Introduction
Histone deacetylase (HDAC) is one of the enzymes
involved in epigenetic modification, removing acetyl groups from
the lysine residues of target proteins. At present, 18 different
HDACs have been identified, and are classified as Class I (HDAC 1–3
and 8), class II (HDAC 4–7, 9, and 10), class III (the sirtuin
family, SIRT1-7) and class IV (HDAC 11) (1). HDACs serve an important role in the
progression of several types of cancer by altering gene expression
for differentiation, the cell cycle and apoptosis (2,3).
HDAC upregulation contributes to tumorigenesis in a
variety of cancer types, including those of the bladder, breast,
lung and colon (4–7). Furthermore, increased expression of
HDAC1 and 6 has been observed in human head and neck squamous cell
carcinoma (HNSCC), where it was correlated with advanced clinical
stage and poor prognosis (8,9). HDAC inhibitors comprise a structurally
diverse class of targeted anti-cancer compounds (10). Trichostatin A (TSA), a known class I
and II HDAC inhibitor, exerts strong antitumor effects (11). In HNSCC, TSA inhibits cell
proliferation by inducing cell cycle arrest, apoptosis and the
downregulation of stemness genes, such as CD44 and ABCG2 (12). Furthermore, TSA induces
G2/M cell cycle arrest as well as upregulating p21
expression in colorectal cancer cells (13) and oral squamous cell carcinoma
(14). The activation of epidermal
growth factor receptor (EGFR) signaling pathways results in
G1/S cell cycle progression in various types of cancer
cell (15,16), and the HDAC inhibitor suberoylanilide
hydroxamic acid (SAHA) downregulates EGFR transcription in
colorectal cancer cells (17).
The p63 gene, a member of the p53 family, is
essential for epithelial development, and the regulation of
epithelial cell proliferation and differentiation (18). The p63 gene exists in two distinct
isoforms, TAp63 (containing an N-terminal transcription domain) and
ΔNp63 (lacking the N-terminal transcription domain) (18). p63 is upregulated in >80% of all
HNSCC tissues (19) and ΔNp63α serves
an important role in HNSCC cell survival, suppressing the
p73-dependent proapoptotic transcriptional program (20,21). The
ΔNp63α/HDAC1/2 complex is also believed to be an essential tumor
maintenance factor in SCC (22).
Junctional adhesion molecule-A (JAM-A) is a tight
junction molecule which belongs to the IgG superfamily (23). Tight junction molecules are associated
with barrier function, but also with cell signaling (24), and the overexpression of JAM-A has
been shown to activate Rap1 and β-1-integrin, inducing cellular
migration in breast cancer (25,26).
Furthermore, increased JAM-A expression was detected in HNSCC
surgical tissues with high expression levels of p63 and ΔNp63
(27). A significant increase in
soluble JAM-A in the sera of patients with HNSCC was also observed,
compared with those of healthy subjects. Furthermore, JAM-A siRNA
knockdown inhibited the proliferation, migration and invasiveness
of HNSCC Detroit 562 cells in vitro; JAM-A overexpression
was also found to be regulated via p63 (27).
The bicellular tight junction molecule claudin-1 is
essential for the barrier function of various normal epithelial
cells (28). Claudin-1 expression is
decreased by transfection of ΔNp63 in primary mouse keratinocytes
via the claudin-1-promoter region (29). Claudin-1 upregulation was also
observed in oral SCC and tonsil SCC tissues, which is associated
with advanced clinical stage and surrounding tissue invasion
(30–32). In oral SCC cells, claudin-1 promotes
cancer cell invasion by upregulating the laminin-5γ2 chain via
matrix metalloproteinase (MMP)-2 and membrane-type MMP-1 (33). Increased expression of claudin-1 was
also observed, as well as that of tricellular tight junction
molecule lipolysis-stimulated lipoprotein receptor (LSR) in HNSCC
surgical tissues (34). In the
present study, to investigate how HDAC inhibitors influence HNSCC,
the HNSCC Detroit 562 cell line and primary cultured HNSCC cells
were treated with HDAC inhibitors. HDAC inhibitors suppress the
proliferation, migration and invasiveness of HNSCC cells via the
downregulation of p63-mediated tight junction molecules, JAM-A and
claudin-1, and induction of p21-mediated growth arrest.
Materials and methods
Reagents and secondary antibodies
Trichostatin A (TSA; cat. no. T8552) was purchased
from Sigma-Aldrich; Merck KGaA. Inhibitors of HDAC1 (cat. no.
sc-3523) and HDAC6 (cat. no. sc-223877) were purchased from Santa
Cruz Biotechnology, Inc., and the EGFR inhibitor AG1478 (cat. no.
658552) was acquired from Merck KGaA. Alexa Flour 488-conjugated
anti-rabbit IgG (cat. no. A32731) and Alexa Flour 594-conjugated
anti-mouse IgG (cat. no. A32'742) antibodies were purchased from
Molecular Probes; Thermo Fisher Scientific, Inc.
Human tissues
A total of 18 paired head and neck cancer (age,
64.1±7.4; sex, 14 men and 4 women) and adjacent healthy tissues
were obtained by tumor resection surgery at the Sapporo Medical
University Department of Otolaryngology between January 2015 and
December 2018). The present study was approved by the ethics
committee of Sapporo Medical University, and written informed
consent was obtained from all patients.
Isolation and culture of human head
and neck cancer cells
Human head and neck cancer tissues were minced into
sections of 2–3 mm3 and washed four times with
phosphate-buffered saline (PBS) containing 100 U/ml penicillin and
100 µg/ml streptomycin. The tissue homogenates were then
resuspended in 10 ml Hanks balanced salt solution (Thermo Fisher
Scientific, Inc.) containing 0.5 µg/ml DNase I (Sigma-Aldrich;
Merck KGaA) and 0.08 mg/ml Liberase Blendzyme (Roche Diagnostics)
in PBS, and then incubated at 37°C for 20 min. The incubated
specimens were subsequently filtered with 300- and 40-µm meshes,
centrifuged at 1,200 × g for 3 min, and then cultured in serum-free
bronchial epithelial growth medium (Clonetics Corp.) supplemented
with 0.5 µg/ml hydrocortisone, 5 µg/ml insulin, 10 µg/ml
transferrin, 0.5 µg/ml epinephrine, 6.5 µg/ml triiodothyronine, 50
µg/ml gentamycin, 50 µg/ml amphotericin B, 0.1 ng/ml retinoic acid,
0.5 ng/ml EGF (Lonza Group, Ltd.) bovine pituitary extract (1%
vol/vol, Pel-Freez, LLC), 100 U/ml penicillin and 100 µg/ml
streptomycin (Sigma-Aldrich; Merck KGaA). The primary cultured
cells were plated onto 60-mm culture dishes (Corning Inc.), which
were precoated with rat tail collagen (500 µg dried tendon/ml 0.1%
acetic acid) in a humidified incubator at 37°C5% (CO2,
95% air).
Cell cultures and treatment
The pharynx carcinoma Detroit 562 cell line (CCL138)
was purchased from the ATCC and cultured in MEM (Sigma-Aldrich;
Merck KGaA) supplemented with 10% fetal bovine serum (FBS;
Invitrogen; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin,
100 µg/ml streptomycin and 50 µg/ml amphotericin B. The cells were
plated onto 60-mm culture dishes (Corning Inc.) precoated with rat
tail collagen (as aforementioned) and cultured in a humidified
incubator at 37°C. The cells were treated with HDAC inhibitors and
an EGFR inhibitor (AG1478) for 24 h.
RNA interference and transfection
An siRNA duplex oligonucleotide against p63 was
synthesized by Santa Cruz Biotechnology, Inc. The sequences were as
follows: Sense, 5′-GGAAUGACUUCAACUUUGA-3′; and antisense,
5′-UCAAAGUUGAAGUCAUUCC-3′. Detroit 562 cells and primary cancer
cells were transfected with 100 nM siRNA against p63 using
Lipofectamine® RNAiMAX Reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) 24 h after plating. A scrambled siRNA
sequence (BLOCK-iT Alexa Fluor fluorescent; Invitrogen; Thermo
Fisher Scientific, Inc.) was employed as a control siRNA.
Experimentation was conducted 48 h after transfection.
Immunohistochemical analysis of human
tissue samples
Human head and neck cancer tissues were fixed with
10% formalin in PBS at room temperature for least 48 h, and then
embedded in paraffin. Briefly, 5-µm-thick sections were dewaxed in
xylene, rehydrated in ethanol, and heated with Vision BioSystems
Bond Max using ER2 solution (Leica Microsystems, Inc.) in an
autoclave for antigen retrieval. Endogenous peroxidase activity was
blocked by incubation with 3% hydrogen peroxide in methanol for 10
min at room temperature. The tissue sections were then washed twice
with Tris-buffered saline (TBS) and pre-blocked with Block Ace
(Bio-Rad Laboratories, Inc.) for 1 h at room temperature. After
washing with TBS, the sections were incubated with anti-p63
(1:200), anti-HDAC1 (1:400), anti-JAM-A (1:1,000) and
anti-claudin-1 (1:400) antibodies (Table
I) for 1 h at room temperature. The sections were then washed
three times in TBS and incubated using the Vision BioSystems Bond
Polymer Refine Detection kit (Leica Microsystems, Inc.). After
three washes in TBS, a diamino-benzidine tetrahydrochloride working
solution was applied at room temperature. Finally, the sections
were counterstained with hematoxylin. A negative control was
performed by replacing the primary antibodies with normal rabbit
serum (cat. no. ab7487, Abcam). The tissue sections were examined
and photographed using an Olympus BX50 microscope with Olympus DP21
digital microscopy camera (Olympus Corporation.
 | Table I.Antibodies. |
Table I.
Antibodies.
|
|
| Dilution
factor |
|
|---|
|
|
|
|
|
|---|
| Antibody | Type | IH | IC | WB | Supplier
details |
|---|
| Claudin-1 | pAb | 1:400 | 1:100 | 1:1,000 | Zymed; Thermo
Fisher Scientific, Inc. (cat. no. 51-9000) |
| Claudin-7 | pAb |
|
| 1:1,000 | Zymed; Thermo
Fisher Scientific, Inc. (cat. no. 34-9100) |
| Tricellulin | pAb |
| 1:100 | 1:1,000 | Zymed; Thermo
Fisher Scientific, Inc. (cat. no. 48-8400) |
| Occludin | pAb |
|
| 1:1,000 | Zymed; Thermo
Fisher Scientific, Inc. (cat. no. 33-1520) |
| JAM-A | pAb | 1:1000 | 1:100 | 1:1,000 | Zymed; Thermo
Fisher Scientific, Inc. |
| LSR | pAb |
|
| 1:1,000 | Novus Biologicals,
LLC (cat. no. NBP1-89631) |
| Cytokeratin7 | mAb |
| 1:200 | 1:1,000 | Sigma-Aldrich;
Merck KGaA (cat. no. 307M-9) |
| Acetylated
tubulin | mAb |
|
| 1:1,000 | Sigma-Aldrich;
Merck KGaA (cat. no. T7451) |
| p63 | pAb |
| 1:100 | 1:1,000 | Abcam (cat. no.
703809) |
| p63 | mAb |
| 1:200 |
| OriGene;
Technologies, Inc. (cat. no. TA802078) |
| p40 (ΔNp63) | mAb | 1:100 |
|
| Nichirei
Biosciences Inc. (cat. no. 418171) |
| p21 | mAb |
|
| 1:1,000 | Invitrogen; Thermo
Fisher Scientific, Inc. (cat. no. PA1-30399) |
| Cyclin D1 | mAb |
|
| 1:1,000 | MBL International
Co. (cat. no. MD-17-3) |
| HDAC1 | pAb | 1:400 | 1:100 |
| Abcam (cat. no.
ab7028) |
| Phospho-EGFR | pAb |
|
| 1:1,000 | Cell Signaling
Technology, Inc. (cat. no. 2234) |
| EGFR | pAb |
|
| 1:1,000 | Cell Signaling
Technology, Inc. (cat. no. 4267) |
| Phospho-ERK1/2 | pAb |
|
| 1:1,000 | Cell Signaling
Technology, Inc. (cat. no. 9101) |
| ERK1/2 | pAb |
|
| 1:1,000 | Promega Corporation
(cat. no. V1141) |
| Actin | pAb |
|
| 1:1,000 | Sigma-Aldrich;
Merck KGaA (cat. no. A2066) |
The degree of positive staining was scored from 0 to
3 according to the percentage of atypical cells with positive IHC
staining in the observed area: i) 0, 0%; ii) 1, 1–25%; iii) 2,
26–50%; and iv) 3, 51–100%. The staining intensity was also graded
from 0 to 3 according to the intensity of immunohistochemical
staining; i) 0, none; ii) 1, low; iii) 2, moderate; and iv) 3,
high.
Western blot analysis
The cultured cells were scraped from a 35-mm dish
containing 400 µl of buffer (1 mM NaHCO3 and 2 mM
phenylmethylsulfonyl fluoride), collected in microcentrifuge tubes,
and then sonicated for 10 sec. The protein concentrations of
samples were determined using a BCA protein assay reagent kit
(Pierce; Thermo Fisher Scientific, Inc.). Aliquots (15 µl of 20 µg
protein/lane for each sample) were separated by electrophoresis
using 5–20% SDS-PAGE gels (FUJIFILM Wako Pure Chemical
Corporation), and electrophoretically transferred onto
nitrocellulose membranes (EMD Millipore) The membranes were
saturated with blocking buffer (25 mM Tris, pH 8.0, 125 mM NaCl,
0.1% Tween 20 and 4% skim milk) for 30 min at room temperature, and
then incubated with anti-p63, anti- JAM-A, anti-EGFR,
anti-phosphorylated-EGFR, anti-ERK1/2 anti-phosphorylated-ERK1/2,
anti-LSR, anti-tricellulin, anti-claudin-1, −4 and −7,
anti-acetylated-tubulin, anti-cyclin D1, anti-p21 and anti-actin
antibodies (all 1:1,000) at room temperature overnight. The
antibody details are displayed in Table
I. The membranes were then incubated with HRP-conjugated
anti-mouse and anti-rabbit IgG antibodies at room temperature for 1
h. The immunoreactive bands were detected using an ECL prime
Western blot system (Cytiva), and quantitated by densitometry. The
expression levels were normalized to that of actin, and were
displayed as bar graphs using Scion Image Beta 4.02 Win (Scion
Corporation) (Figs. S2–S6).
Immunocytochemical analysis
Confluent cells cultured in 35-mm glass-bottom
dishes (Iwaki) were fixed with cold acetone and ethanol (1:1) at
−20°C for 10 min. After rinsing in PBS, the cells were incubated
with anti-cytokeratin 7 (1:200), anti-p63 (1:100), anti ΔNp63
(1:100), anti-JAM-A (1:100), anti-claudin-1 (1:100),
anti-HDAC1(1:100) and anti-tricellulin (1:100) antibodies at room
temperature for 1 h (Table I). Alexa
Fluor 488-conjugated anti-rabbit IgG and Alexa Fluor 594-conjugated
anti-mouse IgG (Invitrogen; Thermo Fisher Scientific, Inc.) were
used as the secondary antibodies (1:200) at room temperature. The
specimens were examined using an epifluorescence microscope
(Olympus Corporation) and a confocal laser scanning microscope
(magnification, ×63; LSM5; Zeiss GmbH).
Cell counting
Detroit 562 cells (1×105) were plated
into 35-mm dishes and cultured for 72 h until subconfluent at 37°C.
The cultured cells were harvested by trypsinization (0.25%
Trypsin-EDTA) and resuspended in 2 ml PBS. Subsequently, 450 µl
Muse® Count & Viability reagent (Merck Millipore,
MA, USA) was added to 50 µl from the 2 ml resuspended cells, and
incubated for 5 min at room temperature. The cell counts were
determined using the Muse® Cell Analyzer (EMD Millipore)
according to the manufacturer's protocol.
Matrigel invasion assay
For invasion assays, Transwell cell culture inserts
were used (pore size, 8-µm; Becton Dickinson and Company). Detroit
562 cells (1×104) were seeded at 37°C into the upper
Matrigel pre-coated chamber (1:50, Becton Dickinson and Company)
with serum-free medium at 4°C for 30 min, and the lower chamber was
filled with human fibroblast conditioned medium containing 10 nM
EGF as an adhesive substrate. The cells were incubated for 24 h at
37°C. The upper chamber was the fixed with 100% methanol for 10 min
and stained with Giemsa for 20 min. The number of invading cells
was assessed using a microscope imaging system (Olympus
Corporation).
Wound-healing assay
Detroit 562 cells and primary cultured HNSCC cells
were seeded into 35-mm dishes and cultured in MEM supplemented with
5% FBS to confluence. A wound was created in each of the cell
monolayers using a plastic 200-µl pipette tip, and the wound size
was measured at 0 and 24 h using a microscope imaging system
(magnification, ×4; Olympus Corporation).
Cell cycle assay
Detroit 562 cells cultured in 35-mm dishes were
harvested using 0.25% Trypsin-EDTA and washed once with PBS. The
cells were slowly added to 1 ml ice cold ethanol (70%) and
incubated for ≥3 h at −20°C. The cells were washed once with PBS,
and 200 µl Muse® Cell Cycle reagent (EMD Millipore) was
added, prior to further incubation for 30 min at room temperature
in the dark. The Muse® Cell Analyzer (EMD Millipore) was
then used to assess the cell cycle according to the manufacturer's
instructions.
Statistical analysis
Each set of results represents ≥3 separate
experiments, and is presented as the mean ± SEM. Significant
differences were determined by one-way ANOVA) with the Tukey-Kramer
method. *P<0.05 was considered to indicate a statistically
significant difference.
Results
Expression and localization of p63 and
HDAC1 in HNSCC tissues
The expression and localization of p63, HDAC1, JAM-A
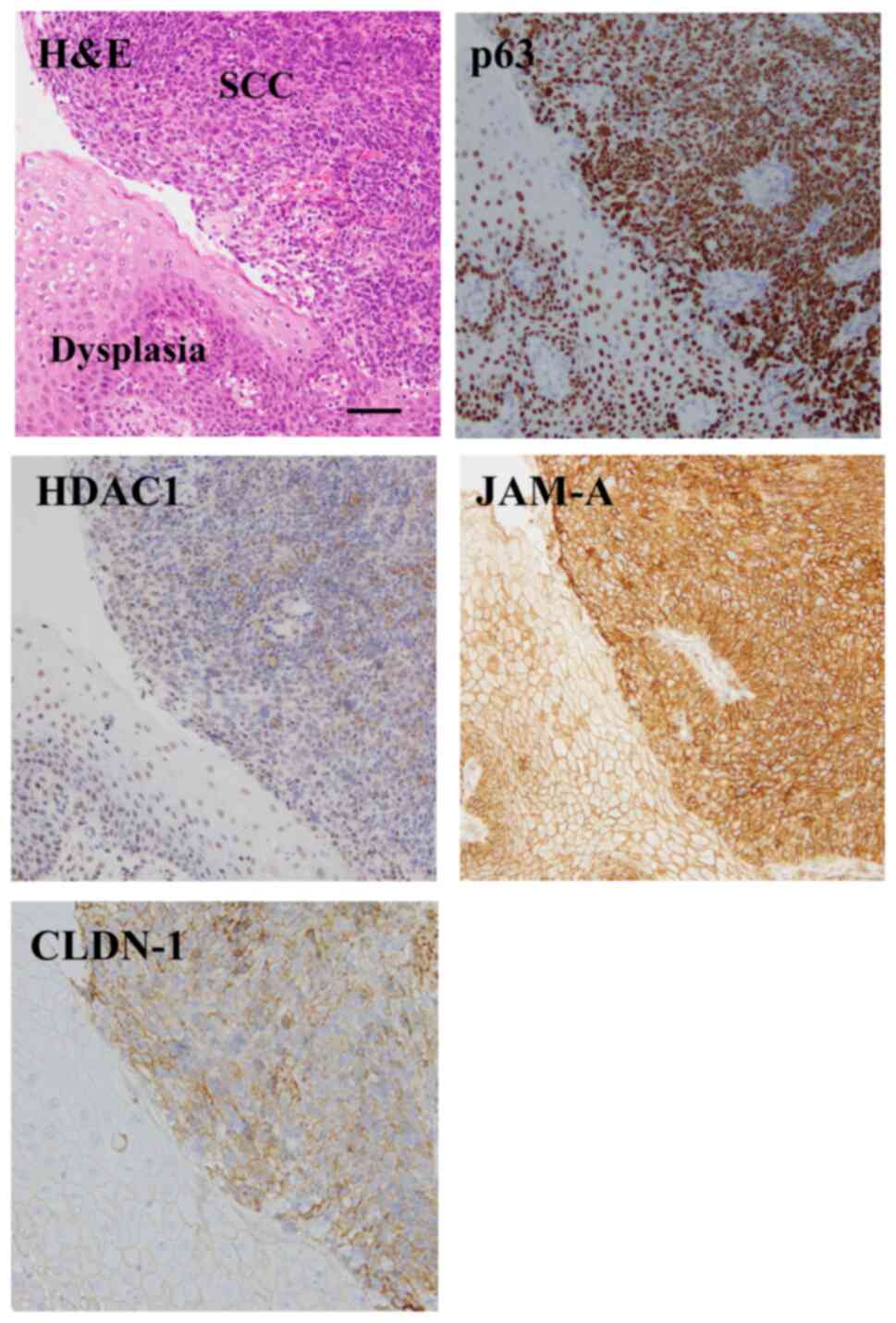
and claudin-1 in HNSCC surgical tissues was investigated. Using
immunohistochemistry, higher expression of p63, HDAC1, JAM-A and
claudin-1 was observed in 83% of the HNSCC tissue samples (15/18)
than in the adjacent dysplastic region (Fig. 1). Furthermore, JAM-A and claudin-1
were expressed at the membranes of cancer cells, whereas p63 and
HDAC1 were expressed in the nuclei.
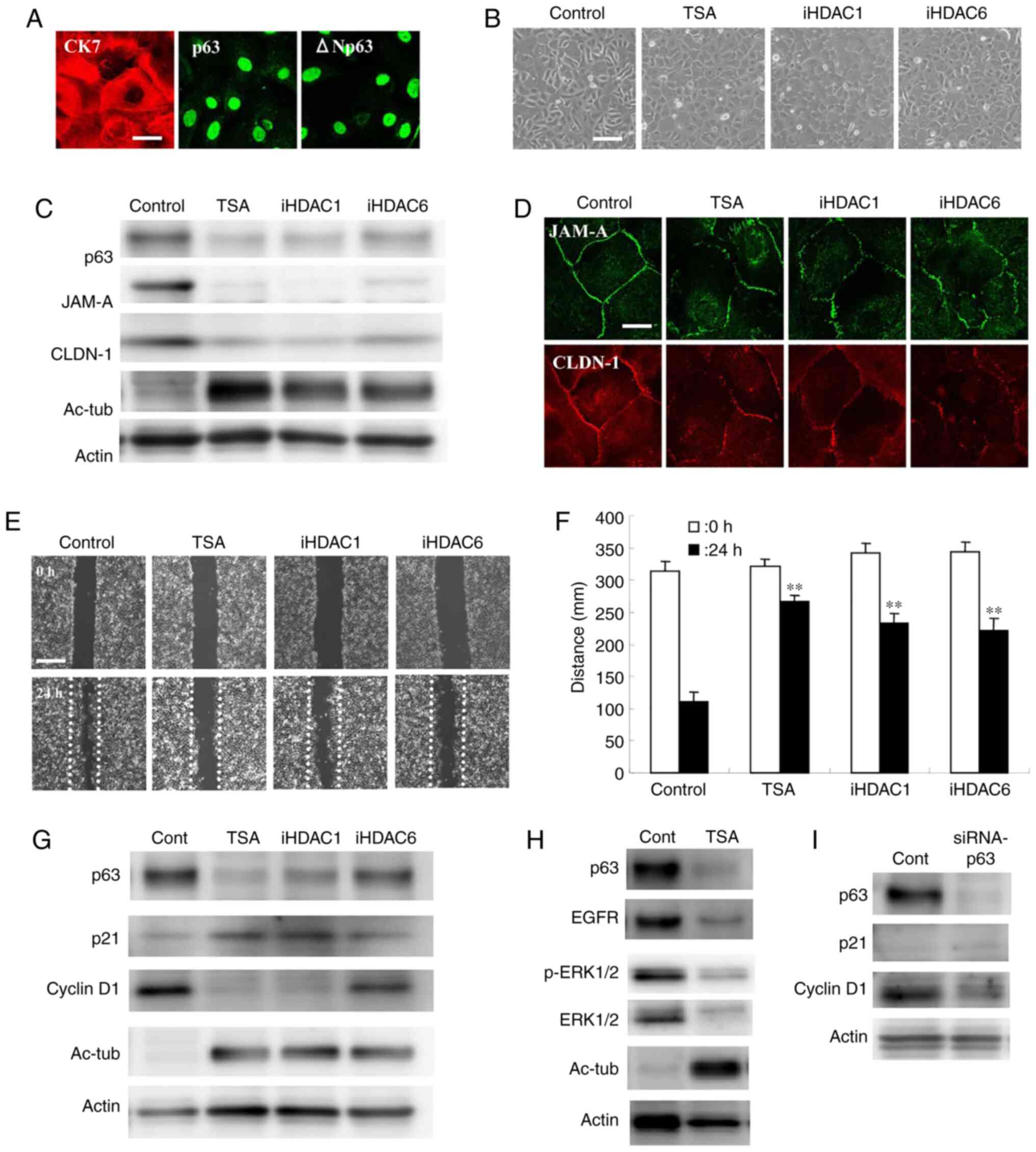
HDAC inhibitors downregulate the
expression of JAM-A, claudin-1 and tricellulin in Detroit 562 cells
via p63
Western blot analysis revealed that p63 knockdown in
Detroit 562 cells resulted in decreased expression of JAM-A and
claudin-1, which is in line with previously reported data (27) (Figs. 2A
and S2).
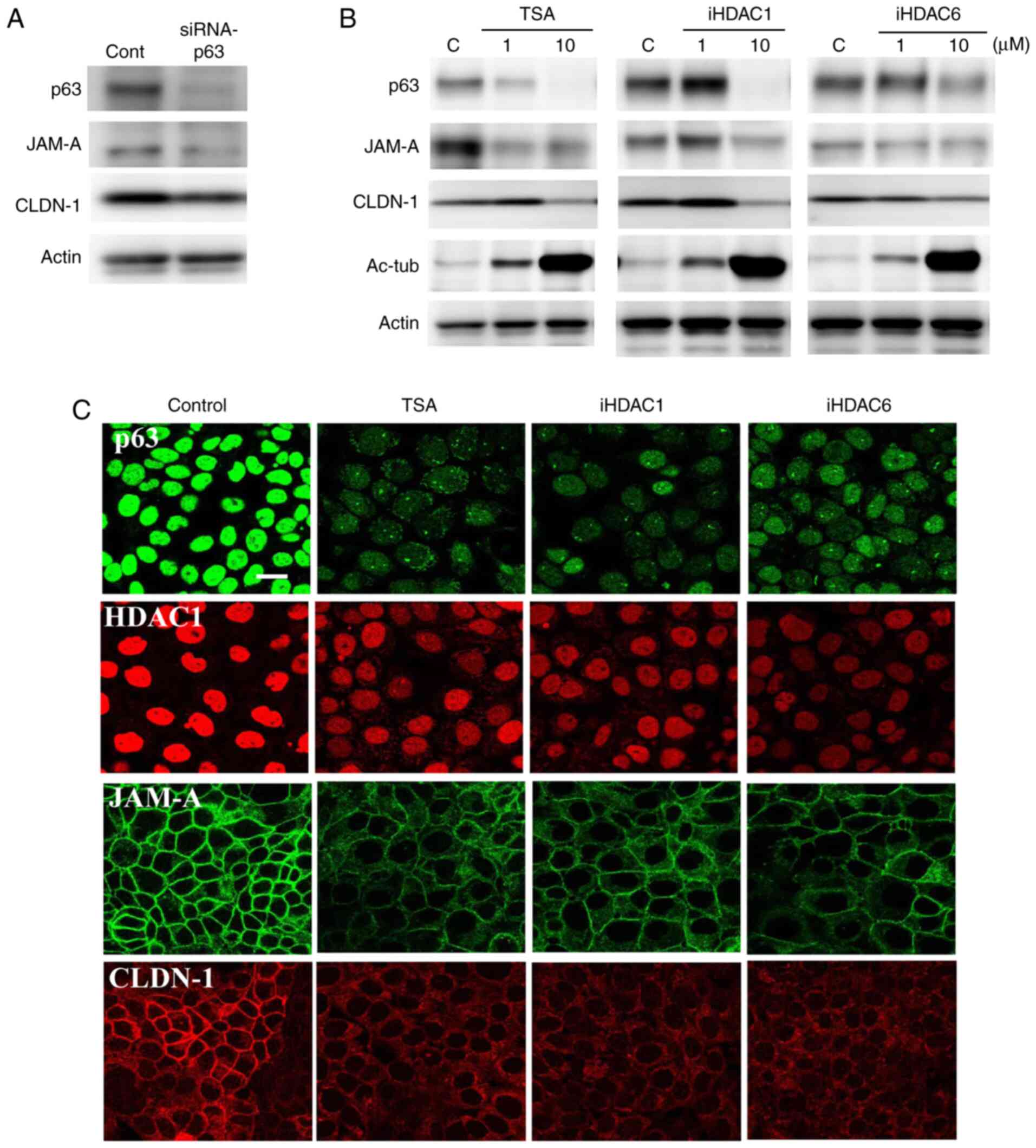 | Figure 2.Western blot analysis of p63, JAM-A
and claudin-1 expression in Detroit 562 cells (A) transfected with
p63 siRNA and (B) treated with HDAC inhibitor TSA, or HDAC1 and 6
inhibitors; 1 and 10 µM. (C) Immunocytochemical staining for p63,
HDAC1, JAM-A and claudin-1 in Detroit 562 cells treated with HDAC
inhibitors at 10 µM. Scale bar, 10 µm. HDAC, histone deacetylase;
iHDAC, HDAC inhibitor; JAM-A, junctional adhesion molecule-A;
CLDN-1, claudin-1; Ac-tub, acetylated tubulin; TSA, trichostatin A;
siRNA, small interfering RNA; cont, control. |
To investigate the association between HDAC and
p63-mediated tight junction molecules JAM-A and claudin-1 in HNSCC,
Detroit 562 cells were treated with the HDAC inhibitor TSA, as well
as HDAC1 and 6 inhibitors. As revealed by western blot analysis,
expression of p63, HDAC1, JAM-A and claudin-1 was decreased
following treatment with all of the HDAC inhibitors in a
dose-dependent manner, whereas expression of acetylated tubulin, an
effecter of HDAC inhibitors, was increased (Figs. 2B and S2). Immunocytochemical analysis revealed
that treatment with 10 µM of all HDAC inhibitors decreased the
expression of p63, HDAC1, JAM-A and claudin-1 in the nuclei and the
cell membrane (Fig. 2C).
The effects of the HDAC inhibitors on the
tricellular tight junction molecules tricellulin and LSR in HNSCC
were also investigated by western blot analysis. Tricellulin
expression was decreased by treatment with all of the HDAC
inhibitors in a dose-dependent manner, whereas LSR expression was
decreased only by treatment with the HDAC1 inhibitor (Figs. S1A and S6). With immunocytochemistry, tricellulin
expression was found to be decreased at the cell membranes
following treatment with all of the HDAC inhibitors at 10 µM
(Fig. S1B).
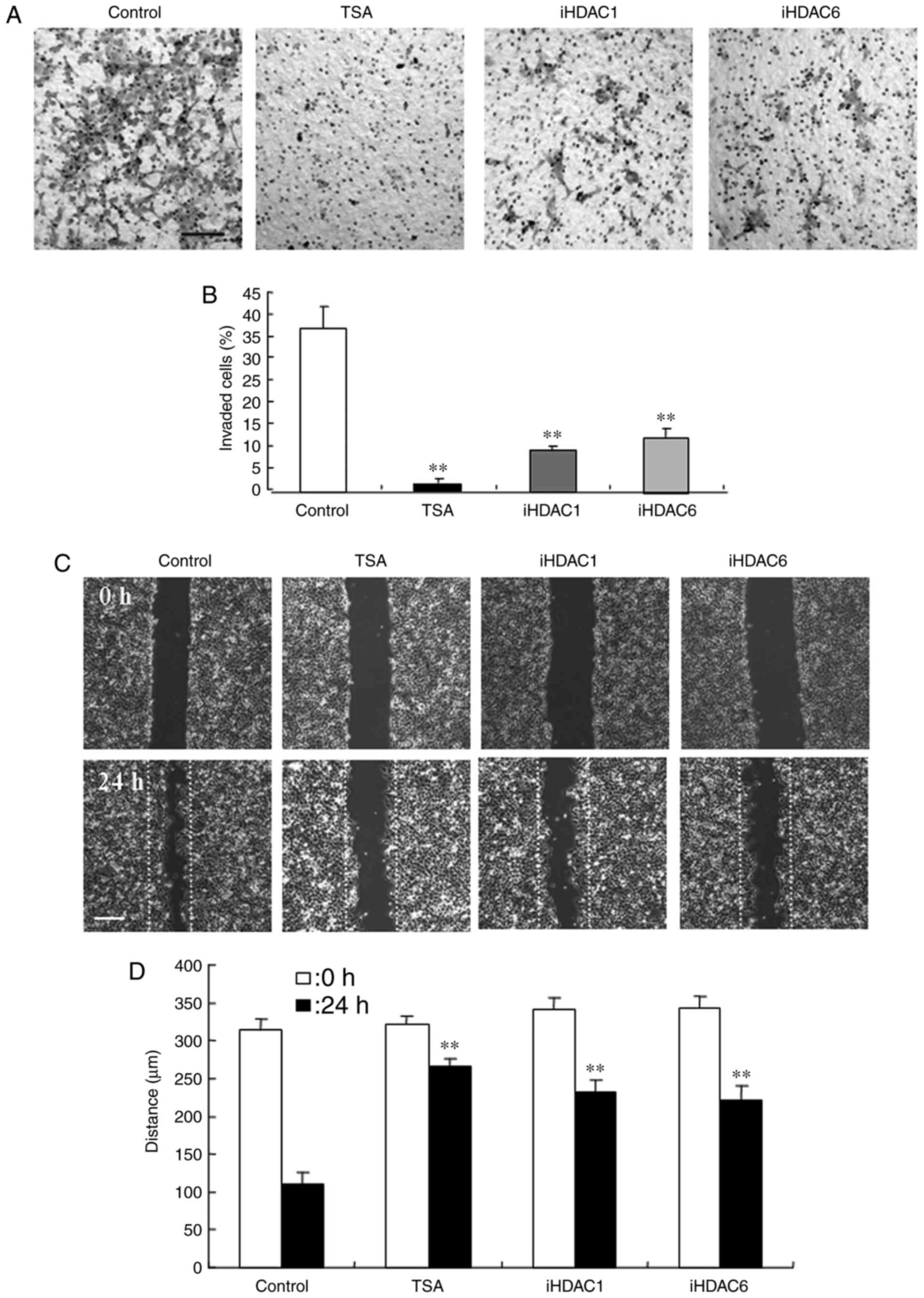
HDAC inhibitors suppress the invasion
and migration capacities of Detroit 562 cells
the effects of the HDAC inhibitor TSA, HDAC1
inhibitor, and HDAC6 inhibitor on invasion and migration were
investigated using Detroit 562 cells. In the cells treated with 10
µM, HDAC inhibitors, the numbers of invasive cells were
significantly decreased compared with those of the control cells
(Fig. 3A and B). For the
wound-healing assay, all of the HDAC inhibitors (10 µM each)
significantly decreased the wound closure distance, compared with
those of the untreated control cells (Fig. 3C and D).
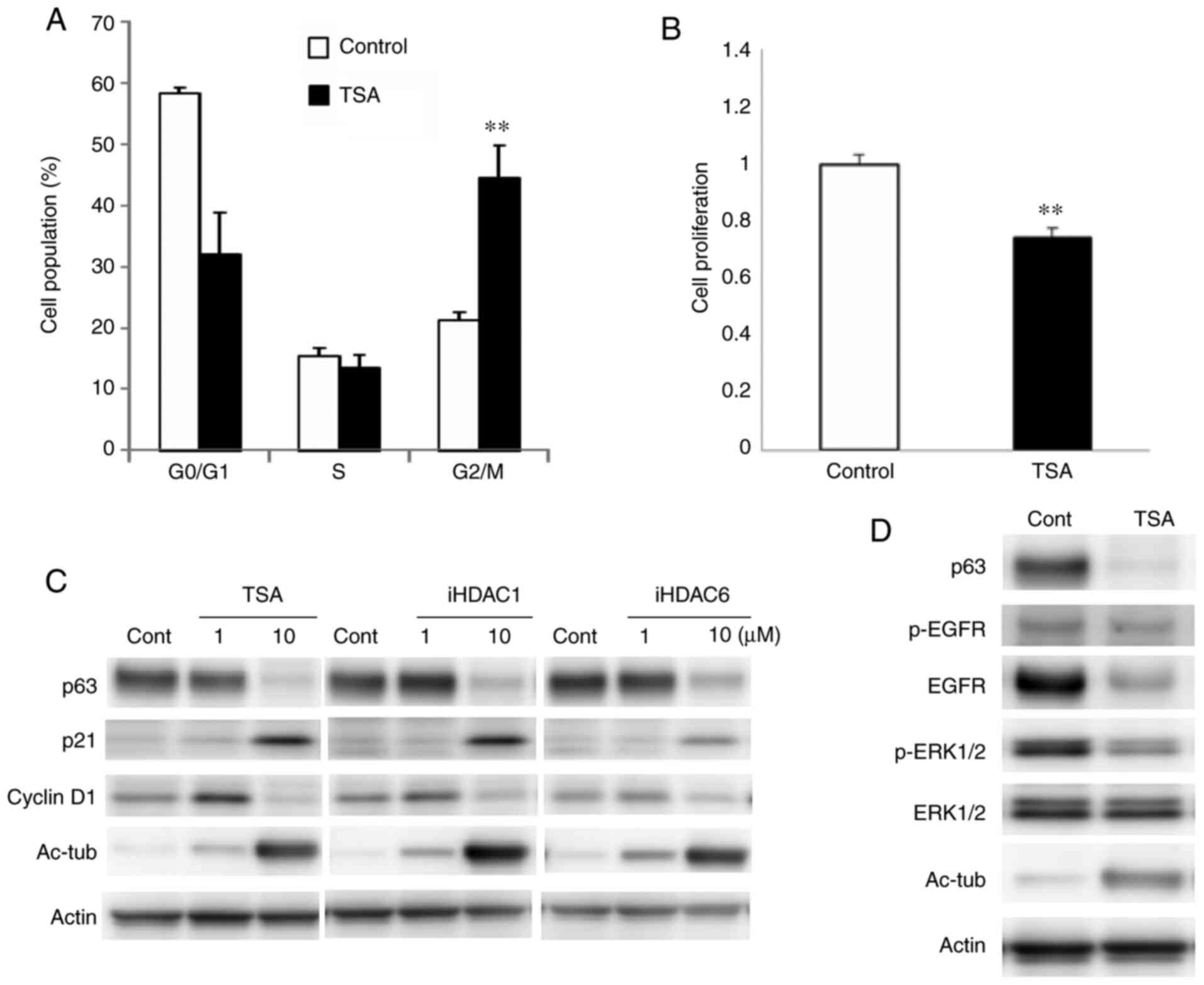
HDAC inhibitors induce G2/M
arrest via p21 in Detroit 562 cells
To investigate the cell cycle inhibitory mechanisms
of HDAC inhibitors in HNSCC, Detroit 562 cells were treated with
TSA, and HDAC1 and 6 inhibitors at 10 µM each. Cell cycle analysis
revealed that the proportion of cells in the G2/M phase
was markedly increased by treatment with TSA (Fig. 4A). Cellular proliferation was
significantly decreased by TSA (Fig.
4B). In western blot analysis, expression of p21 and acetylated
tubulin were markedly upregulated and expression of p63 and
cyclinD1 were decreased by all of the HDAC inhibitors (Figs. 4C and S3). Furthermore, expression of EGFR and
phosphorylated-ERK1/2 was decreased with downregulation of p63 by
treatment with TSA (Figs. 4D and
S3).
Downregulation of p63 by TSA induces
G1 arrest via EGFR in Detroit 562 cells
The effects of p63 expression on cell cycle
progression in Detroit 562 cells (via EGFR) were also investigated.
Transfection with p63 siRNA increased the proportion of cells in
the G0/G1 phase (Fig. 5A), and the proliferative capacity of
these cells was decreased, compared with the control (Fig. 5B). Western blot analysis revealed that
p63 knockdown downregulated EGFR and cyclin D1 expression (Figs. 5C and S4). Treatment with the EGFR inhibitor
AG1478 increased the number of cells in the
G0/G1 phase and inhibited the rate of
cellular proliferation (Fig. 5D and
E). AG1478 was also found to decrease the level of
phosphorylated-ERK1/2 and cyclin D1 (Figs. 5F and S4).
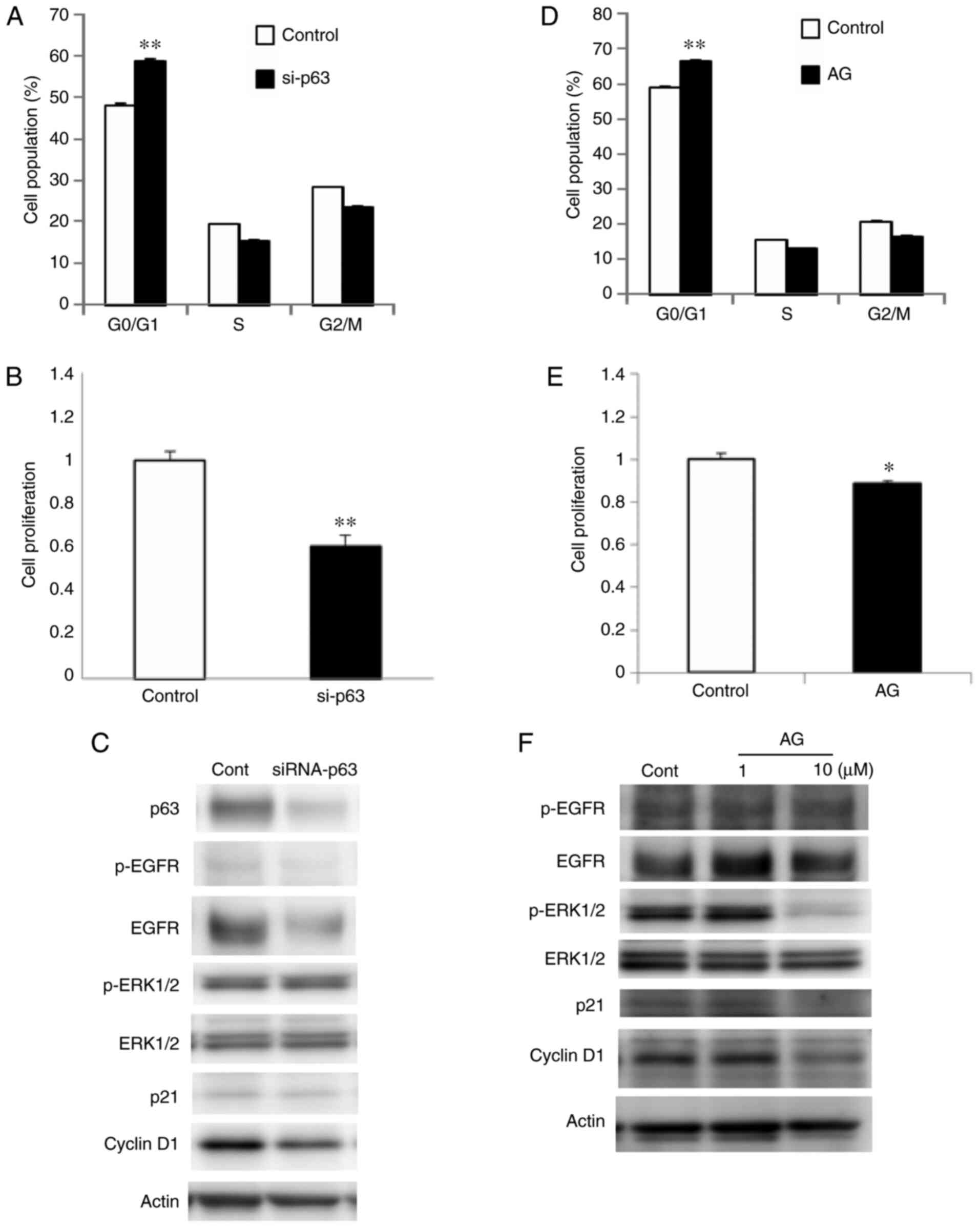 | Figure 5.(A) Cell cycle and (B) cell counting
assays, (C) and western blot analysis for p63, EGFR,
phosphorylated-EGFR, ERK1/2, phosphorylated-ERK1/2, p21 and cyclin
D1 in Detroit 562 cells transfected with p63 siRNA. (D) Cell cycle
assay, (E) cell counting assay and (F) western blotting for EGFR,
phosphorylated-EGFR, ERK1/2, phosphorylated-ERK1/2, p21 and cyclin
D1 in Detroit 562 cells treated with the EGFR inhibitor AG1478.
*P<0.05 and **P<0.01 vs. the control. EGFR, epidermal growth
factor receptor; siRNA, small interfering RNA; AG, AG1478; cont,
control. |
Effects of HDAC inhibitors on primary
cultured HNSCC cells
In primary cultured HNSCC cells derived from
surgical tissues, the expression of CK7, p63 and ΔNp63 was detected
by immunocytochemical analysis (Fig.
6A). Primary cells were treated with TSA, and HDAC1 and 6
inhibitors at 10 µM each. Per the phase contrast images, the shape
of HNSCC cells changed to an epithelial-like form following
treatment with all of the HDAC inhibitors (Fig. 6B). Furthermore, expression of JAM-A
and claudin-1 was decreased following treatment with all of the
HDAC inhibitors (Figs. 6C, D and
S5). Wound-healing assays revealed
that HDAC inhibition decreases the wound closure rate, compared
that for the untreated control (Fig. 6E
and F). Western blot analysis showed that treatment with all of
the HDAC inhibitors decreased the expression levels of p63 and
cyclin D1, and increased p21 expression (Figs. 6G and S5). Furthermore, treatment with TSA not
only decreased p63 expression, but also that of EGFR,
phosphorylated-ERK1/2 and ERK1/2 (Figs.
6H and S5). When primary
cultured HNSCC cells were transfected with p63 siRNA, the
expression of p63 and cyclin D1 was also decreased, as evidenced by
western blot analysis (Figs. 6I and
S5).
 | Figure 6.(A) Immunocytochemical staining for
CK7, p63 and ΔNp63 in primary cultured cancer cells derived from
human head and neck squamous cell carcinoma tissues. Scale bar, 10
µm. (B) Phase-contrast images. Scale bar, 50 µm. (C) Western
blotting for p63, JAM-A and claudin-1, and (D) immunocytochemical
staining for JAM-A and claudin-1 in primary cultured cancer cells
treated with 10 µM HDAC inhibitors. Scale bar, 10 µm. (E)
Wound-healing assays and (G) western blotting for p63, p21 and
cyclin D1 in primary cultured cancer cells treated with HDAC
inhibitors at 10 µM. Scale bar, 200 µm. (F) Quantification of the
results in (E). (H) Western blotting for p63, EGFR,
phosphorylated-EGFR, ERK1/2 and phosphorylated-ERK1/2 in primary
cultured cancer cells treated with 10 µM TSA. (I) Western blotting
for p63, p21 and cyclin D1 in primary cultured cancer cells
transfected with p63siRNA of. **P<0.01 vs. the control. CK7,
anti-cytokeratin 7; HDAC, histone deacetylase; iHDAC, HDAC
inhibitor; JAM-A, junctional adhesion molecule-A; CLDN-1,
claudin-1; Ac-tub, acetylated tubulin; TSA, trichostatin A; siRNA,
small interfering RNA; cont, control. |
Discussion
There is growing interest in the potential clinical
use of HDAC inhibitors as a novel class of targeted cancer
therapeutics. In the present study, the effects of HDAC inhibitors
were investigated in HNSCC; higher expression levels of p63, HDAC1,
JAM-A and claudin-1 were observed in HNSCC tissues than in the
adjacent dysplastic regions. The HDAC inhibitor TSA, and specific
inhibitors of HDAC1 and 6 suppressed the proliferation, migration
and invasiveness of HNSCC by downregulating p63-mediated tight
junction molecules JAM-A and claudin-1, and inducing p63- and
p21-mediated proliferation arrest. In the present study,
wound-healing assays were used to indicate the migration of HNSCC
cells. Although wound-healing assays are generally conducted with
serum-free medium, Detroit 562 cells are sensitive to changes in
serum concentration. Accordingly, 5% FBS was used, which was
acknowledged as a study limitation.
The upregulation of JAM-A and claudin-1 correlates
with tumor progression in various cancer types (35). JAM-A upregulation induces cancer cell
migration by activating Rap1 and β-1-integrin in breast cancer
(25). Furthermore, high expression
levels of JAM-A positively correlate with poor prognosis in
patients with nasopharyngeal carcinoma (NPC), and induces the
epithelial-to-mesenchymal transition of NPC cells via the PI3K/Akt
pathway (36). JAM-A expression also
promotes proliferation and inhibits apoptosis in both gastric and
lung cancer cells (37,38).
JAM-A is dysregulated via p63/GATA-3 in HNSCC
(27). Furthermore, downregulation of
JAM-A by p63 knockdown inhibits the migratory and invasive
capacities of HNSCC cells (27). On
the other hand, increased expression of claudin-1 is associated
with advanced clinical stage and invasive pathologic
characteristics of oral SCC (30–32);
claudin-1 overexpression upregulates the laminin-5γ2 chain via
MMP-2 and membrane-type MMP-1, and induces cancer cell invasion in
oral SCC (33). Claudin-1 is also a
target gene of p63 in epithelial cells (29), and HDAC inhibitors decrease the
stability and expression of claudin-1 mRNA and reduce colonic
cancer cell invasiveness (39). HDAC1
and 2 directly mediate the repressive functions of p63 (40). HDAC inhibitors are also believed to
partially inhibit the migration and invasiveness of HNSCC cells via
JAM-A and claudin-1 downregulation in p63-dependent and
-independent manners.
JAM-A is a reovirus receptor that controls the entry
of the virus into cells (41). The
reovirus-based formulation Reolysin is an oncolytic virus that is
naturally genetically modified to selectively infect and kill
cancer cells, and is being evaluated as an anticancer therapy for
advanced multiple myeloma via the overexpression of JAM-A (42). Notably, HDAC inhibitors increase the
entry and replication of the reovirus into HNSCC cells, and
combination treatment with HDAC inhibitors and Reolysin increases
both the cytotoxic effects of the reovirus and anti-tumor immunity;
however, the mechanisms involving JAM-A remain unclear (43).
Tricellulin and LSR are tricellular tight junction
molecules (44). In HNSCC,
tricellulin is weakly expressed, as is claudin-7, whereas LSR and
claudin-1 are highly expressed (32,34). siRNA
knockdown of LSR, but not tricellulin, markedly increases the
invasiveness of Detroit 562 cells and primary cultured HNSCC cells
(34). In the present study,
tricellulin expression was decreased by all of the HDAC inhibitors
tested in a dose-dependent manner, as were the levels of p63 and
acetylated tubulin, whereas LSR expression was decreased only by
treatment with the HDAC1 inhibitor (Fig.
S1A). In immunocytochemistry, tricellulin was demonstrated to
be decreased at the membranes by treatment with all of the HDAC
inhibitors (Fig. S1B). However, the
roles of tricellular tight junction molecules remain unclear in
HNSCC.
HDAC inhibitors promote proliferative arrest,
differentiation and apoptosis in tumor cells with minimal effects
on normal tissue (45). Moreover, TSA
has been shown to induce the upregulation of p21 and
G2/M cell cycle arrest in colorectal cancer and oral SCC
(13,14). Class 1 HDACs inhibit p21 expression,
and knockdown of HDAC1, 2 and 3 upregulates p21 promoter activity
in human colon cancer cells (37).
The cell cycle of the HNSCC Detroit 562 cell line is affected by
various factors (46,47). Furthermore, the HDAC inhibitor SAHA
causes cell cycle arrest by upregulating cyclin-dependent kinase
inhibitor 1 and downregulating G1/S-specific cyclin-D1 (encoding
p21 and cyclin D1 proteins), as well as inhibiting larynx cancer
cell proliferation (48).
The EGFR-ERK signaling pathway is associated with
G1/S cell cycle progression in cancer cells (15,16). In
the present study, treatment with TSA also downregulated the
expression of p63, EGFR and phospho-ERK1/2 in HNSCC cells.
Furthermore, p63 knockdown or treatment with the EGFR inhibitor
AG1478 induced G1 arrest and the downregulation of p63,
EGFR, phospho-ERK1/2 and cyclin D1, without upregulating of p21.
These findings indicate that TSA suppresses cancer cell
proliferation via p63-mediated G1 arrest in HNSCC.
In conclusion, the HDAC inhibitor TSA, as well as
specific inhibitors of HDAC1 and 6, suppresses the proliferation,
migration and invasiveness of HNSCC cells by downregulating
p63-mediated tight junction molecules JAM-A and claudin-1, and
inducing p63- or p21-mediated growth arrest with single-agent
activity. HDAC inhibitors are considered to be potential
immunomodulatory agents for cancer therapy. HDAC6 is a unique
member of the HDAC family that not only participates in histone
acetylation and deacetylation, but also targets several nonhistone
substrates and upregulates several critical immune factors, such as
programmed death receptor-1 and programmed death receptor ligand-1
(which are major targets for cancer immunotherapy) (49). HDAC inhibitors are an important
emerging therapy with single-agent activity against multiple cancer
types and have significant potential in combination therapy. Thus
the findings of the present study may provide an improved
understanding of the mechanisms by which HDAC inhibition suppresses
head and neck cancers, and thus promote a novel form of cancer
therapy.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Ministry of
Education, Culture, Sports, Science, and Technology of Japan (grant
no. 19K07464).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
AKa, KOh, TH, KT and TKoj designed the study. TKa,
MK, AKo, YM KOba, KN, RM, YK, TKon and TKoh analyzed the data. AKa,
KOha, TH, KT and TKoj wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the ethics
committee of Sapporo Medical University, and informed consent was
obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HNSCC
|
human head and neck squamous cell
carcinoma
|
|
JAM-A
|
junctional adhesion molecule-A
|
|
HDAC
|
histone deacetylase
|
|
TSA
|
torichostatin A
|
|
SAHA
|
suberoylanilide hydroxamic acid
|
|
MMP
|
matrix metalloproteinase
|
|
EGFR
|
epidermal growth factor receptor
|
|
PBS
|
phosphate-buffered saline
|
|
TBS
|
tris-buffered saline
|
|
LSR
|
lipolysis-stimulated lipoprotein
receptor
|
References
|
1
|
Seto E and Yoshida M: Erasers of Histone
Acetylation. The Histone Deacetylase Enzymes. Cold Spring Harb
Perspect Biol. 6:a0187132014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marks P, Rifkind RA, Richon VM, Breslow R,
Miller T and Kelly WK: Histone deacetylases and cancer: Causes and
therapies. Nat Rev Cancer. 1:194–202. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ropero S and Esteller M: The role of
histone deacetylases (HDACs) in human cancer. Mol. Oncol. 1:19–25.
2007.
|
|
4
|
Juengel E, Meyer dos Santos S, Schneider
T, Makarevic J, Hudak L, Bartsch G, Haferkamp A, Wiesner C and
Blaheta RA: HDAC inhibition suppresses bladder cancer cell adhesion
to collagen under flow conditions. Exp Biol Med (Maywood).
238:1297–1304. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Munster PN, Troso-Sandoval T, Rosen N,
Rifkind R, Marks PA and Richon VM: The histone deacetylase
inhibitor suberoylanilide hydroxamic acid induces differentiation
of human breast cancer cells. Cancer Res. 61:8492–8497.
2001.PubMed/NCBI
|
|
6
|
Komatsu N, Kawamata N, Takeuchi S, Yin D,
Chien W, Miller CW and Koeffler HP: SAHA, a HDAC inhibitor, has
profound anti-growth activity against non-small cell lung cancer
cells. Oncol Rep. 15:187–191. 2006.PubMed/NCBI
|
|
7
|
Wilson AJ, Byun DS, Popova N, Murray LB,
L'Italien K, Sowa Y, Arango D, Velcich A, Augenlicht LH and
Mariadason JM: Histone deacetylase 3 (HDAC3) and other class I
HDACs regulate colon cell maturation and p21 expression and are
deregulated in human colon cancer. J Biol Chem. 281:13548–13558.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sakuma T, Uzawa K, Onda T, Shiiba M, Yokoe
H, Shibahara T and Tanzawa H: Aberrant expression of histone
deacetylase 6 in oral squamous cell carcinoma. Int J Oncol.
29:117–124. 2006.PubMed/NCBI
|
|
9
|
Zhao R, Chen K, Cao J, Yu H, Tian L and
Liu M: A correlation analysis between HDAC1 over-expression and
clinical features of laryngeal squamous cell carcinoma. Acta
Otolaryngol. 136:172–176. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
de Ruijter AJ, van Gennip AH, Caron HN,
Kemp S and van Kuilenburg AB: Histone deacetylases (HDACs):
Characterization of the classical HDAC family. Biochem J.
370:737–749. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Khan N, Jeffers M, Kumar S, Hackett C,
Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, et al:
Determination of the class and isoform selectivity of
small-molecule histone deacetylase inhibitors. Biochem J.
409:581–589. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chikamatsu K, Ishii H, Murata T, Sakakura
K, Shino M, Toyoda M, Takahashi K and Masuyama K: Alteration of
cancer stem cell-like phenotype by histone deacetylase inhibitors
in squamous cell carcinoma of the head and neck. Cancer Sci.
104:1468–1475. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Meng J, Zhang HH, Zhou CX, Li C, Zhang F
and Mei QB: The histone deacetylase inhibitor trichostatin A
induces cell cycle arrest and apoptosis in colorectal cancer cells
via p53-dependent and -independent pathways. Oncol Rep. 28:384–388.
2012.PubMed/NCBI
|
|
14
|
Anh TD, Ahn MY, Kim SA, Yoon JH and Ahn
SG: The histone deacetylase inhibitor, Trichostatin A, induces G2/M
phase arrest and apoptosis in YD-10B oral squamous carcinoma cells.
Oncol Rep. 27:455–460. 2012.PubMed/NCBI
|
|
15
|
Massagué J: G1 cell-cycle control and
cancer. Nature. 432:298–306. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang W and Liu HT: MAPK signal pathways
in the regulation of cell proliferation in mammalian cells. Cell
Res. 12:9–18. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chou CW, Wu MS, Huang WC and Chen CC: HDAC
inhibition decreases the expression of EGFR in colorectal cancer
cells. PLoS One. 6:e180872011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Koster MI, Kim S, Mills AA, DeMayo FJ and
Roop DR: p63 is the molecular switch for initiation of an
epithelial stratification program. Genes Dev. 18:126–131. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weber A, Bellmann U, Bootz F, Wittekind C
and Tannapfel A: Expression of p53 and its homologues in primary
and recurrent squamous cell carcinomas of the head and neck. Int J
Cancer. 99:22–28. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rocco JW, Leong CO, Kuperwasser N, DeYoung
MP and Ellisen LW: p63 mediates survival in squamous cell carcinoma
by suppression of p73-dependent apoptosis. Cancer Cell. 9:45–56.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
DeYoung MP, Johannessen CM, Leong CO,
Faquin W, Rocco JW and Ellisen LW: Tumor-specific p73 up-regulation
mediates p63 dependence in squamous cell carcinoma. Cancer Res.
66:9362–9368. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ramsey MR, He L, Forster N, Ory B and
Ellisen LW: Physical association of HDAC1 and HDAC2 with p63
mediates transcriptional repression and tumor maintenance in
squamous cell carcinoma. Cancer Res. 71:4373–4379. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Martìn-Padura I, Lostaglio S, Schneemann
M, Williams S, Romano M, Fruscella P, Panzeri C, Stoppacciaro A,
Ruco L, Villa A, et al: Junctional adhesion molecule, a novel
member of the immunoglobulin superfamily that distributes at
intercellular junctions and modulates monocyte transmigration. J
Cell Biol. 142:117–127. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tsukita S, Furuse M and Itoh M:
Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol.
2:285–293. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McSherry EA, Brennan K, Hudson L, Hill ADK
and Hopkins AM: Breast cancer cell migration is regulated through
junctional adhesion molecule-A-mediated activation of Rap1 GTPase.
Breast Cancer Res. 13:R312011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Severson EA, Lee WY, Capaldo CT, Nusrat A
and Parkos CA: Junctional adhesion molecule A interacts with Afadin
and PDZ-GEF2 to activate Rap1A, regulate beta1 integrin levels, and
enhance cell migration. Mol Biol Cell. 20:1916–1925. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kakuki T, Kurose M, Takano K, Kondoh A,
Obata K, Nomura K, Miyata R, Kaneko Y, Konno T, Takahashi S, et al:
Dysregulation of junctional adhesion molecule-A via p63/GATA-3 in
head and neck squamous cell carcinoma. Oncotarget. 7:33887–33900.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Furuse M, Hata M, Furuse K, Yoshida Y,
Haratake A, Sugitani Y, Noda T, Kubo A and Tsukita S: Claudin-based
tight junctions are crucial for the mammalian epidermal barrier: A
lesson from claudin-1-deficient mice. J Cell Biol. 156:1099–1111.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lopardo T, Lo Iacono N, Marinari B,
Giustizieri ML, Cyr DG, Merlo G, Crosti F, Costanzo A and Guerrini
L: Claudin-1 is a p63 target gene with a crucial role in epithelial
development. PLoS One. 3:e27152008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dos Reis PP, Bharadwaj RR, Machado J,
Macmillan C, Pintilie M, Sukhai MA, Perez-Ordonez B, Gullane P,
Irish J and Kamel-Reid S: Claudin 1 overexpression increases
invasion and is associated with aggressive histological features in
oral squamous cell carcinoma. Cancer. 113:3169–3180. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sappayatosok K and Phattarataratip E:
Overexpression of Claudin-1 is associated with advanced clinical
stage and invasive pathologic characteristics of oral squamous cell
carcinoma. Head Neck Pathol. 9:173–180. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kondoh A, Takano K, Kojima T, Ohkuni T,
Kamekura R, Ogasawara N, Go M, Sawada N and Himi T: Altered
expression of claudin-1, claudin-7, and tricellulin regardless of
human papilloma virus infection in human tonsillar squamous cell
carcinoma. Acta Otolaryngol. 131:861–868. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Oku N, Sasabe E, Ueta E, Yamamoto T and
Osaki T: Tight junction protein claudin-1 enhances the invasive
activity of oral squamous cell carcinoma cells by promoting
cleavage of laminin-5 gamma2 chain via matrix metalloproteinase
(MMP)-2 and membrane-type MMP-1. Cancer Res. 66:5251–5257. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Takano K, Kakuki T, Obata K, Nomura K,
Miyata R, Kondo A, Kurose M, Kakiuchi A, Kaneko Y, Kohno T, et al:
The behavior and role of lipolysis-stimulated lipoprotein receptor,
a component of tricellular tight junctions, in head and neck
squamous cell carcinomas. Anticancer Res. 36:5895–5904. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Leech AO, Cruz RG, Hill AD and Hopkins AM:
Paradigms lost-an emerging role for over-expression of tight
junction adhesion proteins in cancer pathogenesis. Ann Trans Med.
3:1842015.
|
|
36
|
Tian Y, Tian Y, Zhang W, Wei F, Yang J,
Luo X, Zhou T, Hou B, Qian S, Deng X, et al: Junctional adhesion
molecule-A, an epithelial-mesenchymal transition inducer,
correlates with metastasis and poor prognosis in human
nasopharyngeal cancer. Carcinogenesis. 36:41–48. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zang M, Luo W, Huang B, Liu Z, Sun L,
Zhang Q, Qiu X, Xu K and Wang E: Overexpression of JAM-A in
non-small cell lung cancer correlates with tumor progression. PLoS
One. 8:e791732013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ikeo K, Oshima T, Shan J, Matsui H, Tomita
T, Fukui H, Watari J and Miwa H: Junctional adhesion molecule-A
promotes proliferation and inhibits apoptosis of gastric cancer.
Hepatogastroenterology. 62:540–545. 2015.PubMed/NCBI
|
|
39
|
Krishnan M, Singh AB, Smith JJ, Sharma A,
Chen X, Eschrich S, Yeatman TJ, Beauchamp RD and Dhawan P: HDAC
inhibitors regulate claudin-1 expression in colon cancer cells
through modulation of mRNA stability. Oncogene. 29:305–312. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
LeBoeuf M, Terrell A, Trivedi S, Sinha S,
Epstein JA, Olson EN, Morrisey EE and Millar SE: Hdac1 and Hdac2
act redundantly to control p63 and p53 functions in epidermal
progenitor cells. Dev Cell. 19:807–818. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Barton ES, Forrest JC, Connolly JL,
Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA and Dermody TS:
Junction adhesion molecule is a receptor for reovirus. Cell.
104:441–451. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kelly KR, Espitia CM, Zhao W, Wendlandt E,
Tricot G, Zhan F, Carew JS and Nawrocki ST: Junctional adhesion
molecule-A is overexpressed in advanced multiple myeloma and
determines response to oncolytic reovirus. Oncotarget.
6:41275–41289. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jaime-Ramirez AC, Yu JG, Caserta E, Yoo
JY, Zhang J, Lee TJ, Hofmeister C, Lee JH, Kumar B, Pan Q, et al:
Reolysin and histone deacetylase inhibition in the treatment of
head and neck squamous cell carcinoma. Mol Ther Oncolytics.
5:87–96. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Masuda S, Oda Y, Sasaki H, Ikenouchi J,
Higashi T, Akashi M, Nishi E and Furuse M: LSR defines cell corners
for tricellular tight junction formation in epithelial cells. J
Cell Sci. 124:548–555. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lane AA and Chabner BA: Histone
deacetylase inhibitors in cancer therapy. J Clin Oncol.
27:5459–5468. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang H, Xue W and Jiang X: Overexpression
of TRIM24 stimulates proliferation and glucose metabolism of head
and neck squamous cell carcinoma. Biomed Res Int.
2018:61428432018.PubMed/NCBI
|
|
47
|
Dziedzic A, Kubina R, Kabała-Dzik A and
Tanasiewicz M: Induction of cell cycle arrest and apoptotic
response of head and neck squamous carcinoma cells (Detroit 562) by
caffeic acid and aaffeic acid phenethyl ester derivative. Evid
Based Complement Alternat Med. 2017:67934562017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Grabarska A, Łuszczki JJ, Nowosadzka E,
Gumbarewicz E, Jeleniewicz W, Dmoszyńska-Graniczka M, Kowalczuk K,
Kupisz K, Polberg K and Stepulak A: Histone deacetylase inhibitor
SAHA as potential targeted therapy agent for larynx cancer cells. J
Cancer 2017. 8:19–28. 2017.
|
|
49
|
Li T, Zhang C, Hassan S, Liu X, Song F,
Chen K, Zhang W and Yang J: Histone deacetylase 6 in cancer. J
Hematol Oncol. 11:1112018. View Article : Google Scholar : PubMed/NCBI
|