. Leukemia Res. 64:17–23. 2018.
View Article : Google Scholar
|