Introduction
Non-small cell lung cancer (NSCLC), the most common
lung cancer, is one of the leading causes of cancer-related deaths
in patients of developed countries (1,2). In
general, the long-term survival of patients with NSCLC is markedly
poor because the cancer cells are resistant to chemotherapy and
radiotherapy (3). The molecular
mechanisms of cancer cell resistance to anticancer drugs and the
pathogenesis of NSCLC progression remain unclear. Therefore, the
elucidation of lung cancer pathogenesis, the study of molecular
mechanisms of NSCLC progression, and the identification of specific
biomarkers and therapeutic targets are required for development of
improved strategies for NSCLC diagnosis, prognosis and
treatment.
E26 transformation-specific (ETS) transcription
factors are known to play key roles in cell differentiation, tissue
development and oncogenesis (4–6). Little is
known about how these transcription factors govern the
proliferation, migration, and apoptosis of lung cancer cells. It
has been revealed that ETS factors regulate the expression of their
target genes by binding to ETS response elements in the promoters,
and promote cancer development in part by regulating expression of
matrix metalloproteinases, thus influencing normal tissue growth
and promoting cell invasion, metastasis, and angiogenesis in
cancers (7). Some of the ETS family
members including polyoma enhancer activator 3 (PEA3) and
epithelial-specific expression-1 (ESE-1) have been implicated in
the regulation of HER2 oncogene transcription and promotion of
breast cancer growth (8,9). Expression of ETS-homologous factor
(EHF), also known as ESE-1, is mainly restricted to the glandular
organs (10). Loss of EHF expression
has been revealed in several epithelial carcinomas, including
bladder, oral squamous, and breast ductal carcinomas (11), and a downregulation of EHF expression
is associated with prostate cancers (12,13). While
overexpression of EHF in breast cancers is not associated with poor
prognosis of progression-free survival, the expression level of EHF
has been revealed to be significantly higher in primary lung cancer
tissues compared with adjacent non-tumor tissues of the same
patients (14,15). Data obtained from public datasets
indicate that a high level of EHF expression is associated with
poor prognosis of patients with NSCLC, and that loss of EHF
expression impairs the invasion and the metastasis of lung cancer
cells (16,17). These studies suggest that EHF may have
distinct tissue-specific functions in various cancers (12–17). EHF
has been revealed to promote lung cancer cell progression but
inhibit epithelial carcinomas and prostate cancer growth. The
different signaling pathways of EHF in lung cancer cells vs.
epithelial carcinomas remain unknown.
In the present study, the expression level of EHF in
primary NSCLC was compared with that found in adjacent non-tumor
lung tissue and the role of EHF in NSCLC progression in
vitro and in vivo was investigated to explore the
mechanism of EHF action in lung cancer cells.
Material and methods
Human lung cancer cell lines and
antibodies
Cell lines A549, LTEP-s, and 95D were obtained from
the Chinese Academy of Sciences Cell Bank of Type Culture
Collection. Anti-EHF (product code ab220113) and anti-pan-ERK1/2
(product code ab17942) antibodies were purchased from Abcam.
Anti-phospho-AKTSer473 (cat. no. BS4006), anti-total-AKT
(cat. no. BS1810), and anti-phospho-ERK1/2 (cat. no. BS4621)
antibodies were obtained from Bioworld Technology, Inc. Anti-Ki67
antibody (cat. no. 550609) was a product of BD Biosciences.
Anti-GAPDH antibody (cat. no. AM8539b) was obtained from Abcepta,
Inc. The sources and dilutions of the primary antibodies used for
western blot analysis are listed in Table SI.
Human lung cancer and control
tissues
Twenty-one NSCLC and adjacent non-tumor tissues were
surgically collected from patients who had not undergone
chemotherapy or radiotherapy before the surgery at the First
Affiliated Hospital of Xi'an Jiaotong University, Xi'an, China from
October 2013 to April 2014. The detailed background data of the
studied subjects including sex, age medical history, histologic
grade, tumor stage location were listed in a previous study
(18). None of the patients enrolled
received local or systemic treatment before operation and all of
them provided informed written consent for participation in the
present study. The study was approved by the Institutional Review
Board and Human Ethics Committee of the First Affiliated Hospital
of Xi'an Jiaotong University, and written informed consent was
obtained from each patient.
Bioinformatics
Gene expression data (RNA-seq) from 513 lung
adenocarcinomas and 58 matched normal tissues, and from 509
patients with lung squamous cell carcinoma including 44 paired
tumor and normal tissues were downloaded on July 15, 2014 from The
Cancer Genome Atlas (TCGA; http://cancergenome.nih.gov/). The median of gene
expression was used for the Kaplan-Meier plot to analyze the
association of marker gene expression with the survival of
patients. The hazard ratio, 95% confidence intervals, and log rank
P-value were calculated for each gene automatically using web-based
software tools at www.kmplot.com/lung.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted with TRIzol®
reagent (Takara Bio, Inc.) from clinical samples, cancer cell lines
and grafted tumors. An aliquot of total RNA (1 µg) was reverse
transcribed into cDNA in 20 µl volume of reaction by random
primers. Quantitative PCR (qPCR) contained 0.5 µl template cDNA, 1X
SYBR® Premix Ex Taq™ with a qPCR kit from Takara Bio,
Inc. (Takara Bio, Inc.), and 100 nM of specific forward and reverse
primers in 25 µl volume of reaction. Primers used for real-time PCR
are listed in Table I. The PCR
thermocycling conditions were as follows: 95°C for 5 min, 1 cycle,
followed by 40 cycles at 95°C for 45 sec, 59°C for 45 sec and 72°C
for 45 sec. The final PCR products were extended for 5 min.
Housekeeping gene, GAPDH was used as a reference. The
2−ΔΔCq method was used to analyze the data (19).
 | Table I.Primer sequences used for
RT-qPCR. |
Table I.
Primer sequences used for
RT-qPCR.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| EHF |
TGATTCTGGAAGGAGGTGGT |
ATGTCGAACTCTTGGAAAGGG |
| EGFR |
GGGCTCTGGAGGAAAAGAAA |
AAATTCCCAAGGACCACCTC |
| ERBB2 |
ATCAACTGCACCCACTCCTG |
TGATGAGGATCCCAAAGACCAC |
| ERBB3 |
AGTCATGAGGGCGAACGAC |
TCACACTCAGGCCATTCAGA |
| ERBB4 |
ACGGGATCTGAGACTTCCAA |
TTATTCTCCGTTCCTGCACA |
| MET |
CTCCAGCATTTTTACGGACC |
GCTGCAAAGCTGTGGTAAACT |
| 18S |
CGCCGCTAGAGGTGAAATTC |
CTTTCGCTCTGGTCCGTCTT |
| GAPDH |
GCACAGAGCCTCGCCTT |
AGGGGCCATCCACAGTCTTC |
Gene knockdown, transfection and
plasmid construction
Cancer cells (1×105) were cultured in
6-well plates in OPTI-MEM medium (Invitrogen; Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS)
(Hyclone; Cytiva), 100 µg/ml penicillin, 100 units/ml streptomycin
(Hyclone; Cytiva) at 37°C in a humidified incubator containing 5%
CO2 for 18 h. The following day, the cells were
transfected with 400 µl of antibiotic-free and serum-free medium
OPTI-MEM containing 2 µg of plasmid DNA and 4 µl of Lipofectamine
2000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to
manufacturer's instructions. Six hours later, the medium was
removed, and fresh growth medium was added. The cells were cultured
at 37°C for another 48 h prior to harvesting for total cellular
protein or RNA extraction. Gene expression in cancer cells was
knocked down by transfection with 50 nM small interfering (si)RNAs
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). siRNAs against EHF (si-EHF-309 and si-EHF-979)
and negative control siRNA (si-NC) were obtained from Shanghai
GenePharma Co., Ltd. Full-length EHF cDNA was amplified by PCR and
cloned into the pcDNA3.1 plasmid (Invitrogen; Thermo Fisher
Scientific, Inc.). The promoters of Erb-B2 receptor tyrosinekinase
2/3 (ERBB2 and ERBB3), as well as MET genes were generated by PCR
and inserted into the pGL3-basic plasmid (Promega Corporation). The
sequences were verified by DNA sequencing. Primers used for
generation of reporter constructs and ChIP assays are listed in
Tables II and III. The sequences of siRNAs used are
listed in Table IV.
| Table II.Primer sequences for ChIP assays. |
Table II.
Primer sequences for ChIP assays.
| Genes | Sets | Forward primer
(5′-3′) | Reverse primer
(5′-3′) | Product length
(bp) |
|---|
| ERBB2 | P1:-604- −484 |
TCCTTTCGATGTGACTGTCTCC |
TGTGTTTACCTTGTGGCTTCC | 121 |
|
| P2:-274- −155 |
TGCATTTAGGGATTCTCCGA |
ACTCCCAGCTTCACTTTCTC | 120 |
|
| P3: −147- −37 |
CCCAGACTTGTTGGAATGCAG |
ATTCTTATACTTCCTCAAGCAGCC | 111 |
| ERBB3 | P4:-203- −81 |
TTCGAGTCTGGGAGAAACTGAG |
TAGCCGGTTGGTTCACTTGG | 123 |
|
| P5:-77- +43 |
GAGTTGAGTGATTTGGTTAATGGG |
GAGGTCGAGATTCCGAAAGC | 120 |
| MET | P6:-258- −141 |
GACTCGGTCCCGCTTATCTC |
GCCCACGACAAGGTGAAAC | 118 |
|
| P7:+249- +387 |
AGCGCTTTGTGAGCAGATG |
CCAGGAACCAGTGGAGAAGT | 139 |
| Table III.Primer sequences for generation of
luciferase reporter constructs. |
Table III.
Primer sequences for generation of
luciferase reporter constructs.
| Plasmids | Sets | Forward primer
(5′-3′) | Reverse primer
(5′-3′) | Restriction
sites |
|---|
|
pcDNA™3.1/myc-His(−)A-EHF |
|
CCGCTCGAGGCCACCA | CCCAAGCTTGTTT | XhoI and
HindIII |
|
|
|
TGATTCTGGAAGGAGGTGGTG |
TCATTTTCTCTCCATCCTCG |
|
| pGL3-ERBB2-Luc | −607- +11 |
CGGGGTACCAAGTCCT | CCGCTCGAGCTGG | KpnI and
XhoI |
|
|
|
TTCGATGTGACTGTC |
TTTCTCCGGTCCCAAT |
|
| pGL3-ERBB3-Luc | −997- +440 |
CGGGGTACCAGGTTG |
CCCAAGCTTGACTCC | KpnI and
HindIII |
|
|
|
CATATCAATAGGGAGC | GCAGAGGGTGAAG |
|
| pGL3-MET-Luc | −223- +60 |
CGAGCTCCAGACTGC |
CCCAAGCTTGCGACC | SacI and
HindIII |
|
|
| CTGAGCTGGGGGA | AGACTGAGGCGCTC |
|
| Table IV.siRNA sequences used in the present
study. |
Table IV.
siRNA sequences used in the present
study.
| siRNAs | Sequences
(5′-3′) |
|---|
| si-EHF-309
(Sense) |
GCCAGUGGCAUGAAAUUCATT |
| si-EHF-309
(Antisense) |
UGAAUUUCAUGCCACUGGCTT |
| si-EHF-979
(Sense) |
CAGCCGAGCUAUGAGAUAUTT |
| si-EHF-979
(Antisense) |
AUAUCUCAUAGCUCGGCUGTT |
| si-NC (Sense) |
UUCUCCGAACGUGUCACGUTT |
| si-NC
(Antisense) |
ACGUGACACGUUCGGAGAATT |
Cytotoxicity and colony formation
assays
The
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay from Sigma-Aldrich; Merck KGaA was used to determine cell
viability/cytotoxicity as reported (20,21).
Briefly, 20 µl MTT solution (5 g/l) was added to a well of a
96-well plate and the NSCLC cells were cultured at 37°C for an
additional 4 h. After removal of the supernatant, 150 µl dimethyl
sulfoxide was added to each well and the plates were oscillated for
15 min. Absorbance of insoluble formazan, which has a purple color
was measured at a test wavelength of 570 nm and a reference
wavelength of 670 nm by a spectrometer. For colony formation
assays, 1,000 cells/well were seeded into 6-well plates, and the
media were changed every 3 days. After 10–14 days of culture, cells
were fixed with 100% methanol at room temperature for 20 min,
stained with 0.5% crystal violet at room temperature for 10 min,
and the colonies (>150 cells) were counted under a light
microscope with a magnification of ×100.
Migration and invasion assays
Cell migration and invasion assays were performed
using Millicell® chambers (8-µm pore size; EMD
Millipore). For cell invasion assays, chambers were coated with
Matrigel® (4X dilution; 15 µl/well; BD Biosciences). For
cell invasion and migration, cells were transfected with siRNA for
48 h and then seeded in the upper chamber at a density of
1×105 cells/ml in 200 µl of medium containing 0.5% FBS.
Medium containing 20% FBS was added to the lower chamber. After 24
or 48 h of incubation, non-invading cells in the upper chamber were
removed, and invading cells were fixed in 100% methanol at room
temperature for 20 min and stained with 0.5% crystal violet at room
temperature for 10 min. Images were captured randomly from 5 fields
of each membrane. The number of invasive/migrated cells was counted
and the average number of cells per light microscopic field with a
magnification of ×200 was presented.
Cell cycle and apoptosis assays
Lung cancer cells (2×106) were
transfected with siRNA for 48 h, and then synchronized by serum
starvation for 16 h. Cells were harvested at 24, 36 and 48 h,
respectively, and fixed in 70% ethanol on ice for 30 min, followed
by propidium iodide (PI) staining in a PBS buffer containing 50
µg/ml PI, 50 µg/ml RNase A, 0.1% Triton™ X-100, and 0.1 mM
ethylenediaminetetraacetic acid. Stained cells were then examined
by fluorescence-activated cell sorting (FACS) using a flow
cytometer (FACSCalibur; BD Biosciences). The cell-cycle populations
were determined by FlowJo 7.6 software analyses. For apoptosis
assays, cells were harvested and stained in the dark with an
Annexin V-FITC/PI solution (Roche Applied Science) at room
temperature for 15 min, followed by flow cytometric analyses. Thus,
the early apoptotic cells are Annexin V-positive and PI-negative
whereas the late apoptotic cells are Annexin V- and
PI-positive.
Tumor xenografted mice
Ten four-week-old female BALB/c mice (20 g) were
purchased from the Animal Experimental Center of the College of
Medicine, Xi'an Jiaotong University. Mice were under the care of a
qualified veterinarian at the approved animal facility of this
institution under standard approved laboratory conditions with
controlled illumination (14-h light and 10-h dark cycles) and
temperature (22°C) and unrestricted food and water. Ten mice were
randomly divided into two groups (N=5) and anesthetized with 2.5%
isoflurane prior to procedures. Depth of anesthesia was monitored
by tracking respiration rate, and testing for a toe-pinch reflex
every 15 min. A total of 5×106 cancer cells was injected
subcutaneously into the right dorsum of the mouse. Mice were
monitored for discomfort and ease of movement every 12 h in the 2
days post-injection and frequently afterward. Discomfort was
recognized as a reluctance to move, a hunched posture and poor
grooming (a mottled face). To avoid mouse suffering, severely sick
mice would be euthanized, however, in the present study, no sick
mice were observed and euthanized. Tumor size was measured every 2
days, and the volume was calculated by length ×
width2/2. Mice were euthanized with a 30% flow rate of
CO2 prior to cervical dislocation on day 21 after
injection. Sacrifice was verified by lack of pulse and respiration
over a period of at least 10 min followed by decapitation. The
tumors were surgically excised, weighed, fixed in 10% formalin at
4°C for 72 h, washed with 3X PBS, embedded in paraffin, and
sectioned into 4-µm slices. Ki67 immuno-staining was carried out
with a primary antibody at 1:100 dilution at 4°C overnight,
followed by incubation with biotin-labeled secondary antibody (cat
no. SP-9002; ZSGB-BIO; OriGene Technologies, Inc.) at room
temperature for 1.5 h, washing 3 times, and another incubation with
HRP-conjugated avidin working solution (cat. no. SP-9001; ZSGB-BIO;
OriGene Technologies, Inc.) at room temperature for 20 min. After
washing 3 times with PBS, the cells were stained with DAB working
solution (1:20 dilution) for 5 min, followed by hematoxylin
staining for 30 sec. The mean proportion of positive cells was
calculated from 10 randomized microscopic fields at a magnification
of ×200. A Ki67-labeling index was calculated as the number of
positive nuclei divided by the total number of nuclei. The
experiments were approved by the Institutional Animal Care and Use
Committee of Xi'an Jiaotong University.
Luciferase reporter and chromatin
immunoprecipitation (ChIP) assays
A dual luciferase reporter assay system was used to
measure the promoter activity according to manufacturer's
instruction (Promega Corporation). The Renilla luciferase
driven by TK minimal promoter was used as an internal control to
normalize the transfection efficiency. Luciferase activity was
measured 48 h post-transfection by using an EnSpire®
multimode plate reader (Perkin Elmer, Inc.). ChIP assay was carried
out according to the manufacturer's instructions (Pierce™ magnetic
ChIP kit; Thermo Fisher Scientific, Inc. cat. no. 25157). Briefly,
2×106 cells were transfected with Myc-tag EHF expression
construct and cross-linked in 1% formaldehyde at 37°C for 10 min
and lysed in SDS lysis buffer (Pierce magnetic ChIP kit; Thermo
Fisher Scientific, Inc.) supplemented with protease inhibitor
cocktail (Sigma-Aldrich; Merck KGaA) for 10 min on ice. The lysate
was sonicated and precleared with protein A agarose/salmon sperm
DNA prior to immunoprecipitation with 1 µg of anti-Myc Tag antibody
(cat. no. 05-724; Clone 4A6; EMD Millipore) at 4°C overnight.
Following wash and elution steps, cross-links were reversed at 65°C
for 4 h. The DNA in the immunoprecipitated samples was purified by
proteinase K digestion followed by DNA purification. An aliquot of
purified DNA was analyzed by quantitative PCR with specific
primers.
Western blot analysis
The lung cancer cell lines and tumor tissues were
homogenized and lysed with lysis buffer [20 mM Tris-HCl (pH 7.5),
150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1 mM
phenylmethylsulfonyl fluoride, 1X protease inhibitor cocktail and
1X phosphatase inhibitor cocktail] on ice for 20 min, followed by
centrifugation at 9,000 × g for 10 min at 4°C. Total cellular
protein was measured by bicinchoninic acid (BCA) assay. An aliquot
of total cellular protein (30 µg) was resolved on 8%
SDS-polyacrylamide gel electrophoresis, transferred to membranes,
blocked in 5% of fat free milk at room temperature for 4 h, and
immunoblotted with validated primary antibodies at 1:500-1,000
dilutions at 4°C overnight. Specific protein was detected using
appropriate HRP-conjugated secondary anti-mouse (cat. no. ASR1037)
or anti-rabbit IgG (cat. no. ASR1089; both from Abcepta, Inc.)
antibody at a dilution of 1:15,000 at 22°C and western blotting
detection system (Immobilon® Western Chemiluminescent
HRP Substrate; EMD Millipore). After exposure to chemiluminescence
film, the same membrane was stripped in a stripping buffer
containing 62.5 mM Tris-HCl (pH 6.8), 2% SDS and 0.8%
β-mercaptoethanol at 50°C for 45 min, followed by washing 5 times.
The membrane was then re-probed with anti-GAPDH antibody.
Statistical analysis
SPSS13.0 (SPSS, Inc.) was used for statistical
analyses. Experiments were repeated 3 times, and the data were
presented as the mean ± standard deviation (SD). Student's t-tests
were used for two group comparisons and AVOVA, followed by post hoc
Tukey's test was used for multiple group comparisons. All
statistical tests were two-sided. P<0.05 or P<0.01 indicated
a statistically significant difference.
Results
Increased level of EHF expression in
NSCLC is associated with poor prognosis
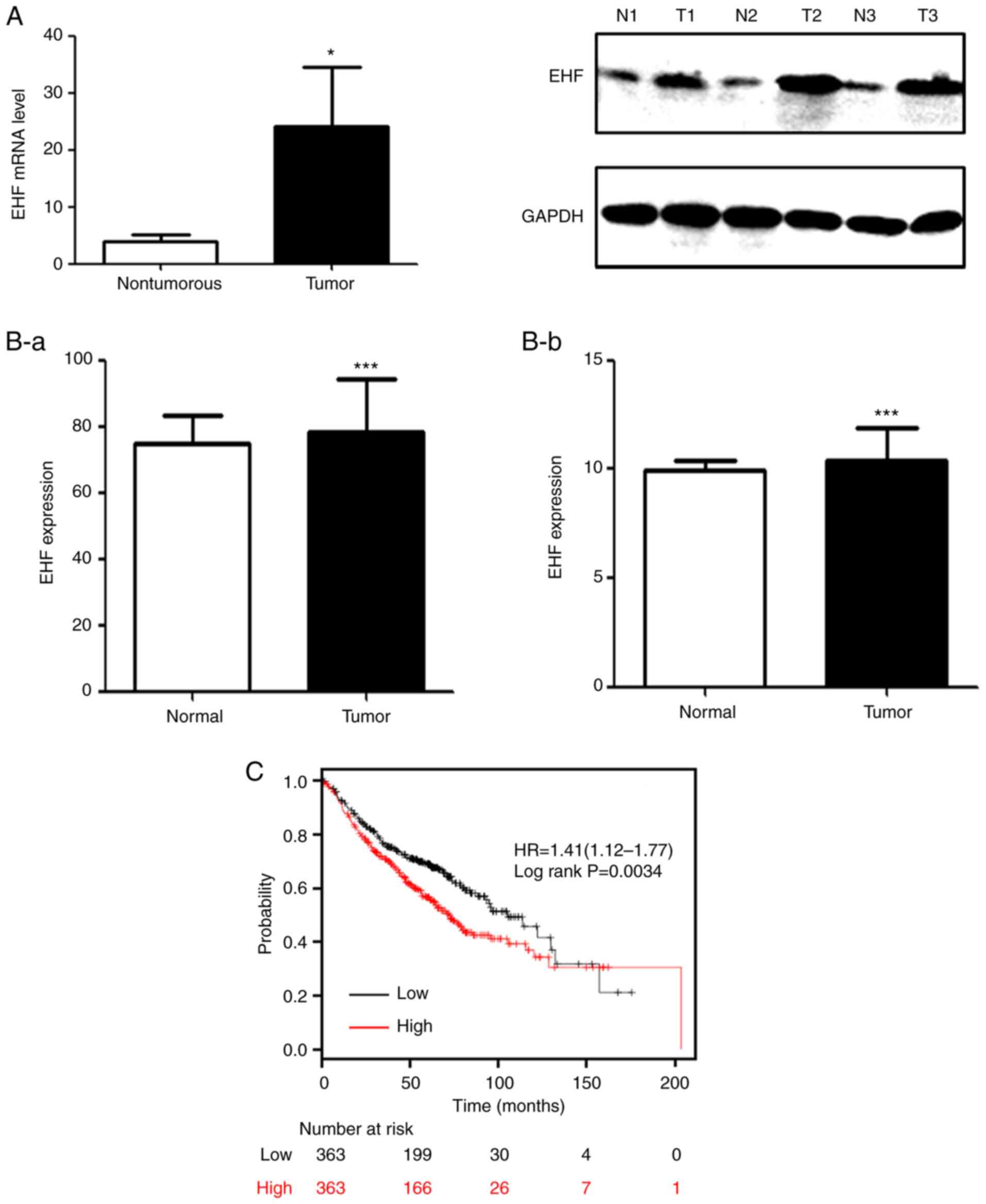
As revealed in Fig. 1A
(left panel), the level of EHF transcript was 4-fold higher in
NSCLC tissues compared to that in adjacent non-tumor tissues.
Consistent with the real-time RT-qPCR data, the western blot
analyses also detected increased levels of EHF expression in 3
tumors as compared to the corresponding control tissues (Fig. 1A, right panel). The data obtained from
TCGA database was also analyzed and it was revealed that EHF was
significantly higher in patients with lung adenocarcinoma and
squamous cancer as compared to the corresponding controls (Fig. 1B). Notably, a Kaplan-Meier plot
exhibited an association of the median of EHF expression level with
the overall survival. An increased level of EHF expression was
significantly associated with poor prognosis in patients with lung
cancer at early stages (Fig. 1C).
Overexpression of EHF promotes lung
cancer cell proliferation, tumor growth, cell migration and
invasion
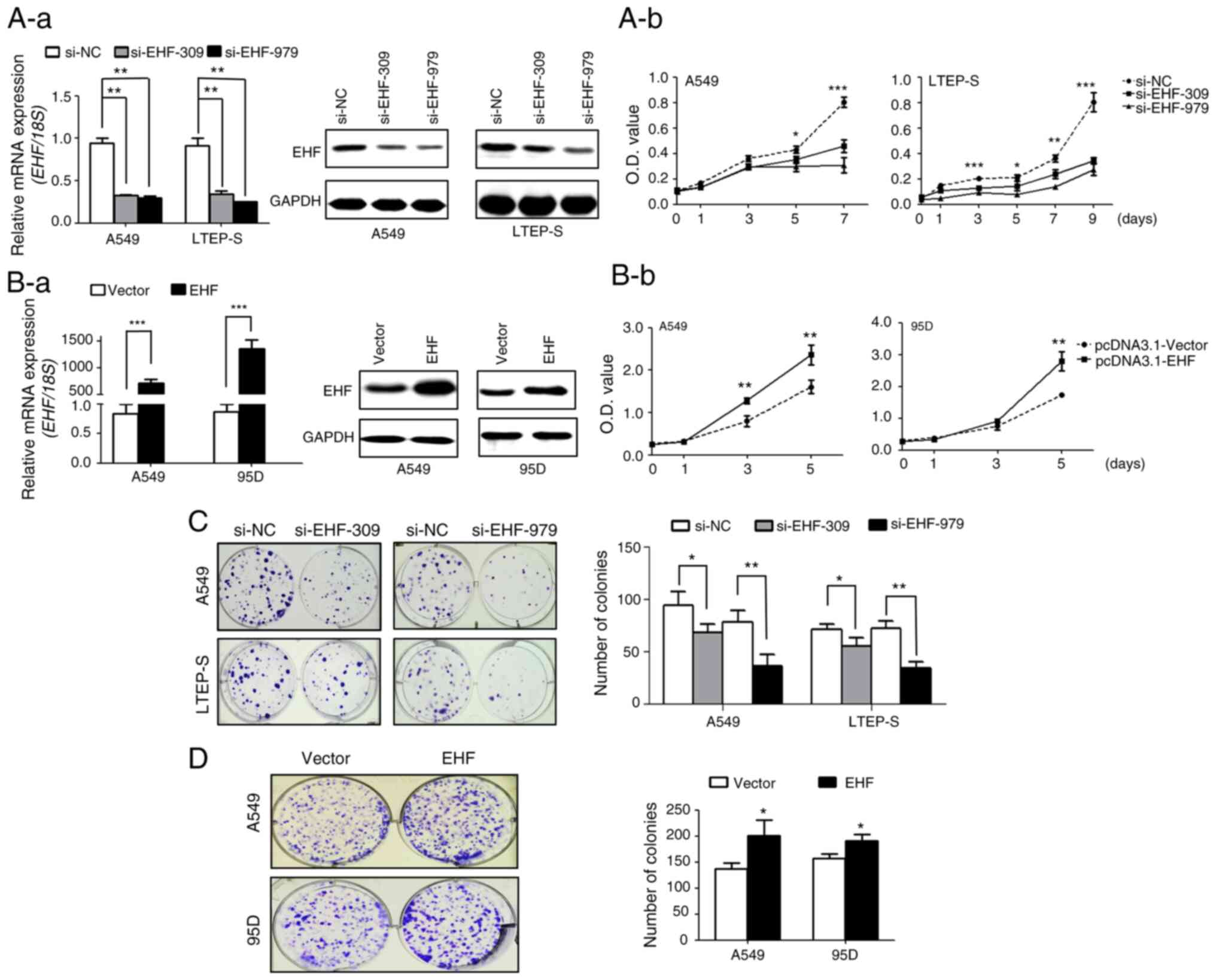
To study the role of EHF overexpression in lung
cancer growth, EHF expression we first knocked down in cancer cells
with siRNA. As revealed in Fig. 2A-a,
transfection of cancer cells with siRNAs against different regions
of EHF mRNA, knocked down EHF expression at both transcript and
protein levels by 60–70% in lung cancer cell lines. In addition,
knockdown of EHF expression significantly diminished cancer cell
proliferation (Fig. 2A-b). In
contrast, overexpression of EHF increased lung cancer cell
proliferation by 50% in both cell lines (Fig. 2B-a and B-b). In agreement with the
cell proliferation data, it was revealed that downregulation of EHF
expression also caused a 20–50% reduction in cell colony formation
whereas overexpression of EHF resulted in an increase in colony
formation (Fig. 2C and D).
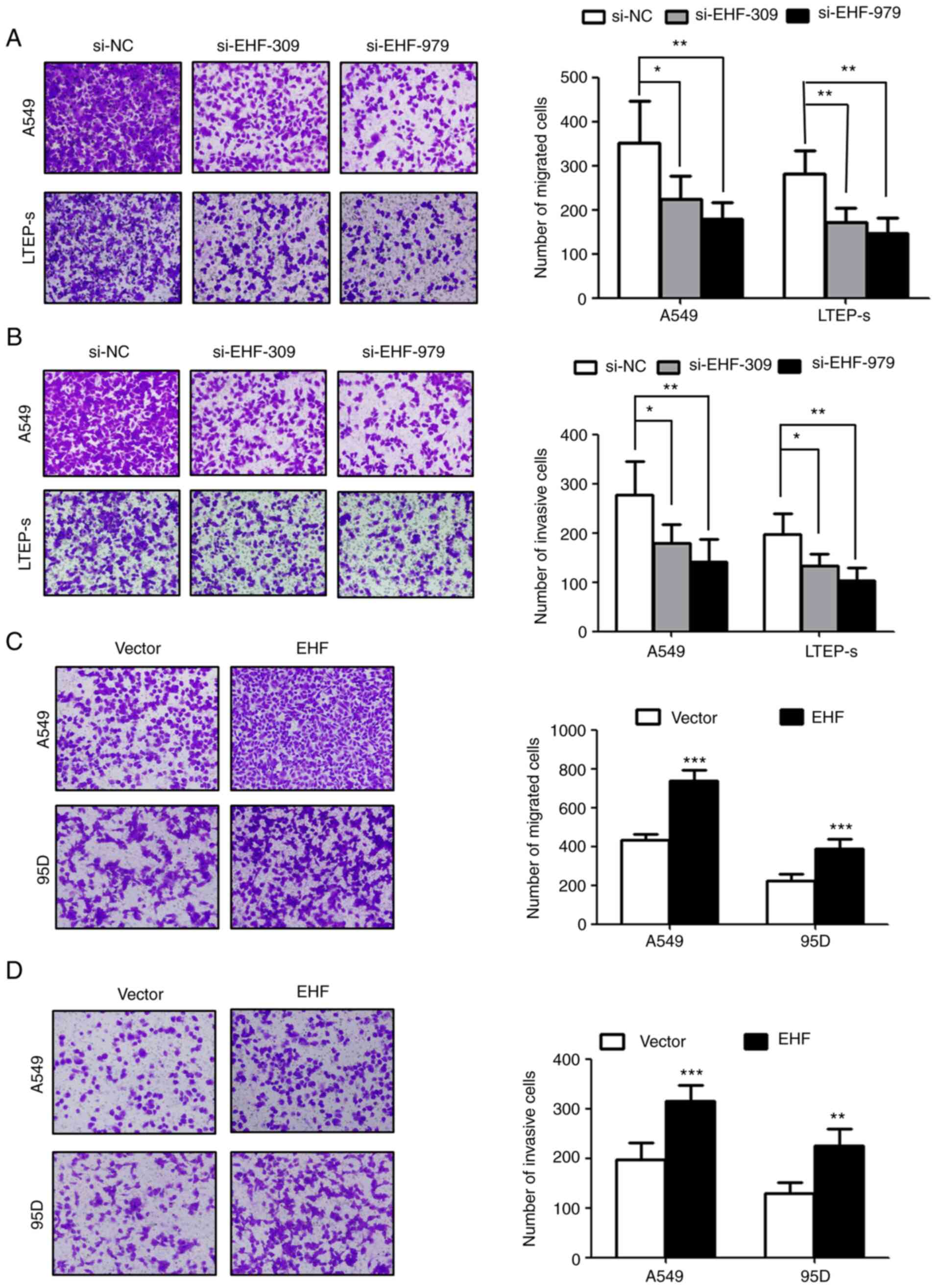
The effects of EHF expression on lung cancer cell
migration and invasion were next evaluated. It was observed that
the cells transfected with siRNA against EHF migrated and invaded
markedly less than the control cells transfected with scramble
siRNA (Fig. 3A and B). Knockdown of
EHF expression in lung cancer cells caused a 30–50% reduction in
cell migration and invasion. In contrast, overexpression of EHF in
lung cancer cells increased cell migration and invasion by 30–40%
as compared to corresponding controls (Fig. 3C and D).
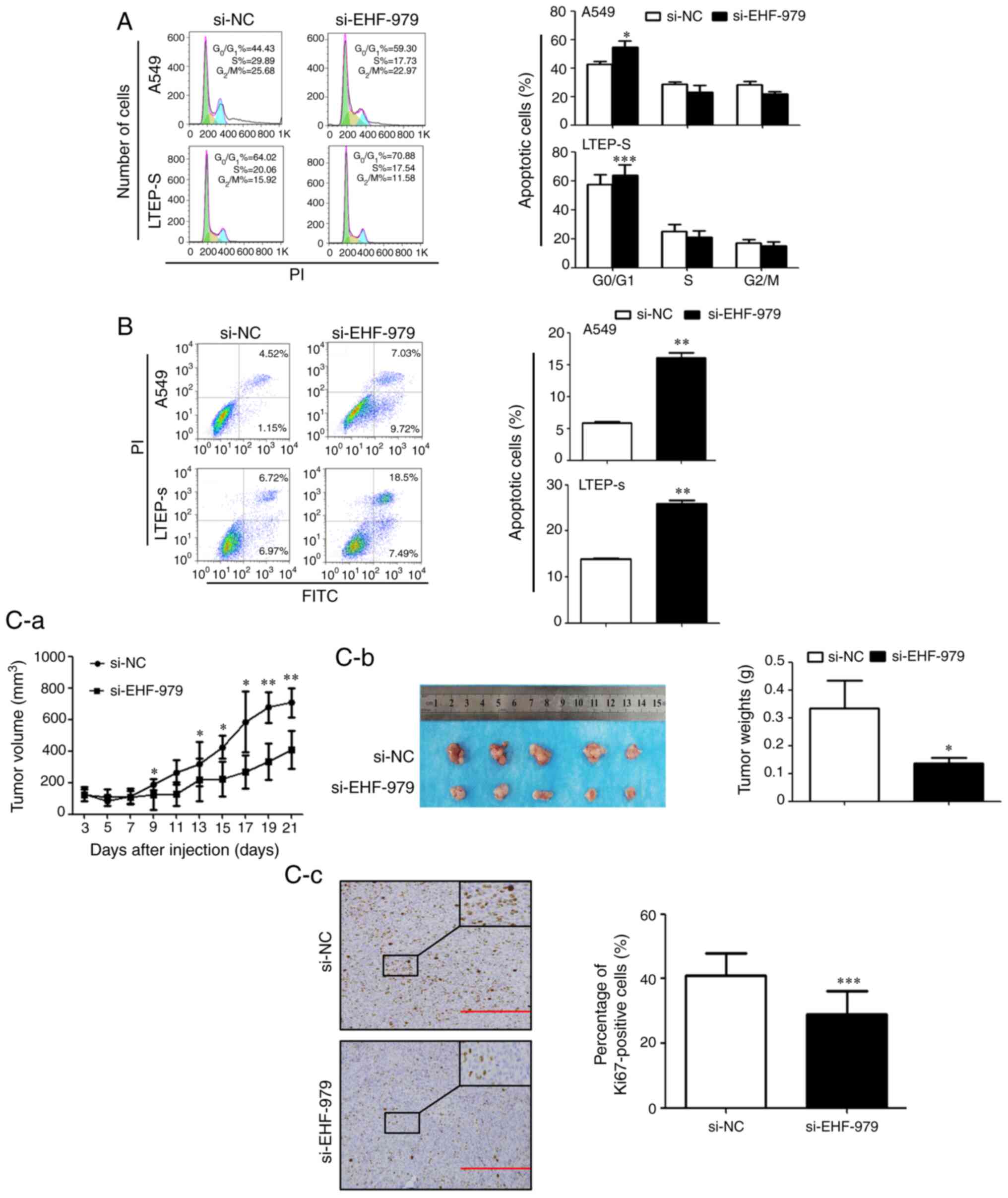
EHF downregulation arrests NSCLC cell
cycle and induces NSCLC cell apoptosis
To study whether EHF downregulation arrests the cell
cycle, growth inhibition of NSCLC cells transfected with si-EHF-979
or control siRNA was determined by flow cytometric analyses. As
revealed in Fig. 4A, knockdown of EHF
expression by si-EHF-797 arrested NSCLC cells at the
G0/G1 phase. The percentage of cells in the
G0/G1 phase was increased by 12% in A549
cells transfected with siEHF-979 as compared with cells transfected
with control scramble siRNA (54.85±4.01 vs. 42.61±2.19%, P=0.026),
and 16% in LTEP-s cells transfected with siEHF-979 (73.95±6.97 vs.
57.59±7.75%, P<0.001). It was also investigated whether EHF
downregulation induced NSCLC cell apoptosis. As revealed in
Fig. 4B, knockdown of EHF expression
by siRNA resulted in an 11% increase in apoptotic A549 cells
(16.03±0.89 vs. 5.89±0.23%, P=0.003) and a 12% increase in
apoptotic LTEP-s cells (25.89±0.77 vs. 13.88±0.17%, P=0.002).
EHF downregulation suppresses tumor
growth in A549 cell xenografted mice
To evaluate the effect of EHF downregulation on
tumor growth in vivo, xenografted mice bearing A549 lung
cancer cells were generated. Consistent with the in vitro
experiments, knockdown of EHF expression significantly decreased
xenografted tumor growth in vivo (Fig. 4C-a and C-b). The volume of xenografted
tumors derived from si-EHF-979 A549 cells at day 21 was reduced by
38% as compared to the control xenografts (P<0.05). To quantify
the proliferation index in the xenograft tumors,
immunohistochemistry was performed. The tumor sections were probed
with an antibody specific to Ki67 or control IgG. As revealed in
Fig. 4C-c, knockdown of EHF
expression by siRNA (si-EHF-979) significantly decreased the
population of Ki67-positive cells in tumors as compared with the
tumors transfected with control siRNA (si-NC). The percentage of
Ki67-positive cells was reduced by 25% in si-EHF-979 transfected
tumors (P<0.001).
EHF promotes tumor growth by
regulating ERBB2, ERBB3 and MET expression and activating the
AKT/ERK signaling pathways in NSCLC cells
To study the mechanism of EHF regulation of tumor
growth, the expression of potential EHF target genes in EHF knocked
down NSCLC cells in vitro was first examined. As revealed in
Fig. 5A, the expression levels of
ERBB2, ERBB3, and MET were reduced by 20–30% in both EHF knocked
down cell lines compared with the controls. It was revealed that
ERBB4 expression was very low in LTEP-s cells and undetectable by
qPCR. No changes in the levels of EGFR and ERBB4 expression were
detectable in A549 cells or in LTEP-s cells (Fig. 5A). To further study the regulation of
EHF on the promoter activity of EHF target genes, proximal
promoters of ERBB2, ERBB3 and MET genes were inserted into
promoter-less pGL3-basic plasmid and luciferase reporter assays in
co-transfected cancer cells were performed. The Renilla
luciferase activity driven by TK minimal promoter in co-transfected
cells was used as an internal control to normalize the transfection
efficiency. The results revealed that overexpression of EHF in lung
cancer cells enhanced luciferase reporter activity by 40% in the
cells co-transfected with EHF expression plasmid and the luciferase
reporters driven by ERBB2, ERBB3 and MET proximal promoters,
respectively, as compared to the control cells co-transfected with
corresponding reporters with a control plasmid (Fig. 5B). In addition, the promoter
activities were also increased in the lung cancer cells transfected
with luciferase reporters under the control of ERBB2, ERBB3 and MET
proximal promoters as compared with the cells transfected with
promoter-less reporter (Fig. 5B). To
determine whether EHF regulates endogenous ERBB2, ERBB3 and MET
expression in cancer cells, we carried out ChIP assays. As revealed
in Fig. 5C, the ChIP assays detected
EHF binding to the promoters of endogenous ERBB2, ERBB3 and MET
genes, respectively. The interactions of EHF with ERBB2, ERBB3 and
MET proximal promoters were increased by more than 2-fold in the
EHF-overexpressing cells as compared to the straight control
cells.
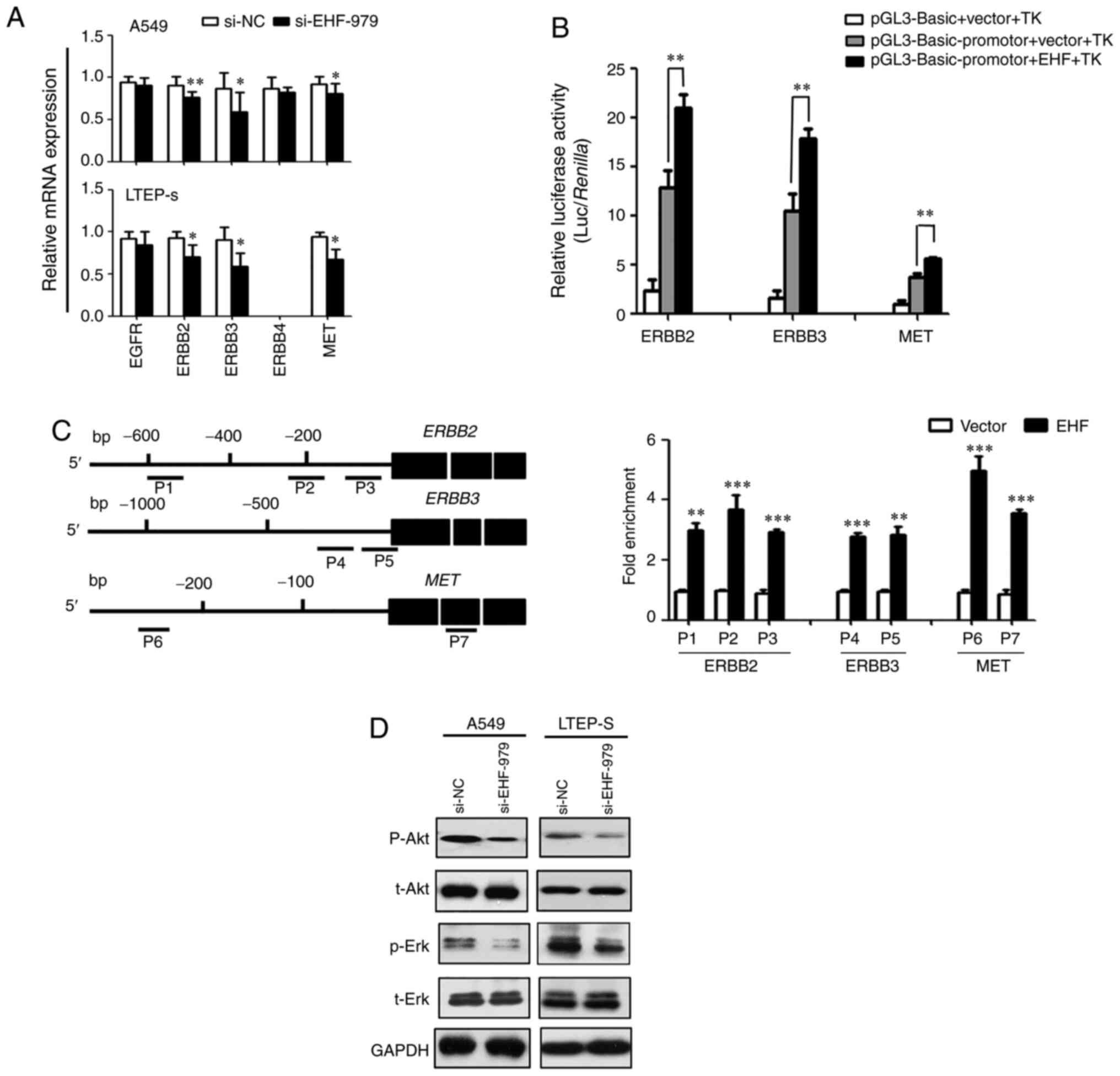 | Figure 5.EHF regulates the expression of
ERBB2, ERBB3 and MET in NSCLC cells via binding to EHF response
elements in the promoter regions. (A) EHF knockdown reduced the
expression of ERBB family members and MET, as assessed by real-time
RT-qPCR. Data are presented as the mean ± SD. (B) EHF regulated
promoter activities of ERBB2, ERBB3 and MET by binding to EHF
response elements, as determined by luciferase assays. Proximal
promoter regions of ERBB2 (−607/+11), ERBB3 (−997/+440) and MET
(−223/+60) were inserted into the pGL3-basic plasmid to generate
luciferase reporters pGL3-ERBB2-Luc, pGL3-ERBB3-Luc, and
pGL3-MET-Luc. 95D cells were co-transfected with reporters
indicated with either pcDNA3.1/Myc-His (−) A-EHF or empty control
vector. pRL-TK reporter was co-transfected as an internal control
to normalize the transfection efficiency. Fold-enrichment data are
presented as the mean ± SD of three independent assays. (C) EHF
binds to EHF response elements in the proximal promoter regions of
ERBB2, ERBB3, and MET, as determined by ChIP analyses. P1-P7
represent the regions analyzed by ChIP assays for ERBB2, ERBB3 and
MET. 95D cells were transiently transfected with pcDNA3.1/Myc-His
(−) A-EHF or empty control vector and were subjected to ChIP-qPCR
assays using an anti-Myc tag antibody. ChIP-PCR products arere
presented in right panel. (D) EHF knockdown reduced phosphorylation
of AKT and ERK in NSCLC cells, as assessed by western blot
analyses. GAPDH was used as a loading control. *P<0.05,
**P<0.01 and ***P<0.001. ERBB2, Erb-B2 receptor
tyrosinekinase 2; ERBB3, Erb-B2 receptor tyrosine kinase 3; ERBB4,
Erb-B2 receptor tyrosine kinase 4; MET, mesenchymal-epithelial
transition factor; EHF, ETS-homologous factor; NSCLC, non-small
cell lung cancer; RT-qPCR, reverse transcription-quantitative
polymerase chain reaction; SD, standard deviation; ChIP, chromatin
immunoprecipitation; si-, small interfering; NC, negative
control. |
It was next determined whether inhibition of tumor
growth by knockdown of EHF expression with siRNA was mediated by
modulating the EGF receptor-mediated AKT and ERK signaling pathways
in lung cancer cells. As revealed in Fig.
5D, downregulation of EHF expression resulted in a reduction in
AKT and ERK phosphorylation while total protein expression of AKT
or ERK in the transfected cells was not altered.
Discussion
ETS transcription factors play crucial roles in
several biological processes including cell proliferation,
differentiation, organ development, and tumorigenesis by modulating
the target gene expression via binding to their cis-elements
of GGAA/T in the regulatory regions (4,5,22). In the present study, we performed a
computer-assisted EST library screening and determined that an
epithelium-specific ETS factor, EHF/ESE-3 contained a putative DNA
binding domain similar to a known consensus motif of ESE-1 that is
known to bind to the promoter regions of EGFR family members
(10). In agreement with our
prediction, the ChIP and promoter/reporter assays revealed that EHF
was able to bind to the promoters of ERBB2, ERBB3 and MET genes and
regulate the promoter activities in NSCLC cells. Because EHF
expression was increased in NSCLC tissue compared with adjacent
non-tumor tissue and overexpression of EHF was associated with poor
prognosis in patients with NSCLC, it was surmised that changes in
EHF expression in cancers would alter tumor behaviors. As
anticipated, knockdown of EHF expression in NSCLC cells with
specific siRNA inhibited cell proliferation and invasion, arrested
the cell cycle at the G0/G1 phase, and
induced cancer cell apoptosis whereas overexpression of EHF in
NSCLC cells promoted cell proliferation as well as tumor growth and
migration in vitro. The in vivo experiments also
confirmed that knockdown of EHF expression in NSCLC cells
significantly inhibited cancer progression in xenografted nude mice
as compared to control mice grafted with NSCLC cells transfected
with control siRNA. It was demonstrated that EHF overexpression in
NSCLC cells could stimulate the expression of ERBB2, ERBB3 and MET
tyrosine kinase receptors and as such activate the AKT and ERK
downstream signaling pathways to promote tumor growth. The present
study indicated that EHF overexpression could be used as a
prognostic marker for NSCLC, and tyrosine kinase receptors of
ERBB2, ERBB3 and MET could be new drug targets for treatment of
patients with NSCLC.
The ERBB family consists of EGFR family members
ERBB1(HER1), ERBB2 (HER2), ERBB3 (HER3), and ERBB4 (HER4) (23). It is well known that the increased
expression of EGFR family members are associated with the poor
overall survival of patients with NSCLC (16). ERBB receptor activation initiates
tyrosine kinase receptor signaling and activates downstream
multiple signal cascades such as RAS/MAPK, PI3K/AKT, and signal
transducer and activator of transcription (STAT), thus regulating
tumor growth, survival, and angiogenesis (24,25). While
MET is another heterodimeric transmembrane receptor tyrosine kinase
(26) composed of an extracellular
α-chain and a transmembrane-spanning β-chain linked via disulfide
bonds, activation of the tyrosine kinase domain also mediates
downstream PI3K/AKT, MAPK, and phospholipase C signaling pathways.
Mutations of MET in patients with cancers have been suggested to
contribute to oncogenic progression (27–30). A
recent study has demonstrated that approximately 40% of patients
with small-cell lung cancer or NSCLC carry the tumor cells in which
MET receptor is overexpressed (17).
These studies raised a question as whether there is crosstalk
between MET and ERBB2 or ERBB3 family members that contributes to
cancer cell resistance to RTK-targeted inhibitors (31). If there is, then inhibition of EGFR or
MET signaling alone may result in a functional compensation of
other signaling. In supporting this assumption, previous studies
revealed that the amplification of the MET gene caused gefitinib
resistance due to activation of ERBB3-dependent PI3K signaling
(32), a pathway specific to
EGFR/ERBB family receptors activated via switching to an
alternative bypass survival signaling (33). In the present study, it was
demonstrated that expression levels of ERBB2/3 and MET were
regulated by EHF; inhibition of one signaling of EHF target genes
would not inactivate others. It is possible that suppression of one
signaling pathway would activate others by a feedback mechanism.
While we do not have direct evidence suggesting that the
interaction of ERBB2/3 with MET would result in a synergized
function on cancer cell migration and/or invasion, it is theorized
they may crosstalk and have additional or synergized cellular
functions. Further studies are required to confirm this theory.
Nevertheless, the present findings provide experimental evidence
that there may be crosstalk between MET and EGFR and a combination
of MET and EGFR inhibitors is required to sufficiently block
downstream EGFR/RTK signaling pathways for NSCLC treatment
(34).
EHF/ESE3 is located at 11p12 which is a hotspot for
loss of heterozygosity in lung, breast, and prostate carcinomas
(11). In contrast to our findings in
lung cancers, a previous study revealed that the loss of EHF
expression in prostate epithelial cells due to DNA methylation and
gene silencing resulted in epithelial-mesenchymal transition,
tumor-initiation, and cancer cell metastasis (14). Re-expression of EHF in prostate cancer
cells induced cancer cell apoptosis (35). A previous study also revealed that
knockdown of EHF expression in ovarian cancer cells stimulated the
expression of cyclin B1, cyclin D1, and PCNA, causing an increase
in cell proliferation and cancer progression (36). In airway epithelial cells including
A549 and Calu-3 cell lines, EHF-regulated protein expression was
involved in intercellular and cell-matrix adhesion in response to
wound healing (37). The significant
difference of EHF functions in different cells or tissues suggests
that EHF could be a tissue-specific repressor or enhancer, and its
function in cancer cells may depend on interaction with other
transcription factor/co-activators/co-repressors on the promoter
region of EHF target genes and chromatin remodeling. Further
studies are required to determine how EHF interacts with other
regulatory factors and remodels chromatin architectures to induce
EGFR and MET expression in NSCLC as well as in prostate cancer
cells.
In conclusion, the role of EHF in NSCLC progression
in vitro and in vivo was investigated and it was
demonstrated that EHF expression was increased in NSCLC. Knockdown
of EHF expression with specific siRNA in NSCLC cells significantly
inhibited cell proliferation and invasion, arrested the cell cycle
at the G0/G1 phase, and induced apoptosis
whereas overexpression of EHF in NSCLC cells promoted cell
proliferation, tumor growth, and cancer cell migration in
vitro. Knockdown of EHF expression in NSCLC cells also
suppressed tumor growth in xenografted nude mice. It was also
demonstrated that EHF promoted NSCLC growth by directly regulating
transcripts of ERBB2, ERBB3 and MET tyrosine kinase receptors and
modulating AKT and ERK signals via binding to the promoter of EHF
target genes. The present findings indicated that EHF could be used
as a prognostic marker for NSCLC, and tyrosine kinase receptors of
ERBB2, ERBB3 and MET could be new drug targets for NSCLC
treatment.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
Not applicable.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LG and MC conceived and supervised the study. LG, TY
and MC designed the experiments. LG, TY, SZ and YL performed
experiments. TY, YL, PS and HR contributed to the collection and
analysis of data. LG and PH drafted the manuscript. PH and MC made
manuscript revisions. All authors have read the final version and
have approved the submission of this study.
Ethics approval and consent to
participate
The study was approved by the Institutional Review
Board and Human Ethics Committee of the First Affiliated Hospital
of Xi'an Jiaotong University, and written informed consent was
obtained from each patient. The animal experiments were approved by
the Institutional Animal Care and Use Committee of Xi'an Jiaotong
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
NSCLC
|
non-small cell lung cancer
|
|
ETS
|
E26 transformation-specific
|
|
PEA3
|
polyoma enhancer activator 3
|
|
ESE-1
|
epithelial-specific expression-1
|
|
EHF
|
ETS-homologous factor
|
|
qPCR
|
quantitative PCR
|
|
FBS
|
fetal bovine serum
|
|
PI
|
propidium iodide
|
|
ChIP
|
chromatin immunoprecipitation
|
|
SD
|
standard deviation
|
|
STAT
|
signal transducer and activator of
transcription
|
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Devesa SS, Bray F, Vizcaino AP and Parkin
DM: International lung cancer trends by histologic type:
Male:Female differences diminishing and adenocarcinoma rates
rising. Int J Cancer. 117:294–299. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sementchenko VI and Watson DK: Ets target
genes: Past, present and future. Oncogene. 19:6533–6548. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Seth A and Watson DK: ETS transcription
factors and their emerging roles in human cancer. Eur J Cancer.
41:2462–2478. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wasylyk B, Hagman J and Gutierrez-Hartmann
A: Ets transcription factors: Nuclear effectors of the
Ras-MAP-kinase signaling pathway. Trends Biochem Sci. 23:213–216.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Trojanowska M: Ets factors and regulation
of the extracellular matrix. Oncogene. 19:6464–6471. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Benz CC, O'Hagan RC, Richter B, Scott GK,
Chang CH, Xiong X, Chew K, Ljung BM, Edgerton S, Thor A and Hassell
JA: HER2/Neu and the Ets transcription activator PEA3 are
coordinately upregulated in human breast cancer. Oncogene.
15:1513–1525. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Eckel KL, Tentler JJ, Cappetta GJ, Diamond
SE and Gutierrez-Hartmann A: The epithelial-specific ETS
transcription factor ESX/ESE-1/Elf-3 modulates breast
cancer-associated gene expression. DNA Cell Biol. 22:79–94. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kas K, Finger E, Grall F, Gu X, Akbarali
Y, Boltax J, Weiss A, Oettgen P, Kapeller R and Libermann TA:
ESE-3, a novel member of an epithelium-specific ets transcription
factor subfamily, demonstrates different target gene specificity
from ESE-1. J Biol Chem. 275:2986–2998. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kleinbaum LA, Duggan C, Ferreira E, Coffey
GP, Buttice G and Burton FH: Human chromosomal localization,
tissue/tumor expression, and regulatory function of the ets family
gene EHF. Biochem Biophys Res Commun. 264:119–126. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cangemi R, Mensah A, Albertini V, Jain A,
Mello-Grand M, Chiorino G, Catapano CV and Carbone GM: Reduced
expression and tumor suppressor function of the ETS transcription
factor ESE-3 in prostate cancer. Oncogene. 27:2877–2885. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shaikhibrahim Z, Lindstrot A, Langer B,
Buettner R and Wernert N: Differential expression of ETS family
members in prostate cancer tissues and androgen-sensitive and
insensitive prostate cancer cell lines. Int J Mol Med. 28:89–93.
2011.PubMed/NCBI
|
|
14
|
Turcotte S, Forget MA, Beauseigle D,
Nassif E and Lapointe R: Prostate-derived Ets transcription factor
overexpression is associated with nodal metastasis and hormone
receptor positivity in invasive breast cancer. Neoplasia.
9:788–796. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brenne K, Nymoen DA, Hetland TE, Trope CG
and Davidson B: Expression of the Ets transcription factor EHF in
serous ovarian carcinoma effusions is a marker of poor survival.
Hum Pathol. 43:496–505. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hynes NE and MacDonald G: ErbB receptors
and signaling pathways in cancer. Curr Opin Cell Biol. 21:177–184.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gelsomino F, Facchinetti F, Haspinger ER,
Garassino MC, Trusolino L, De Braud F and Tiseo M: Targeting the
MET gene for the treatment of non-small-cell lung cancer. Crit Rev
Oncol Hematol. 89:284–299. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang S, Gao L, Thakur A, Shi P, Liu F,
Feng J, Wang T, Liang Y, Liu JJ, Chen M and Ren H: MiRNA-204
suppresses human non-small cell lung cancer by targeting ATF2.
Tumour Biol. 37:11177–11186. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang P, Henning SM and Heber D:
Limitations of MTT and MTS-based assays for measurement of
antiproliferative activity of green tea polyphenols. PLoS One.
5:e102022010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shi J, Liu W, Sui F, Lu R, He Q, Yang Q,
Lv H, Shi B and Hou P: Frequent amplification of AIB1, a critical
oncogene modulating major signaling pathways, is associated with
poor survival in gastric cancer. Oncotarget. 6:14344–14359. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hollenhorst PC, Jones DA and Graves BJ:
Expression profiles frame the promoter specificity dilemma of the
ETS family of transcription factors. Nucleic Acids Res.
32:5693–5702. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Roskoski R Jr: The ErbB/HER family of
protein-tyrosine kinases and cancer. Pharmacol Res. 79:34–74. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Holbro T, Civenni G and Hynes NE: The ErbB
receptors and their role in cancer progression. Exp Cell Res.
284:99–110. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Roskoski R Jr: The ErbB/HER receptor
protein-tyrosine kinases and cancer. Biochem Biophys Res Commun.
319:1–11. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Petrini I: Biology of MET: A double life
between normal tissue repair and tumor progression. Ann Transl Med.
3:822015.PubMed/NCBI
|
|
27
|
Cappuzzo F, Marchetti A, Skokan M, Rossi
E, Gajapathy S, Felicioni L, Del Grammastro M, Sciarrotta MG,
Buttitta F, Incarbone M, et al: Increased MET gene copy number
negatively affects survival of surgically resected non-small-cell
lung cancer patients. J Clin Oncol. 27:1667–1674. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fu P, Du F, Yao M, Lv K and Liu Y:
MicroRNA-185 inhibits proliferation by targeting c-Met in human
breast cancer cells. Exp Ther Med. 8:1879–1883. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hagman Z, Haflidadottir BS, Ansari M,
Persson M, Bjartell A, Edsjö A and Ceder Y: The tumour suppressor
miR-34c targets MET in prostate cancer cells. Br J Cancer.
109:1271–1278. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Phan LM, Fuentes-Mattei E, Wu W,
Velazquez-Torres G, Sircar K, Wood CG, Hai T, Jimenez C, Cote GJ,
Ozsari L, et al: Hepatocyte Growth Factor/cMET pathway activation
enhances cancer hallmarks in adrenocortical carcinoma. Cancer Res.
75:4131–4142. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maroun CR and Rowlands T: The Met receptor
tyrosine kinase: A key player in oncogenesis and drug resistance.
Pharmacol Ther. 142:316–338. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Karamouzis MV, Konstantinopoulos PA and
Papavassiliou AG: Targeting MET as a strategy to overcome
crosstalk-related resistance to EGFR inhibitors. Lancet Oncol.
10:709–717. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Troiani T, Martinelli E, Napolitano S,
Vitagliano D, Ciuffreda LP, Costantino S, Morgillo F, Capasso A,
Sforza V, Nappi A, et al: Increased TGF-α as a mechanism of
acquired resistance to the anti-EGFR inhibitor cetuximab through
EGFR-MET interaction and activation of MET signaling in colon
cancer cells. Clin Cancer Res. 19:6751–6765. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Luk IY, Reehorst CM and Mariadason JM:
ELF3, ELF5, EHF and SPDEF transcription factors in tissue
homeostasis and cancer. Molecules. 23:21912018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cheng Z, Guo J, Chen L, Luo N, Yang W and
Qu X: Knockdown of EHF inhibited the proliferation, invasion and
tumorigenesis of ovarian cancer cells. Mol Carcinog. 55:1048–1059.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fossum SL, Mutolo MJ, Yang R, Dang H,
O'Neal WK, Knowles MR, Leir SH and Harris A: Ets homologous factor
regulates pathways controlling response to injury in airway
epithelial cells. Nucleic Acids Res. 42:13588–13598. 2014.
View Article : Google Scholar : PubMed/NCBI
|