Introduction
Retinoblastoma is the most common intraocular
malignant tumour that threatens the health of children, affecting
1:15,000-1:20,000 live births (1).
Current treatments for retinoblastoma include chemotherapy, plaque
radiotherapy, external beam radiotherapy, cryotherapy and surgery
(2). However, despite the clinical
response to such comprehensive strategies, tumour metastasis and
extraocular invasion in the advanced stage still cause death in
children (3,4). Therefore, investigation of the
underlying mechanism of retinoblastoma tumourigenesis and
development is necessary to improve therapeutic efficacy.
A growing body of literature has reported that the
microenvironment regulates tumour progression and metastasis
(5,6). Macrophages are plastic cells and have
multiple bioactivities in response to environmental signals.
Interferon and TLR ligands could orient macrophage function toward
the M1 phenotype (high levels of proinflammatory), while IL-4 and
IL-13 activate the M2 phenotype (tissue remodelling and promotion
of tumour progression) (7). The
tumour microenvironment can induce macrophages to adopt a
tumour-promoting state (7). IL-6,
TNF-α and monocyte chemotactic protein-1 (MCP-1) are highly
expressed in resident tumour-associated macrophages (TAMs) or in
macrophages derived from peripheral reservoirs, such as the bone
marrow (BM) and spleen (8,9). TAMs contribute to matrix breakdown and
tumour cell motility (10).
However, the mechanism of activation of these cells by tumour cells
is not well defined.
Exosomes (EXOs) are endocytosis-derived small
membrane vesicles (40–150 nm) that are composed of proteins,
lipids, microRNA (miRNA/miR) and mRNA surrounded by a phospholipid
bilayer and are secreted into the extracellular space (11). A growing number of studies have
demonstrated that EXOs and other extracellular vesicles secreted by
tumour cells play an important role in cell-to-cell communication
(12–16). EXOs can bind to target cells, fuse
with the membrane and transfer their contents to mediate
intercellular communication. Additionally, tumour-derived EXOs can
facilitate tumour malignancy (17).
Moreover, previous reports have verified that tumour-derived EXOs
contribute to the establishment of a premetastatic niche and
generate oncogenic microenvironments at distant metastatic sites by
modulating stromal cells and remodelling the extracellular matrix
(18,19).
Nevertheless, very little is known concerning the
possible functions of retinoblastoma cell EXOs in tumour
progression. Based on the aforementioned evidence, the aim of the
present study was to determine the effects of retinoblastoma cell
EXOs to elucidate the possible underlying mechanisms of
retinoblastoma deterioration.
Materials and methods
Cell culture
WERI-Rb1 human retinoblastoma cells (American Type
Culture Collection) were cultured in RPMI-1640 (Gibco; Thermo
Fisher Scientific, Inc.) with 10% foetal bovine serum (FBS; Gibco;
Thermo Fisher Scientific, Inc.). Murine macrophages RAW264.7 (The
Cell Bank of Type Culture Collection of the Chinese Academy of
Sciences) were cultured in DMEM (Gibco; Thermo Fisher Scientific,
Inc.) with 10% FBS. Bone marrow mesenchymal stem cells (BMSCs) were
acquired from C57 mice and BMSCs from passages 4–6 were cultured in
DMEM with 10% FBS for use (8×104/ml). These cells were
incubated in a humidified atmosphere with a mixture of 1%
O2, 5% CO2 and 94% N2 at 37°C.
Primary BMSC isolation
BMSCs were isolated from the bone marrow of C57 BL/6
mice using previously described methods (20). Briefly, 4–6 week old C57 BL/6 mice
were anesthetized by intraperitoneal injection of Nembutal (30
mg/kg; cat. no. P3761, Sigma-Aldrich; Merck KGaA) and then
sacrificed by cervical dislocation, and their tibias and femurs
were separated aseptically. DMEM was used to flush the marrow out
with a sterile 25-guage needle and the extruded BM cells were
filtered through a 70-µm cell strainer (BD Biosciences) and
centrifuged at 1,000 × g for 5 min at 37°C. The BM cells were
resuspended with DMEM containing 10% FBS and plated into flasks.
The cells were treated with 0.25% trypsin/1 mM EDTA for 2 min until
the cells reached 90% confluence. The culture medium was changed
every 3 days. After 3–4 passages, isolated cells were used for
experiments.
EXO isolation, characterization and
treatment
EXOs were purified from WERI-Rb1-derived conditioned
medium (collected from 48 h cell cultures with RPMI-1640 and 10%
exosome-free FBS) by ultracentrifugation, as outlined previously
(21). The FBS was depleted of EXOs
through ultracentrifugation at 110,000 × g overnight at 4°C. After
48 h, the conditioned media were centrifuged at 1,000 × g for 5
min, followed by 10,000 × g for 30 min, and then
ultracentrifugation of the supernatants was performed at 100,000 ×
g for 90 min. All centrifugation was performed at 4°C. The EXOs
were washed with phosphate-buffered saline (PBS), and then
ultracentrifugation was carried out at 100,000 × g for 90 min at
4°C, following which, EXOs were resuspended in PBS. The size and
number of EXOs were counted by Nano-Flow cytometry, according to a
previous method (22), which is the
imaging of individual fluorescently labelled EXOs passing through
nanochannels in a pressure-driven flow (23). Lastly, the protein concentration was
measured using a Bicinchoninic Acid Protein Assay Kit (Beyotime
Institute of Biotechnology).
EXOs were viewed by transmission electron microscopy
(FEI Tecnai Spirit G2; Thermo Fisher Scientific, Inc.), according
to a previous method (21).
Briefly, freshly isolated EXO pellets were transferred to a copper
grid coated with carbon in a 30-µl drop of 1% glutaraldehyde for 5
min and then the grid was washed with distilled water. The grids
were stained with 4% uranyl-acetate solution for 10 min at 37°C,
and then treated with a 50-µl drop of methyl cellulose for 5 min on
ice. Then, the grid was washed with distilled water and observed
under transmission electron microscopy at 80 Kv.
For EXO labelling, EXOs were fluorescently labelled
using the PKH26 membrane dye (Sigma-Aldrich; Merck KGaA), according
to a previous method (24).
Briefly, EXOs were resuspended into 1 ml Diluent C and 6 µl PKH26
dye was added for 5 min at room temperature. Then, the reaction was
quenched by adding 2 ml 10% BSA (cat. no. 0332; Amresco, LLC)
followed by ultracentrifugation at 100,000 × g for 2 h to remove
excess dye.
BMSCs and WERI-Rb1 co-culture
For the co-culture experiments, BMSCs
(4×104) treated with PBS or EXOs were plated in 24-well
plates. WERI-Rb1 cells (4×104) were suspended in
EXO-free medium and seeded into Transwell chambers with 0.4-µm pore
size inserts (BD Biosciences). The viability of BMSCs and WERI-Rb1
cells were assessed using a CCK-8 assay after 24 h. BMSC RNA was
extracted for RT-qPCR analysis.
Cell viability assay
The viabilities of WERI-Rb1 cells and RAW264.7
macrophages treated with EXOs or PBS were detected using a Cell
Counting Kit-8 (CCK-8) assay (Dojindo Molecular Technologies,
Inc.). Each group of cells was incubated with CCK-8 reagent for 3 h
at 37°C. Finally, the absorbance was assessed at 450 nm and
recorded.
Reverse transcription-quantitative
(RT-q)PCR analysis
Total RNA from WERI-Rb1 cells, RAW264.7 cells and
BMSCs was extracted with TRIzol® reagent (Invitrogen;
Thermo Fisher Scientific, Inc.). Total RNA (1 µg) was subjected to
reverse transcription using a PrimeScript™ RT Reagent kit (Takara
Biotechnology Co., Ltd.), following the manufacturer's protocol.
Total RNA from exosomes was extracted with TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and Dr GenTLE™
Precipitation Carrier (Takara Biotechnology Co., Ltd.). Total RNA
(1 µg) was subjected to reverse transcription using a Mir-X™ miRNA
First-Strand Synthesis Kit (Takara Biotechnology Co., Ltd.),
following the manufacturer's protocol. RT-qPCR was performed with a
SYBR Prime Script RT-PCR Kit on a Roche 480 system (Roche
Diagnostics) following the manufacturer's protocols. PCR conditions
were as follows: 94°C for 5 min, followed by 40 cycles of 94°C for
30 sec, 62°C for 30 sec, and 72°C for 30 sec. The relative target
gene expression was quantitated using the 2−ΔΔCq method
(25) and normalized to the
endogenous expression of b-actin, GAPDH or miR-16. The primers for
mRNA and miRNA detection are listed in Tables SI and SII, respectively.
Immunofluorescence assay
Cells or tissue sections (tumour, spleen and liver)
were fixed with 4% paraformaldehyde (10 min at 37°C), and then
blocked with 1% BSA and 0.2% Triton X-100 for 30 min at 37°C.
Samples were incubated with primary antibodies overnight at 4°C,
including anti-CD49b for natural killer (NK) cells (1:100; cat. no.
14-5971-85; Invitrogen; Thermo Fisher Scientific, Inc.), anti-CD45
for leukocytes (1:100; cat. no. sc-1178; Santa Cruz Biotechnology,
Inc.), anti-CD68 for macrophages (1:100; cat. no. BA3638; Wuhan
Boster Biological Technology Co., Ltd.), anti-Ki-67 for
proliferative cells (1:500; cat. no. MA5-14520; Thermo Fisher
Scientific, Inc.), anti-Vimentin for invasive tumour cells (1:100;
cat. no. sc-6260; Santa Cruz Biotechnology, Inc.). After washing
with PBS, samples were stained with the appropriate Alexa
Fluor-conjugated secondary antibody for 1 h at 37°C (Alexa Fluor
555 anti-rabbit lgG, 1:500, cat. no. 4413; Alexa Fluor 488
anti-rabbit lgG, 1:500, cat. no. 4412; Alexa Fluor 488 anti-mouse
lgG, 1:500, cat. no. 4408; all purchased from Cell Signaling
Technology, Inc.). Images were captured using fluorescence
microscopy (Axio Imager Z1; Carl Zeiss AG; magnification,
×100).
Immunofluorescence staining for cells was quantified
by analysing the fraction or number of positively-stained pixels
per total field using ImageJ software (version 1.51; National
Institutes of Health). This protocol was used in a previous study
(26). Briefly, the images were
binarized to black and white with a common threshold level such
that white pixels represented positive cells and were quantified by
histogram analysis using GraphPad Prism system 7 (GraphPad
Software, Inc.). At least five random sections per mouse were used
for quantification.
Western blotting
Whole proteins of cells and EXOs were extracted
using a RIPA lysate kit (Beyotime Institute of Biotechnology). The
protein concentration was determined with a BCA assay (Beyotime
Institute of Biotechnology). Total protein (30 µg) from each sample
was separated via 10% sodium dodecyl sulfate-polyacrylamide gel by
electrophoresis, and subsequently transferred to a polyvinylidene
difluoride membrane. The membranes were blocked with 5% BSA for 2 h
at 37°C, then incubated with primary antibodies overnight at 4°C.
The following primary antibodies were used: Anti-GAPDH (1:10,000;
cat. no. 10494-1-AP; ProteinTech Group, Inc.), anti-Tubulin
(1:1,000; cat. no. sc-5274; Santa Cruz Biotechnology, Inc.),
anti-CD63 (1:1,000; cat. no. EXOAB-CD63A-1; Systems Biosciences,
LLC), anti-tumour susceptibility gene 101 protein (1:1,000; cat.
no. ab125011; Abcam), anti-CD9 (1:1,000; cat. no. 13403; Cell
Signalling Technology, Inc.), anti-heat shock 70 kDa protein 1A
(1:1,000; cat. no. 4873; Cell Signalling Technology, Inc.),
anti-proliferating cell nuclear antigen (PCNA; 1:1,000; cat. no.
13110; Cell Signalling Technology, Inc.), anti-Bax (1:1,000; cat.
no. 2772; Cell Signalling Technology, Inc.), anti-Bcl-2 (1:1,000;
cat. no. A00040-1; Wuhan Boster Biological Technology, Ltd.),
anti-Caspase-3 (1:1,000; cat. no. 9662; Cell Signalling Technology,
Inc.), anti-C-X-C chemokine receptor type 4 (CXCR4; 1:1,000; cat.
no. ab124824; Abcam), anti-MMP2 (1:1,000; cat. no. 10373-2-AP;
ProteinTech Group, Inc.) and anti-thrombospondin-1 (TSP-1; 1:1,000;
cat. no. ab1823; Abcam). Membranes were then incubated with
HRP-conjugated anti-mouse IgG (1:3,000; cat. no. 7076s; Cell
Signalling Technology, Inc.) or anti-rabbit IgG (1:10,000; cat. no.
7074s; Cell Signalling Technology, Inc.) for 1 h at 37°C, following
which the blots were visualized with an enhanced chemiluminescence
system (Thermo Fisher Scientific, Inc.). GAPDH or Tubulin were used
as loading controls. The band intensity was semi-quantified by
ImageJ software (version 1.51; National Institutes of Health).
Murine xenograft model of
retinoblastoma
A total of 20 female athymic nude mice (4–6 weeks
old) were subcutaneously injected in the left submaxillary region
with 300 µl WERI-Rb1 cells (1×107) mixed with Matrigel
(18 mg/ml, vol/vol, 1:1). One week after subcutaneous WERI-Rb1 cell
injection, successfully transplanted mice (tumour volume, >200
mm3) were randomly divided into two groups (n=10) and
subcutaneously injected with an equal volume of PBS or EXOs (75 µg)
into the site of primary tumours every 3 days for 15 days. The
tumour sizes of the mice were measured and the tumour volume was
calculated as the length × width2/2 every 5 days. Mice
were anesthetized by intraperitoneal injection of Nembutal (30
mg/kg) and then sacrificed by cervical dislocation 15 days after
PBS or EXO injection. Tumours, spleens and livers were harvested
and stored at −80°C. Sham surgery that did not include the
injection of tumour cells, PBS and EXOs was performed on three
female athymic nude mice (4–6 weeks old).
Ethics statement
A total of 23 female nude mice (age, 4–6 weeks;
weight, 16–18 g) and four C57 BL/6 mice (age, 4–6 weeks; weight,
16–20 g) were obtained from the Ophthalmic Animal Laboratory,
Zhongshan Ophthalmic Center, Sun Yat-sen University (Guangzhou,
China). All animal experiments adhered to the ARVO Statement for
the Use of Animals in Ophthalmic and Vision Research (27) and were approved and monitored by the
Institutional Animal Care and Use Committee of Zhongshan Ophthalmic
Center [approval no. SYXK (YUE) 2018-101, 2018-168, 2019-009]. The
mice had free access to food and water and were maintained under a
12-h light/dark cycle in an air-conditioned room (16-26°C and
40–70%).
Statistical analysis
Data are presented as the mean ± standard deviation,
and the differences between mean values were evaluated using a
Student's two-tailed t-test (for two groups) or analysis of
variance followed by Tukey's post hoc (for multiple group
comparisons). P<0.05 was considered to indicate a statistically
significant difference.
Results
EXOs derived from WERI-Rb1 cells
increases cell viability
WERI-Rb1 cells grew in loose grape-like clusters and
were cultured in EXO-free media (Fig.
1A). EXOs were isolated from cells, as described in the
materials and methods section, and observed under a transmission
electron microscopy. The diameter of vesicles was ~30–100 nm
(Fig. 1B). The NanoFCM tracking
analysis also indicated that the size distribution and number of
exosomes derived from WERI-Rb1 cells, ranged from 50 to 150 nm with
a mean size of ~77 nm (Fig. 1C).
Additionally, known exosome putative markers, including HSP70,
TSG101, CD9 and CD63, were identified by western blotting (Fig. 1D). To determine whether EXOs could
affect the growth of retinoblastoma cells, WERI-Rb1 cells were
incubated with EXOs labelled with PKH26 (red). As presented in
Fig. 1E, EXOs were taken up by
WERI-Rb1 cells at 24 h after incubation. The CCK-8 assay indicated
that the viability of WERI-Rb1 cells treated with EXOs derived from
the same cells was significantly increased compared with that of
the control (PBS, 0.122±0.002; EXO, 0.145±0.005; *P<0.05;
Fig. 1F). However, the mRNA
expression levels of VEGF, PCNA and CXCR4, which can be used to
predict cancer cell malignancy (28–30),
were not affected by EXOs [no significance (ns); Fig. 1G]. These data suggested that EXOs
derived from WERI-Rb1 cells have no significant influence on
malignancy in vitro.
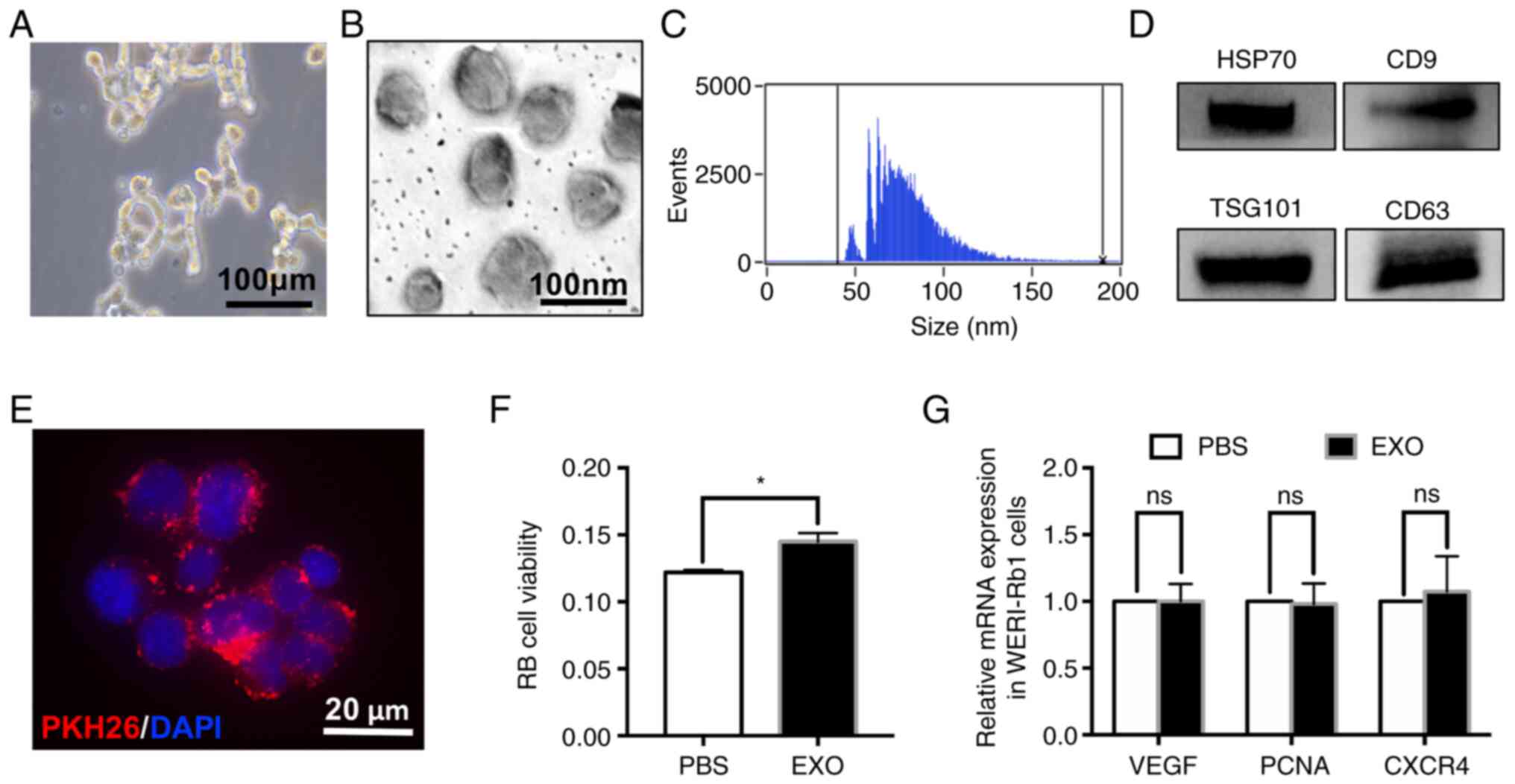 | Figure 1.EXOs derived from WERI-Rb1 cells
slightly increase cell viability. (A) Loose grape-like clusters of
WERI-Rb1 cells. Scale bar, 100 µm. (B) Representative transmission
electron microscopy images of WERI-Rb1 EXOs. The diameter of
vesicles was between 30 and 100 nm. Scale bar, 100 nm. (C)
Nano-Flow cytometry tracking analysis indicated that the size
distribution and number of EXOs derived from WERI-Rb1 cells ranged
from 50 to 150 nm with a mean size of ~77 nm. (D) Western blot
analysis of different protein markers (HSP70, CD9, TSG101 and CD63)
of EXOs collected from WERI-Rb1 cells. (E) EXOs labelled with PKH26
were engulfed by WERI-Rb1 cells. Scale bar, 20 µm. (F) PBS or EXOs
were added to WERI-Rb1 cells, and WERI-Rb1 cell viability was
measured using a Cell Counting Kit-8 assay (red, PKH26; blue,
DAPI). (G) Reverse transcription-quantitative PCR showed that there
was no significant difference between the mRNA levels of VEGF, PCNA
and CXCR4 in the PBS and EXO groups. Data are from three
independent experiments. *P<0.05. EXO, exosome; ns, no
significance; HSP70, heat shock 70 kDa protein 1A; PCNA,
proliferating cell nuclear antigen; CXCR4, C-X-C chemokine receptor
type 4; RB, retinoblastoma. |
EXOs derived from WERI-Rb1 cells
affect the antitumour activity of macrophages and BMSCs in
vitro
To reveal the effect of EXOs on immune cells,
RAW264.7 cells, which are well-characterized with regard to their
macrophage-specific immune functions (31), were incubated with EXOs from
WERI-Rb1 cells labelled with PKH26 (red). At 6 h after incubation,
EXOs could be taken up by RAW264.7 cells. PKH26 was partially
colocalized with CD68 (green, a specific marker of macrophages;
Fig. 2A). Moreover, the cells
changed from round to spindle-shaped with ramified morphology
(Fig. 2B), which indicated that
macrophages were activated. However, EXOs significantly increased
the levels of IL-6 and MCP-1 (relative fold-change: IL-6,
2.30±1.11-fold; MCP-1, 1.43±0.43-fold; *P<0.05). The increase in
IL-6 and MCP-1 expression implied that the antitumour activity of
macrophages was inhibited by EXOs, which was consistent with a
previous study (6). There was no
significant difference in TNF-α expression between the PBS and EXO
groups (relative fold-change: 1.16±0.28-fold; ns; Fig. 2C). It has been speculated that TNF-α
expression does not change in the early response of RAW 264.7 cells
to EXOs (24 h). Accordingly, treatment with EXOs also resulted in
decreased proliferation of RAW264.7 cells (PBS, 0.949±0.271; EXO,
0.303±0.124; *P<0.05; Fig.
2D).
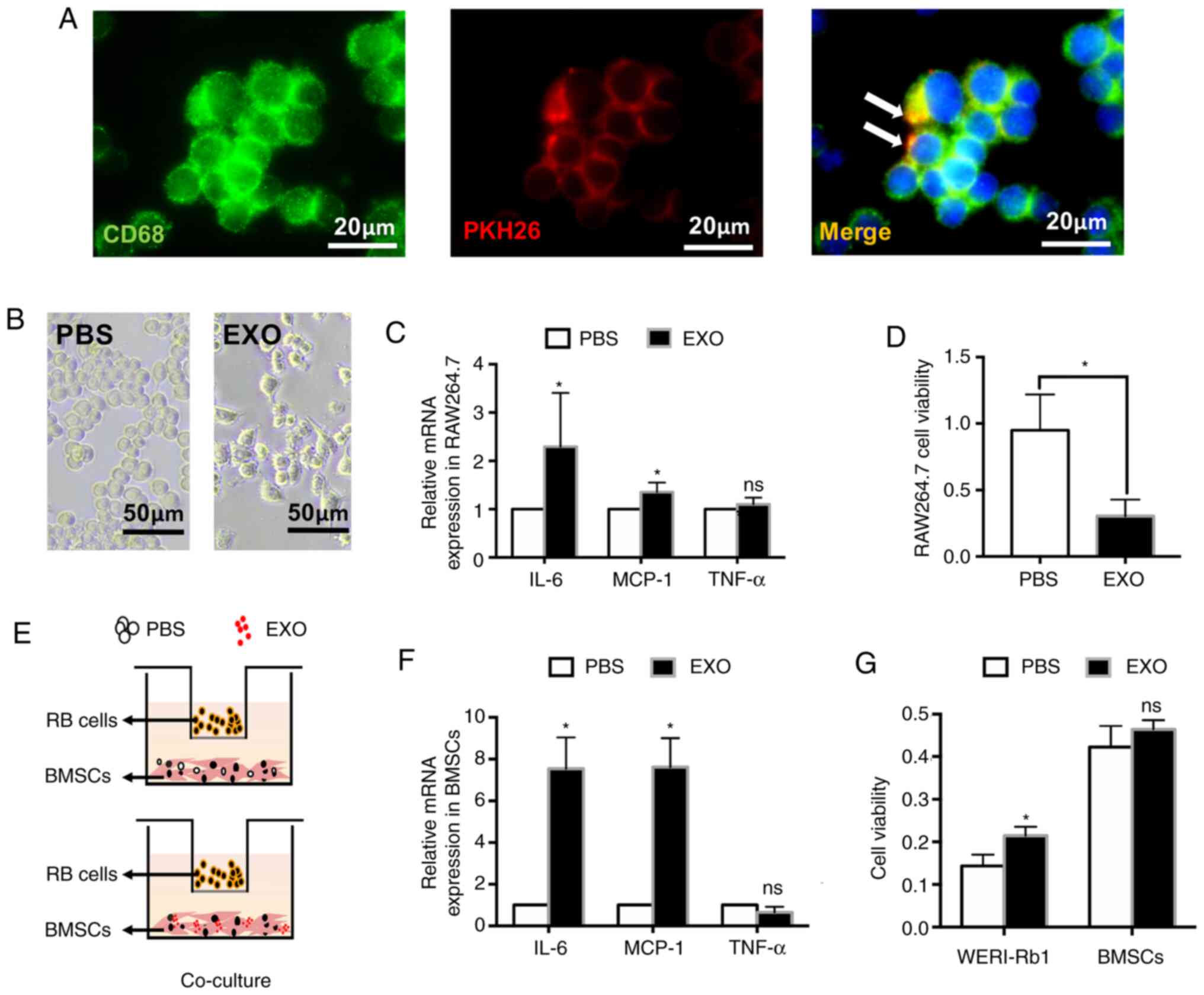 | Figure 2.EXOs derived from WERI-Rb1 cells
affect the antitumour activity of macrophages and BMSCs in
vitro. (A) EXOs labelled with PKH26 were engulfed by RAW264.7
cells. (green, CD68; red, PKH26). Scale bar, 20 µm. (B) Morphology
changes of RAW264.7 cells following treatment with EXOs. Scale bar,
50 µm. (C) RT-qPCR analysis of IL-6, MCP-1 and TNF-α expression in
RAW264.7 cells incubated with EXOs derived from WERI-Rb1 cells for
24 h. (D) Cell Counting Kit-8 assay showed that RAW264.7 cell
viability was significantly decreased by EXOs. (E) WERI-Rb1 cells
and BMSCs co-culture system, BMSCs treated with PBS or EXOs were
co-cultured with WERI-Rb1 cells for 24 h. (F) RT-qPCR analysis of
IL-6, MCP-1 and TNF-α expression in BMSCs from the co-culture
system. (G) Cell viability of WERI-Rb1 cells and BMSCs from the
co-culture system. Data are from three independent experiments.
*P<0.05 vs. PBS group. EXO, exosome; ns, no significance;
RT-qPCR, reverse transcription-quantitative PCR; BMSC, bone marrow
mesenchymal stem cells; MCP-1, monocyte chemotactic protein-1; RB,
retinoblastoma. |
To assess the effect of EXOs on the bioactivity of
BMSCs and WERI-Rb1 cells, a co-culture system was used to mimic
retinoblastoma physiological conditions. The primary BMSCs were
incubated with PBS or EXOs and co-cultured with WERI-Rb1 cells for
24 h (Fig. 2E). As shown in
Fig. 2F, EXOs could significantly
increase the expression levels of IL-6 and MCP-1 in BMSCs (relative
fold-change: IL-6, 7.54±1.50-fold; MCP-1, 7.62±1.39-fold;
*P<0.05), which could promote tumour growth and metastasis
(32,33). Similarly, there was no significant
difference in TNF-α expression between the PBS and EXO groups in
BMSCs (relative fold-change: 0.66±0.26-fold; ns). Moreover, the
viability of WERI-Rb1 cells was significantly increased (PBS,
0.14±0.03; EXO, 0.21±0.02; *P<0.05) in the co-culture system,
whereas the viability of BMSCs was not affected by EXOs (PBS,
0.42±0.05; EXO, 0.46±0.02; ns) (Fig.
2G).
Taken together, these findings demonstrated that
EXOs derived from WERI-Rb1 cells could inhibit the antitumour
activity of macrophages and induce the BMSCs to promote
retinoblastoma cell growth in vitro.
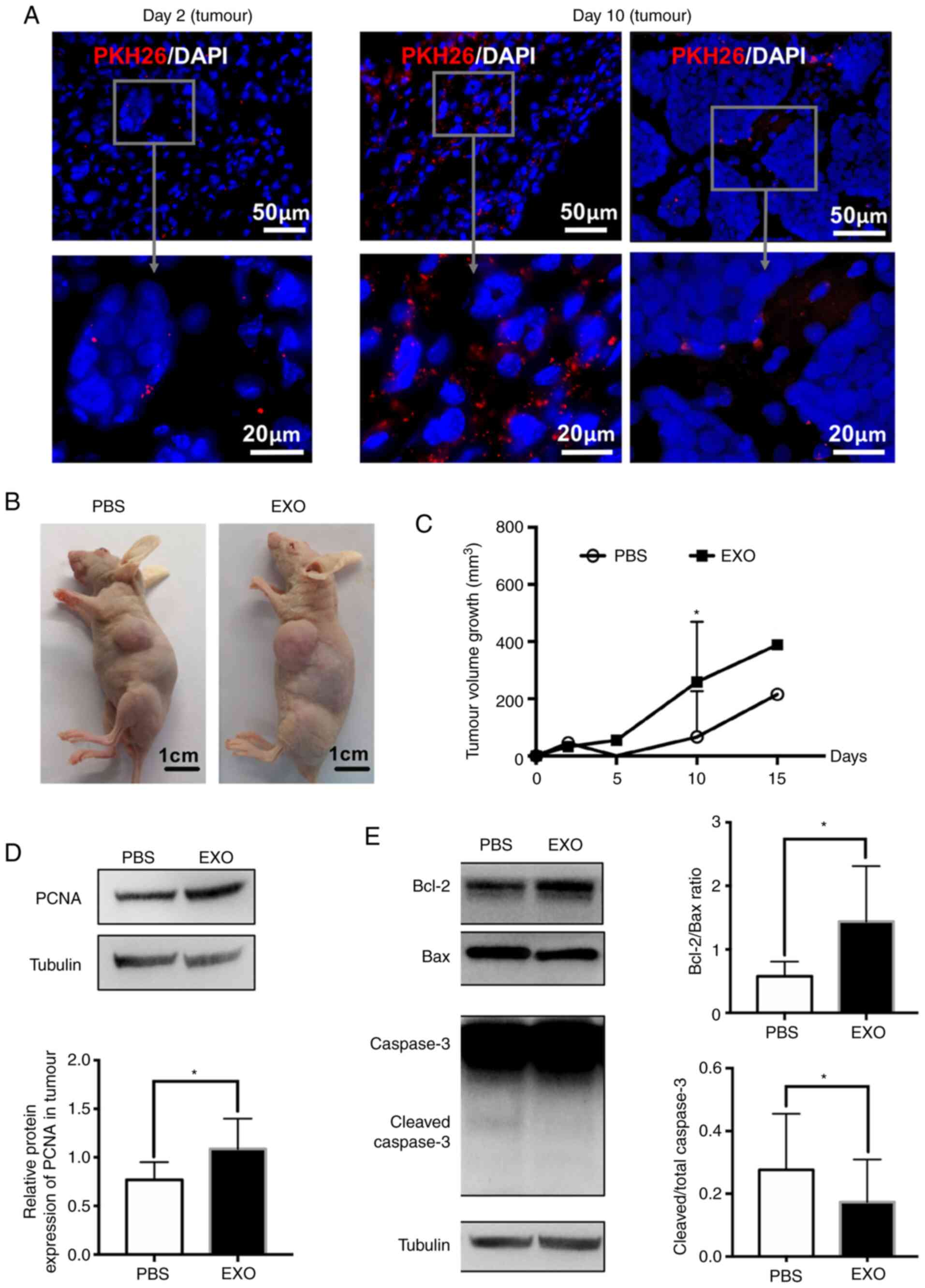
EXOs derived from WERI-Rb1 cells
increase retinoblastoma growth
To confirm the in vitro results, a
xenotransplantation model was established and then mice were
regularly injected with EXOs or PBS around the tumour. EXOs in
tumour tissues were observed after 2 days and were markedly
increased in the loose tumour tissue at day 10. It was observed
that EXOs were mainly located in loose tissue outside the tumour,
and few were detected in the dense tissue within the tumour
(Fig. 3A). However, quantification
of tumour volume growth showed that EXO treatment significantly
increased retinoblastoma tumour volume 10 days after
transplantation (PBS, 66±172 mm3; EXO, 259±224
mm3; *P<0.05; Fig. 3B and
C). Moreover, the protein levels of PCNA were significantly
elevated by EXOs in the tumour (PBS, 0.77±0.18; EXO, 1.08±0.31;
*P<0.05; Fig. 3D). Furthermore,
the protein levels of apoptosis-related Bcl-2, Bax and caspase-3
were detected. As presented in Fig.
3E, the ratio of Bcl-2 to Bax was significantly increased (PBS,
0.58±0.23; EXO, 1.44±0.87; *P<0.05), whereas the relative
expression of cleaved caspase-3 was decreased (PBS, 0.28±0.18; EXO,
0.17±0.14; *P<0.05) in the EXO group. In summary, EXOs derived
from WERI-Rb1 cells could inhibit tumour apoptosis and promote
tumour growth in vivo.
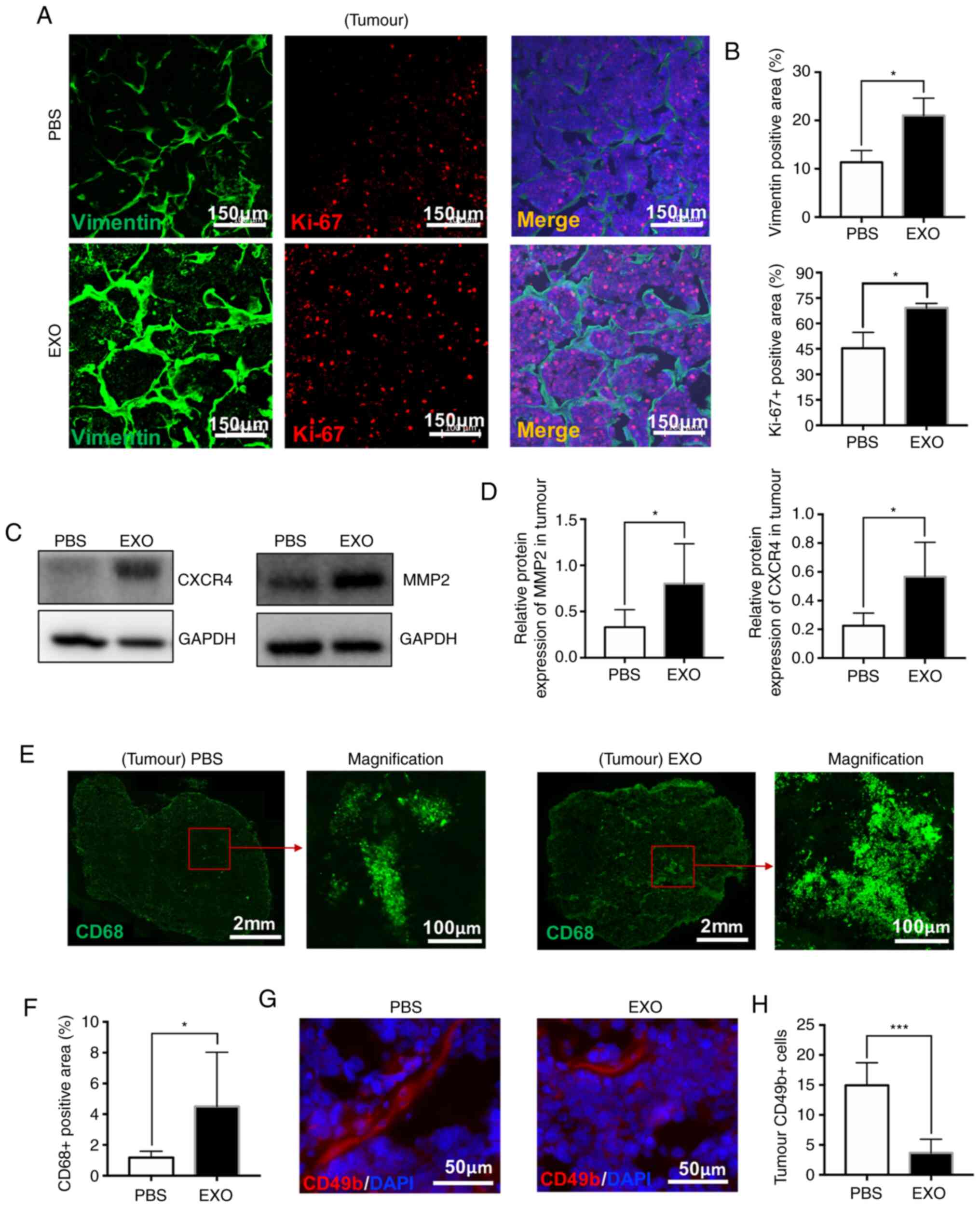
EXOs derived from WERI-Rb1 cells could
promote the malignancy of retinoblastoma
Ki-67 is widely used to measure tumour proliferation
and assess the prognosis of patients with cancer (34). Vimentin, an intermediate filament
protein, is often used as a marker of invasive tumour cells due to
its expression during activation of the epithelial-mesenchymal
transition (35). Therefore, tissue
sections were double stained with antibodies against Ki-67 and
Vimentin. As shown in Fig. 4A and
B, the relative Vimentin-positive area (PBS, 11.35±2.43%; EXO,
21.04±3.58%; *P<0.05) and Ki-67-positive rate in cells were
significantly increased (PBS, 45.41±9.39; EXO, 69.20±2.62;
*P<0.05) in tumour tissues injected with EXOs. Moreover, the
protein levels of CXCR4 and MMP2, which play a crucial role in
malignancy and metastasis during tumour progression (36,37),
were significantly increased (CXCR4: PBS, 0.225±0.088; EXO,
0.567±0.238; MMP2: PBS, 0.330±0.189; EXO, 0.802±0.433; *P<0.05;
Fig. 4C and D).
Furthermore, TAMs, a significant subset of
tumour-infiltrating immune cells, play an important role in tumour
growth and metastasis (38,39). Therefore, TAMs were evaluated in
whole tumour sections using staining with an anti-CD68 antibody
(Fig. 4E). Accordingly, the
CD68-positive area was significantly increased in tumour tissues
treated with EXOs compared with that in control tissue (PBS,
1.18±0.41%; EXO, 4.50±3.53%; *P<0.05; Fig. 4F). NK cells labelled with CD49b
(red) surrounded tumour cells and were decreased in the EXO-treated
group (PBS, 14.94±3.77; EXO, 3.66±2.29; ***P<0.001; Fig. 4G and H). Therefore, these results
also partially supported the aforementioned results that
demonstrated that exosomes derived from WERI-Rb1 cells promoted
tumour growth and malignancy.
EXOs derived from WERI-Rb1 cells
inhibit innate immunity in vivo
Although athymic nude mice with immunodeficiency
were used in the present study, the mice still exhibited intact
innate and adaptive immunity. Thus, the spleen was analysed to
determine whether EXOs affect immune processes. Of note, it was
found that EXOs could diffuse into the spleen after 2 days and
continue to accumulate at day 10 (Fig.
5A and B). EXOs were also observed in CD68-positive macrophages
in spleen tissues (Fig. 5C).
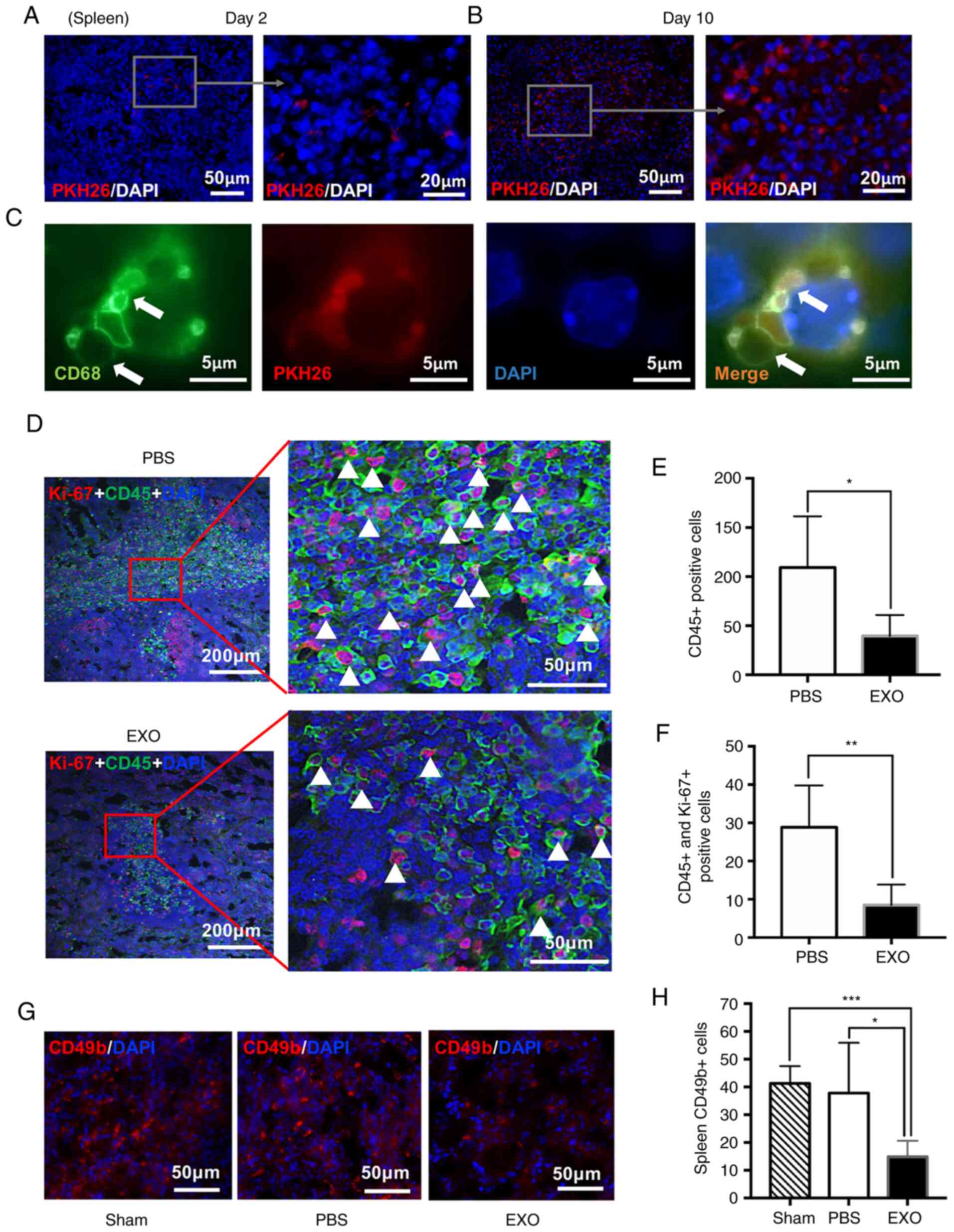 | Figure 5.EXOs derived from WERI-Rb1 cells
inhibit innate immunity in vivo. PKH26 EXOs (red) were
internalized by cells from spleen tissues (A) 2 or (B) 10 days
following EXO injection. Scale bar, 50 or 20 µm. (C) EXOs labelled
by PKH26 were engulfed by CD68-positive macrophages in the spleen
(green, CD68; red, PKH26; blue, DAPI). Scale bar, 5 µm. (D) Ki-67
(red) and CD45 (green) were labelled by immunofluorescence in
spleens from the PBS-injected or EXO-injection groups. Scale bar,
200 or 50 µm. (E) Quantification data showed CD45-positive cells in
spleens from the PBS-injected and EXO-injected groups (n=5). (F)
Quantification of CD45 and Ki-67 double-positive cells in spleens
from the PBS-injected and EXO-injected groups (n=5). (G) Staining
of CD49b (red) in spleens from the Sham (normal mice), PBS-injected
and EXO-injected groups. Scale bar, 50 µm. (H) Quantification of
CD49b-positive cells in spleens from the Sham (n=3), PBS-injected
(n=5) and EXO-injected groups (n=5). *P<0.05, **P<0.01 and
***P<0.001. EXO, exosome. |
Additionally, NK cells can limit tumourigenesis in
the absence of functioning T-lymphocytes (40,41).
Therefore, spleen tissues were double stained with antibodies
against Ki-67 and CD45 to determine the number and proliferative
activity of leukocytes (Fig. 5D).
CD45-positive leukocytes, which include granulocytes, monocytes and
lymphocytes, control the innate immune response (42). The present data showed that EXOs
significantly decreased the number of leukocytes in the spleen
(PBS, 109.30±52.11; EXO, 37.54±21.42; *P<0.05; Fig. 5E). More specifically, EXOs also
significantly decreased the ratio of cells co-expressing Ki-67 and
CD45 (white triangle), which indicated that compared with the
control, EXOs inhibited leukocyte proliferation in the spleen (PBS,
28.87±10.85; EXO, 8.47±5.39; **P<0.01; Fig. 5F). Similarly, immunofluorescence
staining of CD49b, a pan-marker of NK cells (43), indicated that NK cells were
significantly decreased in the spleens of the EXO-injected groups
compared with the sham operation and PBS-injected groups (sham,
41.28±6.24; PBS, 37.81±18.08; EXO, 14.84±5.84; *P<0.05,
***P<0.001; Fig. 5G and H).
Collectively, these data demonstrated that EXOs inhibited the
innate immune response in vivo.
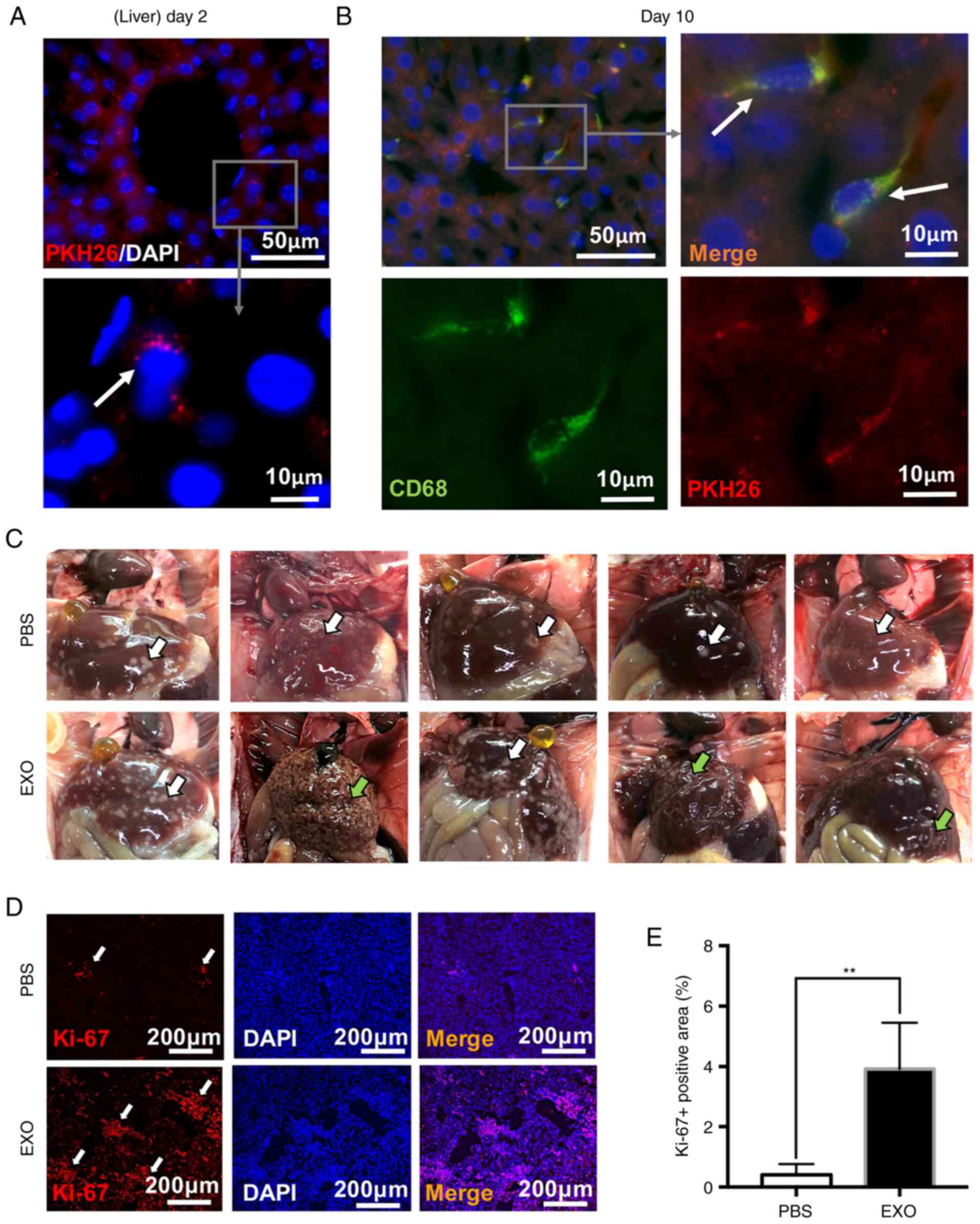
EXOs derived from WERI-Rb1 cells
accelerate retinoblastoma metastasis
To explore the effects of EXOs on retinoblastoma
metastasis, the liver, which was the closest organ to the
subcutaneous tumour, was analysed. As shown in Fig. 6A, EXOs were observed in the cells
around the liver sinusoids 2 days following the EXO injection and
only appeared in the cells near the liver sinusoids. At day 10,
EXOs (red) appeared in distant liver tissues and were mainly
engulfed by Kupffer cells (CD68-positive), demonstrating that EXOs
could spread throughout liver tissues and accumulate in macrophages
(white arrows; Fig. 6B).
Additionally, macroscopic images of liver metastatic nodules shown
in Fig. 6C indicated that EXO
injection led to more metastatic lesions (white arrowheads,
metastatic liver nodules). Three livers from the EXO-injection
group were cirrhotic, which indicated liver deterioration (green
arrowheads, liver cirrhosis). Then, the excised livers were stained
with Ki-67. It was found that livers from the EXO-injected group
exhibited increased Ki-67-positive areas, which was consistent with
metastatic nodules in the liver tissues (white arrowheads; PBS,
0.405±0.353%; EXO, 3.926±1.533%; **P<0.01; Fig. 6D and E). Thus, EXOs derived from
WERI-Rb1 cells may exhibit a promoting effect on tumour
metastasis.
Constitution of EXOs derived from
WERI-Rb1 cells
Since EXOs derived from WERI-Rb1 cells significantly
affected tumour malignancy, innate immunity and metastasis as
aforementioned, the expression of a selection of miRNAs and
proteins involved in tumour deterioration in EXOs were analysed via
RT-qPCR and western blotting. As shown in Fig. 7A, miR-92a, miR-20a, miR-129 and
miR-17 were detected. Moreover, the chemokine receptor CXCR4 was
not only expressed in WERI-Rb1 cells but was also detected in EXOs
(Fig. 7B). TSP-1, a matricellular
protein, plays multiple roles in tumour development and was not
observed in WERI-Rb1 cells, which was consistent with our previous
study (44). However, of note,
TSP-1 was detected in EXOs. CXCR4 and TSP-1 are involved in
proliferation and TAM recruitment (45–51).
Thus, the contents of EXOs may play a key role in EXO-mediated
tumour progression.
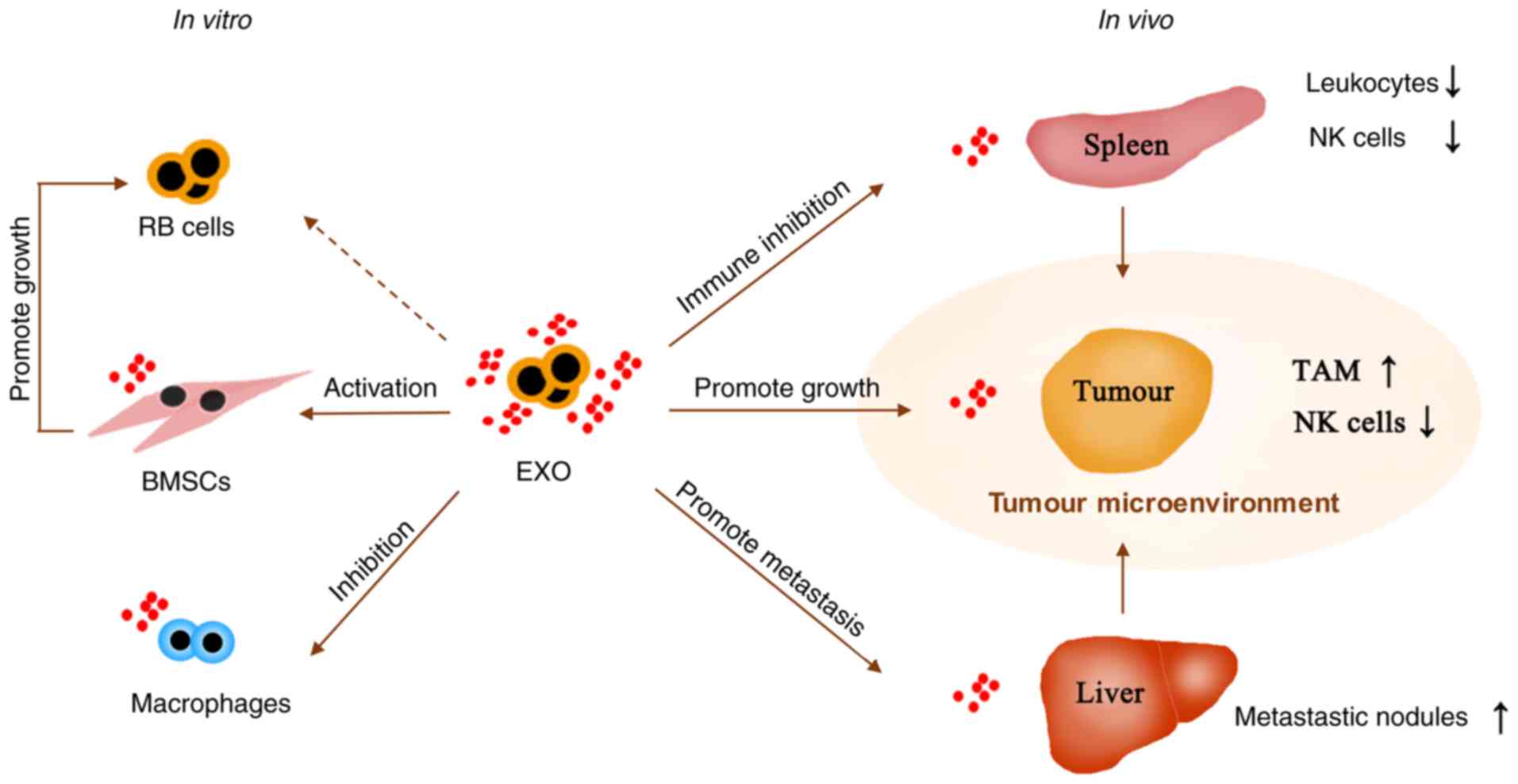
Discussion
A growing body of evidence has suggested that EXOs
play an important role in tumour invasion and metastasis (13,14,52).
EXOs derived from retinoblastoma cells may have multiple effects on
retinoblastoma development. In the present study, it was found that
EXOs could inhibit the antitumour effects of macrophages and induce
BMSCs to promote tumour growth. In vivo, through a
xenotransplantation model in nude mice, the data showed that EXOs
significantly increased tumour growth. Remarkably, EXOs injected
around the tumour infiltrated the tumour tissue, spleen and liver.
The changes in TAMs, leukocytes and NK cells induced antitumour
activity that was inhibited by EXO infiltration of these tissues.
Analysis of the contents of EXOs showed that EXOs were rich in
miRNAs and proteins involved in tumour deterioration. Thus, these
data provided direct evidence, both in vitro and in
vivo, to support the notion that EXOs derived from
retinoblastoma promote tumour progression by infiltrating the
microenvironment.
Notably, the present study showed that EXOs derived
from WERI-Rb1 cells slightly increased the growth of those cells
in vitro (Fig. 1F), but had
a more significant effect in vivo (Fig. 3C). After incubation with EXOs,
WERI-Rb1 cell viability was slightly increased. The expression
levels of PCNA, a specific cell proliferation marker, were the same
in cells treated with EXOs compared with the control (Fig. 1G). However, EXOs extracted from
WERI-Rb1 cells significantly enhanced PCNA protein levels and
inhibited tumour apoptosis in vivo (Fig. 3E and F). The ratio of Ki-67-positive
cells following EXO treatment was significantly higher than that
after control treatment (Fig. 4B).
Tumour growth in the EXO-injected group was increased ~4.0-fold 10
days after treatment (Fig. 3B and
C). Therefore, the inconsistency between tumour proliferative
activity in vitro and in vivo indicated that EXOs
promote tumour growth by affecting the peripheral
microenvironment.
Indeed, it was found that the viability of
macrophages was significantly inhibited by EXOs in vitro.
The inflammatory cytokines IL-6 and MCP-1 were increased in
macrophages incubated with EXOs (Fig.
2). Both IL-6 and MCP-1 could further contribute to tumour
growth and metastasis (6).
Similarly, IL-6 and MCP-1 levels were also increased in BMSCs,
leading to elevated WERI-Rb1 cell viability. Moreover, EXOs could
exacerbate the microenvironment in xenograft tumours. EXOs
infiltrated and accumulated in the tumour, spleen and liver. TAMs
were enhanced and NK cells were decreased in tumours injected with
exosomes (Fig. 4). Similarly,
PKH26-labelled EXOs were directly taken up by macrophages in the
spleen. CD45-positive leukocytes and NK cells, a type of lymphocyte
that predominantly mediates innate antitumour immunity, were
significantly decreased in spleens with EXO treatment (Fig. 5). Moreover, EXOs appeared in distant
liver tissues and were mainly engulfed by Kupffer cells (Fig. 6B). In accordance with the roles of
TAMs, leukocytes and NK cells, EXOs derived from WERI-Rb1 cells
accelerated the proliferation, malignancy and metastasis of
retinoblastoma in vivo. Therefore, the present findings
indicated that EXOs modify the tumour microenvironment and promote
tumour progression (Fig. 8).
The contents of EXOs include small non-coding RNA
molecules, such as miRNAs, piwi-interacting RNAs, tRNA-derived
small RNAs, and a series of proteins (53). It was found that miRNAs (miR-92a,
miR-20a, miR-129 and miR-17), CXCR4 and TSP-1 were detectable in
EXOs derived from retinoblastoma cells. According to previous
studies, miRNAs (miR-92a, miR-20a, miR-129 and miR-17), CXCR4 and
TSP-1 are involved in TAM recruitment and proliferation (45–51).
For example, miR-92a stimulates the secretion of the
proinflammatory cytokine IL-6 in TAMs, which in turn promotes
tumour cell proliferation, invasion and metastasis by interacting
with the surrounding microenvironment (46). CXCR4 accelerates TAM recruitment
(36,48). However, the role of TSP-1 expression
in tumorigenesis is complex and controversial. Some studies have
reported that TSP-1 could inhibit tumour angiogenesis and suppress
tumour growth in various types of cancer, such as cervical cancer,
lung cancer and melanoma (54,55).
Other studies have suggested that TSP-1 can be detected in multiple
types of cancers, including prostate cancer, breast cancer and
cutaneous melanoma, and is associated with tumour proliferation,
migration and metastasis, leading to a poor prognosis (56–59).
Specifically, Xiao et al (49) reported that EXO-transferred TSP-1
can activate TAMs and promote malignant migration in oral squamous
cell carcinoma, which is consistent with the present results. Based
on the aforementioned previous studies, the function of elevated
TSP1 expression in retinoblastoma EXOs may promote retinoblastoma
progression. However, a limitation of the present study was that
only a select few miRNAs and proteins were investigated. The
overall identification of protein and miRNA profiles of EXOs will
be performed in future studies.
Overall, the present study elucidated the role and
function of EXOs derived from retinoblastoma cells in
retinoblastoma growth and metastasis. The findings of this study
suggested that retinoblastoma EXOs may be a therapeutic target that
can be exploited to inhibit retinoblastoma by regulating its
composition; alternatively, inhibitors that interfere with EXO
uptake could be developed. Thus, the present study warrants future
clinical exploration of the suppression of retinoblastoma with EXOs
in the paediatric population.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81670848)
and the Science and Technology Planning Project of Guangzhou City
(grant no. 201803010091).
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
SC and XC designed and performed the study, analysed
the data and drafted the manuscript. JQ, PC, XH and YW assisted in
performing the experiments and analysed the data. JieZ, MY, CW, NW
and YY participated in collecting data and organizing the figures.
JG was involved in designing the study and provided professional
advice. SC and JinZ reviewed the manuscript and organized the
figures. KY and JinZ designed the study, analysed the data,
administered the project and supervised the manuscript. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments adhered strictly to the ARVO
Statement for the Use of Animals in Ophthalmic and Vision Research
and were approved and monitored by the Institutional Animal Care
and Use Committee of the Zhongshan Ophthalmic Center [approval no.
SYXK (YUE) 2018-101, 2018-168, 2019-009].
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jain M, Rojanaporn D, Chawla B, Sundar G,
Gopal L and Khetan V: Retinoblastoma in Asia. Eye (Lond). 33:87–96.
2019. View Article : Google Scholar
|
|
2
|
Francis JH, Roosipu N, Levin AM, Brodie
SE, Dunkel IJ, Gobin YP and Abramson DH: Current treatment of
bilateral retinoblastoma: The impact of intraarterial and
intravitreous chemotherapy. Neoplasia. 20:757–763. 2018. View Article : Google Scholar
|
|
3
|
Abramson DH, Shields CL, Munier FL and
Chantada GL: Treatment of retinoblastoma in 2015: Agreement and
disagreement. JAMA Ophthalmol. 133:1341–1347. 2015. View Article : Google Scholar
|
|
4
|
Xu L, Li W, Shi Q, Wang M, Li H, Yang X
and Zhang J: MicroRNA-936 inhibits the malignant phenotype of
retinoblastoma by directly targeting HDAC9 and deactivating the
PI3K/AKT pathway. Oncol Rep. 43:635–645. 2020.
|
|
5
|
Quail DF and Joyce JA: Microenvironmental
regulation of tumor progression and metastasis. Nat Med.
19:1423–1437. 2013. View
Article : Google Scholar
|
|
6
|
Kitamura T, Qian BZ and Pollard JW: Immune
cell promotion of metastasis. Nat Rev Immunol. 15:73–86. 2015.
View Article : Google Scholar
|
|
7
|
Sica A and Mantovani A: Macrophage
plasticity and polarization: In vivo veritas. J Clin Invest.
122:787–795. 2012. View
Article : Google Scholar
|
|
8
|
Kim J and Bae JS: Tumor-associated
macrophages and neutrophils in tumor microenvironment. Mediators
Inflamm. 2016:60581472016. View Article : Google Scholar
|
|
9
|
Saunderson SC, Dunn AC, Crocker PR and
McLellan AD: CD169 mediates the capture of exosomes in spleen and
lymph node. Blood. 123:208–216. 2014. View Article : Google Scholar
|
|
10
|
DeNardo DG, Brennan DJ, Rexhepaj E,
Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD,
Junaid SA, et al: Leukocyte complexity predicts breast cancer
survival and functionally regulates response to chemotherapy.
Cancer Discov. 1:54–67. 2011. View Article : Google Scholar
|
|
11
|
Théry C, Zitvogel L and Amigorena S:
Exosomes: Composition, biogenesis and function. Nat Rev Immunol.
2:569–579. 2002. View
Article : Google Scholar
|
|
12
|
Mathieu M, Martin-Jaular L, Lavieu G and
Théry C: Specificities of secretion and uptake of exosomes and
other extracellular vesicles for cell-to-cell communication. Nat
Cell Biol. 21:9–17. 2019. View Article : Google Scholar
|
|
13
|
Peinado H, Alečković M, Lavotshkin S,
Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M,
Williams C, García-Santos G, Ghajar CM, et al: Melanoma exosomes
educate bone marrow progenitor cells toward a pro-metastatic
phenotype through MET. Nat Med. 18:883–891. 2012. View Article : Google Scholar
|
|
14
|
Maji S, Chaudhary P, Akopova I, Nguyen PM,
Hare RJ, Gryczynski I and Vishwanatha JK: Exosomal annexin II
promotes angiogenesis and breast cancer metastasis. Mol Cancer Res.
15:93–105. 2017. View Article : Google Scholar
|
|
15
|
Messenger SW, Woo SS, Sun Z and Martin
TFJ: A Ca2+-stimulated exosome release pathway in cancer cells is
regulated by Munc13-4. J Cell Biol. 217:2877–2890. 2018. View Article : Google Scholar
|
|
16
|
Haga H, Yan IK, Takahashi K, Wood J,
Zubair A and Patel T: Tumour cell-derived extracellular vesicles
interact with mesenchymal stem cells to modulate the
microenvironment and enhance cholangiocarcinoma growth. J Extracell
Vesicles. 4:249002015. View Article : Google Scholar
|
|
17
|
Boyiadzis M and Whiteside TL: The emerging
roles of tumor-derived exosomes in hematological malignancies.
Leukemia. 31:1259–1268. 2017. View Article : Google Scholar
|
|
18
|
Feng Q, Zhang C, Lum D, Druso JE, Blank B,
Wilson KF, Welm A, Antonyak MA and Cerione RA: A class of
extracellular vesicles from breast cancer cells activates VEGF
receptors and tumour angiogenesis. Nat Commun. 8:144502017.
View Article : Google Scholar
|
|
19
|
Klingeborn M, Dismuke WM, Rickman CB and
Stamer WD: Roles of exosomes in the normal and diseased eye. Prog
Retin Eye Res. 59:158–177. 2017. View Article : Google Scholar
|
|
20
|
Soleimani M and Nadri S: A protocol for
isolation and culture of mesenchymal stem cells from mouse bone
marrow. Nat Protoc. 4:102–106. 2009. View Article : Google Scholar
|
|
21
|
Théry C, Clayton A, Amigorena S and Raposo
G: Isolation and characterization of exosomes from cell culture
supernatants and biological fluids. Curr Protoc Cell Biol. Chapter
3: Unit 3.22. 2006. View Article : Google Scholar
|
|
22
|
Tian Y, Ma L, Gong M, Su G, Zhu S, Zhang
W, Wang S, Li Z, Chen C, Li L, et al: Protein Profiling and Sizing
of Extracellular Vesicles from Colorectal Cancer Patients via Flow
Cytometry. ACS Nano. 12:671–680. 2018. View Article : Google Scholar
|
|
23
|
Friedrich R, Block S, Alizadehheidari M,
Heider S, Fritzsche J, Esbjörner EK, Westerlund F and Bally M: A
nano flow cytometer for single lipid vesicle analysis. Lab on a
Chip. 17:830–841. 2017. View Article : Google Scholar
|
|
24
|
Hoshino A, Costa-Silva B, Shen T,
Rodrigues G, Hashimoto A, Mark MT, Molina H, Kohsaka S, Giannatale
AD, Ceder S, et al: Tumour exosome integrins determine organotropic
metastasis. Nature. 527:329–335. 2015. View Article : Google Scholar
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
26
|
Paschos GK, Ibrahim S, Song WL, Kunieda T,
Grant G, Reyes TM, Bradfield CA, Vaughan CH, Eiden M, Masoodi M, et
al: Obesity in mice with adipocyte-specific deletion of clock
component Arntl. Nat Med. 18:1768–1777. 2012. View Article : Google Scholar
|
|
27
|
Connor KM, Krah NM, Dennison RJ, Aderman
CM, Chen J, Guerin KI, Sapieha P, Stahl A, Willett KL and Smith LE:
Quantification of oxygen-induced retinopathy in the mouse: A model
of vessel loss, vessel regrowth and pathological angiogenesis. Nat
Protoc. 4:1565–1573. 2009. View Article : Google Scholar
|
|
28
|
Balkwill F: The significance of cancer
cell expression of the chemokine receptor CXCR4. Seminars in cancer
biology. Semin Cancer Biol. 14:171–179. 2004. View Article : Google Scholar
|
|
29
|
Cao Y, Guangqi E, Wang E, Pal K, Dutta SK,
Bar-Sagi D and Mukhopadhyay D: VEGF exerts an
angiogenesis-independent function in cancer cells to promote their
malignant progression. Cancer Res. 72:3912–3918. 2012. View Article : Google Scholar
|
|
30
|
Ortega J, Li JY, Lee S, Tong D, Gu L and
Li GM: Phosphorylation of PCNA by EGFR inhibits mismatch repair and
promotes misincorporation during DNA synthesis. Proc Natl Acad Sci
USA. 112:5667–5672. 2015. View Article : Google Scholar
|
|
31
|
Zhao N, Tsuda H, Murofushi T, Imai K,
Ochiai K, Yang P and Suzuki N: Chaetocin inhibits RANKL-induced
osteoclast differentiation through reduction of Blimp1 in Raw264.7
cells. Life Sci. 143:1–7. 2015. View Article : Google Scholar
|
|
32
|
Ara T, Song L, Shimada H, Keshelava N,
Russell HV, Metelitsa LS, Groshen SG, Seeger RC and DeClerck YA:
Interleukin-6 in the bone marrow microenvironment promotes the
growth and survival of neuroblastoma cells. Cancer Res. 69:329–337.
2009. View Article : Google Scholar
|
|
33
|
Tang Z, Li D, Hou S and Zhu X: The cancer
exosomes: Clinical implications, applications and challenges. Int J
Cancer. 146:2946–2959. 2020. View Article : Google Scholar
|
|
34
|
Yerushalmi R, Woods R, Ravdin PM, Hayes MM
and Gelmon KA: Ki67 in breast cancer: Prognostic and predictive
potential. Lancet Oncol. 11:174–183. 2010. View Article : Google Scholar
|
|
35
|
Pastushenko I, Brisebarre A, Sifrim A,
Fioramonti M, Revenco T, Boumahdi S, Keymeulen AV, Brown D, Moers
V, Lemaire S, et al: Identification of the tumour transition states
occurring during EMT. Nature. 556:463–468. 2018. View Article : Google Scholar
|
|
36
|
Chen Z, Pan X, Georgakilas AG, Chen P, Hu
H, Yang Y, Tian S, Xia L, Zhang J, Cai X, et al:
Tetramethylpyrazine (TMP) protects cerebral neurocytes and inhibits
glioma by down regulating chemokine receptor CXCR4 expression.
Cancer Lett. 336:281–289. 2013. View Article : Google Scholar
|
|
37
|
Chen Q, Zhang JJ, Ge WL, Chen L, Yuan H,
Meng LD, Huang XM, Shen P, Miao Y and Jiang KR: YY1 inhibits the
migration and invasion of pancreatic ductal adenocarcinoma by
downregulating the FER/STAT3/MMP2 signaling pathway. Cancer Lett.
463:37–49. 2019. View Article : Google Scholar
|
|
38
|
Takase N, Koma Y, Urakawa N, Nishio M,
Arai N, Akiyama H, Shigeoka M, Kakeji Y and Yokozaki H: NCAM-and
FGF-2-mediated FGFR1 signaling in the tumor microenvironment of
esophageal cancer regulates the survival and migration of
tumor-associated macrophages and cancer cells. Cancer Lett.
380:47–58. 2016. View Article : Google Scholar
|
|
39
|
Esbona K, Yi Y, Saha S, Yu M, Doorn RV,
Conklin MW, Graham DS, Wisinski KB, Ponik SM, Eliceiri KW, et al:
The presence of cyclooxygenase 2, tumor-associated macrophages, and
collagen alignment as prognostic markers for invasive breast
carcinoma patients. Am J Pathol. 188:559–573. 2018. View Article : Google Scholar
|
|
40
|
Shultz LD, Lyons BL, Burzenski LM, Gott B,
Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, et al:
Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R
gamma null mice engrafted with mobilized human hemopoietic stem
cells. J Immunol. 174:6477–6489. 2005. View Article : Google Scholar
|
|
41
|
Shultz LD, Goodwin N, Ishikawa F, Hosur V,
Lyons BL and Greiner DL: Human cancer growth and therapy in
NOD/SCID/IL2Rγnull (NSG) mice. Cold Spring Harbor Protocols.
2014:6942014. View Article : Google Scholar
|
|
42
|
Bartal I, Melamed R, Greenfeld K, Atzil S,
Glasner A, Domankevich V, Naor R, Beilin B, Yardeni IZ and
Ben-Eliyahu S: Immune perturbations in patients along the
perioperative period: Alterations in cell surface markers and
leukocyte subtypes before and after surgery. Brain Behav Immun.
24:376–86. 2010. View Article : Google Scholar
|
|
43
|
Arase H, Saito T, Phillips JH and Lanier
LL: Cutting edge: The mouse NK cell-associated antigen recognized
by DX5 moncoclonal antibody is CD49b (α2 integrin, very late
antigen-2). J Immunol. 167:1141–1144. 2001. View Article : Google Scholar
|
|
44
|
Chen P, Yu N, Zhang Z, Zhang P, Yang Y, Wu
N, Xu L, Zhang J, Ge J, Yu K, et al: Thrombospondin-1 might be a
therapeutic target to suppress RB cells by regulating the DNA
double-strand breaks repair. Oncotarget. 7:6105–6120. 2016.
View Article : Google Scholar
|
|
45
|
Hirschberger S, Hinske LC and Kreth S:
MiRNAs: Dynamic regulators of immune cell functions in inflammation
and cancer. Cancer Lett. 431:11–21. 2018. View Article : Google Scholar
|
|
46
|
Casadei L, Calore F, Creighton CJ,
Guescini M, Batte K, Iwenofu OH, Zewdu A, Braggio DA, Bill KL,
Fadda P, et al: Exosome-derived miR-25-3p and miR-92a-3p stimulate
liposarcoma progression. Cancer Res. 77:3846–3856. 2017. View Article : Google Scholar
|
|
47
|
Xu Z, Zhao L, Zhu LY, He M, Zheng L and Wu
Y: MicroRNA-17, 20a regulates the proangiogenic function of
tumor-associated macrophages via targeting hypoxia-inducible factor
2α. PLoS One. 8:e778902013. View Article : Google Scholar
|
|
48
|
Kim DI, Kim E, Kim YA, Cho SW, Lim JA and
Park YJ: Macrophage densities correlated with CXC chemokine
receptor 4 expression and related with poor survival in anaplastic
thyroid cancer. Endocrinol Metab (Seoul). 31:469–475. 2016.
View Article : Google Scholar
|
|
49
|
Xiao M, Zhang J and Chen W and Chen W:
M1-like tumor-associated macrophages activated by
exosome-transferred THBS1 promote malignant migration in oral
squamous cell carcinoma. J Exp Clin Cancer Res. 37:1432018.
View Article : Google Scholar
|
|
50
|
Bonauer A, Carmona G, Iwasaki M, Mione M,
Koyanagi M, Fischer A, Burchefield J, Fox H, Doebele C, Ohtani K,
et al: MicroRNA-92a controls angiogenesis and functional recovery
of ischemic tissues in mice. Science. 324:1710–1713. 2009.
View Article : Google Scholar
|
|
51
|
Mathiyalagan P, Liang Y, Kim D, Misener S,
Thorne T, Kamide CE, Klyachko E, Losordo DW, Hajjar RJ and Sahoo S:
Angiogenic mechanisms of human CD34+ stem cell exosomes
in the repair of ischemic hindlimb. Circ Res. 120:1466–1476. 2017.
View Article : Google Scholar
|
|
52
|
Tkach M and Théry C: Communication by
extracellular vesicles: Where we are and where we need to go. Cell.
164:1226–1232. 2016. View Article : Google Scholar
|
|
53
|
Huang X, Yuan T, Liang M, Du M, Xia S,
Dittmar R, Wang D, See W, Costello BA, Quevedo F, et al: Exosomal
miR-1290 and miR-375 as prognostic markers in castration-resistant
prostate cancer. Eur Urol. 67:33–41. 2015. View Article : Google Scholar
|
|
54
|
Tsuchida R, Osawa T, Wang F, Nishii R, Das
B, Tsuchida S, Muramatsu M, Takahashi T, Inoue T, Wada Y, et al:
BMP4/Thrombospondin-1 loop paracrinically inhibits tumor
angiogenesis and suppresses the growth of solid tumors. Oncogene.
33:3803–3811. 2014. View Article : Google Scholar
|
|
55
|
Ramchandani D and Mittal V: Thrombospondin
in tumor microenvironment. Adv Exp Med Biol. 1272:133–147. 2020.
View Article : Google Scholar
|
|
56
|
Firlej V, Mathieu JR, Gilbert C, Lemonnier
L, Nakhlé J, Gallou-Kabani C, Guarmit B, Morin A, Prevarskaya N,
Delongchamps NB, et al: Thrombospondin-1 triggers cell migration
and development of advanced prostate tumors. Cancer Res.
71:7649–7658. 2011. View Article : Google Scholar
|
|
57
|
Horiguchi H, Yamagata S, Rong Qian Z,
Kagawa S and Sakashita N: Thrombospondin-1 is highly expressed in
desmoplastic components of invasive ductal carcinoma of the breast
and associated with lymph node metastasis. J Med Invest. 60:91–96.
2013. View Article : Google Scholar
|
|
58
|
Borsotti P, Ghilardi C, Ostano P, Silini
A, Dossi R, Pinessi D, Foglieni C, Scatolini M, Lacal PM, Ferrari
R, et al: Thrombospondin-1 is part of a Slug-independent motility
and metastatic program in cutaneous melanoma, in association with
VEGFR-1 and FGF-2. Pigment Cell Melanoma Res. 28:73–81. 2015.
View Article : Google Scholar
|
|
59
|
Byrne GJ, Hayden KE, McDowell G, Lang H,
Kirwan CC, Tetlow L, Kumar S and Bundred NJ: Angiogenic
characteristics of circulating and tumoural thrombospondin-1 in
breast cancer. Int J Oncol. 31:1127–1132. 2007.
|