Introduction
Breast cancer is one of the most common malignant
tumors and is the leading cause of cancer-related deaths among
women worldwide (1). In developed
countries, one in between 9 and 12 women develop breast cancer in
their lifetime (2). Over the past
two decades, the breast cancer mortality rate has decreased due to
significant progress in its diagnosis and treatment. As breast
cancer is a heterogeneous disease, the response to treatment
depends on its biological subtypes.
Breast cancer is generally divided into distinct
subtypes i.e., luminal A, luminal B, HER2-enriched, basal-like and
normal-like subtypes (3–5). Luminal A- and B-type breast cancers
express estrogen receptor (ER), and their growth is regulated in an
estrogen-dependent manner. In addition, HER2 is a receptor for
epidermal growth factor (EGF) and promotes cell survival through
several oncogenic signaling pathways including RAS, AKT and/or mTOR
(6,7). Anticancer drugs targeting HER2 are
used in the primary treatment of patients with HER2-enriched-type
breast cancer. Breast cancer without ER, progesterone receptor
(PgR), and HER2 expression is designated as triple negative breast
cancer (TNBC). An extensive search for mutations demonstrated that
loss of function mutations are common within TP53, pRb and
BRCA1 loci in TNBC, suggesting that these mutations are
involved in the development of the aggressive phenotypes of TNBC
(8).
E2F transcription factor 5 (E2F5) is a transcription
factor belonging to the E2F family, which is composed of eight
members designated as E2F1-7 and 8 (9). E2F1-3 bind to the hypophosphorylated
forms of pRb, and inhibit their cell cycle-regulatory role. E2F4
and E2F5 interact with pRb-related p107/p130 and p130, respectively
(10–12). Among E2F family members, E2F5 has
been revealed to act as an oncogene for prostate (13), esophageal (14), ovarian (15), and colorectal cancer (16) as well as hepatocellular carcinoma
(HCC) (17). Several lines of
evidence suggest that E2F5 has a tumor-suppressive role in HCC
(18) and non-small cell lung
cancer (19). A study using the
Oncomine and Cancer Genome Atlas databases revealed that E2F5 was
upregulated in breast cancer compared to normal tissue and
overexpression of E2F5 mRNA was related to lower rate of
relapse-free survival and post-progression survival (20). We reported previously that E2F5 was
overexpressed in ER-negative breast cancer, especially in TNBC
(20). Based on our results, the
E2F5-positive subgroup exhibited a higher histological grade,
greater rate of ER and PgR negativity, and a higher Ki-67 labeling
index than those in the E2F5-negative subgroup. Moreover, among the
patients without the lymph node metastasis, the E2F5-positive
subgroup exhibited a significantly shorter disease-free survival
period than the E2F5-negative subgroup. Therefore, it was
speculated that E2F5 may act as an oncogene for breast cancer.
However, it remains unclear how E2F5 could
contribute to the development and/or progression of breast cancer.
The genomic amplification and/or hypomethylation of CpG islands
within the E2F5 promoter region were revealed to result in
the aberrant overexpression of E2F5 (21,22).
Mutant TP53 contributed to inhibition of E2F5 via
microRNA-182-2, and resulted in suppression of
p21WAF1 and cell survival (19).
In the present study, it was revealed that E2F5
participated at least in part in the carcinogenesis of breast
cancer carrying wild-type TP53 through the suppression of
TP53, while E2F5 alone was not sufficient for the development
and/or maintenance of the malignant phenotypes of
TP53-mutant breast cancer.
Materials and methods
Cell lines and culture conditions
Human breast cancer MCF7, MDA-MB-175VII, BT474 and
MDA-MB-231 cells were obtained from American Type Culture
Collection (ATCC). MCF7 and BT474 cells were cultured in RPMI-1640
medium (Nacalai Tesque, Inc.) supplemented with 10%
heat-inactivated fetal bovine serum (FBS; Nichirei Biosciences,
Inc.). MDA-MB-175VII and MDA-MB-231 cells were maintained in
Leibovitz's L-15 medium (Thermo Fisher Scientific, Inc.) containing
10% heat-inactivated FBS. All of the media contained 100 IU/ml of
penicillin and 100 µg/ml of streptomycin (both from Thermo Fisher
Scientific, Inc.). Cells were maintained at 37°C in an incubator
with a controlled humidified atmosphere consisting of 95% air and
5% Co2.
Small interfering (si)RNA-mediated
knockdown of E2F5 and TP53
Cells were seeded at a density of 5×104
cells/ml and allowed to attach on the bottom of culture plate.
Twenty-four hours after seeding, cells were transfected with siRNA
using Lipofectamine 3000 (Thermo Fisher Scientific, Inc.) in
accordance with the manufacturer's instructions. siRNA for E2F5
(siRNA ID s4417; cat. no. 4392420; Thermo Fisher Scientific, Inc.),
siRNA for TP53 (cat. no. sc-29435; Santa Cruz Biotechnology, Inc.),
and control siRNA (cat. no. 4390843; Thermo Fisher Scientific,
Inc.) were used in the present study.
Cell viability and cell cycle
distribution
MCF7 and MDA-MB-231 cells were seeded in 96-well
culture plates at a density of 5×104 cells/ml and
transfected with E2F5 siRNA or control siRNA 24 h after
seeding. At the indicated time-points, viability of the transfected
cells was measured by the standard WST-8 assay using Cell Count
Reagent CF (Nacalai Tesque, Inc.). One tenth volume of WST8
solution was added to culture medium and incubated for 1 h at 37°C
in Co2 incubator. Then absorbance of culture medium at
OD 450 nm was measured.
For fluorescent-activated cell sorting (FACS)
analysis, MCF7 and MDA-MB-231 cells were seeded in 6-well culture
plates at a density of 1×105 cells/ml and transfected
with E2F5 siRNA or control siRNA 24 h after seeding. Cells
were cultured for 4 days, and then both floating and attached cells
were collected for FACS analysis. Cells were washed in PBS and
fixed in 70% ethanol overnight at 4°C. After washing in PBS, cells
were suspended in PBS containing 0.1% FBS, 25 µg/ml propidium
iodide (Sigma-Aldrich; Merck KGaA) and 200 µg/ml of RNase A
(Sigma-Aldrich; Merck KGaA), and incubated for 15 min at room
temperature. Subsequently, the cells were subjected to FACS
analysis (FACSCalibur; BD Biosciences).
Real-time reverse
transcription-quantitative (RT-q)PCR
Total RNA was extracted from cells using RNeasy mini
kits (Qiagen, Inc.) according to the manufacturer's instructions.
For cDNA synthesis, aliquots of 500 ng of total RNA were
reverse-transcribed using iScript cDNA synthesis system (Bio-Rad
Laboratories, Inc.). Real-time RT-qPCR for E2F5 and
β-actin was performed using Premix Ex Taq Perfect Real Time
(Takara Βio, Inc.) with TaqMan Pre-Developed Assay Reagents (Thermo
Fisher Scientific, Inc.), Hs00231092_m1 for E2F5 and Hs99999903_m1
for β-actin. The reaction was carried out at 95°C for 5 sec and
60°C for 20 sec, for total of 40 cycles. Real-time RT-qPCR for
p21WAF1, BAX, NOXA and PUMA was performed
using SYBR Premix Ex Taq™ (Takara Bio, Inc.) according to the
manufacturer's recommendations. The primers used were as follows:
p21WAF1, 5′-GCAGACCAGCATGACAGATTT-3′ (sense) and
5′-GGATTAGGGCTTCCTCTTGGA-3′ (antisense); BAX,
5′-TTGCTTCAGGGTTTCATCCA-3′ (sense) and 5′-AGACACTCGCTCAGCTTCTTG-3′
(antisense); NOXA, 5′-GCAGAGCTGGAAGTCGAGTG-3′ (sense) and
5′-GAGCAGAAGAGTTTGGATATCAG-3′ (antisense); PUMA,
5′-GACGACCTCAACGCACAGTA-3′ (sense) and 5′-AGGAGTCCCATGATGAGATTGT-3′
(antisense). The reaction for p21WAF1 and
BAX was carried out at 95°C for 5 sec, 55°C for 10 sec and
72°C for 10 sec, for total of 40 cycles. The reaction for
NOXA and PUMA was carried out at 95°C for 5 sec and
60°C for 20 sec, for total of 40 cycles. Assessments were performed
three times. A mixture of cDNA generated from total RNA of MCF7 and
MDA-MB-231 cells was used as a reference. A cDNA mixture dilution
series was prepared and used for real-time RT-qPCR as templates to
obtain a standard curve for each gene. The housekeeping gene
β-actin was used as an internal reference.
Immunoblotting
Cells were lysed in RIPA buffer containing protease
inhibitor cocktail (Nacalai Tesque, Inc.) and phosphatase inhibitor
cocktail (Nacalai Tesque, Inc.), followed by a brief sonication
(BIORUPTOR UDC250; Cosmo Bio). Protein concentration of the lysates
was measured using Bio-Rad DC kits (Bio-Rad Laboratories, Inc.).
The lysates containing 15 µg protein/lane were separated by 4–12%
SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and then
electroblotted onto Immobilon-P membranes (EMD Millipore).
Membranes were blocked with Blocking-one (Nacalai Tesque, Inc.)
overnight at 4°C, and incubated with anti-E2F5 (dilution 1:500;
cat. no. GTX129491; GeneTex, Inc.), anti-TP53 (DO1; dilution
1:1,000; cat. no. sc-126; Santa Cruz Biotechnology, Inc.),
anti-phospho-TP53 at Ser-15 (dilution 1:1,000; product no. 9284;
Cell Signaling Technology, Inc.), anti-p21WAF1
(dilution 1:500; cat. no. sc-756; Santa Cruz Biotechnology, Inc.),
anti-BAX (dilution 1:1,000; cat. no. 2772; Cell Signaling
Technology, Inc.), anti-PARP (dilution 1:1,000; product no. 9542;
Cell Signaling Technology, Inc.) or with anti-β-actin antibody
(dilution 1:5,000; product no. A5441; Sigma-Aldrich; Merck KGaA) at
4°C. After 24 h of incubation, the membranes were washed with
Tris-buffered saline containing 0.1% Tween-20 (TBS-T), followed by
incubation with horseradish peroxidase-conjugated secondary
antibodies for mouse (dilution 1:2,000; cat. no. NA931V) or for
rabbit (dilution 1:2,000; cat. no. NA934V; both GE Healthcare Life
Sciences) for 1 h at room temperature. The membranes were washed
extensively with TBS-T, and treated with Chemi-Lumi-One Super
(Nacalai Tesque, Inc.) to visualize immunoreactivity using LAS4000
(Fujifilm). Intensity of the bands for TP53 and phosphorylated TP53
were measured by ImageJ software ver. 1.48 (National Institutes of
Health).
Statistical analysis
Statistical analyses to examine the significance of
differences between paired data were performed by Student's t-test,
and one-way ANOVA followed by post hoc Tukey test was used to
examine significance of differences among multiple data. All of the
statistical analyses were performed using JMP software ver. 11.2
(SAS Institute, Inc.). Data are presented as the means ± SD from at
least three independent experiments. In all analyses, P<0.05 was
considered to indicate a statistically significant difference.
Results
Knockdown of E2F5 suppresses the
proliferation of breast cancer cells
To clarify the potential role of E2F5 in the
development and/or the progression of breast cancer, the expression
level of E2F5 in the triple-negative-type breast cancer
MDA-MB-231 cells, HER2-positive-type breast cancer BT474 cells,
plus the luminal-type breast cancer MCF7 and MDA-MB-175VII cells,
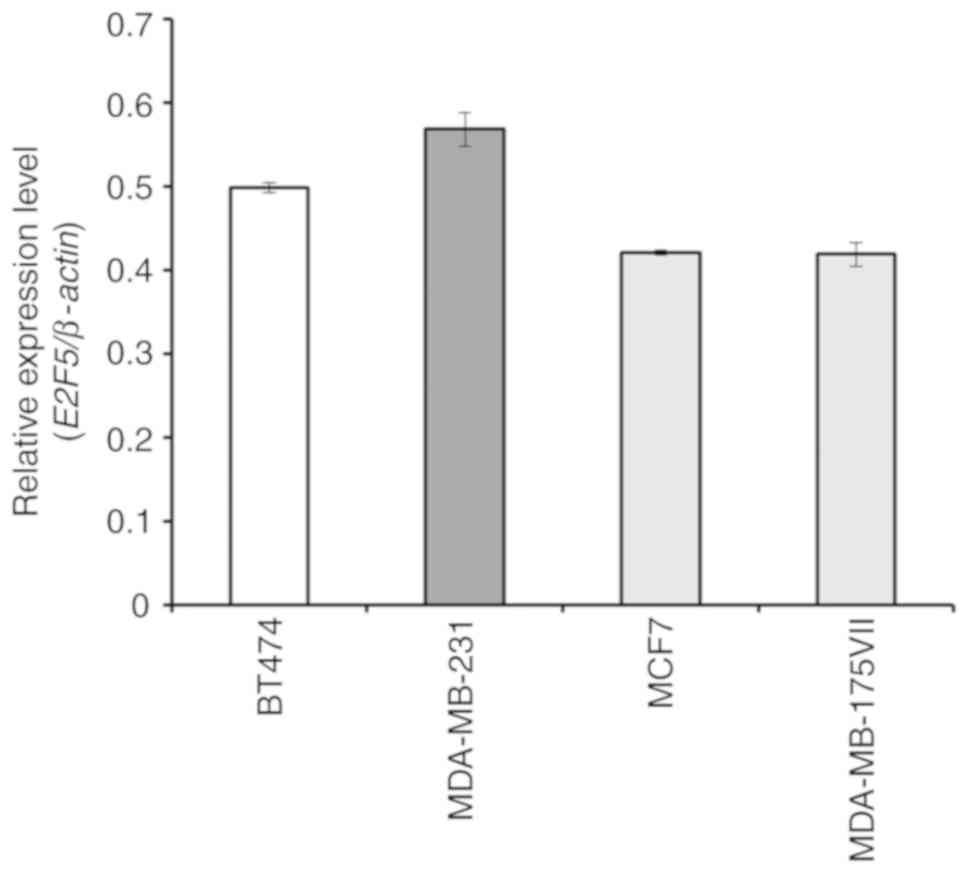
was examined. As revealed in Fig.
1, real-time RT-qPCR analysis demonstrated that there were no
significant differences in the E2F5 expression levels among
the cell lines examined. MDA-MB-231 (triple-negative-type) and MCF7
(luminal-type) cells were used for further experiments.
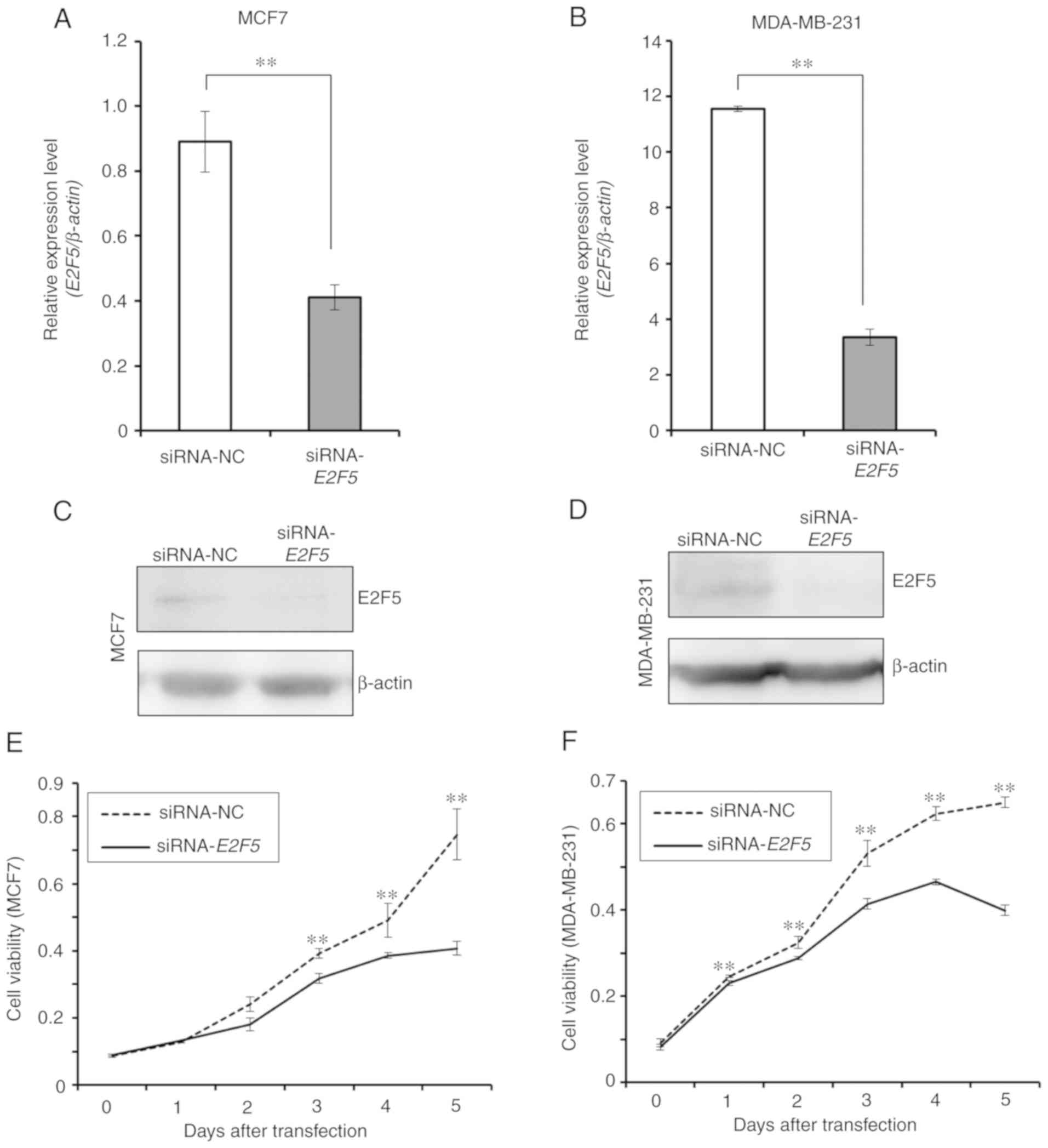
siRNA-mediated knockdown of E2F5 in MCF7 and
MDA-MB-231 cells was then performed. Under the experimental
conditions, E2F5 mRNA and protein expression levels were
efficiently downregulated in MCF7 (Fig.
2A and C) and MDA-MB-231 cells (Fig. 2B and D). Next, the possible effects
of E2F5 downregulation on cell proliferation were examined.
As revealed in Fig. 2E and F,
downregulation of E2F5 expression caused a significant
decrease in the proliferation rates of MCF7 and MDA-MB-231 cells,
indicating that E2F5 may regulate proliferation of breast
cancer cells regardless of their subtype.
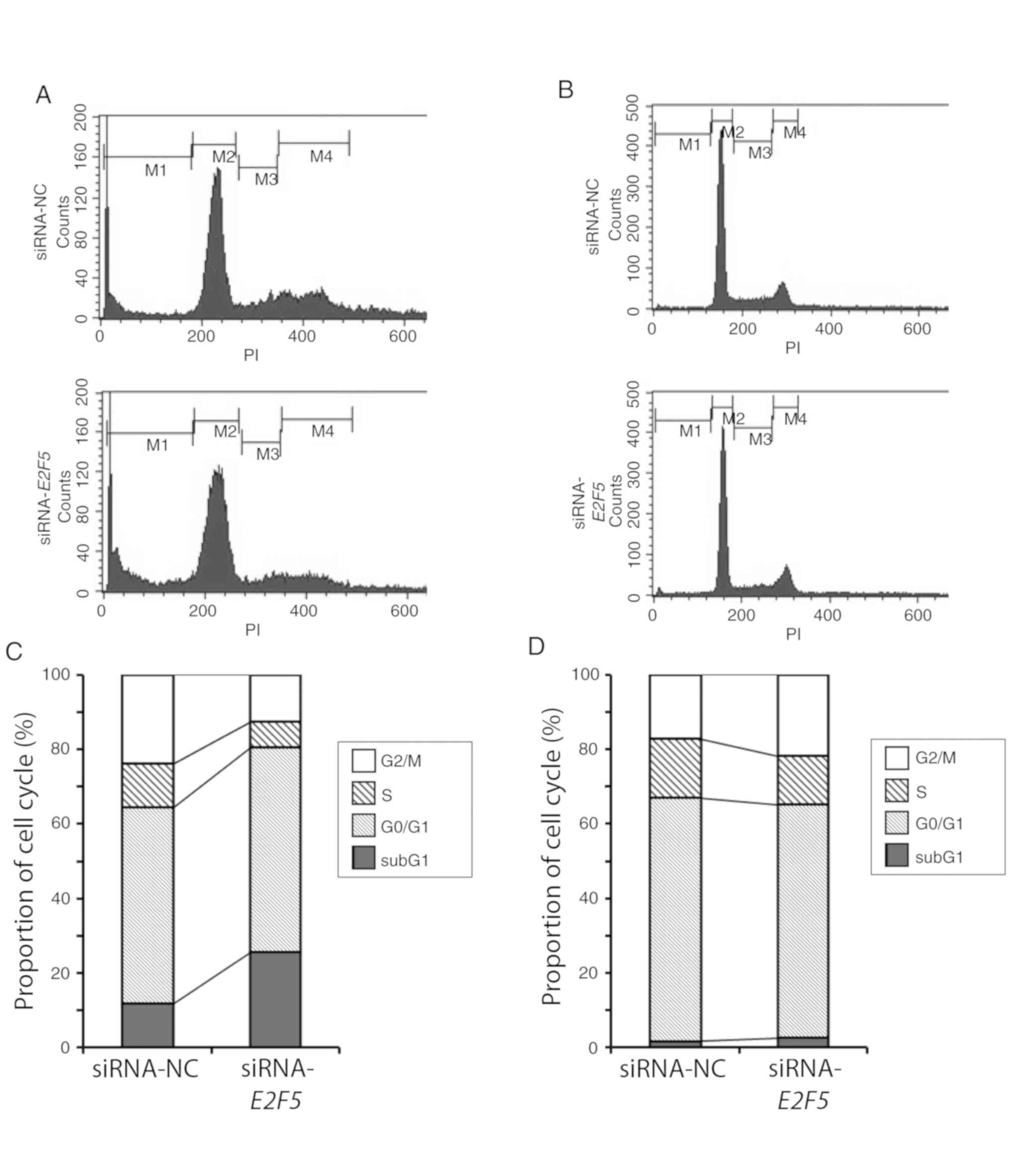
E2F5 downregulation potentiates cell
death in MCF7 cells but not MDA-MB-231 cells
Since E2F5 knockdown resulted in a
significant decrease in the proliferation rates of MCF7 and
MDA-MB-231 cells, the possible effects of E2F5 knockdown on
cell cycle distribution was examined. FACS analysis demonstrated a
clear increase in the proportion of cells with sub-G1 DNA content,
indicating that the proportion of dead cells was increased, in
E2F5-knockdown MCF7 cells in comparison to control cells
(Fig. 3A and C). In contrast,
depletion of E2F5 had no detectable effect on the number of
MDA-MB-231 cells with sub-G1 DNA content (Fig. 3B and D). Further analysis of these
results indicated that the proportion of mitotic cells was
marginally increased in E2F5-knockdown MDA-MB-231 cells
relative to the control cells (Fig.
3D). These observations indicated that E2F5 may regulate
breast cancer cell proliferation through distinct mechanisms in
different types of cells.
E2F5 gene silencing triggers
TP53-mediated cell death in MCF7 cells but not in MDA-MB-231
cells
Finally, the molecular mechanisms underlying the
cell death mediated by E2F5 knockdown in MCF7 cells were
examined. As MCF7 cells carry the wild-type TP53 (23) and MDA-MB-231 cells carry mutant
TP53 (23), we postulated
that knockdown of E2F5 in MCF7 induced cell death via the
TP53-dependent pathway. The observation that the effects of
E2F5 knockdown in BT474 cells carrying mutant TP53 (23) were similar to those in MDA-MB-231
cells (Fig. S1A) supported this
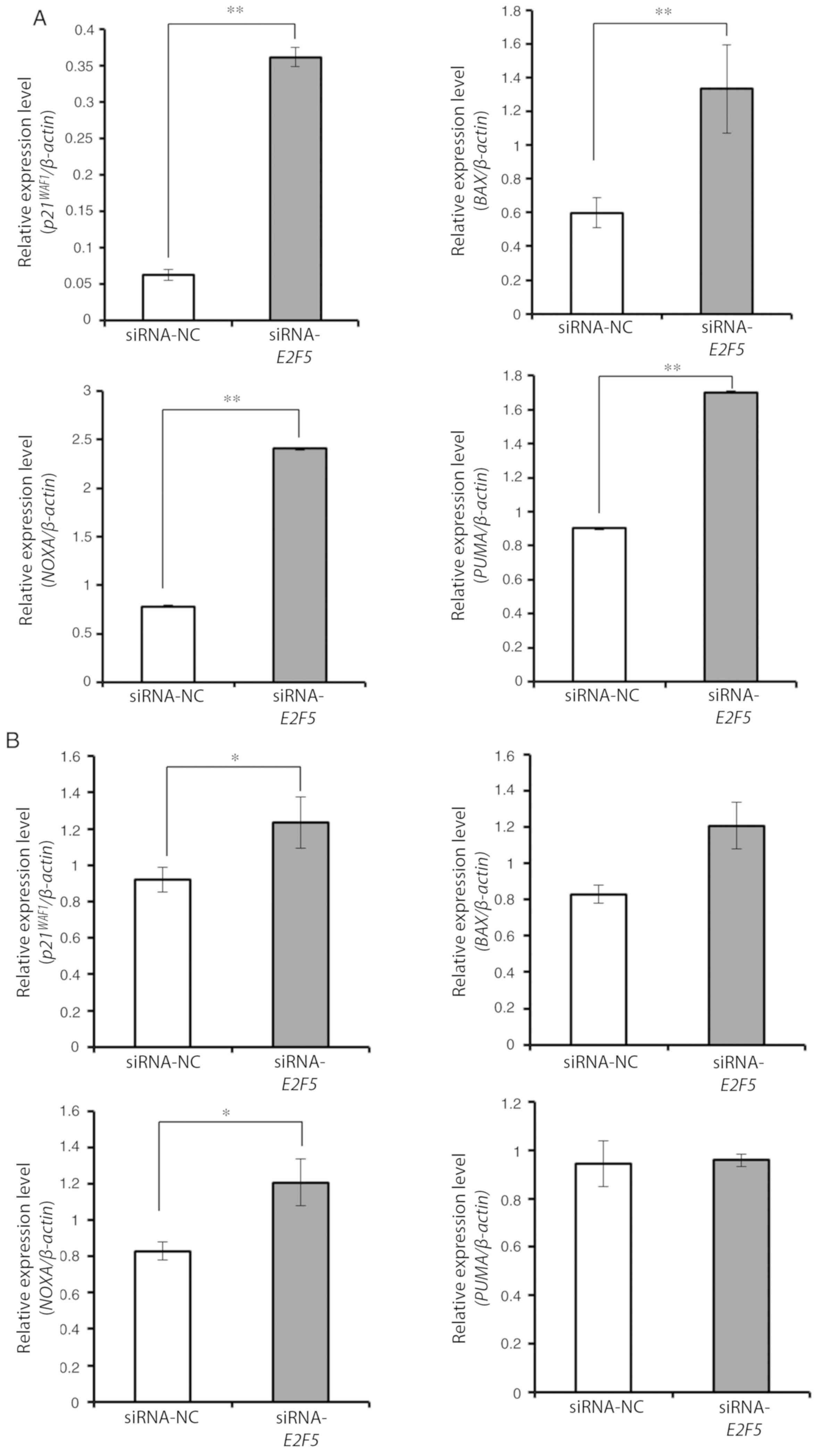
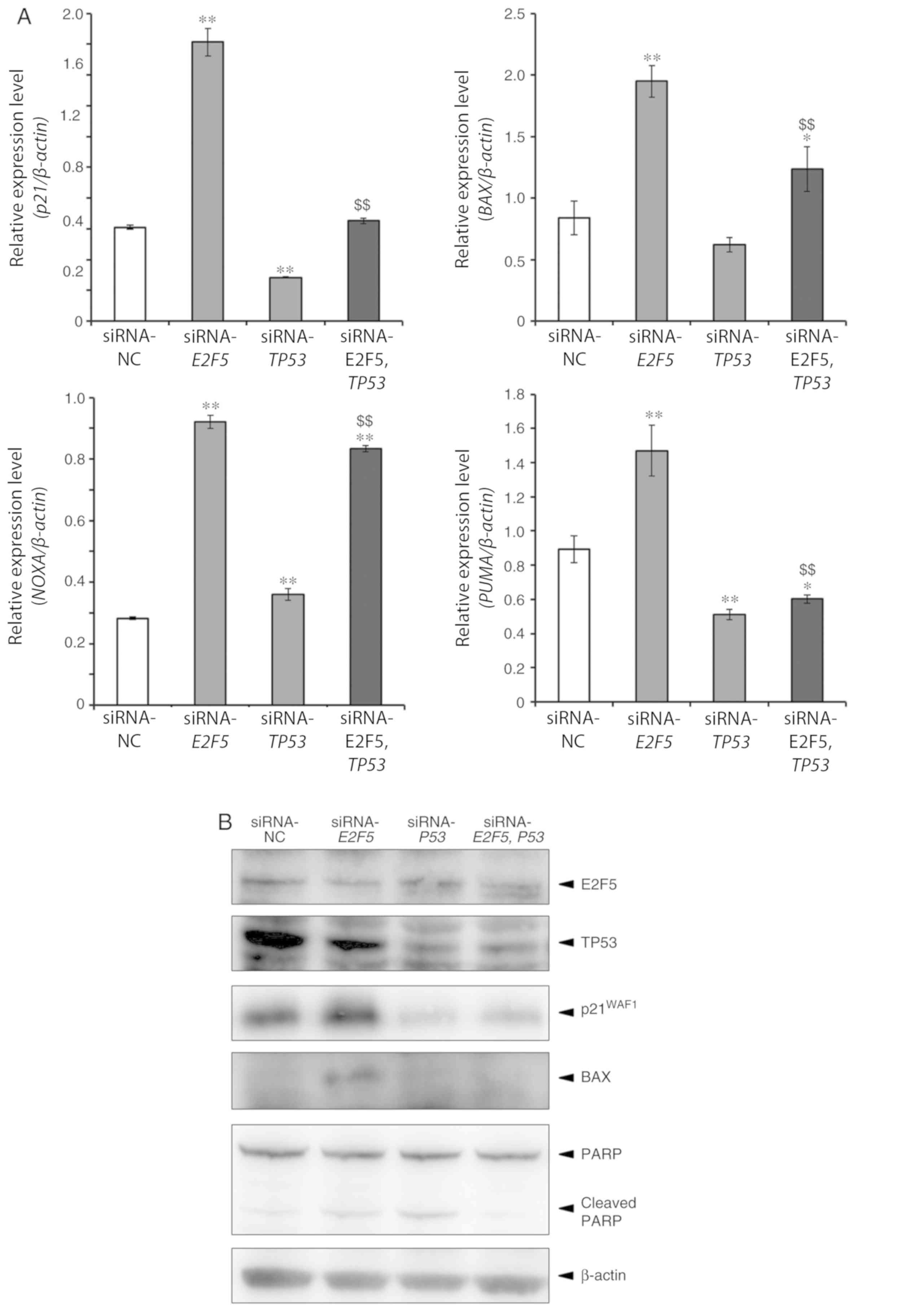
theory. Real-time RT-qPCR experiments revealed that several
TP53-target genes implicated in the induction of cell cycle arrest
and/or cell death including p21WAF1, BAX, NOXA
and PUMA were significantly upregulated in
E2F5-depleted MCF7 cells compared to control cells (Fig. 4A). In contrast, E2F5 gene
silencing resulted in induction of p21WAF1 and
NOXA but not of BAX and PUMA in TP53-mutant
MDA-MB-231 cells (Fig. 4B). In
another TP53-mutant cell line BTB474, silencing of
E2F5 induced all of the TP53 target genes aforementioned
except PUMA, but to a lesser degree (Fig. S2A). To further confirm these
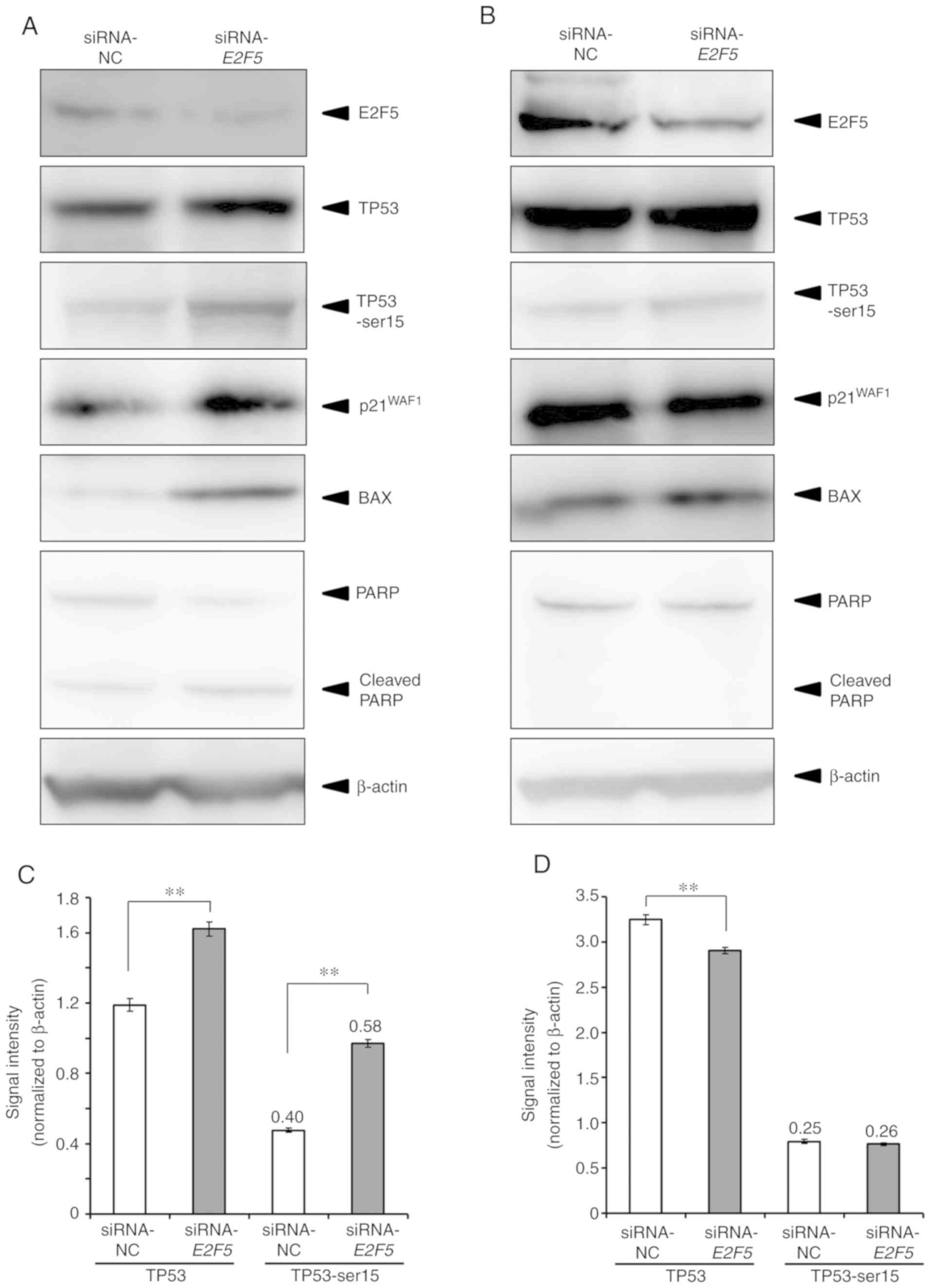
results, immunoblotting analysis was performed. Consistent with the
results of real-time RT-qPCR, E2F5 depletion promoted the
expression of p21WAF1 and BAX in MCF7 cells
(Fig. 5A). The level of cleaved
PARP was clearly increased, and the expression levels of TP53 and
phosphorylated TP53 at Ser-15 were increased in
E2F5-knockdown cells (Fig. 5A
and C). The intensity ratio of phosphorylated TP53 to TP53 was
also higher in E2F5-knockdown cells compared to control
cells (Fig. 5C). In a sharp
contrast to MCF7 cells, knockdown of E2F5 expression in
MDA-MB-231 cells had a negligible effect on the expression levels
of p21WAF1 and BAX, and the expression levels of
TP53 and phosphorylated TP53 at Ser-15 were not altered by
E2F5 knockdown (Fig. 5B and
D). Similar results as in MDA-MB-231 cells were also obtained
in BT474 cells (Fig. S2B and
C).
Silencing of TP53 cancels the effect
of E2F5 silencing in MCF7 cells
Since the results outlined above indicated that E2F5
regulates cellular proliferation and cell death in a TP53-dependent
manner in MCF7 cells, the effects of E2F5 silencing in
TP53-silenced MCF7 cells were next examined (Fig. 6A). As revealed in Fig. 6B, the viability of MCF7 cells
transfected with both E2F5 siRNA and TP53 siRNA was
almost the same as that of control cells. In addition, the
proportion of double-knockdown cells with sub-G1 DNA was also the
same as the control cells (Fig. 6C and
D). Real-time RT-qPCR analysis demonstrated that induction of
p21WAF1, BAX and PUMA by E2F5
depletion was significantly suppressed by co-depletion of TP53 in
comparison to siRNA-E2F5 (Fig. 7A).
The level of NOXA expression was also significantly reduced in the
double-knockdown cells compared to the cells in which only
E2F5 was silenced, however the extent of the reduction was
not as marked as in the other 3 genes (Fig. 7A). Consistent with these results,
induction of p21WAF1 and BAX and increase in the
level of cleaved PARP in E2F5-silenced cells were strongly
suppressed by co-transfection with TP53 siRNA (Fig. 7B).
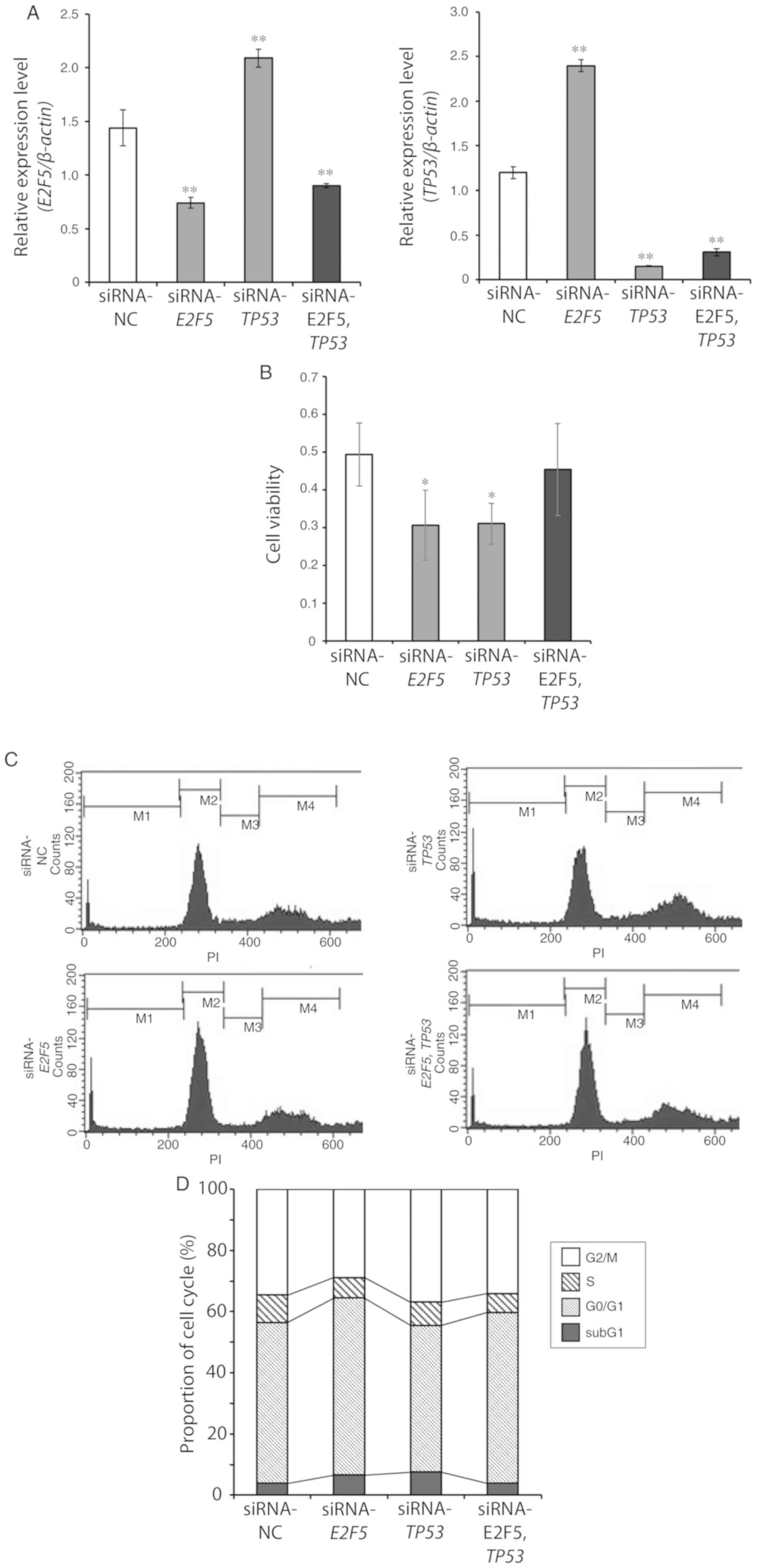 | Figure 6.Knockdown of TP53 expression
in E2F5-silenced MCF7 reveals a similar phenotype to control
cells. (A) MCF7 cells were transfected with E2F5 siRNA, TP53
siRNA, both E2F5 siRNA and TP53 siRNA, or with
control siRNA. Total RNA was extracted 48 h after transfection, and
analyzed for E2F5 and TP53 expression by real-time
RT-qPCR. β-Actin was used as an internal control. Data are
presented as the means ± SD of measurements performed in
triplicate. Significant differences with siRNA-NC were indicated as
**P<0.01. (B) MCF7 cells were transfected as described in A, and
viabilities of the cells were measured by the standard WST8 assay 4
days after transfection. Data are presented as the means ± SD of
measurements performed in triplicate. Significant differences with
siRNA-NC were indicated as *P<0.05. Knockdown of TP53
expression in E2F5-silenced MCF7 reveals a similar phenotype
to control cells. (C and D) MCF7 cells were transfected as in A,
and then floating and adherent cells were harvested, 4 days after
the transfection. Cells were stained with propidium iodide, and
their cell cycle distributions were analyzed by FACS. The
experiments were performed at least three times. Representative
histograms are presented in C. E2F5, E2F transcription factor 5;
RT-qPCR, reverse transcription-quantitative PCR; FACS,
fluorescent-activated cell sorting. |
Discussion
The results in the present study demonstrated that
knockdown of E2F5 expression decreased the proliferation
rate of breast cancer cells, including luminal-type breast
cancer-derived MCF7 cells, TNBC-derived MDA-MB-231 cells, and
HER2-positive-type BT474 cells. Cell cycle analysis revealed that
cell death was induced in E2F5-knockdown MCF7 cells, whereas
E2F5 depletion had only a marginal effect on MDA-MB-231 and
BT474 cells carrying mutant TP53. Notably, E2F5 gene
silencing in MCF7 cells stimulated the transcription of TP53-target
genes, such as p21WAF1, BAX, NOXA and
PUMA. In contrast to MCF7 cells, knockdown of E2F5 in
MDA-MB-231 cells induced the transcription of
p21WAF1 and NOXA but not of BAX and
PUMA. In BT474 cells, E2F5 knockdown induced all of
these TP53 target genes except PUMA, but to a lower extent
compared to MCF7 cells. As MCF7 cells carry the wild-type
TP53, and MDA-MB-231 and BT474 cells carry the mutated
TP53, these results indicated that E2F5 regulates cell death
through a TP53-dependent pathway in MCF7 cells. The observation
that silencing of TP53 ameliorated the effects of
E2F5 silencing in MCF7 supports this theory.
E2F4 and E2F5 are highly expressed in quiescent
cells (21), and their expression
has been revealed to be required for pocket protein-mediated G1
arrest in fibroblasts (22). Based
on these observations, E2F5 is expected to be a tumor-suppressor,
however, there is accumulating evidence that E2F5 may have a
potential pro-oncogenic function in numerous types of human cancer
(13–17). In fact, the aberrant overexpression
of E2F5 has been observed in a variety of malignancies including
breast cancer (20,24,25).
The molecular basis of the aforementioned dual roles of E2F5 has
yet to be elucidated. It has been reported that E2F4 contributes to
the formation of cyclin E repressor complex (CREC), which plays a
pivotal role in the transcription of cyclin E1 during G1 phase, and
thereby blocks cellular proliferation (26). These findings suggest that the
aberrant expression of E2F4 may cause uncontrolled cell division.
Consistent with these observations, unlike E2F4, E2F5 is not
implicated in the complex formation of CREC.
The results presented herein revealed that E2F5
regulated the cellular proliferation and cell death of MCF7 cells
in a wild-type TP53-dependent manner. In support of this theory,
knockdown of E2F5 expression resulted in upregulation of
TP53 target downstream genes involved in the induction of cell
cycle arrest and/or cell death in MCF7 cells carrying wild-type
TP53. The observations of cells with depletion of both
E2F5 and TP53 suggested that at least
p21WAF1, BAX and PUMA were induced by
E2F5 depletion in a TP53-dependent manner. Although it has
been reported that TP53 may regulate the activity of E2F4 and E2F5
(27), complex formation between
TP53 and E2F4/E2F5 was not detected (27). Similar to these previous results,
the TP53/E2F5 complex was also undetectable under our experimental
conditions (data not shown).
In contrast to MCF7 cells, E2F5 depletion in
TP53-mutant MDA-MB-231 and BT474 cells attenuated cellular
proliferation but did not trigger cell death. The expression level
of mutant TP53 is usually markedly higher than that of
wild-type TP53 (28), and
mutant TP53 is known to disturb the pro-apoptotic function
of wild-type TP53 (29). The
results of the present study also confirmed that the expression
levels of TP53 in breast cancer carrying mutant TP53 were
markedly higher than in breast cancer cells with wild-type
TP53. Therefore, it is possible that the induction of cell
death by wild-type TP53 may be suppressed by mutant
TP53 in the MDA-MB-231 and BT474 cells. The present results
revealed that the proportion of cells with G2/M DNA content was
slightly increased in E2F5-depleted MDA-MB-231 cells.
Recently, it has been reported that knockdown of E2F5
expression in prostate cancer cells resulted in activation of p38
MAPK and SMAD3 pathways, and increased the number of cells with G1
DNA content but not of cells with G2/M DNA content (30). In non-small-cell lung cancer cells,
E2F5 may act as a transcriptional repressor of
p21WAF1 (19).
However, a significant change in the expression level of
p21WAF1 by E2F5 depletion in MDA-MB-231
cells was not detected. In addition, no increase in proportion of
the cells in G2/M phase was observed in BT474 cells. Further
analyses are required to clarify the molecular mechanisms
underlying E2F5-mediated regulation of the cell cycle in
TP53-mutant breast cancer cells.
In conclusion, the present study revealed that E2F5
participated in the carcinogenesis of breast cancer carrying
wild-type TP53 through the suppression of TP53, while E2F5
had a pro-proliferative but not anti-apoptotic function in
TP53-mutated breast cancer. At present, the precise
molecular mechanisms of how TP53 could suppress the cellular
proliferation of TP53-mutated breast cancer cells has yet to be
elucidated. To adequately understand these mechanisms, further
study using an increased number of breast cancer cells should be
conducted. It is also well-known that estrogen signaling has a
crucial role in the development of breast cancer, however, we did
not extend our present study to address the functional relationship
of E2F5 and estrogen signaling. Further analysis using numerous
types of breast cancer cell lines with or without estrogen receptor
are required. Moreover, in vivo xenograft experiments may
provide insights into the vital roles of E2F5 in breast cancer
development.
Supplementary Material
Supporting Data
Acknowledgements
We thank Ms. K. Tagata (Division of General
Medicine, Department of Medicine, Nihon University School of
Medicine) for her secretarial assistance.
Funding
The present study was supported in part by
MEXT-Supported Program for the Strategic Research Foundation at
Private Universities (2011–2015) to KF, MS, and NF.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
KF, NF, MS, TO and SM planned the experiments. YIn,
DW, KF, YIs, AO, ToT, TaT and TO performed the experiments. NF
performed statistical analysis. KF, TO and SM wrote the manuscript.
KF, NF, MS, TO and SM critically revised the manuscript and all the
authors provided final approval of the manuscript to be
published.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ginsburg O, Bray F, Coleman MP, Vanderpuye
V, Eniu A, Kotha SR, Sarker M, Huong TT, Allemani C, Dvaladze A, et
al: The global burden of women's cancers: A grand challenge in
global health. Lancet. 389:847–860. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lalloo F and Evans DG: Familial breast
cancer. Clin Genet. 82:105–114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Perou CM, Sørlie T, Eisen MB, van de Rijn
M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA,
et al: Molecular portraits of human breast tumours. Nature.
406:747–752. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sørlie T, Perou CM, Tibshirani R, Aas T,
Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey
SS, et al: Gene expression patterns of breast carcinomas
distinguish tumor subclasses with clinical implications. Proc Natl
Acad Sci USA. 98:10869–10874. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sorlie T, Tibshirani R, Parker J, Hastie
T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, et
al: Repeated observation of breast tumor subtypes in independent
gene expression data sets. Proc Natl Acad Sci USA. 100:8418–8423.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Coussens L, Yang-Feng TL, Liao YC, Chen E,
Gray A, McGrath J, Seeburg PH, Libermann TA, Schlessinger J,
Francke U, et al: Tyrosine kinase receptor with extensive homology
to EGF receptor shares chromosomal location with neu oncogene.
Science. 230:1132–1139. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Akiyama T, Sudo C, Ogawara H, Toyoshima K
and Yamamoto T: The product of the human c-erbB-2 gene: A
185-kilodalton glycoprotein with tyrosine kinase activity. Science.
232:1644–1646. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Perou CM: Molecular stratification of
triple-negative breast cancers. Oncologist. 16 (Suppl 1):S61–S70.
2011. View Article : Google Scholar
|
|
9
|
Xanthoulis A and Tiniakos DG: E2F
transcription factors and digestive system malignancies: How much
do we know? World J Gastroenterol. 19:3189–3198. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lees JA, Saito M, Vidal M, Valentine M,
Look T, Harlow E, Dyson N and Helin K: The retinoblastoma protein
binds to a family of E2F transcription factors. Mol Cell Biol.
13:7813–7825. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dyson N: The regulation of E2F by
pRB-family proteins. Genes Dev. 12:2245–2262. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stevaux O and Dyson NJ: A revised picture
of the E2F transcriptional network and RB function. Curr Opin Cell
Biol. 14:684–691. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao J, Wu XY, Ling XH, Lin ZY, Fu X, Deng
YH, He HC and Zhong W: Analysis of genetic aberrations on
chromosomal region 8q21-24 identifies E2F5 as an oncogene with copy
number gain in prostate cancer. Med Oncol. 30:4652013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ishimoto T, Shiozaki A, Ichikawa D,
Fujiwara H, Konishi H, Komatsu S, Kubota T, Okamoto K, Nakashima S,
Shimizu H, et al: E2F5 as an independent prognostic factor in
esophageal squamous cell carcinoma. Anticancer Res. 33:5415–5420.
2013.PubMed/NCBI
|
|
15
|
Kothandaraman N, Bajic VB, Brendan PN,
Huak CY, Keow PB, Razvi K, Salto-Tellez M and Choolani M: E2F5
status significantly improves malignancy diagnosis of epithelial
ovarian cancer. BMC Cancer. 10:642010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu G, Sun Y, An S, Xin S, Ren X, Zhang D,
Wu P, Liao W, Ding Y and Liang L: MicroRNA-34a targets FMNL2 and
E2F5 and suppresses the progression of colorectal cancer. Exp Mol
Pathol. 99:173–179. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jiang Y, Yim SH, Xu HD, Jung SH, Yang SY,
Hu HJ, Jung CK and Chung YJ: A potential oncogenic role of the
commonly observed E2F5 overexpression in hepatocellular carcinoma.
World J Gastroenterol. 17:470–477. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zou C, Li Y, Cao Y, Zhang J, Jiang J,
Sheng Y, Wang S, Huang A and Tang H: Up-regulated MicroRNA-181a
induces carcinogenesis in hepatitis B virus-related hepatocellular
carcinoma by targeting E2F5. BMC Cancer. 14:972014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Donzelli S, Fontemaggi G, Fazi F, Di
Agostino S, Padula F, Biagioni F, Muti P, Strano S and Blandino G:
MicroRNA-128-2 targets the transcriptional repressor E2F5 enhancing
mutant p53 gain of function. Cell Death Differ. 19:1038–1048. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Umemura S, Shirane M, Takekoshi S,
Kusakabe T, Itoh J, Egashira N, Tokuda Y, Mori K and Osamura YR:
Overexpression of E2F-5 correlates with a pathological basal
phenotype and a worse clinical outcome. Br J Cancer. 100:764–771.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hijmans EM, Voorhoeve PM, Beijersbergen
RL, van't Veer LJ and Bernards R: E2F-5, a new E2F family member
that interacts with p130 in vivo. Mol Cell Biol. 15:3082–3089.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gaubatz S, Lindeman GJ, Ishida S, Jakoi L,
Nevins JR, Livingston DM and Rempel RE: E2F4 and E2F5 play an
essential role in pocket protein-mediated G1 control. Mol Cell.
6:729–735. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bouaoun L, Sonkin D, Ardin M, Hollstein M,
Byrnes G, Zavadil J and Olivier M: TP53 Variations in Human
Cancers: New Lessons from the IARC TP53 Database and Genomics Data.
Hum Mutat. 37:865–876. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Polanowska J, Le Cam L, Orsetti B, Vallés
H, Fabbrizio E, Fajas L, Taviaux S, Theillet C and Sardet C: Human
E2F5 gene is oncogenic in primary rodent cells and is amplified in
human breast tumors. Genes Chromosomes Cancer. 28:126–130. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ali A, Ullah F, Ali IS, Faraz A, Khan M,
Shah ST, Ali N and Saeed M: Aberrant promoter methylation at CpG
cytosines induce the upregulation of the E2F5 gene in breast
cancer. J Breast Cancer. 19:133–141. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Le Cam L, Polanowska J, Fabbrizio E,
Olivier M, Philips A, Ng Eaton E, Classon M, Geng Y and Sardet C:
Timing of cyclin E gene expression depends on the regulated
association of a bipartite repressor element with a novel E2F
complex. EMBO J. 18:1878–1890. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vaishnav YN and Pant V: Differential
regulation of E2F transcription factors by p53 tumor suppressor
protein. DNA Cell Biol. 18:911–922. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Strano S, Dell'Orso S, Mongiovi AM, Monti
O, Lapi E, Di Agostino S, Fontemaggi G and Blandino G: Mutant p53
proteins: between loss and gain of function. Head Neck. 29:488–496.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ozaki T and Nakagawara A: Role of p53 in
cell death and human cancers. Cancers (Basel). 3:994–1013. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Majumder S, Bhowal A, Basu S, Mukherjee P,
Chatterji U and Sengupta S: Deregulated E2F5/p38/SMAD3 circuitry
reinforces the pro-tumorigenic switch of TGFβ signaling in prostate
cancer. J Cell Physiol. 231:2482–2492. 2016. View Article : Google Scholar : PubMed/NCBI
|