Introduction
Acute myeloid leukemia (AML) is a genetically
heterogeneous disease characterized by infiltration of the bone
marrow (BM), blood, and other tissues with proliferative, clonal,
abnormally differentiated cells of the hematopoietic origin
(1). Although the overall survival
rate of AML has increased significantly, relapse after remission is
still the most important issue for AML treatment. Mounting evidence
shows that leukemia stem cells (LSCs) can initiate tumor formation
and lead to chemotherapy failure or resistance and disease relapse
(2,3). LSCs and leukemic blasts ensure their
survival by utilizing autophagy to respond to the specific
metabolic demands during rapid cell proliferation and to counteract
chemotherapeutic stress (4).
Autophagy is an evolutionarily conserved process
during which intracellular components are catabolized to sustain
energy metabolism homeostasis and to safeguard cells against stress
(5). There are many reports
concerning the function of autophagy in AML. Usually, we consider
autophagy as a protective mechanism in AML. Larrue et al
(6) reported that bortezomib could
induce FLT3-ITD degradation through autophagy which resulted in the
death of AML cells. Jin et al (7) believed that granulocytic AML
differentiation relies on noncanonical autophagy pathways and that
restoring autophagic activity may be beneficial in differentiation
therapies. Several studies have shown that low level of basal
autophagy gene expression such as ATG5, ATG7 and LC3 (7,8), and
reduced autophagic flux occur in patient-derived AML blasts, and
the loss of core autophagy genes results in leukemia initiation and
progression in mouse models (9).
Pharmacological or genetic inhibition of autophagy leads to
impaired leukemic cell viability and increased sensitivity to
standard chemotherapy (6,7,10,11).
β-catenin has been demonstrated as a critical
regulator of the self-renewal and therapeutic target for various
cancer stem cells including leukemia-initiating cells (LICs)
(12,13). Casein kinase 1α (CK1α), encoded by
CSNK1A1, is a classical negative regulator for the
Wnt/β-catenin signaling pathway (14). In addition to Wnt/β-catenin
signaling, CK1α also regulates p53, GLI transcription factors and
other important signaling pathways (15–17).
CK1α has been reported to be implicated in apoptosis, survival,
senescence and other cellular physiological processes, thus
regulating the occurrence and development of multiple tumors
(17,18). Recently, CK1α was reported to act as
a key negative regulator of oncogenic RAS-induced autophagy,
suggesting that targeting CK1α-regulated autophagy provides a
promising therapeutic opportunity to treat oncogenic RAS-driven
cancers (19). In contrast to solid
tumors, the role of CK1α in regulating autophagy in hematologic
malignancies is less well defined.
In the present study, we found that CK1α was
upregulated and correlated with prognosis in AML. Furthermore, CK1α
was found to inhibit p53 downstream of murine double minute 2
(MDM2)-mediated autophagy and apoptosis. Pharmacological or genetic
inhibition of CK1α suppressed the proliferation and increased the
autophagy flux as well as apoptosis in multiple AML cell lines and
patient blast cells. Spautin-1, a specific autophagy inhibitor
(20), aggravated the cell death
induced by D4476, a classical inhibitor of CK1α (21), suggesting that CK1α
inhibition-mediated autophagy might be a pro-survival signaling in
AML. Additionally, D4476 upregulated p53 and phosphorylated 5′
AMP-activated protein kinase (AMPK), and reduced the
phosphorylation of mammalian target of rapamycin (mTOR).
Materials and methods
AML cell lines and primary cell
separation
Human AML cell lines HL-60, HEL and THP-1 were
purchased from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China), and cultured in RPMI-1640 medium supplemented
with 7% fetal bovine serum (FBS; both from Gibco; Thermo Fisher
Scientific, Inc.) at 37°C in 5% CO2. A total of 61 newly
diagnosed AML patients and 6 AML patients in complete remission
(CR) in the First Affiliated Hospital of Wenzhou Medical University
from March to December 2018 according to the FAB classification
system were enrolled in this study (Table SI). Eleven age-matched healthy
donors were enrolled as controls. Bone marrow mononuclear cells
(BMMNCs) were isolated using Ficoll-Hypaque (Haoyang Institute of
Biotechnology, China). In addition, BMMNCs of the newly diagnosed
AML patients were not used for the following experiments until the
percentage of CD45dimSSCdim cells was more
than 70% using flow cytometric analyses. All primary blast cells
were cultured in RPMI-1640 medium supplemented with 20% FBS at 37°C
in 5% CO2.
This study was approved by the Institutional Review
Board of the First Affiliated Hospital of Wenzhou Medical
University, and informed consent was obtained from all participants
in accordance with the Declaration of Helsinki protocol.
TCGA data analysis
The relationship of CSNK1A1 expression and
the prognosis in patients with AML was analyzed using UALCAN
(22), an interactive web-portal to
perform to in-depth analyses of TCGA gene expression data. UALCAN
is publicly available at http://ualcan.path.uab.edu.
Quantitative real-time PCR
The total RNA was extracted from cells using Trizol
reagent (Life Technologies, USA) and reverse transcribed into cDNA
with following procedure: 42°C for 30 min, 70°C for 15 min, and
then cDNA was amplified and quantified according to the procedure:
95°C for 30 sec, followed by 40 cycles at 95°C for 5 sec and at
60°C for 34 sec by ABI Prism 7500 using SYBR Green PCR Master Mix
(Takara) with primer pairs. All results were normalized against
GAPDH, RQ=2−ΔΔCq (23).
Primer sequences are listed in Table
SII.
Lentiviral-mediated RNA
interference
Four separate short hairpin RNA (shRNA) constructs
targeting CSNK1A1 (shCSNK1A1-A, shCSNK1A1-B,
shCSNK1A1-C, and shCSNK1A1-D) and one control shRNA
construct targeting lactose gene were synthesized (Hanyin
Biotechnology Co., Ltd.). The sequences of the target sequences for
shRNAs and synthetic oligo information are shown in Table SIII. Each sequence pair was cloned
into the LV3 shuttle plasmid with a RSV and CMV and H1 three
promoter-driven GFP expression cassette, respectively. Each
lentiviral expression plasmid and three corresponding packaging
plasmids were co-transfected into 293T cells with RNAi-Mate reagent
(GenePharma), respectively. Supernatants were collected 72 h after
transfection, and the final virus titer in the supernatant was
1.3×109 TU/ml for each lentivirus. AML cells were
infected with the lentivirus in the presence of 10 µg/ml polybrene,
and 72 h after infection, 3 µg/ml puromycin was used to screen
those successfully transfected cells. The efficiency of gene
silencing was examined using RT-PCR and western blot analysis.
Western blot analysis and
co-immunoprecipitation (Co-IP)
The cells were harvested, and lysed with RIPA lysis
buffer (Beyotime) containing PMSF and protease inhibitor cocktail
(Beyotime). The protein was boiled and subjected to western blot
analysis with different antibodies, including p53 [Cell Signaling
Technology, Inc. (CST), #2527], SQSTM1/p62 (CST, #8025), p-AMPK
(CST, #4184), AMPK (CST, #2532), mTOR (CST, #4517), p-mTOR (CST,
#5536), PARP (CST, #9542), LC3-I/II (CST, #4108), GAPDH (CST,
#2118), CK1α (Abcam, ab206652), ATG-7 (CST, #2631), at 1/1,000
dilution for incubation overnight at 4°C, respectively. For Co-IP,
the cells were lysed with RIPA lysis buffer containing PMSF on ice
for 0.5 h. Protein concentration was determined using a BCA protein
assay kit. Five hundred micrograms of protein per sample was used
and coated with 20 µl protein A/G-agarose beads and 1 µg
mouse-control IgG with protein to eliminate non-specific proteins
for 1 h, and then was centrifugated at 2,400 × g for 5 min. The
supernatants were incubated with beads coated with 10 µg/ml
mouse-source anti-MDM2 or 1 µg control IgG overnight at 4°C. Beads
were washed three times with lysis buffer and boiled for 5 min. The
bead-protein mixture was subjected to western blotting with
antibodies against mouse-source anti-MDM2 (#AF208, Affinity, at
1/1,000 dilution), rabbit-source anti-p53 (#2527, CST, at 1/1,000
dilution), anti-CK1α (ab206652, Abcam, at 1/1,000 dilution),
respectively. Optical densities of the bands were scanned and
analyzed with FluorChem E and AlphaView SA software (Version:
3.4.0.0) (both from ProteinSimple).
Flow cytometry assay and cell
viability assay
Apoptosis assay was measured using the Annexin
V-FITC/PI apoptosis kit and Annexin V-APC/7-AAD apoptosis kit
(MultiSciences Biotech Co., Ltd.) as described previously (24). The cell viability assay was measured
using the Cell Counting Kit-8 (CCK-8, Dojindo, Japan) according to
the manufacturer's protocol.
Colony formation assay
The cells were cultured in methylcellulose medium
(MethoCult™ H4034, Stemcell Technologies) according to the
manufacturer's protocol. Visible colonies, defined to consist of at
least 60 cells were counted. The colonies were photographed by
microscope (BX51, Olympus, Japan) and the total cells were counted
after rinsing with PBS.
Transmission electron microscope
(TEM)
Cell masses were fixed with 2.5% glutaraldehyde and
were cut into 1×1×1 mm pieces. Then, the cell masses were cleaned
by 0.1 M phosphoric acid bleach, dehydrated by 50, 70, 90 and 100%
ethanol and 100% epoxy for 15 min, embedded with epoxypropane +
buried solution (2:1) for 2 h, epoxypropane + burden (1:2) for 3 h,
pure buried liquid overnight and pure buried liquid for 4 h, sliced
by slim slicer (70 nm), and stained by 3% uranium acetate-lead
citrate. Finally, the mitochondrial membrane and typical autophagic
vacuoles of each cell sample were observed under ×20,000
magnification using a transmission electron microscope (Jeol JEM
1230, Japan).
Statistical analysis
Two-tailed unpaired Student's t-tests with 95%
confidence interval (CI) were used to analyze the data involving
direct comparison of an experimental group with a control group.
One-way ANOVA tests with Dunnett's method for multiple comparisons
with 95% CI were used to analyze the data involving 2 or more test
groups and a control group. One-way ANOVA with Tukey test was used
to perform multiple comparisons for up to 3 groups. P-value
<0.05 was considered to indicate a statistically significant
difference. All experiments were performed with three replicates,
unless stated.
Results
CK1α is overexpressed and its
inhibition impairs the proliferation and the colony formation in
AML cells
Mounting evidence indicates that β-catenin is
overexpressed and promotes disease recurrence contributing to a
poor prognosis in AML (25), which
motivated us to investigate whether CK1α, a negative regulator of
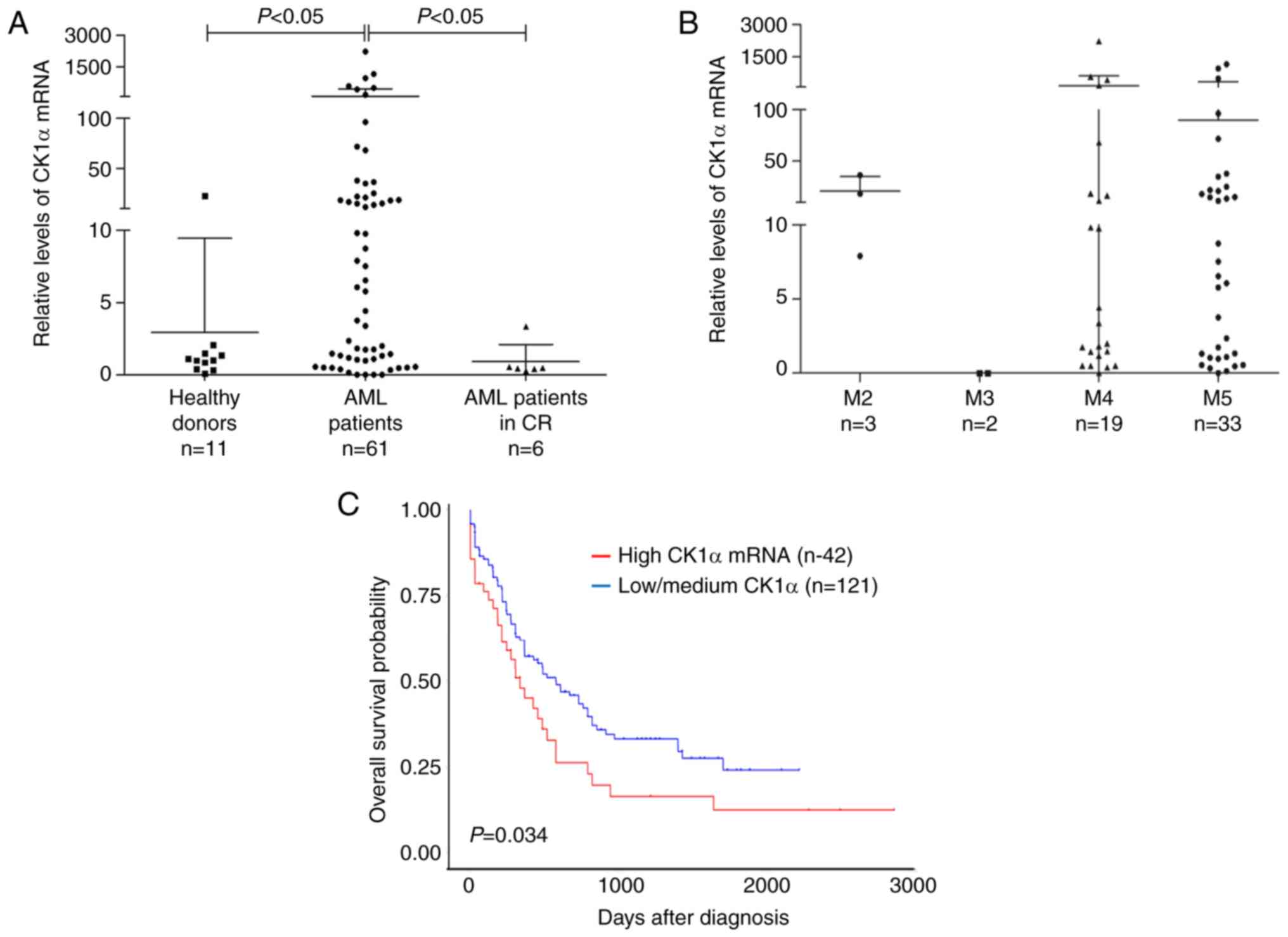
β-catenin, also has a similar role. CK1α mRNA was significantly
higher in BMMNCs from the newly diagnosed AML patients than those
from healthy donors or AML patients who achieved CR (Fig. 1A). There was no difference in the
level of CK1α mRNA among the different types of FAB (Fig. 1B). Furthermore, the patients with a
high level of CK1α mRNA showed a poorer outcome than those with a
low or medium level of CK1α mRNA (P=0.034, Fig. 1C). These findings suggested that
CK1α may be overexpressed and confer a poor prognosis in AML. All
patient information is listed in Table
SI.
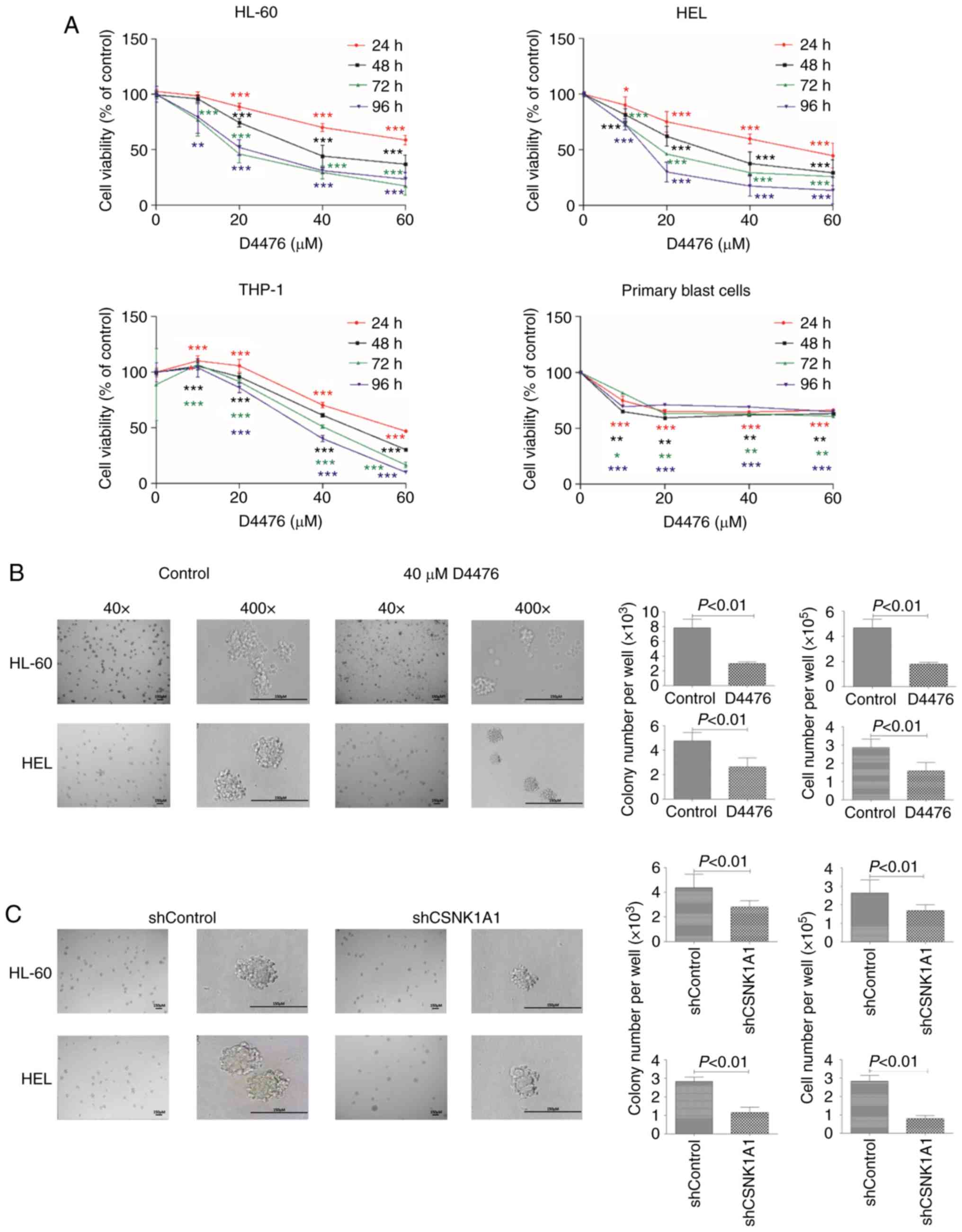
To investigate the role of CK1α in AML cells, we
decreased CK1α using pharmacological or genetic inhibition. We
found that D4476 significantly reduced the cell viability of three
AML cell lines HL-60, HEL and THP-1 in a time- and dose-dependent
manner (Fig. 2A), while only a
dose-dependent manner was shown after treatment with D4476 lower
than 10 µM in patient blast cells. We further investigated whether
D4476 affects the colony formation ability of AML cells, and found
that D4476 at 40 µM dramatically impaired the colony formation, as
indicated with colony number and total cell number, in both HL-60
and HEL cells (Fig. 2B). Notably,
there were many small cell clusters, but did not reach the standard
of a colony in the D4476 treatment group in these two cell lines
(Fig. 2B). Meanwhile, similar
results were also observed in these two AML cell lines with genetic
inhibition of CK1α using lentiviral-mediated shRNA (Figs. 2C and S1). Importantly, we found that the colony
size was decreased in both the D4476 treatment group and the
shCSNK1A1 group (Fig. 2B and
C), suggesting that pharmacological or genetic inhibition of
CK1α affects the colony formation mainly through decreasing the
size of the colony in AML cells.
CK1α inhibition induces apoptosis but
does not influence cell cycle in the AML cells
We found that D4476 dramatically induced apoptosis
in a dose-dependent manner in three AML cell lines tested as well
as patient blast cells (Fig. 3A).
As expected, D4476 treatment promoted the cleavage of PARP, a
classical marker of apoptosis, and decreased the expression of
survivin, a negative marker protein of apoptosis, in HL-60, THP-1
and HEL cells as well as in patient blast cells (Fig. 3A). Similarly, the genetic inhibition
of CK1α also induced a moderate apoptosis in the three AML cell
lines HL-60, THP-1 and HEL (Fig.
3B). Furthermore, combination with cytarabine (Ara-c) and D4476
or gene silencing of CK1α had a synergistic effect on the induction
of apoptosis in THP-1 and HEL cells (Fig. S2). In addition, we found that
pharmacological or genetic inhibition of CK1α had almost no effect
on cell cycle distribution of HL-60 or HEL cells (Fig. S3).
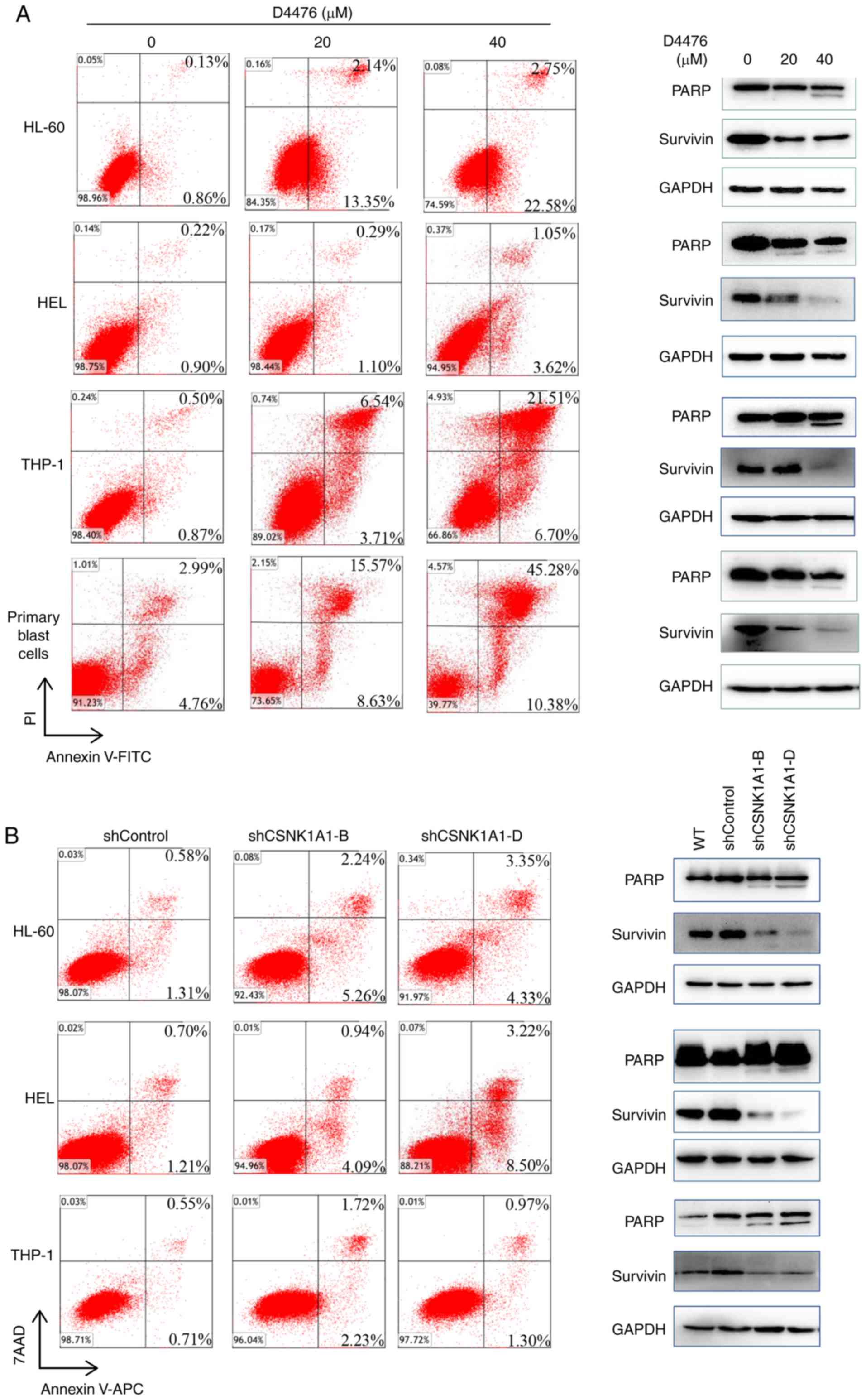 | Figure 3.Inhibition of CK1α induces apoptosis
in AML cells. (A) Three AML cell lines HL-60, THP-1 and HEL as well
as patient blast cells were treated with 0, 20, 40 µM D4476 for 48
h, and the apoptosis was determined by Annexin V-FITC/PI.
Meanwhile, the cleavage of PARP and the expression of
anti-apoptotic protein survivin were also assessed. Images
representing four independent experiment are shown. (B) HL-60,
THP-1 and HEL cells were treated with lentivirus-mediated shRNA for
72 h, and the apoptosis was subsequently determined using Annexin
V-APC/7AAD. The cleavage of PARP and the expression of survivin
were also determined. Images representing four independent
experiment are shown. CK1α, casein kinase 1α; AML, acute myeloid
leukemia; PARP, poly(ADP-ribose) polymerase. |
CK1α inhibition promotes autophagy in
AML cells
Autophagy is a cellular process activated in
response to various forms of stresses such as nutrient deprivation,
hypoxia and endoplasmic reticulum (ER) stress. Apoptosis triggered
by ER stress is often accompanied by increased autophagy. Apoptosis
induced by CK1α inhibition prompted us to investigate whether CK1α
inhibition also activates autophagy. We used TEM to observe
autophagosomes and found that pharmacological or genetic inhibition
of CK1α promoted the autophagy flux of HL-60 and HEL cells with
increased autophagosomes (Fig. 4A).
We found that D4476 treatment upregulated the expression of ATG-7
and LC3-II in all three AML cell lines tested as well as patient
blast cells (Fig. 4B), which
further confirmed the occurrence of autophagy. SQSTM1/p62, a
substrate of LC3-II, is usually reduced when autophagy occurs.
However, we found that D4476 treatment concomitantly increased the
levels of SQSTM1/p62 and LC3-II protein in HL-60 or HEL cells in a
dose-dependent manner within 48 h (Fig. S4). It has been reported that in
nutrient-rich conditions, β-catenin can inhibit autophagy and the
generation of SQSTM1/p62 mRNA by regulating TCF4/LEF, but when
autophagy occurs, β-catenin can be degraded by autophagy and p62
becomes derepressed and dramatically increased (26). We therefore determined the mRNA
expression of SQSTM1/p62 and found that D4476 treatment
significantly increased the level of SQSTM1/p62 mRNA, suggesting
that enhanced autophagy reduced the inhibition of β-catenin on p62,
resulting in greater p62 generation than degradation, ultimately
leading to the accumulation of p62 (Fig. 4C). Furthermore, combination with
Ara-c and D4476 resulted in the upregulation of autophagic flux in
THP-1 and HEL cells (Fig.
S2A).
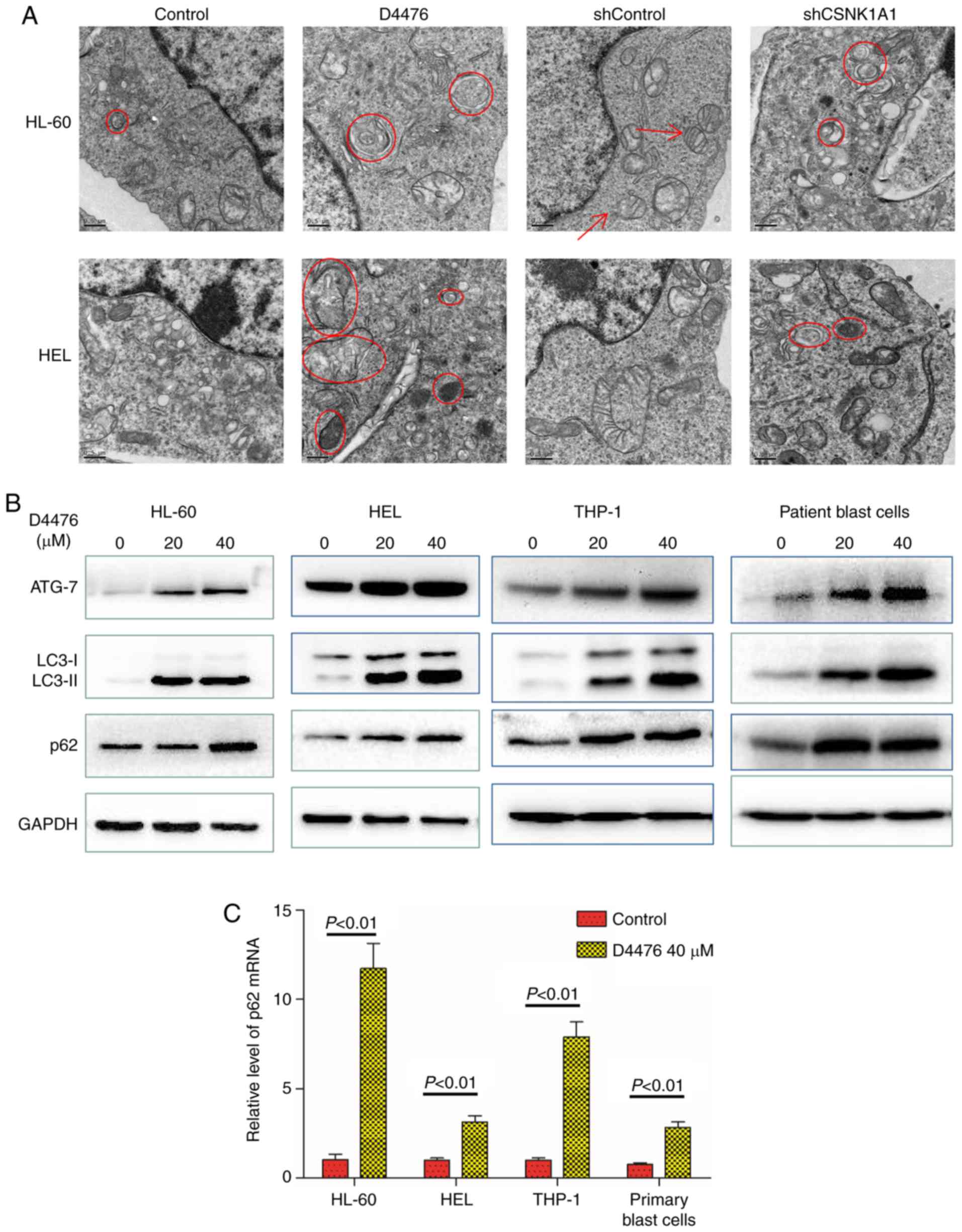 | Figure 4.Inhibition of CK1α promotes autophagy
in AML cells. (A) HL-60 and HEL cells were treated with or without
40 µM D4476 for 48 h, or transfected with shControl or shCSNK1A1
for 72 h. The cell masses were cleaned, dehydrated, embedded,
sliced, stained, and subsequently observed using a transmission
electron microscope. Images representing at least 10 views of each
group are shown. The red arrows indicate mitochondria, and the red
circles indicate autophagosomes. (B) Three AML cell lines (HL-60,
HEL, and THP-1) as well as patient blast cells were treated with or
without 40 µM D4476 for 48 h and the autophagy marker proteins p62,
ATG-7 and LC3-I/II were determined. Images representing at least 3
independent experiment are shown. (C) The AML cell lines (THP-1,
HL-60, and HEL) as well as patient blast cells were treated with or
without 40 µM D4476 for 48 h and the level of p62 mRNA was
determined by RT-qPCR. Results are expressed as mean ± SD,
representing at least three independent experiments. CK1α, casein
kinase 1α; AML, acute myeloid leukemia; ATG-7, autophagy related 7;
LC3, microtubule-associated protein 1A/1B-light chain 3. |
CK1α inhibition-mediated autophagy
exerts protective effects on AML cells
Once confirmed, autophagy and apoptosis occurring
together in the D4476-treated AML cells, the underlying
relationship aroused our curiosity. Firstly, we combined Spautin-1
with D4476 to unravel the veil of mystery. Spautin-1 aggravated the
cell growth inhibition induced by D4476 on all three AML cell lines
tested as well as in the patient blast cells (Fig. 5A). Furthermore, we determined the
apoptosis when the autophagy was inhibited. Spautin-1 alone did not
induce apoptosis in AML cells, and Spautin-1 potently aggravated
D4476-induced apoptosis on HEL cells but merely moderately
exacerbated D4476-induced apoptosis in HL-60 cells (Fig. 5B). These data suggested that D4476
combined with Spautin-1 exerted a synergistic antitumor effect on
AML cells.
 | Figure 5.Autophagy inhibitor Spautin-1
aggravates apoptosis induced by D4476, and CK1α inhibits autophagy
by targeting the p53/AMPK/mTOR signaling pathway downstream of MDM2
in AML cells. (A) After treatment with 40 µM D4476 in the presence
or absence of 10 µM autophagy inhibitor Spautin-1 for 48 h, the
cell viability was measured by CCK-8 assay. Data are expressed as
mean ± SD of at least three independent experiments, and
statistical analysis was performed by one-way ANOVA with Tukey
test. (B) After treatment with 40 µM D4476 in the presence or
absence of 10 µM Spautin-1 for 48 h, apoptosis was determined by
Annexin V-FITC/PI. (C) Co-immunoprecipitation showed that CK1α
interacted with MDM2 and p53 in HEL cells. Ten percent whole cell
lysate was used as input samples. (D) Treatment with D4476
upregulated the level of p53 and MDM2 and phosphorylated AMPK, and
inhibited the phosphorylation of mTOR in both HL-60 and HEL cells.
(E) Treatment with D4476 did not significantly alter the level of
MDM2 when CK1α was genetically inhibited in HEL cells. Images
representing at least three independent experiments are shown.
CK1α, casein kinase 1α; AML, acute myeloid leukemia; MDM2, murine
double minute 2; mTOR, mammalian target of rapamycin; AMPK, 5′
AMP-activated protein kinase. |
CK1α interacts with MDM2 and mediates
autophagy by targeting p53/AMPK/mTOR signaling pathway in AML
A number of signaling pathways including
PI3K/Akt/mTOR, p53/AMPK/mTOR, and MAPKs, are implicated in
autophagic cell survival and cell death (27,28).
We found that CK1α had a relation with MDM2 and p53 (Fig. 5C). It has been recognized that MDM2
is a negative regulator of p53 (29,30)
and p53 regulates AMPK/mTOR signaling to activate autophagy
(31). We found that CK1α
inhibition by D4476 increased the expression of p53 and MDM2,
promoted the phosphorylation of AMPK, and inhibited the
phosphorylation of mTOR in a dose-dependent manner in HL-60 and HEL
cells (Fig. 5D). Furthermore, we
found that D4476 did not affect the level of MDM2 when CK1α was
genetically inhibited in HEL cells (Fig. 5E), indicating that D4476 may
influence MDM2 through CK1α. These data suggested that CK1α may
regulate p53 through MDM2, and inhibit autophagy by the AMPK/mTOR
signaling pathway downstream of p53.
Discussion
Casein kinase 1α (CK1α) is able to regulate several
important signaling molecules in different types of tumors
(32). Especially its ability to
regulate the Wnt signaling, p53, and apoptosis induction makes it a
potential target in tumor therapy (32,33).
In the present study, we found that CK1α was overexpressed in newly
diagnosed acute myeloid leukemia (AML) patient samples, which
further confirmed the concept of overexpression of CK1α in AML
based on its expression in multiple AML cell lines detected by gene
chip (34). When AML patients
achieve complete remission (CR), the level of CK1α was decreased.
Then we analyzed the data of TCGA via UALCAN and found that AML
patients with high expression of CK1α mRNA had poor prognosis
compared with those with low or medium expression of CK1α mRNA.
This finding was in accordance with the role of CK1α in colorectal
cancer (35).
The relationship between autophagy and tumors is of
current research interest. To the best of our knowledge, this is
the first study to reveal the relationship of CK1α and autophagy in
AML. Pharmacological or genetic inhibition of CK1α induced
apoptosis and autophagy in AML cells, which were not completely
consistent with other cancers, such as multiple myeloma (36). Autophagy is responsible for the
degradation of p62 and p62 knockout cells possess higher basal
levels of LC3-II, followed by an increase in autophagic function
(37). However, in our present
study, inhibition of CK1α induced autophagy and increased p62 mRNA
and protein. When CK1α was inhibited by D4476, autophagy occurred
and the inhibitory function via β-catenin regulating-TCF4 on p62
was attenuated, p62 became derepressed and dramatically increased
[21]. We also found that inhibition of CK1α did not affect cell
cycle progression in AML cells, inconsistent with the results of a
previous study (32), suggesting
that the role of CK1α on cell cycle progression is cell
type-specific.
The relationship between autophagy and apoptosis is
two-sided. In some situations, autophagy weakens the apoptotic
effect and acts as a pro-survival role (38), but in other situations this is
absolutely opposed (24). In the
present study, pharmacological inhibition of CK1α by D4476 induced
obvious apoptosis with increased cleavage of PARP and decreased
survivin in a dose-dependent manner in AML cells. Furthermore,
autophagy inhibitor Spautin-1 exacerbated D4476-mediated apoptosis,
suggesting that CK1α inhibition-mediated autophagy may be a
pro-survival element, and co-targeting CK1α and autophagy may
confer a more effective anti-leukemia effect.
Murine double minute 2 (MDM2), a negative regulator
of the tumor-suppressor p53, functions as an inhibitor of apoptosis
in AML (39). CK1α plays a central
role in mediating MDM2 control of p53 protein stability (40). Inhibition of CK1α has p53-dependent
therapeutic efficacy in AML (32).
The activation of p53 and the AMPK/mTOR signaling pathway (27) plays a critical role in regulating
autophagy. In our present study, CK1α inhibitor D4476 upregulated
the levels of MDM2 and p53 in AML cells, similarly with the results
of melanoma cell line A375 as described previously (41). In normal conditions, CK1α promotes
p53 degradation and inhibition by MDM2. Therefore, when CK1α was
inhibited by D4476, the complex of CK1α and MDM2 was disrupted,
which subsequently resulted in the upregulation of p53. It has been
reported that p53 can bind the MDM2 P2 promoter and
transcriptionally upregulate MDM2 expression (32). D4476 did not increase the level of
MDM2 when CK1α was genetically inhibited in HEL cells, indicating
that CK1α may influence p53 through MDM2.
In conclusion, our findings demonstrated that CK1α
is overexpressed in AML patients and acts as a negative element in
the prognosis of AML patients. CK1α inhibits p53 downstream of
MDM2-mediated autophagy and apoptosis, and the targeting of CK1α
and autophagy may offer a potential therapeutic opportunity to
treat AML.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This research was supported by the Zhejiang
Provincial Natural Science Foundation of China (no. LY20H080005),
and the Health Commission of Zhejiang Province (no. 2020KY179).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
SZ and KY designed the research. WX, YG, ZH, RC, YH,
and BX performed the experiments. WX, SJ, ZY, SZ and KY analyzed
the data; and WX and SZ wrote the paper. All authors read and
approved the manuscript and agree to be accountable for all aspects
of the research in ensuring that the accuracy or integrity of any
part of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
This study was approved by the Institutional Review
Board of the First Affiliated Hospital of Wenzhou Medical
University (Wenzhou, China), and informed consent was obtained from
all participants in accordance with the Declaration of Helsinki
protocol.
Patient consent for publication
No applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Döhner H, Weisdorf DJ and Bloomfield CD:
Acute myeloid leukemia. N Engl J Med. 373:1136–1152. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thomas D and Majeti R: Biology and
relevance of human acute myeloid leukemia stem cells. Blood.
129:1577–1585. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ho TC, LaMere M, Stevens BM, Ashton JM,
Myers JR, O'Dwyer KM, Liesveld JL, Mendler JH, Guzman M,
Morrissette JD, et al: Evolution of acute myelogenous leukemia stem
cell properties after treatment and progression. Blood.
128:1671–1678. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rothe K, Porter V and Jiang X: Current
outlook on autophagy in human leukemia: Foe in cancer stem cells
and drug resistance, friend in new therapeutic interventions. Int J
Mol Sci. 20:4612019. View Article : Google Scholar
|
|
5
|
Hansen M, Rubinsztein DC and Walker DW:
Autophagy as a promoter of longevity: Insights from model
organisms. Nat Rev Mol Cell Biol. 19:579–593. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Larrue C, Saland E, Boutzen H, Vergez F,
David M, Joffre C, Hospital MA, Tamburini J, Delabesse E, Manenti
S, et al: Proteasome inhibitors induce FLT3-ITD degradation through
autophagy in AML cells. Blood. 127:882–892. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jin J, Britschgi A, Schläfli AM, Humbert
M, Shan-Krauer D, Batliner J, Federzoni EA, Ernst M, Torbett BE,
Yousefi S, et al: Low autophagy (ATG) gene expression is associated
with an immature AML blast cell phenotype and can be restored
during AML differentiation therapy. Oxid Med Cell Longev.
2018:14827952018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mohamadimaram M, Allahbakhshian Farsani M,
Mirzaeian A, Shahsavan S, Hajifathali A, Parkhihdeh S and Mohammadi
MH: Evaluation of ATG7 and light chain 3 (LC3) autophagy genes
expression in AML patients. Iran J Pharm Res. 18:1060–1066.
2019.PubMed/NCBI
|
|
9
|
Rudat S, Pfaus A, Cheng YY, Holtmann J,
Ellegast JM, Bühler C, Marcantonio DD, Martinez E, Göllner S,
Wickenhauser C, et al: RET-mediated autophagy suppression as
targetable co-dependence in acute myeloid leukemia. Leukemia.
32:2189–2202. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hu X, Mei S, Meng W, Xue S, Jiang L, Yang
Y, Hui L, Chen Y and Guan MX: CXCR4-mediated signaling regulates
autophagy and influences acute myeloid leukemia cell survival and
drug resistance. Cancer Lett. 425:1–12. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Heydt Q, Larrue C, Saland E, Bertoli S,
Sarry JE, Besson A, Manenti S, Joffre C and Mansat-De Mas V:
Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid
leukemia. Oncogene. 37:787–797. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Siriboonpiputtana T, Zeisig BB, Zarowiecki
M, Fung TK, Mallardo M, Tsai CT, Lau PNI, Hoang QC, Veiga P, Barnes
J, et al: Transcriptional memory of cells of origin overrides
β-catenin requirement of MLL cancer stem cells. EMBO J.
36:3139–3155. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang Y, Xia F, Liu X, Yu Z, Xie L, Liu L,
Chen C, Jiang H, Hao X, He X, et al: JAM3 maintains
leukemia-initiating cell self-renewal through
LRP5/AKT/β-catenin/CCND1 signaling. J Clin Invest. 128:1737–1751.
2018. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thorne CA, Hanson AJ, Schneider J, Tahinci
E, Orton D, Cselenyi CS, Jernigan KK, Meyers KC, Hang BI, Waterson
AG, et al: Small-molecule inhibition of Wnt signaling through
activation of casein kinase 1α. Nat Chem Biol. 6:829–836. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chang CH, Kuo CJ, Ito T, Su YY, Jiang ST,
Chiu MH, Lin YH, Nist A, Mernberger M, Stiewe T, et al: CK1α
ablation in keratinocytes induces p53-dependent, sunburn-protective
skin hyperpigmentation. Proc Natl Acad Sci USA. 114:E8035–E8044.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Elyada E, Pribluda A, Goldstein RE,
Morgenstern Y, Brachya G, Cojocaru G, Snir-Alkalay I, Burstain I,
Haffner-Krausz R, Jung S, et al: CKIα ablation highlights a
critical role for p53 in invasiveness control. Nature. 470:409–413.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rodriguez-Blanco J, Li B, Long J, Shen C,
Yang F, Orton D, Collins S, Kasahara N, Ayad NG, McCrea HJ, et al:
A CK1α activator penetrates the brain and shows efficacy against
drug-resistant metastatic medulloblastoma. Clin Cancer Res.
25:1379–1388. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Minzel W, Venkatachalam A, Fink A, Hung E,
Brachya G, Burstain I, Shaham M, Rivlin A, Omer I, Zinger A, et al:
Small molecules co-targeting CKIα and the transcriptional kinases
CDK7/9 control AML in preclinical models. Cell. 175:171–185.e125.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheong JK, Zhang F, Chua PJ, Bay BH,
Thorburn A and Virshup DM: Casein kinase 1α-dependent feedback loop
controls autophagy in RAS-driven cancers. J Clin Invest.
125:1401–1418. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L,
Cai Y, Norberg HV, Zhang T, Furuya T, et al: Beclin1 controls the
levels of p53 by regulating the deubiquitination activity of USP10
and USP13. Cell. 147:223–234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rena G, Bain J, Elliott M and Cohen P:
D4476, a cell-permeant inhibitor of CK1, suppresses the
site-specific phosphorylation and nuclear exclusion of FOXO1a. EMBO
Rep. 5:60–65. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chandrashekar DS, Bashel B, Balasubramanya
SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK and
Varambally S: UALCAN: A portal for facilitating tumor subgroup gene
expression and survival analyses. Neoplasia. 19:649–658. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qian S, Han Y, Shi Y, Xu W, Zhu Y, Jiang
S, Chen Y, Yu Z, Zhang S, Yang Y, et al: Benzene induces
haematotoxicity by promoting deacetylation and autophagy. J Cell
Mol Med. 23:1022–1033. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li XX, Guo H, Zhou JD, Wu DH, Ma JC, Wen
XM, Zhang W, Xu ZJ, Lin J and Jun Q: Overexpression of CTNNB1:
Clinical implication in Chinese de novo acute myeloid leukemia.
Pathol Res Pract. 214:361–367. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Petherick KJ, Williams AC, Lane JD,
Ordóñez-Morán P, Huelsken J, Collard TJ, Smartt HJ, Batson J, Malik
K, Paraskeva C and Greenhough A: Autolysosomal β-catenin
degradation regulates Wnt-autophagy-p62 crosstalk. EMBO J.
32:1903–1916. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hardie DG, Scott JW, Pan DA and Hudson ER:
Management of cellular energy by the AMP-activated protein kinase
system. FEBS Lett. 546:113–120. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang F, Song W, Zhao H, Ma Y, Li Y, Zhai
D, Pi J, Si Y, Xu J, Dong L, et al: The RNA-binding protein QKI5
regulates primary miR-124-1 processing via a distal RNA motif
during erythropoiesis. Cell Res. 27:416–439. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huang X, Wu Z, Mei Y and Wu M: XIAP
inhibits autophagy via XIAP-Mdm2-p53 signalling. EMBO J.
32:2204–2216. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chae YB and Kim MM: Activation of p53 by
spermine mediates induction of autophagy in HT1080 cells. Int J
Biol Macromol. 63:56–63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sui X, Jin L, Huang X, Geng S, He C and Hu
X: p53 signaling and autophagy in cancer: A revolutionary strategy
could be developed for cancer treatment. Autophagy. 7:565–571.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Järås M, Miller PG, Chu LP, Puram RV, Fink
EC, Schneider RK, Al-Shahrour F, Peña P, Breyfogle LJ, Hartwell KA,
et al: Csnk1a1 inhibition has p53-dependent therapeutic efficacy in
acute myeloid leukemia. J Exp Med. 211:605–612. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen L, Li C, Pan Y and Chen J: Regulation
of p53-MDMX interaction by casein kinase 1 alpha. Mol Cell Biol.
25:6509–6520. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schittek B and Sinnberg T: Biological
functions of casein kinase 1 isoforms and putative roles in
tumorigenesis. Mol Cancer. 13:2312014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Richter J, Kretz AL, Lemke J, Fauler M,
Werner JU, Paschke S, Leithäuser F, Henne-Bruns D, Hillenbrand A
and Knippschild U: CK1α overexpression correlates with poor
survival in colorectal cancer. BMC Cancer. 18:1402018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Carrino M, Quotti Tubi L, Fregnani A,
Canovas Nunes S, Barilà G, Trentin L, Zambello R, Semenzato G,
Manni S and Piazza F: Prosurvival autophagy is regulated by protein
kinase CK1 alpha in multiple myeloma. Cell Death Discov. 5:982019.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Duran A, Amanchy R, Linares JF, Joshi J,
Abu-Baker S, Porollo A, Hansen M, Moscat J and Diaz-Meco MT: p62 is
a key regulator of nutrient sensing in the mTORC1 pathway. Mol
Cell. 44:134–146. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ma R, Zhang Y, Wang W, Wu J, Yang Q, Xu W,
Jiang S, Han Y, Yu K and Zhang S: Inhibition of autophagy enhances
the antitumour activity of tigecycline in multiple myeloma. J Cell
Mol Med. 22:5955–5963. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kojima K, Konopleva M, Samudio IJ, Schober
WD, Bornmann WG and Andreeff M: Concomitant inhibition of MDM2 and
Bcl-2 protein function synergistically induce mitochondrial
apoptosis in AML. Cell Cycle. 5:2778–2786. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huart AS, MacLaine NJ, Meek DW and Hupp
TR: CK1alpha plays a central role in mediating MDM2 control of p53
and E2F-1 protein stability. J Biol Chem. 284:32384–32394. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Perry ME, Piette J, Zawadzki JA, Harvey D
and Levine AJ: The mdm-2 gene is induced in response to UV light in
a p53-dependent manner. Proc Natl Acad Sci USA. 90:11623–11627.
1993. View Article : Google Scholar : PubMed/NCBI
|