Introduction
Lung cancer is the leading cause of cancer-related
death in the United States and China, with an approximate 5-year
survival rate of ~18% (1). The most
common type of lung cancer is non-small cell lung cancer (NSCLC),
which accounts for >80% of all cases (2). NSCLC can be further divided into three
predominant histological subtypes: Adenocarcinoma, squamous cell
carcinoma and large cell carcinoma (2). A total of 70% of patients with lung
cancer are diagnosed with advanced stage disease, which poses more
challenges for therapy (3). Over the
past two decades, the application of various targeted therapies,
including the EGFR inhibitor afatinib, and the anaplastic lymphoma
kinase inhibitor ceritinib, and immunotherapies, such as the immune
checkpoint blocker nivolumab, have resulted in remarkable progress
in treating patients with advanced NSCLC; however, drug resistance
and a poor response prevented the therapeutic efficacy (4). Therefore, the identification and
characterization of novel molecules associated with NSCLC
progression is of critical importance for improving both the early
diagnosis and treatment of the disease.
Transmembrane protein 100 (TMEM100) transcript was
first identified in the mouse genome in 2001 (5). TMEM100 is a 134-amino acid protein,
which contains two transmembrane domains at amino acid residues
53–75 and 85–107 (6). It is highly
conserved in vertebrates and mammals; however, there is no sequence
homology between TMEM100 and any other proteins (6), therefore the biological functions of
TMEM100 were further explored in the present study. Previous
in-depth studies have revealed that TMEM100 was involved in
arterial endothelial differentiation and vascular integrity
(6–8),
as well as apoptosis (9), enteric
nervous system development (10) and
persistent pain (11). Interestingly,
TMEM100 was demonstrated to be mainly expressed in arterial
endothelial cells during the embryonic developmental stage, whereas
during adulthood, TMEM100 was most abundantly expressed in the
lung, with lower expression in the brain, heart and muscle tissues
(7), suggesting that TMEM100 may
serve important roles in lung development.
In our previous study, Gene Expression Omnibus
datasets (GSE19804, GSE18842, GSE27262 and GSE43458) were
downloaded and analyzed to screen differentially expressed genes
(DEGs) in NSCLC. Among these DEGs, TMEM100 expression level was
significantly decreased in NSCLC tissues compared with normal
tissues, which suggested that TMEM100 may serve as a tumor
suppressor in NSCLC (data not shown). Recently, TMEM100 has been
reported to inhibit tumor progression of NSCLC and hepatocellular
carcinoma (12–14). Although the tumor suppressor role of
TMEM100 in NSCLC has been revealed, the mechanism of its function
is still poorly understood.
Autophagy is an evolutionarily conserved and highly
regulated degradation pathway, in which a cell digests its damaged
cytoplasmic proteins, macromolecules and organelles to maintain
cellular homeostasis (15). The
dysregulation of autophagy has been associated with a number of
human diseases, including cancer (16). In hepatocellular carcinoma, the levels
of autophagy in both aggressive malignant cell lines and tissues
from patients with recurrent disease were significantly decreased
compared with those of less aggressive cell lines or tissues
(17). In addition, Zhang et
al (18) indicated that DEAD box
protein 5 inhibited liver tumor progression by inducing autophagy
via interacting with p62. However, other previous studies have
reported that the metabolic recycling role of autophagy promoted
cancer cell survival under nutrient-deprived conditions (19,20).
Furthermore, the deletion of essential autophagy genes, such as
Atg7 or Atg5, was demonstrated to impair tumor growth (21). These paradoxical reports have
indicated that the role of autophagy in tumor progression may vary
depending on the pathological conditions. For example, in NSCLC,
nuclear factor erythroid 2-related factor 2 (Nrf2) has been
indicated to promote tumorigenic processes by activating autophagy
(22), whereas the tumor suppressor
casein kinase 1α has been revealed to function as an autophagy
inducer to inhibit tumor growth (23), indicating that autophagy serves a
contradictory role in NSCLC. However, it remains unknown whether
TMEM100, as a potential tumor suppressor, can affect the
progression of NSCLC by regulating autophagy.
Therefore, the present study aimed at validating the
functions of TMEM100 in different NSCLC cell lines and exploring
the potential mechanism of TMEM100 in NSCLC progression. TMEM100
expression level was observed to be decreased in NSCLC tissues and
cell lines, and its expression level was negatively associated with
the clinical staging and positively associated with the prognosis
of patients. Moreover, the overexpression of TMEM100 inhibited
proliferation, whilst promoting apoptosis and autophagy in lung
cancer cells. Mechanistically, TMEM100-induced autophagy was, at
least partially, mediated by inhibiting the PI3K/AKT signaling
pathway, and the inhibition of TMEM100-induced autophagy promoted
apoptosis to compensate for the cell death. These findings
suggested that TMEM100 may serve essential roles as a tumor
suppressor in NSCLC by promoting autophagy via the inhibition of
the PI3K/AKT signaling pathway. The findings of the present study
may provide novel insights for developing new targeted therapy
strategies for NSCLC.
Materials and methods
Analysis of the cancer genome atlas
(TCGA) database
Gene expression data and the relevant clinical
information of 533 cases with lung adenocarcinoma and 59 cases with
normal lung tissue were downloaded from TCGA database (http://cancergenome.nih.gov/).
Analysis of the cancer cell line
encyclopedia (CCLE) database
Relative gene expression levels in different NSCLC
cell lines, including HCC4006, H1734, H460, H1581, H1975, HCC15,
H1092, SW900, H1944, H520, A549, SK-MES-1 and H1299, were
downloaded from the online CCLE database (https://portals.broadinstitute.org/ccle).
Analysis of Kaplan-Meier Plotter
database
The survival rate of 1,926 patients with lung cancer
was analyzed using the online database Kaplan-Meier Plotter
(http://kmplot.com/analysis/index.php?p=background).
All patients were divided into high and low expression groups
according to the median level of TMEM100.
Cell culture
The human NSCLC cell lines, A549 and H460, were
purchased from Shanghai GeneChem Co., Ltd. (http://www.genechem.com.cn/). H1299 (cat. no.
CL-0165), SK-MES-1 (cat. no. CL-0213) and human bronchial
epithelial (HBE) cells (cat. no. CL-0346) from Procell Life Science
& Technology Co., Ltd. (https://www.procell.com.cn/) were kindly provided by
Professor Bu (School of Basic Medicine, Chongqing Medical
University, Chongqing, China). The 293T cell line was purchased
from American Type Culture Collection (https://www.atcc.org/). A549 cells were cultured in
F-12K medium (HyClone; Cytiva), whereas the other cell lines were
cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific,
Inc.), except for 293T cells, which were cultured in DMEM (HyClone;
Cytiva). All culture mediums were supplemented with 10% FBS
(Biological Industries), 100 U/ml penicillin and 100 µg/ml
streptomycin (HyClone; Cytiva), and cells were maintained at 37°C
in a humidified atmosphere containing 5% CO2.
Cell transfection
The overexpression plasmid pHBLV-CMV-TMEM100 and the
corresponding empty vector were purchased from Hanbio Biotechnology
Co., Ltd., and TMEM100 small interfering (si)RNAs were obtained
from Shanghai Transheep Bio-Tech Co., Ltd. (http://www.transheep.com/). The following TMEM100
siRNA sequences were used: si-TMEM100-1,
5′-GGGAGUGCACAGCAUUAGATT−3′; and si-TMEM100-2,
5′-CCUACAGUCUUAAGAUGUATT−3′. A non-targeting siRNA oligonucleotide
(5′-UUCUCCGAACGUGUCACGUTT−3′) was used as a control. A total of
3.5×105 A549 and H460 cells were seeded into six-well
plates, followed by transient transfection with 3 µg plasmids and
100 pmol siRNAs at 37°C using Lipofectamine® 2000
reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to
the manufacturer's instructions. Cells were collected 24 and 48 h
after transfection for RNA and protein extraction,
respectively.
Lentiviral production
A 2nd generation system was used for lentiviral
production. Lentiviruses were produced by co-transfecting 293T
cells in 60 mm dishes with 1.5 µg pHBLV-CMV-TMEM100 and two
packaging plasmids (1.5 µg psPAX2 and 1.5 µg pMD2.G) using
Lipofectamine 2000 reagent. Lentiviruses were harvested following
48 h of transfection, centrifuged at 500 × g at 4°C for 5 min to
remove the cell debris, and filtered through a 0.45-µm membrane
(EMD Millipore). A549 cells were infected with lentiviruses at a
MOI of 50. A549 cells stably overexpressing TMEM100 were selected
by treating the lentivirus-infected cells with 1.5 µg/ml puromycin
(Beijing Solarbio Science & Technology Co., Ltd.) for 14
days.
Cell viability assay
A total of 2×103 A549 cells were seeded
into 96-well plates with six replicates/group, and cultured for 0,
24, 48 or 72 h continuously. At each time point, 10 µl of Cell
Counting Kit-8 (Beijing Solarbio Science & Technology Co.,
Ltd.) reagent was added into each well and incubated for 2 h at
37°C. The optical density at 450 nm was measured using a microplate
reader. The experiments were repeated in triplicate.
Colony formation assay
A549 cells were plated in a six-well plate at a
density of 600 cells/well and incubated for 10 days. The plate was
washed with PBS, fixed with 4% paraformaldehyde at room temperature
for 15 min and the colonies were stained with 0.5% crystal violet
at room temperature for 20 min. The number of colonies containing
>50 cells was manually counted under an optical microscope
(magnification, ×400).
Cell cycle analysis
A total of 3.5×105 A549 cells were seeded
into a six-well plate and transfected with 3 µg pHBLV-CMV-TMEM100
plasmid or empty vector for 24 h. A total of 4×106 cells
from each group were washed with cold PBS, collected and fixed in
70% alcohol at 4°C overnight. Cells were treated with RNase A for
30 min at 37°C and subsequently stained with PI at room temperature
for 15 min in the dark. Cell cycle analysis was performed using a
CytoFLEX flow cytometer (Beckman Coulter, Inc.) and the results
were analyzed by CytExpert software v1.0 (Beckman Coulter, Inc.).
The assay was repeated in triplicate.
Flow cytometric analysis of
apoptosis
A549 and H460 cells transfected with 3 µg
pHBLV-CMV-TMEM100 plasmid or 100 pmol siRNAs were collected and
washed twice with cold PBS, then resuspended in 100 µl 1X binding
buffer. A volume of 5 µl Annexin V-APC (Tianjin Sanjian
Biotechnology Co., Ltd.) and 5 µl DAPI were added, mixed well and
incubated at room temperature for 10 min in the dark. Apoptotic
cells were analyzed using a CytoFLEX flow cytometer and the results
were analyzed by CytExpert software v1.0 (Beckman Coulter, Inc.).
The lower right (early apoptosis stage) and upper right quadrants
(late apoptosis stage) were counted to assess the apoptosis rate.
The assay was repeated in triplicate.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA of H460, HBE, A549, SK-MES-1 and H1299
cells was extracted using RNAiso Plus (Takara Bio, Inc.). The RNA
concentration and purity were measured using a Nanodrop ND-100
spectrophotometer (NanoDrop Technologies; Thermo Fisher Scientific,
Inc.). A total of 1 µg RNA was reverse transcribed into cDNA using
the PrimeScript™ RT Reagent kit (Takara Bio, Inc.), according to
the manufacturer's instructions. qPCR was subsequently performed
using the CFX96 real-time PCR detection system (Bio-Rad
Laboratories, Inc.) and a TB Green™ Premix Ex Taq™ II kit (Takara
Bio, Inc.). The following primer sequences were used for the qPCR:
GAPDH forward, 5′-TGCACCACCAACTGCTTAGC-3′ and reverse,
5′-GGCATGGACTGTGGTCATGA-3′; and TMEM100 forward,
5′-TGCTGTGGTTGTCTTCATCG-3′ and reverse,
5′-CTCTCCCGTCTCTTGGCTTTC-3′. The following thermocycling conditions
were used for the qPCR: Initial denaturation at 95°C for 5 min; 42
cycles of denaturation at 95°C for 30 sec, annealing at 55°C for 30
sec and extension at 72°C for 30 sec. Relative expression levels
were determined using the 2−ΔΔCq method (24) and normalized to GAPDH.
Immunofluorescence analysis
A549 cells were fixed with 4% paraformaldehyde at
room temperature for 10 min and washed thrice with PBS.
Subsequently, cells were permeabilized with 0.5% Triton X-100 at
room temperature for 10 min and blocked with 5% BSA (Amresco, LLC)
at 37°C for 1 h. Cells were incubated with anti-LC3A/B (monoclonal;
rabbit anti-human; 1:200; cat. no. 4108; Cell Signaling Technology,
Inc.) at 4°C overnight and with anti-rabbit IgG (H + L) (Alexa
Fluor® 594 Conjugate; 1:500; cat. no. 8889S; Cell
Signaling Technology, Inc.) at 37°C for 1 h. Nuclei were stained
with DAPI at room temperature for 15 min and stained cells were
visualized using a fluorescent microscope (magnification, ×400;
Nikon Corporation).
Immunohistochemistry (IHC)
Paraffin-embedded sections, including 15 cases of
NSCLC tissues and benign adjacent non-cancerous tissues, were
obtained from the Laboratory of Pathology, The First Affiliated
Hospital of Chongqing Medical University (Chongqing, China) from
2016 to 2018. The average age of the patients was 58.87±11.92
years, and males accounted for 66.7% and females for 33.3%. The use
of these tissue samples was approved by the Ethics and Research
Committees of Chongqing Medical University. Written informed
consent was obtained from all participants. The slides were
routinely deparaffinized, rehydrated and boiled for antigen
retrieval in citric acid buffer (pH 6.0) for 10 min. Subsequently,
sections were treated with a mouse/rabbit Streptomyces
vitellogenin-Biotin Detection system (cat. no. SP-9000; Beijing
Zhongshan Golden Bridge Biotechnology Co., Ltd.), according to the
manufacturer's instructions. Finally, the slides were mounted and
photographed using an optical microscope (magnification, ×400).
Western blotting
Total protein was extracted from A549 and H460 cells
using RIPA lysis buffer, containing the protease inhibitor PMSF and
a phosphatase inhibitor (all from Beyotime Institute of
Biotechnology). Protein concentrations were determined using a BCA
Protein Assay kit (Beyotime Institute of Biotechnology) according
to the manufacturer's instructions. A total of 40–50 µg
protein/lane were separated via 12% SDS-PAGE. The separated
proteins were transferred onto PVDF membranes (EMD Millipore) and
blocked with 5% BSA (Amresco, LLC) at 37°C for 2 h. The membranes
were incubated with the following primary antibodies overnight at
4°C: Anti-TMEM100 (monoclonal; mouse anti-human; 1:500; cat. no.
MA5-24949; Invitrogen; Thermo Fisher Scientific, Inc.), anti-BAX
(monoclonal; rabbit anti-human; 1:1,000; cat. no. AF1270; Beyotime
Institute of Biotechnology), anti-BCL2 (monoclonal; rabbit
anti-human; 1:1,000; cat. no. AF1915; Beyotime Institute of
Biotechnology), anti-LC3A/B (monoclonal; rabbit anti-human;
1:1,000; cat. no. 4108; Cell Signaling Technology, Inc.), anti-p62
(monoclonal; rabbit anti-human; 1:5,000; cat. no. CY5546; Abways
Technology), anti-GAPDH (monoclonal; rabbit anti-human; 1:5,000;
cat. no. AF1186; Beyotime Institute of Biotechnology),
anti-phosphorylated (p)-PI3K (monoclonal; rabbit anti-human;
1:1,000; cat. no. 4228S; Cell Signaling Technology, Inc.),
anti-total (t)-PI3K (monoclonal; rabbit anti-human; cat. no. 4257S;
1:1,000; Cell Signaling Technology, Inc.), anti-p-AKT (monoclonal;
rabbit anti-human; 1:1,000; cat. no. 2965S; Cell Signaling
Technology, Inc.), anti-t-AKT (monoclonal; rabbit anti-human;
1:1,000; cat. no. 4691S; Cell Signaling Technology, Inc.).
Following the primary antibody incubation, membranes were incubated
with HRP-conjugated goat anti-rabbit IgG (H + L) (1:10,000; cat.
no. 04-15-06; KPL) or HRP-conjugated goat anti-mouse IgG (H + L)
(1:10,000; cat. no. 04-18-06; KPL) at 37°C for 1 h. Protein bands
were visualized using Immobilon™ Western HRP Substrate Peroxide
solution (EMD Millipore) and a Bio-Rad electrophoresis
documentation system.
Agonist and inhibitor treatment
A549 cells were pretreated with 30 µM 740Y-P
(MedChemExpress) or 20 µM SC79 (Beyotime Institute of
Biotechnology) for 1.5 h at 37°C, and subsequently transfected with
3 µg pHBLV-CMV-TMEM100 plasmid or empty vector for 24 h. H460 cells
were pretreated with 20 µM LY294002 (Beyotime Institute of
Biotechnology) for 1.5 h at 37°C, followed by transfection with 100
pmol siRNA or si-NC for 24 h. A total of 50 nM Bafilomycin A1
(Selleck Chemicals) was added into A549 cells for 2 h at 37°C prior
to transfection of 3 µg pHBLV-CMV-TMEM100 plasmid or empty vector.
Control cells received an equal volume of DMSO (Beijing Solarbio
Science & Technology Co., Ltd.) and the final concentration of
DMSO was <0.1%. Subsequently, protein expression levels were
determined by western blotting.
Mouse xenograft experiment
The animal studies were approved by the
Institutional Animal Care and Use Committee of Chongqing Medical
University (Chongqing, China). A total of 10 female athymic BALB/c
nude mice (age, 4–5 weeks old; weight, 16–20 g; n=5 per group) were
purchased from Chongqing Medical University and housed in specific
pathogen-free conditions at constant temperature (22°C) and
humidity (50–60%) on a 12 h light/dark cycle with free access to
food and water. A549 cells transfected with empty vector or the
TMEM100 overexpression plasmid in the exponential growth phase were
diluted to a density of 5×106 cells/100 µl PBS and the
cell suspension was injected subcutaneously into the left axilla of
each mouse. The mice were monitored every 3 days. Tumors were
allowed to grow to 12 mm diameter. The protocol continued until 30
days following tumor cell injection. All experimental mice were
sacrificed by cervical dislocation simultaneously, and tumor sizes
and weights were measured.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism version 5.0 software (GraphPad Software, Inc.) and data are
presented as the mean ± SD. Student's t-test was used to determine
the differences between two groups. One-way ANOVA followed by
Bonferroni's post hoc test was used for multiple group comparisons.
Overall survival was calculated using SPSS version 17 (SPSS, Inc.)
with Kaplan-Meier estimate and log-rank test. All experiments were
carried out at least three times. P<0.05 was considered to
indicate a statistically significant difference.
Results
TMEM100 expression levels are
decreased in NSCLC specimens and cell lines
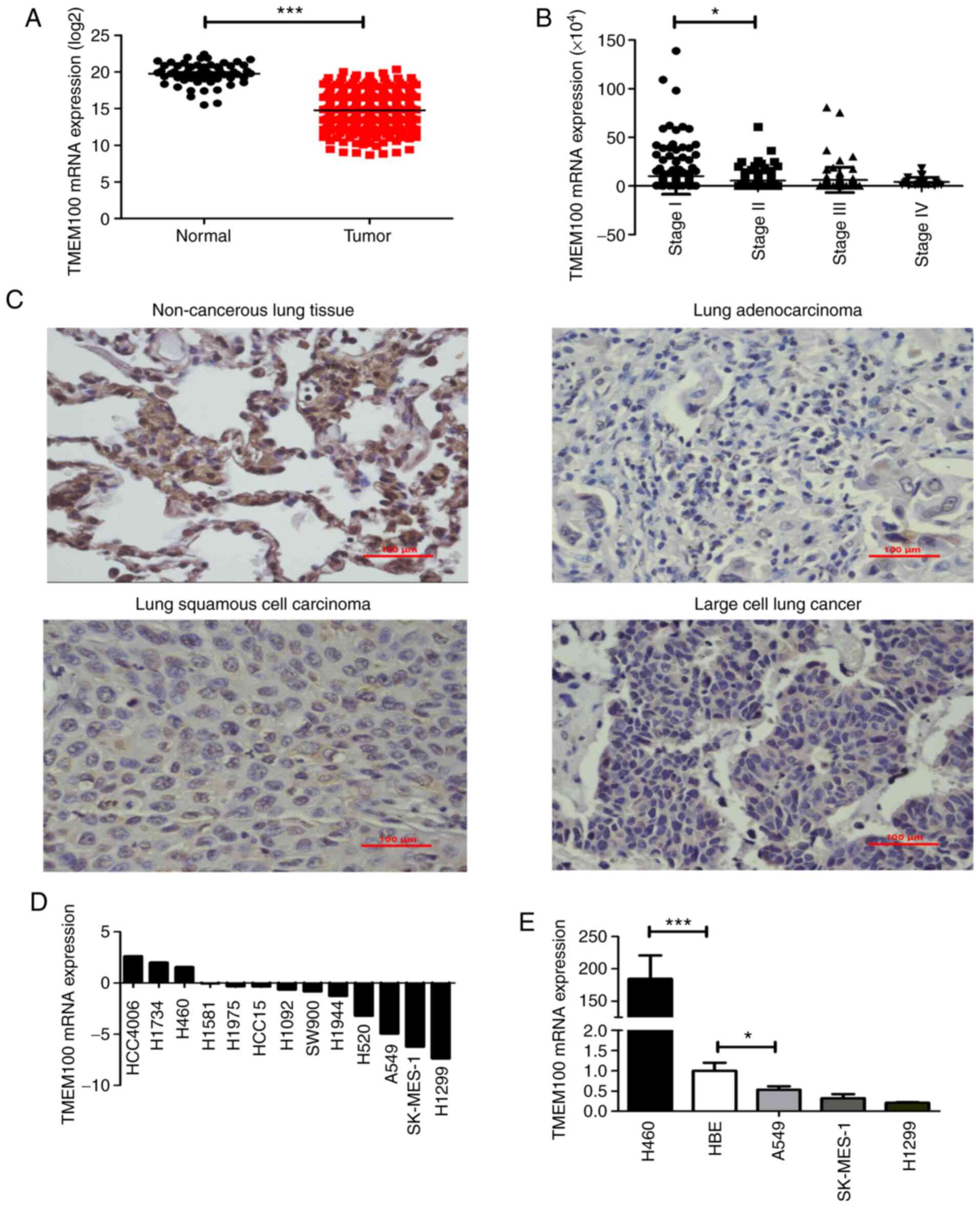
To identify the potential role of TMEM100 in NSCLC,
the expression levels of TMEM100 were analyzed in human lung cancer
tissues from TCGA database. TMEM100 expression level was
significantly decreased in NSCLC specimens (n=533) compared with
normal lung tissues (n=59; Fig. 1A).
Additionally, it was observed that the expression level of TMEM100
exhibited a decreasing trend at more advanced TNM stages of NSCLC
(Fig. 1B), demonstrating that the
expression level of TMEM100 was negatively associated with the
tumor stage. In addition, the association between TMEM100
expression level and the survival of patients in TCGA database was
analyzed using SPSS software. It was indicated that the patients
with lower TMEM100 expression level had worse overall survival, but
there was no statistical significance (Fig. S1A). However, the result from the
online database Kaplan-Meier Plotter demonstrated that the survival
rate of patients with high expression of TMEM100 was significantly
higher than those with low expression of TMEM100 (Fig. S1B), which suggested that the
expression level of TMEM100 was positively associated with the
prognosis of patients with NSCLC. Furthermore, the expression level
of TMEM100 in NSCLC tissues (n=15) and the matched benign adjacent
non-cancerous tissues were detected using IHC. The expression level
of TMEM100 was indicated to be markedly decreased in NSCLC tissues
compared with the adjacent non-cancerous tissues (Fig. 1C), which was consistent with the
results of TCGA database. In addition, data from the CCLE database
were used to examine the relative expression level of TMEM100 in
various NSCLC cell lines. It was revealed that the expressions
level of TMEM100 was relatively low in 10 NSCLC cell lines,
particularly in the A549, SK-MES-1 and H1299 cell lines, whereas it
was relatively high in three NSCLC cell lines, including H460
(Fig. 1D). These results were
validated in four NSCLC cell lines (A549, SK-MES-1, H1299 and H460)
and one normal lung epithelial cell line (HBE) by RT-qPCR (Fig. 1E). Altogether, these findings
suggested that TMEM100 may serve an antitumor role in NSCLC.
TMEM100 inhibits the proliferation of
NSCLC cells
To investigate the role of TMEM100 in NSCLC
progression, TMEM100 was overexpressed in A549 cells and knocked
down in H460 cells using a TMEM100 overexpression plasmid and
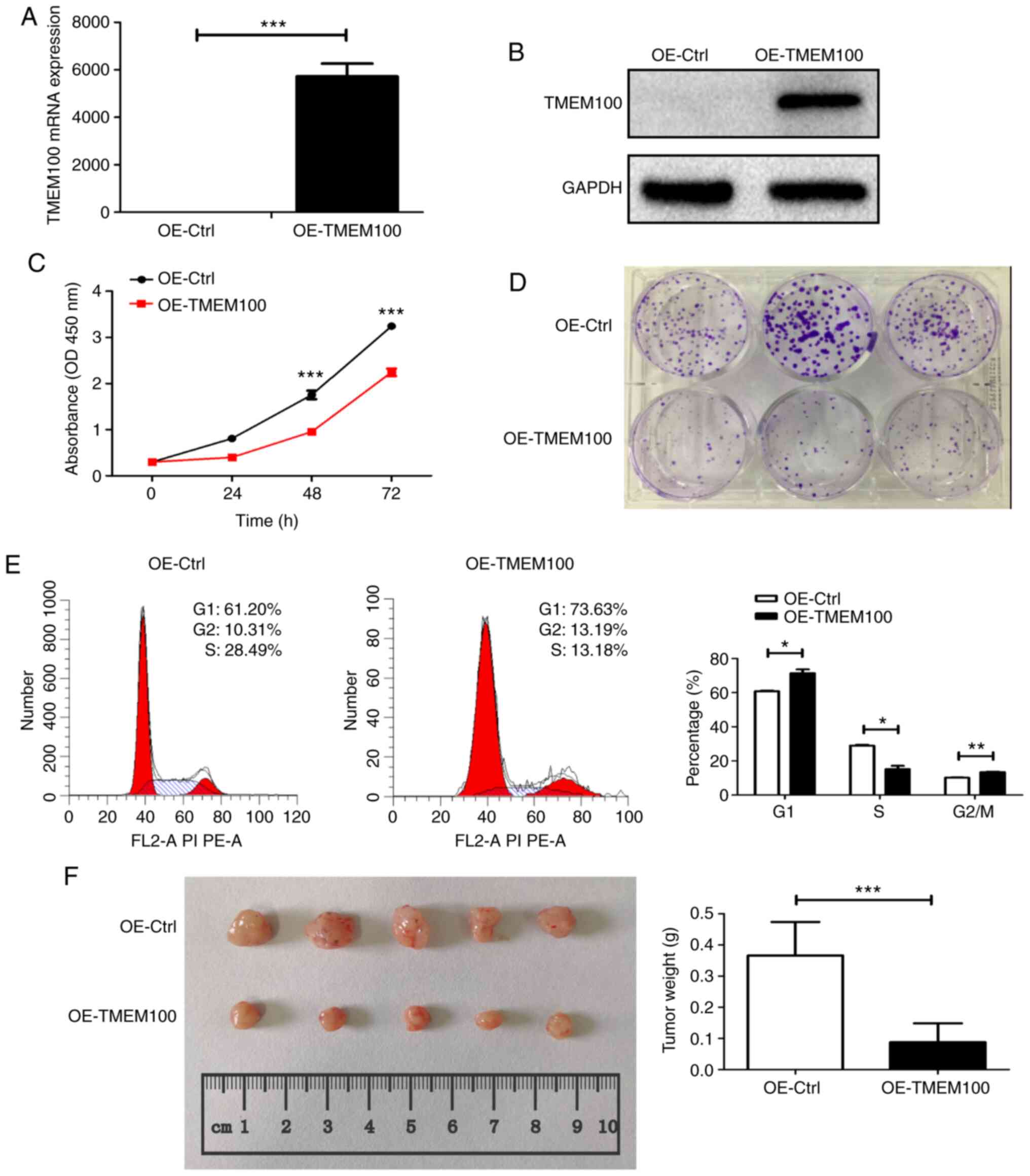
TMEM100 siRNAs, respectively. The transfection efficiency of the
overexpression (Fig. 2A and B) and
knockdown (Fig. S2A and B) was
confirmed by RT-qPCR and western blotting. Subsequently, the effect
of TMEM100 on cell proliferation was investigated. As expected,
overexpression of TMEM100 significantly inhibited A549 cell
proliferation and colony formation (Fig.
2C and D). Furthermore, cell cycle analysis demonstrated that
TMEM100 induced cell cycle arrest in the G1 phase, as
ectopic overexpression of TMEM100 significantly increased the
percentage of A549 cells in the G1 phase from 61.20 to
73.63%, but decreased the percentage of cells in the S phase from
28.49 to 13.18% (Fig. 2E). By
contrast, TMEM100 knockdown enhanced the proliferation of H460
cells (Fig. S2C). To assess the
impact of TMEM100 on tumor growth in vivo, A549 cells with
or without ectopic overexpression of TMEM100 were subcutaneously
injected into athymic nude mice. At day 30, tumor size and weight
were notably decreased in the TMEM100 overexpression group compared
with the control group (Fig. 2F).
Thus, both in vitro and in vivo experiments
demonstrated a marked antitumor role of TMEM100 in NSCLC.
TMEM100 promotes apoptosis in NSCLC
cells
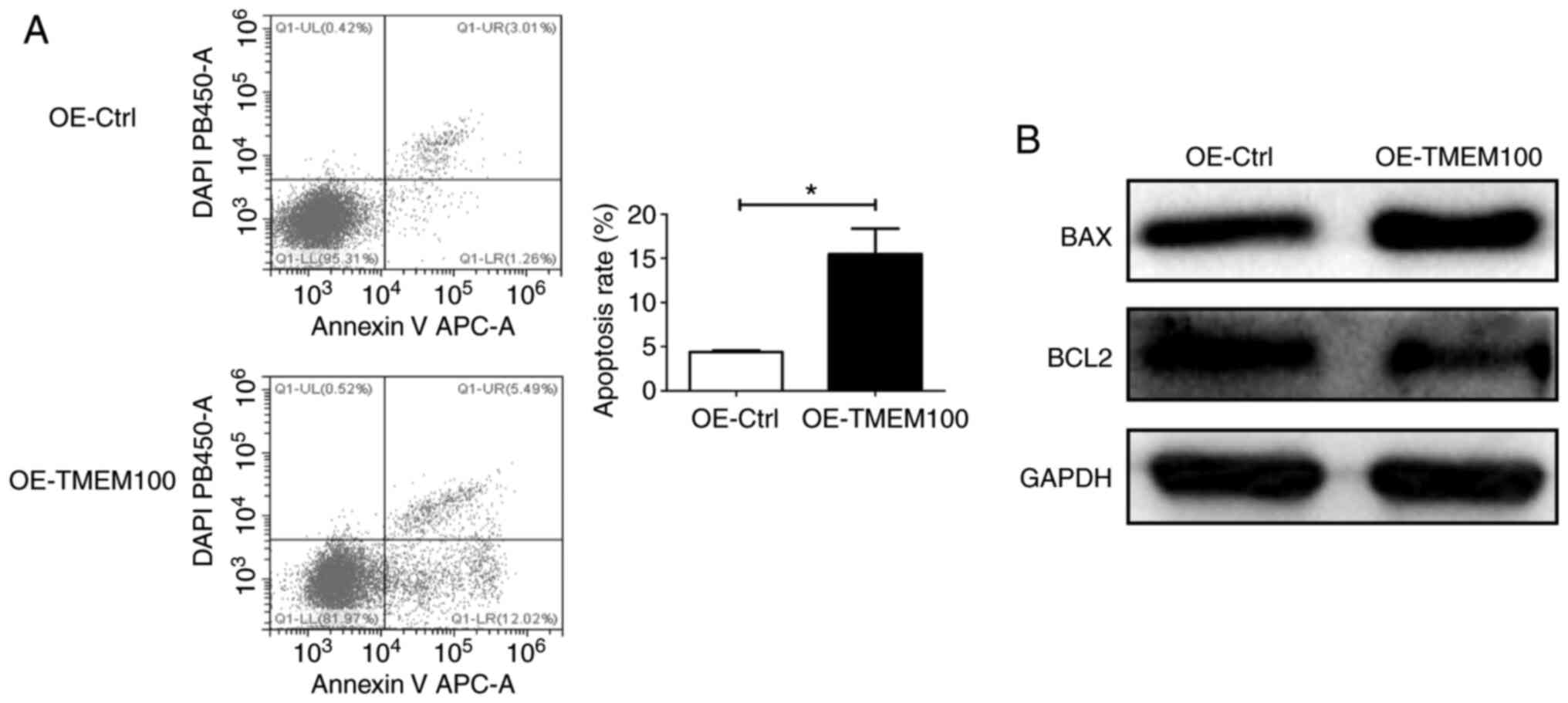
To investigate the effect of TMEM100 on cell
apoptosis, an Annexin V-DAPI apoptosis assay and western blotting,
to determine the expression levels of BAX/BCL2, were performed. The
early-stage and late-stage apoptotic rate was significantly
increased in TMEM100-overexpressing cells compared with control
cells (Fig. 3A), suggesting that
ectopic overexpression of TMEM100 may promote apoptosis in A549
cells. Consistently, the expression level of the pro-apoptotic
protein BAX was increased and that of the anti-apoptotic protein
BCL2 was decreased following TMEM100 overexpression (Fig. 3B). By contrast, knocking down TMEM100
resulted in the opposite effects in H460 cells (Fig. S3A and B). Altogether, these findings
indicated that TMEM100 may act as a tumor suppressor that is
involved in the promotion of cell apoptosis.
TMEM100 induces autophagy in NSCLC
cells via inhibiting the PI3K/AKT signaling pathway
Accumulating evidence has emphasized the importance
of autophagy in lung cancer progression (25,26),
therefore the effect of TMEM100 on cell autophagy was investigated.
The expression of p62 and the transformation of LC3 I to LC3 II,
which are essential for autophagy and mainly used as protein
markers of autophagy (27), were
examined. The results revealed a notable increase in the ratio of
LC3 II/LC3 I and a decrease in the expression level of p62 in A549
cells following TMEM100 overexpression (Fig. 4A). Furthermore, knocking down TMEM100
in H460 cells yielded the opposite results (Fig. S4A). In addition, it was observed that
TMEM100 overexpression promoted the accumulation of LC3 puncta in
A549 cells (Fig. 4B). These results
indicated that TMEM100 may induce autophagy in NSCLC cells.
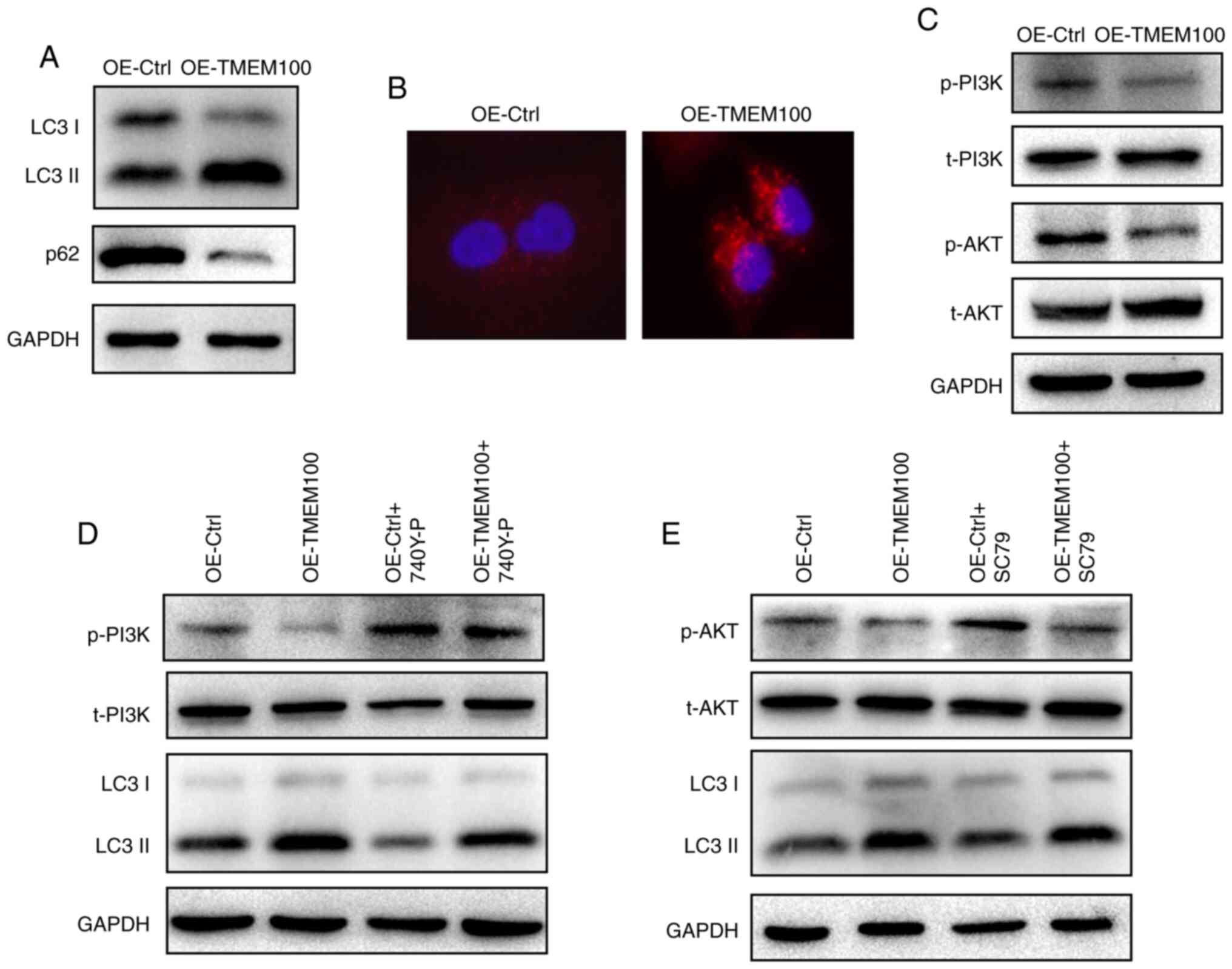 | Figure 4.TMEM100 promotes autophagy in
non-small cell lung cancer cells via inhibiting the PI3K/AKT
signaling pathway. (A) Expression level of the autophagy markers
LC3 and p62 in A549 cells upon TMEM100 overexpression. (B)
Immunofluorescence staining of LC3 puncta in A549 cells transfected
with TMEM100 overexpression plasmid. Red represents the LC3 puncta
and blue the cell nucleus. Magnification, ×400. (C) Phosphorylation
level of PI3K and AKT in A549 cells after TMEM100 overexpression.
(D) Expression level of LC3 and phosphorylation level of PI3K in
A549 cells transfected with control or TMEM100 overexpression
plasmid, with or without treatment with 740Y-P, a PI3K activator.
(E) Expression level of LC3 and phosphorylation level of AKT in
A549 cells transfected with control or TMEM100 overexpression
plasmid, with or without the treatment with SC79, an AKT activator.
Data are representative of three independent experiments. TMEM100,
transmembrane protein 100; OE, overexpression; Ctrl, control; p,
phosphorylated; t, total. |
It is well established that the PI3K/AKT signaling
pathway is a key signal transduction pathway of autophagy (28,29),
therefore the effect of TMEM100 on the PI3K/AKT signaling pathway
was investigated. As hypothesized, the level of p-PI3K and p-AKT
was decreased after TMEM100 overexpression in A549 cells (Fig. 4C), whereas the expression level of
both proteins was markedly increased following knockdown of TMEM100
in H460 cells (Fig. S4B), suggesting
that TMEM100 may inhibit the PI3K/AKT signaling pathway.
Subsequently, 740Y-P, a PI3K activator, and SC79, an AKT agonist,
were used to further examine the association between TMEM100 and
the PI3K/AKT signaling pathway and verify whether TMEM100-induced
autophagy was associated with the PI3K/AKT signaling pathway. As
demonstrated in Fig. 4D and E,
pretreatment of A549 cells with 740Y-P and SC79 resulted in an
increased activation of PI3K and AKT, respectively, and TMEM100 was
observed to attenuate the activatory effects of 740Y-P and SC79. In
addition, the ratio of LC3II/LC3I was suppressed after pretreatment
with 740Y-P and SC79, and TMEM100 reversed these inhibitory effects
(Fig. 4D and E). Pretreatment of H460
cells with LY294002, a PI3K inhibitor, notably reduced the p-PI3K
level and increased the LC3 II/LC3 I ratio, whereas TMEM100
knockdown partially reversed the effect of LY294002 (Fig. S4C). Collectively, these findings
suggested that TMEM100 may induce autophagy via inhibiting the
PI3K/AKT signaling pathway.
Baf-A1 prevents TMEM100-induced
autophagy and promotes TMEM100-induced apoptosis
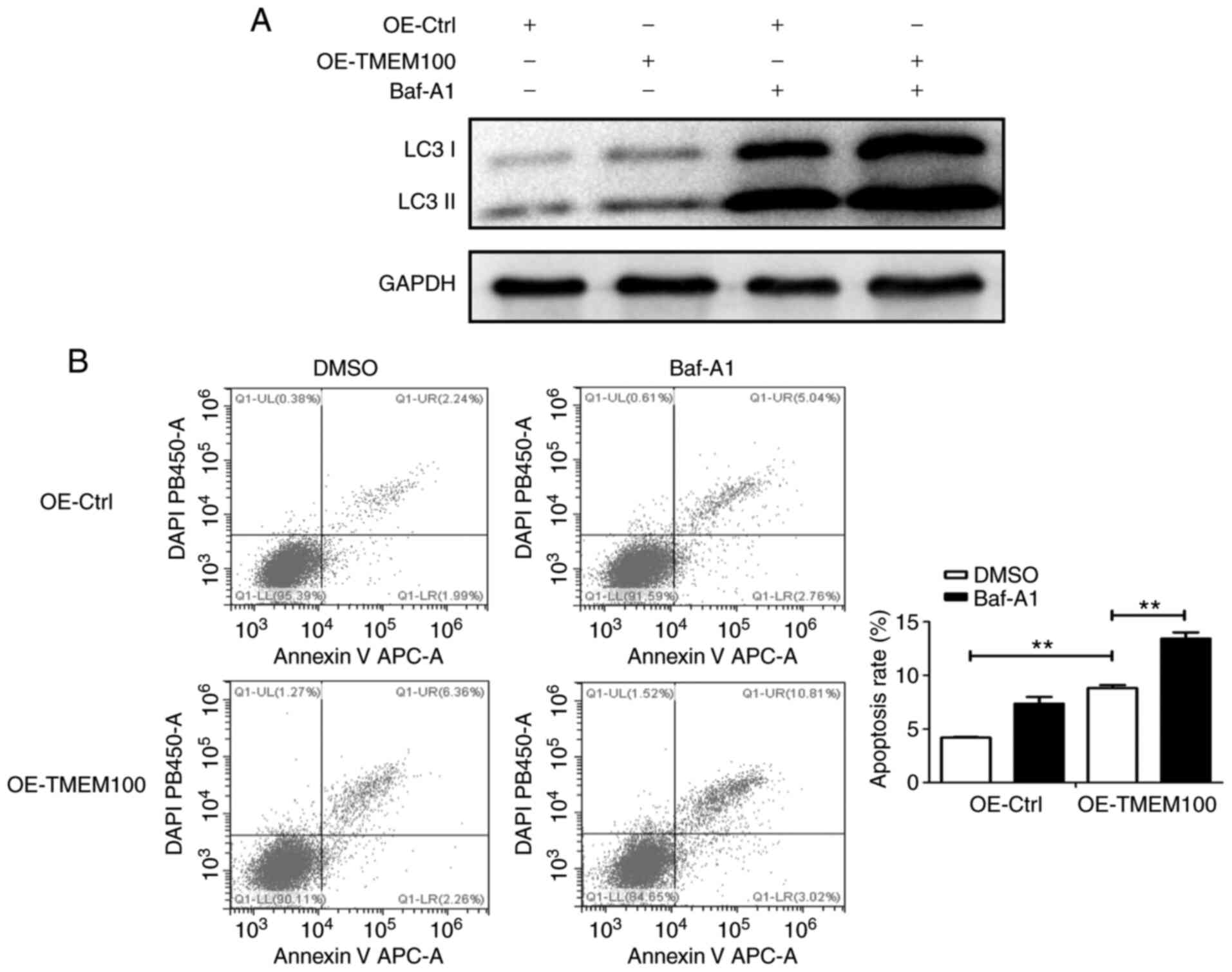
The aforementioned results revealed that TMEM100
induced both apoptosis and autophagy in NSCLC cells. To further
explore the relationship between TMEM100-induced autophagy and
apoptosis, A549 cells were pretreated with Bafilomycin A1 (Baf-A1),
a late autophagy inhibitor, and subsequently transfected with
TMEM100 overexpression plasmid or control plasmid. The combination
of Baf-A1 treatment and TMEM100 overexpression resulted in
increased LC3 II/LC3 I ratio compared with that observed in
response to treatment with Baf-A1 or TMEM100 overexpression alone
(Fig. 5A), suggesting that TMEM100
may induce autophagy, while Baf-A1 prevented autophagy in A549
cells. In addition, pretreatment with Baf-A1 significantly enhanced
apoptosis induced by TMEM100 in A549 cells (Fig. 5B), suggesting that the inhibition of
TMEM100-induced autophagy may promote cell death by inducing
apoptosis, thereby providing a compensatory mechanism.
Discussion
TMEM100 has been initially identified in the mouse
genome and it has been reported to be mainly associated with
arterial endothelial differentiation and angiogenesis (5,7). The
findings of the present study suggested that TMEM100 may serve as a
potential tumor suppressor in NSCLC. The datasets from TCGA
database were analyzed to determine the expression level of TMEM100
in 533 NSCLC specimens and 59 normal lung tissues. TMEM100
expression level was indicated to be decreased in NSCLC tissues,
and it was negatively correlated with the TNM stage and positively
correlated with the survival rate of patients, suggesting that
TMEM100 may be used as a marker of clinical TNM staging and
prognosis in NSCLC. Consistently, the IHC results demonstrated that
the expression level of TMEM100 was decreased in NSCLC tissues
compared with adjacent non-cancerous tissues. At the cellular
level, the expression of TMEM100 in the normal lung epithelial cell
line HBE was higher than that in NSCLC cell lines, such as A549,
SK-MES-1 and H1299, which was consistent with the results obtained
from the CCLE database. Han et al (14) compared the expression of TMEM100 in
H460, H522, PC9 and A549 cell lines and observed that the
expression level of TMEM100 was highest in H460 cells and lowest in
A549 cells. However, they selected A549, PC9 and H522 cell lines to
demonstrate the antitumor role of TMEM100. Based on the
aforementioned results, the present study compared the expression
of TMEM100 in H460, HBE, A549, SK-MES-1 and H1299 cell lines and
revealed that the expression level of TMEM100 was the highest in
H460 and lower in A549 cells. It is worth mentioning that lung
adenocarcinoma is the most common type of NSCLC (2) and A549 cell line is a commonly used lung
adenocarcinoma cell line. Therefore, H460, HBE and A549 cell lines
were selected to demonstrate the antitumor mechanism of TMEM100.
Similar to the results of Han et al (14), the in vitro and in vivo
experiments of the current study using different cell lines
demonstrated the tumor suppressive effects of TMEM100, including
the inhibition of cell proliferation and promotion of apoptosis.
Collectively, these results revealed that TMEM100 may serve a role
as a novel tumor suppressor in NSCLC.
By performing functional studies for TMEM100, the
mechanisms underlying the antitumor role of TMEM100 were explored.
Autophagy is a highly conserved and finely regulated pathway for
maintaining cell homeostasis, which is achieved via inducing
lysosomal degradation and recycling of dysfunctional proteins and
organelles (30,31). The autophagic process can be divided
into four steps: Induction, elongation, transportation to lysosomes
and degradation (32). Autophagy can
be activated by various type of stress, such as starvation, hypoxia
and radiation exposure (33).
Notably, the dysregulation of autophagy has been identified as a
pathogenic feature of cancer (34,35).
Previous studies have reported a dual role for autophagy in
tumorigenesis. For instance, cancer cells have been demonstrated to
induce autophagy under stressful or nutrition-deprived conditions
to promote cell survival (36), but
excessive or constitutive activation of autophagy has been
indicated to result in cancer cell death (37). In NSCLC, Nrf2 has been demonstrated to
promote tumorigenesis via activating autophagy (22). In Ras-transformed cells, autophagy has
been indicated to facilitate tumor cell survival via maintaining
aerobic glycolysis and the tricarboxylic acid cycle for cellular
energy demands (38). The antitumor
effect of autophagy has also been reported in NSCLC; for instance,
autophagy induced by Licarin A from Myristica fragrans has
been revealed to promote cell death in NSCLC cell lines (32). Interestingly, the deletion of Atg7 has
been indicated to accelerate tumor development initially, but
suppress tumor progression in the later stages (20), demonstrating the paradoxical role of
autophagy in cancer even within the same model. To date, there have
been no reports elucidating the association between TMEM100 and
autophagy, to the best of our knowledge. In the present study, the
effect of TMEM100 on the autophagy of NSCLC cells was investigated.
The results revealed that TMEM100 could induce autophagy by
increasing the ratio of LC3 II /LC3 I and decreasing the expression
level of p62. Moreover, LC3 puncta were observed to increase upon
TMEM100 overexpression. Previous studies have demonstrated that a
tumor suppressor, such as p53 or PTEN, inhibited tumor growth by
inducing autophagy (39,40), which are in line with the results of
the present study.
There are several signaling pathways underlying the
regulation of cell autophagy, including the PI3K/AKT, Beclin-1 and
ERK1/2 signaling pathways (41–44).
Accumulating evidence has suggested that the PI3K/AKT signaling
pathway is a critical mediator in the regulation of cell autophagy
(41–44). It has been demonstrated that the
deactivation of the PI3K/AKT signaling pathway may result in
autophagy induction (45). In
addition, the dysregulation of the PI3K/AKT signaling pathway has
been closely associated with tumor development, metastasis and
apoptosis in lung cancer (45). Based
on these findings, it was hypothesized that TMEM100 may stimulate
autophagy via blocking the PI3K/AKT signaling pathway. This
hypothesis was supported by the decreased level of p-PI3K and p-AKT
upon TMEM100 overexpression. Conversely, the activation of PI3K and
AKT by 740Y-P and SC79 significantly reduced the expression level
of LC3 II, which was increased by the overexpression of TMEM100.
These findings validated that TMEM100 may activate autophagy via
inhibiting the PI3K/AKT signaling pathway. To further investigate
the mechanisms of TMEM100 function associated with NSCLC
progression, whole transcriptome sequencing analysis was performed
in A549 cells transfected with TMEM100 overexpression plasmid and
empty plasmid. The sequencing results demonstrated that CHOP,
GADD34, GRP78, XBP1 and other genes involved in endoplasmic
reticulum stress (ERS) were downregulated after TMEM100
overexpression, which was confirmed by RT-qPCR and western blotting
(data not shown). Future studies will be focused on the role and
mechanism of function of TMEM100 in the regulation of ERS and the
association between ERS and TMEM100-induced autophagy.
Recently, autophagy was termed type II programmed
cell death (46). In certain cases,
autophagy and apoptosis are interconnected, but their association
has not been fully investigated. Previous studies have supported
the relevance of the interplay between autophagy and apoptosis
(37,47). It has been reported that both
autophagy and apoptosis served important roles in lung cancer
progression (48), and their
synergistic actions promoted cell death. For instance, Licarin A,
which was derived from Myristica fragrans, activated
autophagy and apoptosis, thereby resulting in the death of NSCLC
cells (32). HSP90 inhibitor DPB has
been also indicated to inhibit A549 cell growth by inducing
apoptosis and autophagy (49). In the
present study, it was revealed that TMEM100 could promote both
apoptosis and autophagy, and inhibition of autophagy by Baf-A1
notably enhanced TMEM100-induced apoptosis. The results were
consistent with a previous study in gastric cancer, which
demonstrated that the tumor suppressor XIAP-associated factor 1
(XAF1) could induce autophagic cell death, whilst 3-MA treatment
enhanced XAF1-induced apoptosis (43). These results indicated that
TMEM100-induced autophagy and apoptosis may be complementary events
to ensure cell death.
In conclusion, the present study validated the role
of TMEM100 as a tumor suppressor in NSCLC. TMEM100 was demonstrated
to inhibit cell proliferation and promote cell death by inducing
apoptosis and autophagy. To the best of our knowledge, the current
study was the first to demonstrate that TMEM100 could activate
autophagy in NSCLC cells and to indicate that the inhibition of the
PI3K/AKT signaling pathway may be one of the mechanisms underlying
TMEM100-induced autophagy. Furthermore, the inhibition of autophagy
by Baf-A1 was indicated to increase the proportion of apoptotic
cells, suggesting a complementary role of TMEM100-induced apoptosis
and autophagy. Therefore, the present study revealed a novel
mechanism by which TMEM100 may inhibit tumor progression.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by National Natural
Science Foundation of China (grant no. csfc81373151) and Chongqing
Science and Technology Commission (grant no.
cstc2018jcyjAX0257).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QH and YH conceived and designed the research. QH,
YD, YZ, ZD, XZ, ZW, RA performed the experiments and analyzed the
data. QH and YH wrote the manuscript. ZD and YH interpreted the
data and corrected the manuscript. QH and YH confirm the
authenticity of the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study was approved by Ethics and
Research Committees of Chongqing Medical University (Chongqing,
China). All animal experiments were approved by the Institutional
Animal Care and Use Committee of Chongqing Medical University
(Chongqing, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
Baf-A1
|
bafilomycin A1
|
|
CCLE
|
cancer cell line encyclopedia
|
|
DEG
|
differentially expressed gene
|
|
HBE
|
human bronchial epithelial
|
|
IHC
|
immunohistochemistry
|
|
Nrf2
|
nuclear factor erythroid 2-related
factor 2
|
|
NSCLC
|
non-small cell lung cancer
|
|
TCGA
|
The Cancer Genome Atlas
|
|
TMEM100
|
transmembrane protein 100
|
|
XAF1
|
XIAP-associated factor 1
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Herbst RS, Heymach JV and Lippman SM: Lung
cancer. N Engl J Med. 359:1367–1380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lemjabbar-Alaoui H, Hassan OU, Yang YW and
Buchanan P: Lung cancer: Biology and treatment options. Biochim
Biophys Acta. 1856:189–210. 2015.PubMed/NCBI
|
|
4
|
Herbst RS, Morgensztern D and Boshoff C:
The biology and management of non-small cell lung cancer. Nature.
533:446–454. 2018. View Article : Google Scholar
|
|
5
|
Kawai J, Shinagawa A, Shibata K, Yoshino
M, Itoh M, Ishii Y, Arakawa T, Hara A, Fukunishi Y, Konno H, et al:
Functional annotation of a full-length mouse cDNA collection.
Nature. 409:685–690. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moon EN, Kim MJ, Ko KS, Kim YS, Seo J, Oh
SP and Lee YJ: Generation of mice with a conditional and reporter
allele for Tmem100. Genesis. 48:673–678. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Somekawa S, Imagawa K, Hayashi H, Sakabe
M, Ioka T, Sato GE, Inada K, Iwamoto T, Mori T, Uemura S, et al:
Tmem100, an ALK1 receptor signaling-dependent gene essential for
arterial endothelium differentiation and vascular morphogenesis.
Proc Natl Acad Sci USA. 109:12064–12069. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Moon EH, Kim YS, Seo J, Lee YJ and Oh SP:
Essential role for TMEM100 in vascular integrity but limited
contributions to the pathogenesis of hereditary haemorrhagic
telangiectasia. Cardiovasc Res. 105:353–360. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yamazaki T, Muramoto M, Okitsu O, Morikawa
N and Kita Y: Discovery of a novel neuroprotective compound,
AS1219164, by high-throughput chemical screening of a newly
identified apoptotic gene marker. Eur J Pharmacol. 669:7–14. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eisenman ST, Gibbons SJ, Singh RD, Bernard
CE, Wu J, Sarr MG, Kendrick ML, Larson DW, Dozois EJ, Shen KR and
Farrugia G: Distribution of TMEM100 in the mouse and human
gastrointestinal tract-a novel marker of enteric nerves.
Neuroscience. 240:117–128. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weng HJ, Patel KN, Jeske NA, Bierbower SM,
Zou W, Tiwari V, Zheng Q, Tang Z, Mo GC, Wang Y, et al: Tmem100 is
a regulator of TRPA1-TRPV1 complex and contributes to persistent
pain. Neuron. 85:833–846. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Frullanti E, Colombo F, Falvella FS,
Galvan A, Noci S, De Cecco L, Incarbone M, Alloisio M, Santambrogio
L, Nosotti M, et al: Association of lung adenocarcinoma clinical
stage with gene expression pattern in noninvolved lung tissue. Int
J Cancer. 131:E643–E648. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ou D, Yang H, Hua D, Xiao S and Yang L:
Novel roles of TMEM100: Inhibition metastasis and proliferation of
hepatocellular carcinoma. Oncotarget. 6:17379–17390. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Han Z, Wang T, Han S, Chen Y, Chen T, Jia
Q, Li B, Li B, Wang J, Chen G, et al: Low-expression of TMEM100 is
associated with poor prognosis in non-small-cell lung cancer. Am J
Transl Res. 9:2567–2578. 2017.PubMed/NCBI
|
|
15
|
Yang ZJ, Chee CE, Huang S and Sinicrope
FA: The role of autophagy in cancer: Therapeutic implications. Mol
Cancer Ther. 10:1533–1541. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Arroyo DS, Gaviglio EA, Peralta Ramos JM,
Bussi C, Rodriguez-Galan MC and Iribarren P: Autophagy in
inflammation, infection, neurodegeneration and cancer. Int
Immunopharmacol. 18:55–65. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ding ZB, Shi YH, Zhou J, Qiu SJ, Xu Y, Dai
Z, Shi GM, Wang XY, Ke AW, Wu B and Fan J: Association of autophagy
defect with a malignant phenotype and poor prognosis of
hepatocellular carcinoma. Cancer Res. 68:9167–9175. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang H, Zhang Y, Zhu X, Chen C, Zhang C,
Xia Y, Zhao Y, Andrisani O and Kong L: DEAD box protein 5 inhibits
liver tumorigenesis by stimulating autophagy via interaction with
p62/SQSTM1. Hepatology. 69:1046–1063. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guo JY, Teng X, Laddha SV, Ma S, Van
Nostrand SC, Yang Y, Khor S, Chan CS, Rabinowitz JD and White E:
Autophagy provides metabolic substrates to maintain energy charge
and nucleotide pools in Ras-driven lung cancer cells. Genes Dev.
30:1704–1717. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Strohecker AM, Guo JY, Karsli-Uzunbas G,
Price SM, Chen GJ, Mathew R, McMahon M and White E: Autophagy
sustains mitochondrial glutamine metabolism and growth of
BrafV600E-driven lung tumors. Cancer Discov. 3:1272–1285. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Poillet-Perez L, Xie X, Zhan L, Yang L,
Sharp DW, Hu ZS, Su X, Maganti A, Jiang C, Lu W, et al: Autophagy
maintains tumour growth through circulating arginine. Nature.
563:569–573. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang J, Liu Z, Hu T, Han L, Yu S, Yao Y,
Ruan Z, Tian T, Huang T, Wang M, et al: Nrf2 promotes progression
of non-small cell lung cancer through activating autophagy. Cell
Cycle. 16:1053–1062. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cai J, Li R, Xu X, Zhang L, Lian R, Fang
L, Huang Y, Feng X, Liu X, Li X, et al: CK1α suppresses lung tumour
growth by stabilizing PTEN and inducing autophagy. Nat Cell Biol.
20:465–478. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Karsli-Uzunbas G, Guo JY, Price S, Teng X,
Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD and
White E: Autophagy is required for glucose homeostasis and lung
tumor maintenance. Cancer Discov. 4:914–927. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rao S, Yang H, Penninger JM and Kroemer G:
Autophagy in non-small cell lung carcinogenesis: A positive
regulator of antitumor immunosurveillance. Autophagy. 10:529–531.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fan S, Zhang B, Luan P, Gu B, Wan Q, Huang
X, Liao W and Liu J: PI3K/AKT/mTOR/p70S6K pathway is involved in
Aβ25-35-induced autophagy. Biomed Res Int. 2015:1610202015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hou X, Hu Z, Xu H, Xu J, Zhang S, Zhong Y,
He X and Wang N: Advanced glycation endproducts trigger autophagy
in cadiomyocyte via RAGE/PI3K/AKT/mTOR pathway. Cardiovasc
Diabetol. 13:782014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dikic I, Johansen T and Kirkin V:
Selective autophagy in cancer development and therapy. Cancer Res.
70:3431–3434. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Janku F, McConkey DJ, Hong DS and Kurzrock
R: Autophagy as a target for anticancer therapy. Nat Rev Clin
Oncol. 8:528–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Maheswari U, Ghosh K and Sadras SR:
Licarin A induces cell death by activation of autophagy and
apoptosis in non-small cell lung cancer cells. Apoptosis.
23:210–225. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kimura S, Noda T and Yoshimori T:
Dissection of the autophagosome maturation process by a novel
reporter protein, tandem fluorescent-tagged LC3. Autophagy.
3:452–460. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
White E: The role for autophagy in cancer.
J Clin Invest. 125:42–46. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lavandero S, Chiong M, Rothermel BA and
Hill JA: Autophagy in cardiovascular biology. J Clin Invest.
125:55–64. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guo XL, Li D, Sun K, Wang J, Liu Y, Song
JR, Zhao QD, Zhang SS, Deng WJ, Zhao X, et al: Inhibition of
autophagy enhances anticancer effects of bevacizumab in
hepatocarcinoma. J Mol Med (Berl). 91:473–483. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang P, Zheng Z, Ling L, Yang X, Zhang N,
Wang X, Hu M, Xia Y, Ma Y, Yang H, et al: w09, a novel autophagy
enhancer, induces autophagy-dependent cell apoptosis via activation
of the EGFR-mediated RAS-RAF1-MAP2K-MAPK1/3 pathway. Autophagy.
13:1093–1112. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Abida WM and Gu W: p53-dependent and
p53-independent activation of autophagy by ARF. Cancer Res.
68:352–357. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Arico S, Petiot A, Bauvy C, Dubbelhuis PF,
Meijer AJ, Codogno P and Ogier-Denis E: The tumor suppressor PTEN
positively regulates macroautophagy by inhibiting the
phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol
Chem. 276:35243–35246. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu J, Wang X, Zheng M and Luan Q:
Lipopolysaccharide from Porphyromonas gingivalis promotes autophagy
of human gingival fibroblasts through the PI3K/Akt/mTOR signaling
pathway. Life Sci. 211:133–139. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Qi HY, Qu XJ, Liu J, Hou KZ, Fan YB, Che
XF and Liu YP: Bufalin induces protective autophagy by Cbl-b
regulating mTOR and ERK signaling pathways in gastric cancer cells.
Cell Bio Int. 43:33–43. 2019. View Article : Google Scholar
|
|
43
|
Sun PH, Zhu LM, Qiao MM, Zhang YP, Jiang
SH, Wu YL and Tu SP: The XAF1 tumor suppressor induces autophagic
cell death via upregulation of Beclin-1 and inhibition of Akt
pathway. Cancer Lett. 310:170–180. 2011.PubMed/NCBI
|
|
44
|
Li YC, He SM, He ZX, Li M, Yang Y, Pang
JX, Zhang X, Chow K, Zhou Q, Duan W, et al: Plumbagin induces
apoptotic and autophagic cell death through inhibition of the
PI3K/Akt/mTOR pathway in human non-small cell lung cancer cells.
Cancer Lett. 344:239–259. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Schuurbiers OC, Kaanders JH, van der
Heijden HF, Dekhuijzen RP, Oyen WJ and Bussink J: The PI3-K/AKT-
pathway and radiation resistance mechanisms in non-small cell lung
cancer. J Thorac oncol. 4:761–767. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cao L, Walker MP, Vaidya NK, Fu M, Kumar S
and Kumar A: Cocaine-mediated autophagy in astrocytes involves
sigma 1 receptor, PI3K, mTOR, Atg5/7, Beclin-1 and induces type II
programmed cell death. Mol Neurobiol. 53:4417–4430. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kaminskyy VO, Piskunova T, Zborovskaya IB,
Tchevkina EM and Zhivotovsky B: Suppression of basal autophagy
reduces lung cancer cell proliferation and enhances
caspase-dependent and -independent apoptosis by stimulating ROS
formation. Autophagy. 8:1032–1044. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu G, Pei F, Yang F, Li L, Amin AD, Liu
S, Buchan JR and Cho WC: Role of autophagy and apoptosis in
non-small-cell lung cancer. Int J Mol Sci. 18:3672017. View Article : Google Scholar
|
|
49
|
Zhao Y, Li K, Zhao B and Su L: HSP90
inhibitor DPB induces autophagy and more effectively apoptosis in
A549 cells combined with autophagy inhibitors. In Vitro Cell Dev
Biol Anim. 55:349–354. 2019. View Article : Google Scholar : PubMed/NCBI
|