Introduction
Chronic myeloid leukemia (CML) is a hematopoietic
stem cell disorder that affects the blood and bone marrow and
accounts for about 15% of newly diagnosed adult leukemia cases
(1). CML is a myeloproliferative
disease comprising three stages, including a chronic phase,
accelerated phase, and lymphatic/myeloid blast phase, and is
characterized by the Philadelphia chromosome. The Ph chromosome is
a specific CML diagnostic marker and is present in more than 95% of
patients (2,3). This chromosome is the result of a
translocation between chromosomes 9 and 22, leading to the
generation of a chimeric gene product of the breakpoint cluster
region (BCR) and the Abelson murine leukemia (ABL), a fusion gene
known as BCR-ABL (4,5). The BCR-ABL fusion gene can form a
chimeric protein, resulting in a constitutively active ABL kinase,
which can transform hematopoietic stem cells into leukemic stem
cells and actuate the overproduction and expansion of leukocytes in
the bone marrow (6,7). Thus, the BCR-ABL fusion protein is an
important molecular basis for CML pathogenesis.
The oncogenic BCR-ABL fusion protein has persistent
kinase activity, resulting in the uncontrolled proliferation of
myeloid cells through multiple downstream pathways (2). BCR-ABL phosphorylates several substrates
that activate a number of aberrant kinase-dependent pathways,
including the Ras-mitogen-activated protein kinase (MAPK) pathway,
which results in increased proliferation; the Janus-activated
kinase/signal transducer and activator of transcription (JAK/STAT)
pathway, which impairs transcriptional activity, and the
phosphatidylinositide 3-kinase/protein kinase B (PI3K/AKT) pathway,
which leads to enhanced survival (8–10). Thus,
BCR-ABL has been recognized as the most important target for CML
treatment.
CML therapy has seen impressive advances with the
development of tyrosine kinase inhibitors (TKIs) against the
BCR-ABL pathway (11),
revolutionizing CML management and resulting in a 10-year overall
survival rate of more than 90% (12).
In terms of CML treatment, nilotinib (a second-generation TKI) has
certain advantages over the first-generation TKI imatinib, such as
improved response kinetics, a significant progression rate
reduction, and deeper molecular responses (13,14).
Although existing TKIs are highly effective in CML patients, many
patients develop drug resistance. Therefore, the development of
novel kinase inhibitors in an effort to overcome drug resistance is
crucial (15).
ND-09
(4-methoxyl-3-[[4-(3-pyridyl)-2-pyrimidinyl]amino] benzoic acid
pyrimidin-2-ylamino-benzamides) is a novel compound developed by
our group (16). The aim of the
present study was to investigate whether ND-09 could regulate
BCR-ABL signaling to suppress CML growth.
Materials and methods
Chemicals and reagents
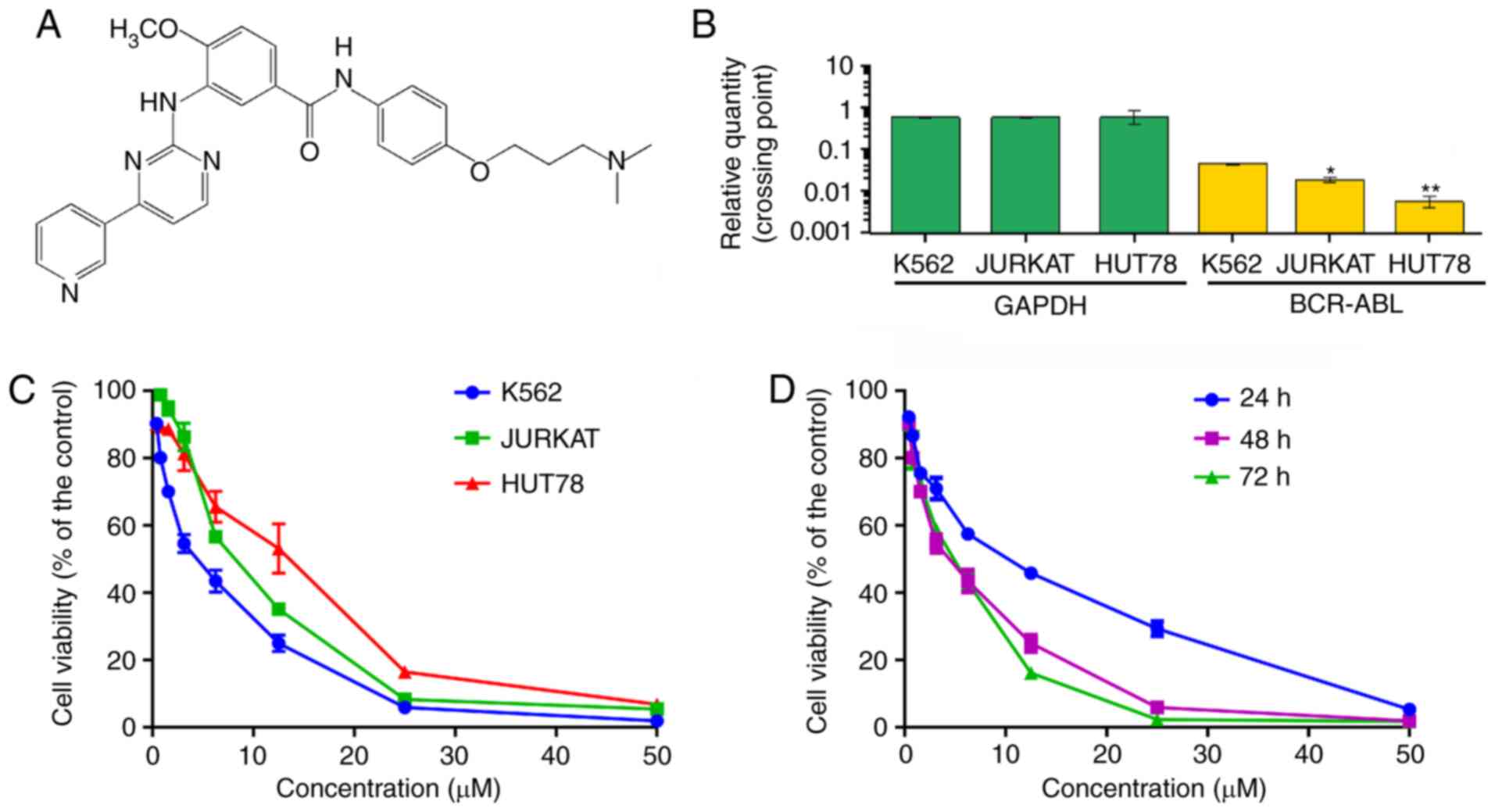
ND-09 (Fig. 1A) was
developed in the Research and Engineering Center for Natural
Medicine, Xi'an Jiaotong University (Shaanxi, China). K562, HUT78
and JURKAT cells were obtained from the Shanghai Institute of Cell
Biology in the Chinese Academy of Sciences (Shanghai, China).
Iscove's modified Dulbecco's medium (IMDM) and RPMI-1640 medium
were purchased from Sigma-Aldrich.
3-(4,5-Dimethylthiazol-2-yl)-2.5-diphenyl-2H-tetrazolium brom-ide
(MTT) were purchased from Sigma-Aldrich. Fetal bovine serum (FBS)
was obtained from ExCell Bio Co., Ltd.
Thermal Cycle Dice Real time system, PrimeScript RT
Master Mix Perfect Real Time kit, and SYBR® Premix Ex
Taq™ II were obtained from Takara Biotechnology. Annexin V-FITC
cell apoptosis detection kit was obtained from Pioneer
Biotechnology Co., Ltd. RNase and propidium iodide (PI) were
obtained from Sigma-Aldrich. Opti-MEM medium was purchased from
Gibco. Lipofectamine 2000 reagent was obtained from Invitrogen.
Phosphatase and protease inhibitors were obtained from Roche Tech.
The BCA kit, RNAfast200 kit, and RIPA lysis buffer were obtained
from Pioneer Biotechnology Co., Ltd. BCR-ABL and control
siRNA were obtained from Shanghai GenePharma Co., Ltd.
The mcl-1 (monoclonal, 66026-1-Ig), Bad (monoclonal,
67830-1-Ig), Bcl-2 (monoclonal, 60178-1-Ig), Bak (polyclonal,
14673-1AP), Bax (monoclonal, 60267-1-Ig), CDC2 (monoclonal,
67575-1-Ig), Cyclin D1 (monoclonal, 60186-1-Ig), Cyclin E
(polyclonal, 11554-1-AP), CDK2 (monoclonal, 60312-1-Ig), Cyclin A2
(monoclonal, 66391-1-Ig), and Cyclin B1 (monoclonal, 67686-1-Ig)
antibodies were purchased from Protein Technology Group. The
phospho-BCR-ABL (polyclonal, Tyr177), BCR-ABL (monoclonal, L99H4),
phospho-ABL (monoclonal, 73E5), ABL (polyclonal, 2862), phospho-BCR
(polyclonal, Tyr177), and BCR (polyclonal, 3902) antibodies were
obtained from Cell Signaling Technology. The JAK2 (monoclonal,
D2E12), phospho-JAK2 (monoclonal, D15E2), JAK3 (monoclonal, D1H3),
phospho-JAK3 (monoclonal, D44E3), STAT3 (monoclonal, D3Z2G),
phospho-STAT3 (monoclonal, D3A7), STAT5 (monoclonal, D3N2B), and
phospho-STAT5 (monoclonal, D47E7) antibodies were obtained from
Cell Signaling Technology. The p53 (polyclonal, 10442-1-AP), p38
(monoclonal, 66234-1-Ig), and PTEN (monoclonal, 60300-1-Ig)
antibodies were obtained from the Protein Technology Group. The
phospho-p38 (monoclonal, D3F9), PI3K-p110α (monoclonal, C73F8),
PI3K-p110β (monoclonal, C33D4), PI3K-p110γ (monoclonal, D55D5),
PI3K-p85 (monoclonal, 19H8), and phospho-PI3K-p85/p55 (monoclonal,
E3U1H) antibodies were obtained from Cell Signaling Technology. The
AKT (monoclonal, C67E7), phospho-AKT (monoclonal, D9E), PLCγ
(monoclonal, D9H10), phospho-PLCγ (monoclonal, D6M9S), Erk1/2
(monoclonal, 137F5), phospho-Erk1/2 (monoclonal, Tyr202/204),
MEK1/2 (monoclonal, D1A5), phospho-MEK1/2 (monoclonal, 166F8), mTOR
(monoclonal, 7C10), and phospho-mTOR (monoclonal, D9C2) antibodies
were obtained from Cell Signaling Technology. Rabbit anti-GAPDH
(monoclonal, 60004-1-lg) was obtained from Protein Technology
Group. The working solution for the primary antibodies was 1:1000,
except for the Bad and Cyclin E antibodies, which was 1:500.
HRP-conjugated goat anti-rabbit IgG (secondary antibody, dilution:
1:20,000) was obtained from Pierce Biotechnology. Enhanced
Chemiluminescent Plus Reagent was obtained from Biotech Co.,
Ltd.
Cell culture
K562 and HUT78 cells were cultured in IMDM medium
containing 10% FBS, 100 U/ml penicillin, and 100 U/ml streptomycin.
JURKAT cells were cultured in RPMI-1640 medium containing 10% FBS,
100 U/ml penicillin, and 100 U/ml streptomycin. All cell lines were
grown at 37°C in a 95% humidified atmosphere with 5%
CO2.
Cell viability assay
K562, JURKAT and HUT78 cells were seeded in 96-well
plates and cultivated in complete medium for 24 h. Then cells were
treated with ND-09 (0.39, 0.78, 1.56, 3.12, 6.25, 12.5, 25, and 50
µM) for 48 h. Then, 20 µl MTT solution (5 mg/ml) was added to each
well and incubated at 37°C for 4 h. Subsequently, the cells were
enriched at the bottom of each well using a plate spinner (4,192.5
× g for 5 min at 25°C) and the medium was slowly removed from each
well. Then, 150 µl DMSO was added in each well for 15 min. The
absorbance was recorded by a microplate reader (Bio-Rad) at 490
nm.
Flow cytometric analysis of cell
apoptosis
Cells were seeded in 6-well plates and treated with
ND-09 for 48 h, after which the cells were collected by
centrifugation (4,192.5 × g for 5 min at 25°C), washed, and
resuspended in PBS. According to the instructions, Annexin V-FITC
and PI double staining were used to detect the apoptotic rate.
Then, Cell Quest software was used for flow cytometry and data
analysis.
Determination of mitochondrial
transmembrane potential (Δψm)
Cells were seeded in 6-well plates and treated with
ND-09 for 48 h, after which the cells were washed with complete
medium, followed by incubation with 1 mM Rhodamine 123 at 37°C in
the dark for 30 min. Then BD FACSCalibur Flow cytometer was used to
perform flow cytometric analysis and analyze data.
Flow cytometric analysis of cell
cycle
Cells were seeded in 6-well plates, treated with
ND-09 for 48 h, and then fixed in ice-cold 70% ethanol at 4°C
overnight. Then, the cells were washed with cold PBS and stained
with RNase and PI for 30 min in the dark. Cell Quest software was
used to perform flow cytometric analysis and analyze data.
RNA extraction and RT-PCR assay
Cells were seeded in 6-well plates and treated with
ND-09 for 48 h. According to the manufacturer's protocol, total RNA
from K562, JURKAT and HUT78 cells was extracted using RNAfast200
kit (Pioneer Biotechnology Co., Ltd). RT-PCR was performed using
PrimeScript RT Master Mix Perfect Real Time kit and was performed
using SYBR® Premix Ex Taq™ II and Thermal Cycle Dice
Real time system. GAPDH was considered the reference gene.
The primer sequences used were: GAPDH forward:
5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse: 5′-TGGTGAAGACGCCAGTGGA-3′;
BCR-ABL forward: 5′-CATTCCGCTGACCATCAATAAG-3′ and reverse:
5′-GATGCTACTGGCCGCTGAAG-3′.
Threshold cycle (Ct) values of BCR-ABL in
each sample were normalized with the GAPDH expression. The
ratio of BCR-ABL versus the corresponding GAPDH of
each sample was determined on the basis of the equation
BCR-ABL/GAPDH=2Cq(GAPDH)-Cq(BCR-ABL).
SiRNA transfection
K562 cells were seeded in cell culture dishes at a
density of approximately 50%. According to the manufacturer's
protocol, Lipofectamine 2000 reagent was used to transfect
anti-BCR-ABL siRNA or control siRNA (100 nM) into cells for 24 h.
Then, transfected cells were immediately seeded to perform
subsequent assays.
Plasmid transfection
K562 cells were seeded into 6-well plates at the
density of 2×105 per well. After 24 h, transfection of
EphB4 plasmid into BCR-ABL-depleted K562 cells was performed
using Lipofectamine 2000 reagent. The ratio of Lipofectamine 2000
(µl) to BCR-ABL plasmid (µg) was 2:1.
Molecular docking (MD)
SYBYL-X 1.1 was used to conduct docking studies to
understand the binding mode of ND-09 and BCR-ABL domain (PDB ID:
1IEP). The substrate was constructed by Sybyl/Sketch modul and
optimized by Powell's method. Tripos force field and
Gasteiger-Hückel charges were used to minimize energy, the
convergence criterion was set at 0.005 kcal/(Å mol), and the
maximum value was set at 1,000 iterations. To explore
intramolecular interactions, the non-bonded cut-off distance was
set to 8 Å.
Kinase assay
Firstly, BCR-ABL kinase (2 µl, 5 ng/µl) and
substrate (2 µl, 10 µM) were added to 384-well plates, and then
drugs (4 µl) were added to the test plate at different
concentrations (0.016, 0.008, 0.040, 0.20, 1.02, 5.12, 25.60,
128.00, and 640 nM). ATP solution (2 µl, 1 mM) was added and the
system was incubated at 37°C for 30 min. TK-Antibody (5 µl, 1,000
tests, reconstituted with 5 ml of HTRF® detection
buffer) and streptavidin-XL665 (5 µl, 125 nM) were then added to
the test plate at room temperature for 1 h.
Western blot analysis
Cells treated with ND-09 were seeded in 6-well
plates. RIPA buffer with protease and phosphatase inhibitor were
used to extract protein from cells. The cell lysate was then
concentrated at 12,000 × g for 10 min at 4°C. Protein (40 µg)
quantified by using BCA kit was then loaded to 10% SDS-PAGE gel,
after which protein was transferred to polyvinylidene difluoride
membranes. The membranes with protein were blocked with
Tris-buffered saline for 1 h at room temperature. Subsequently, the
primary antibody was incubated overnight at 4°C, and the secondary
antibody was incubated at room temperature for 1 h. The blot was
then exposed to the Enhanced Chemiluminescent substrate. Band
intensity was quantified by densitometric analysis using an image
quantitative analysis system (Image-Pro Plus 5.1; Media Cybernetics
Inc.).
Statistical analysis
Statistical analyses were performed using SPSS 18.0.
All the experiments were performed at least three times, and the
results are expressed as mean ± SD. Statistical analyses of
differences between the groups were performed with ANOVA followed
by the Tukey post-hoc test. P<0.05 was considered statistically
significant, and P<0.01 was considered extremely
significant.
Results
Inhibitory effect of ND-09 on the
proliferation of hematologic cancer cells
In order to evaluate the inhibitory effect of ND-09
on hematologic tumor cell growth, CML K562 cells, cutaneous T-cell
lymphoma HUT78 cells, and acute T-cell lymphoma JURKAT cells were
treated with ND-09. As BCR-ABL is a key factor in the
myeloproliferative disorder of CML, BCR-ABL expression was
investigated in all three cell lines. The results indicated that
the expression of BCR-ABL in K562 cells was higher than that
in JURKAT and HUT78 cells (Fig. 1B).
As seen in Fig. 1C, K562 cells were
more sensitive to ND-09 than JURKAT and HUT78 cells. The
IC50 values of ND-09 for K562, JURKAT, and HUT78 cells
were 6.08, 7.81, and 14.43 µM, respectively. Furthermore, ND-09
inhibited the proliferation of K562 cells in a dose- and
time-dependent manner (Fig. 1D).
Thus, the inhibitory effect of ND-09 on K562 cell growth may be
related to BCR-ABL.
ND-09 induces cell apoptosis
Following ND-09 treatment of 1.56, 3.12, and 6.25
µM, the percentage of apoptotic K562 cells increased to 10.17±1.03,
16.40±1.41, and 49.50±1.25%, respectively, compared to control
cells (5.55±0.93%) (Fig. 2A). In
addition, ND-09 could induce JURKAT and HUT78 cell apoptosis
(Fig. 2B and C). The percentage of
apoptotic JURKAT cells increased to 7.27±0.95, 9.03±0.83, and
54.39±1.79%, respectively, compared to control cells (7.29±1.02%).
The percentage of apoptotic HUT78 cells increased to 12.14±1.15,
17.10±1.34, and 23.98±1.67%, respectively, compared to control
cells (11.02±0.96%). The above data indicated that the
apoptosis-inducing effect of ND-09 in the two cell lines was
relatively weaker than that in K562 cells.
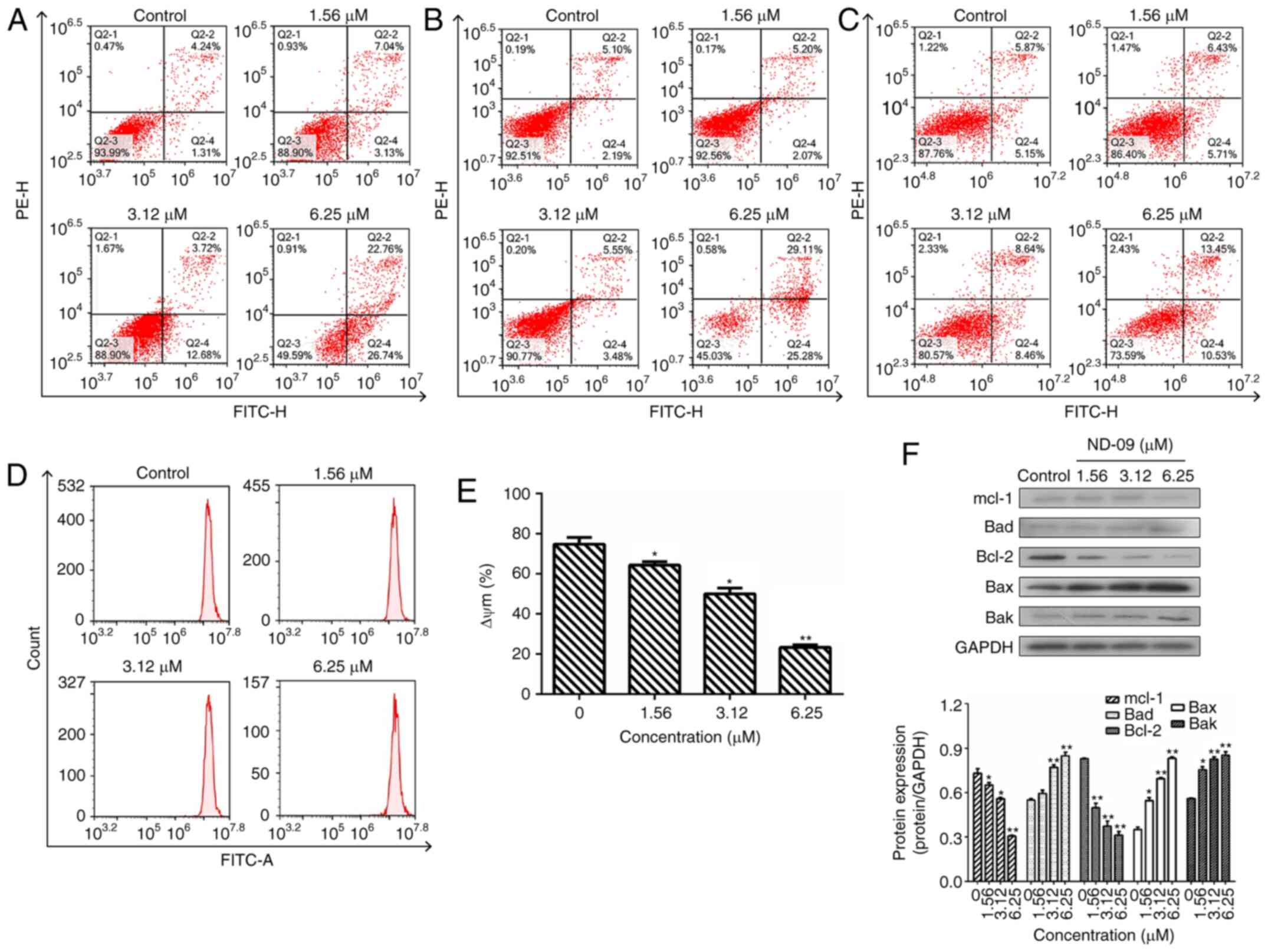 | Figure 2.Effect of ND-09 treatment on cell
apoptosis. The proportion of apoptotic cells was determined by
double staining with Annexin V/FITC and PI in (A) K562, (B) JURKAT,
and (C) HUT78 cells after treatment with ND-09 (0, 1.56, 3.12, and
6.25 µM). (D) Effect of ND-09 on mitochondrial membrane potential
(Δψm). Δψm was assessed through flow cytometry following treatment
of K562 cells with ND-09 (0, 1.56, 3.12, and 6.25 µΜ) for 48 h. (E)
Quantitative analysis of flow cytometry data. (F) Effects of ND-09
on apoptosis-related protein expression in K562 cells. All results
were quantified by densitometric analysis of the bands and were
normalized to GAPDH (internal control). Samples were derived from
the same experiment, and blots were processed in parallel. Values
represent the average of three independent experiments. Data are
presented as mean ± SEM (n=3). *P<0.05, **P<0.01 compared to
the untreated control group. |
Subsequently, the effect of ND-09 on mitochondrial
transmembrane potential was examined. Rhodamine 123 fluorescence
intensity of K562 cells exposed to ND-09 decreased significantly
from 74.82±2.58% in the control group to 64.26±2.15% (1.56 µΜ),
49.98±1.69% (3.12 µΜ), and 23.25±1.76% (6.25 µΜ) (Fig. 2D and E).
To explore the mechanism of ND-09-induced cell
apoptosis, western blot analysis was performed to detect the levels
of apoptosis-related proteins. Results indicated that Bak, Bax, and
Bad levels significantly increased after ND-09 treatment, while
Bcl-2 and Mcl-1 levels decreased in a dose-dependent manner
(Fig. 2F).
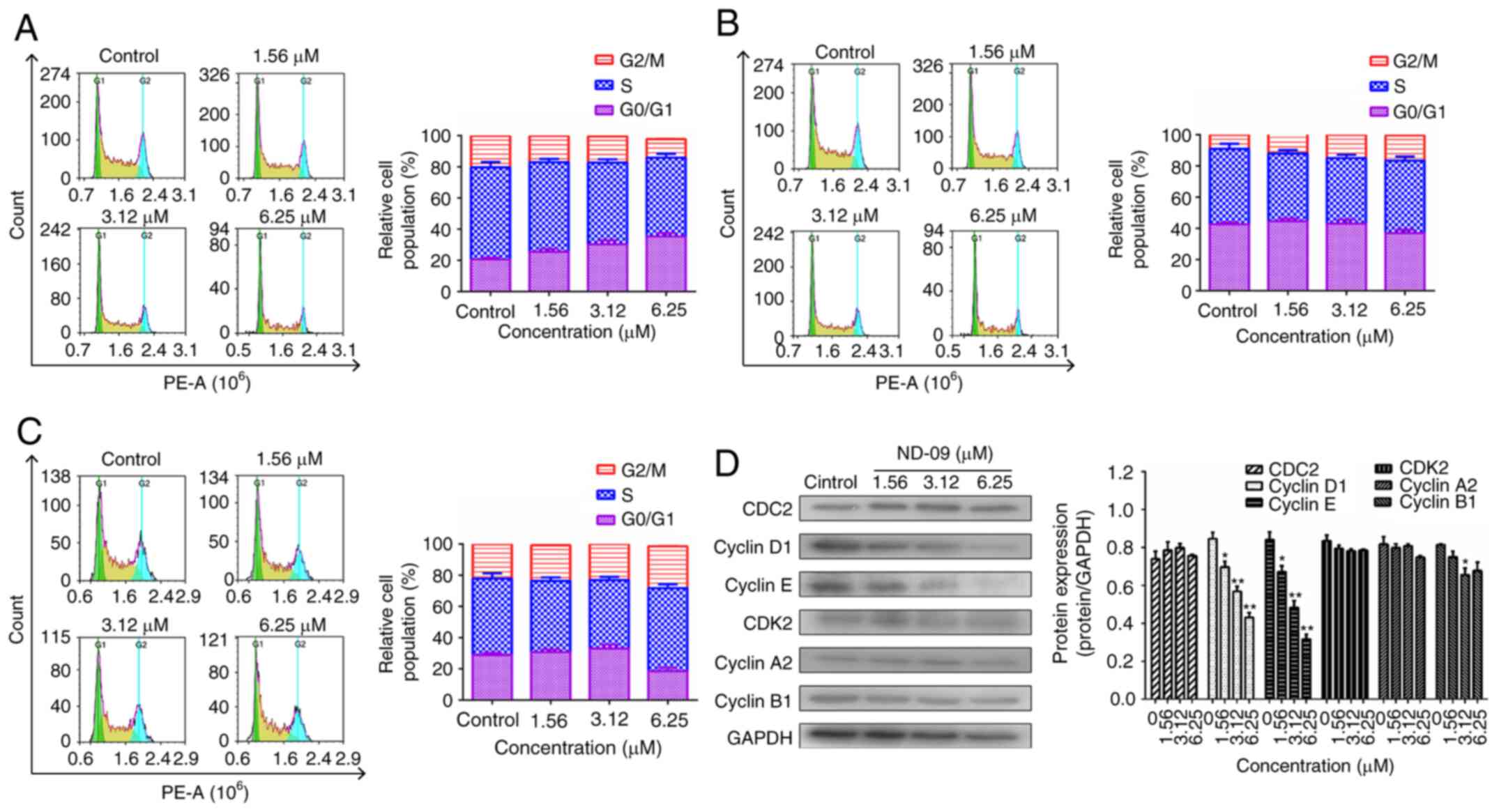
ND-09 induces G0/G1 phase arrest in
K562 cells
ND-09 treatment led to a significant increase in
G0/G1 phase cells (Fig. 3A). At
increasing ND-09 concentrations of 1.56, 3.12 and 6.25 µM, the K562
cell population in G0/G1 phase increased by 25.56±1.15, 30.29±1.43,
and 35.31±1.37% compared to the untreated cells (20.61±1.26%). A
different mode of cell cycle arrest induction was observed in
JURKAT and HUT78 cells, as these were arrested in the G2/M phase
(Fig. 3B and C).
As ND-09 arrested the K562 cell cycle in the G0/G1
phase, levels of CDK/cyclin proteins were evaluated through western
blot analysis. Fig. 3D revealed that
cyclin E and cyclin D1 levels decreased in ND-09-treated K562
cells.
Role of BCR-ABL on the effects of
ND-09 on cell growth
A siRNA assay was carried out to determine whether
BCR-ABL was a key factor for the ND-09-induced inhibition of
K562 cell growth. First, BCR-ABL knockdown and
BCR-ABL rescue of K562 cells were successfully established
(Fig. 4A and B).
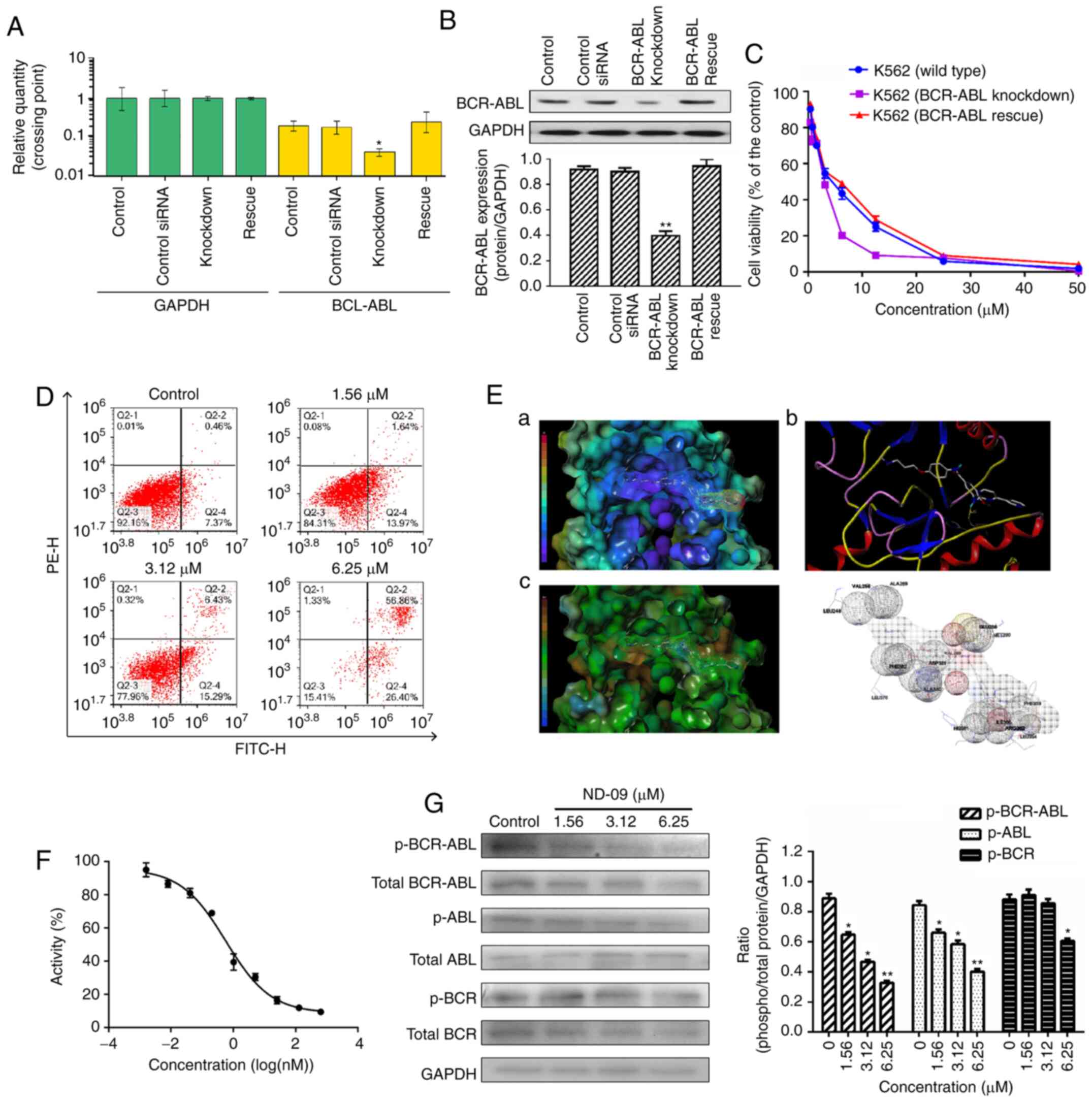 | Figure 4.The role of BCR-ABL in the biological
activity of ND-09 treatment. (A) BCR-ABL mRNA expression and
(B) protein expression in wild-type, BCR-ABL knockdown, and
BCR-ABL rescue K562 cells were determined by RT-PCR analysis
and western blot analysis, respectively. Cells transfected with a
control siRNA construct served as negative controls. Samples are
derived from the same experiment, and blots were processed in
parallel. (C) An MTT assay was used to evaluate the effect of ND-09
on cell proliferation in wild-type, BCR-ABL knockdown, and BCR-ABL
rescue cells. (D) Effect of ND-09 on apoptosis in BCR-ABL-depleted
K562 cells. Values represent the average of three independent
experiments. Data are presented as mean ± SEM (n=3). *P<0.05,
**P<0.01 compared to the untreated control group. (E) The
binding mode of ND-09 to BCR-ABL (PDB ID: 1IEP). a, ND-09 in the
crystal structure of BCR-ABL with electrostatic coloring; b,
Hydrogen bonds are depicted by dashed yellow lines; c, ND-09 in the
crystal structure of BCR-ABL with hydrophobic coloring. (F) Effect
of ND-09 on BCR-ABL kinase activity. Values represent the average
of three independent experiments. Values are presented as mean ±
SEM (n=3). (G) Levels of BCR-ABL, p-BCR-ABL, ABL, p-ABL, BCR, and
p-BCR in K562 cells treated with ND-09 were examined by western
blot analysis. Results were quantified by densitometric analysis of
the bands and were normalized to GAPDH (internal control). Samples
were derived from the same experiment, and blots were processed in
parallel. Values represent the average of three independent
experiments. Data are presented as the mean ± SEM (n=3).
*P<0.05, **P<0.01 compared to the untreated control
group. |
Then, to assess the role of BCR-ABL in ND-09-induced
cell growth inhibition, wild-type and BCR-ABL-depleted K562
cells were treated with ND-09. ND-09 had a dose-dependent
inhibitory effect on the proliferation of the two groups, and
combined ND-09 and siRNA treatment resulted in an enhanced
inhibitory effect (Fig. 4C). The
IC50 values of ND-09 in wild-type and
BCR-ABL-depleted K562 cells were 6.08 and 3.61 µM,
respectively. Furthermore, this growth effect of BCR-ABL
siRNA could be fully rescued by transfection with BCR-ABL.
In addition, BCR-ABL knockdown by siRNA led to increased K562 cell
apoptosis, and combined ND-09 and siRNA treatment enhanced this
effect (Fig. 4D). These observations
suggested that BCR-ABL was a key target for ND-09.
Effect of ND-09 on BCR-ABL
An MD assay was performed to evaluate the affinity
characteristics of ND-09 binding to the active site of BCR-ABL. The
binding energy of ND-09 with BCR-ABL was −7.07. ND-09 bound to the
BCR-ABL ATP-binding pocket through an electrostatic interaction
(Fig. 4E-a), hydrogen-bond
interaction (Fig. 4E-b), and
hydrophobic interaction (Fig. 4E-c).
Furthermore, ND-09 may interact with VAL256, ALA269, LEU248,
GLU288, VET290, VAL299, ASP381, PHE382, ALA380, LEU370, PHE359,
ILE360, ARG362, LEU354, and HIS361 amino acid residues of BCR-ABL
(Fig. 4E-d). The docking assay
results demonstrated that ND-09 fit well within BCR-ABL.
Accordingly, we examined the inhibitory effect of
ND-09 on BCR-ABL kinase activity. ND-09 inhibited BCR-ABL kinase
activity with an IC50 value of 0.57 nM (Fig. 4F). Furthermore, ND-09 markedly
inhibited the phosphorylation of BCR-ABL and ABL in K562 cells
(Fig. 4G). These data indicate that
ND-09 could inhibit BCR-ABL phosphorylation and kinase
activity.
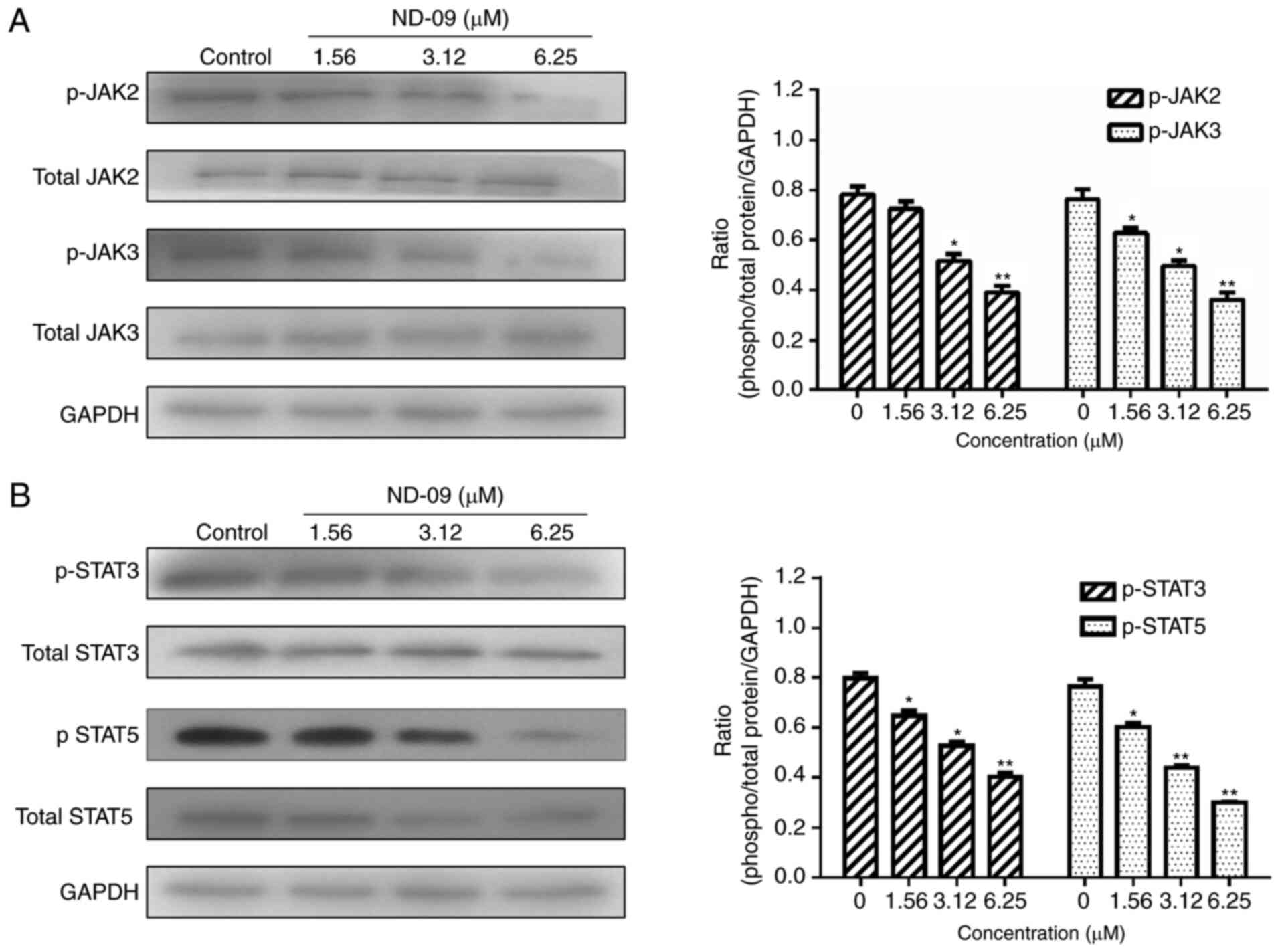
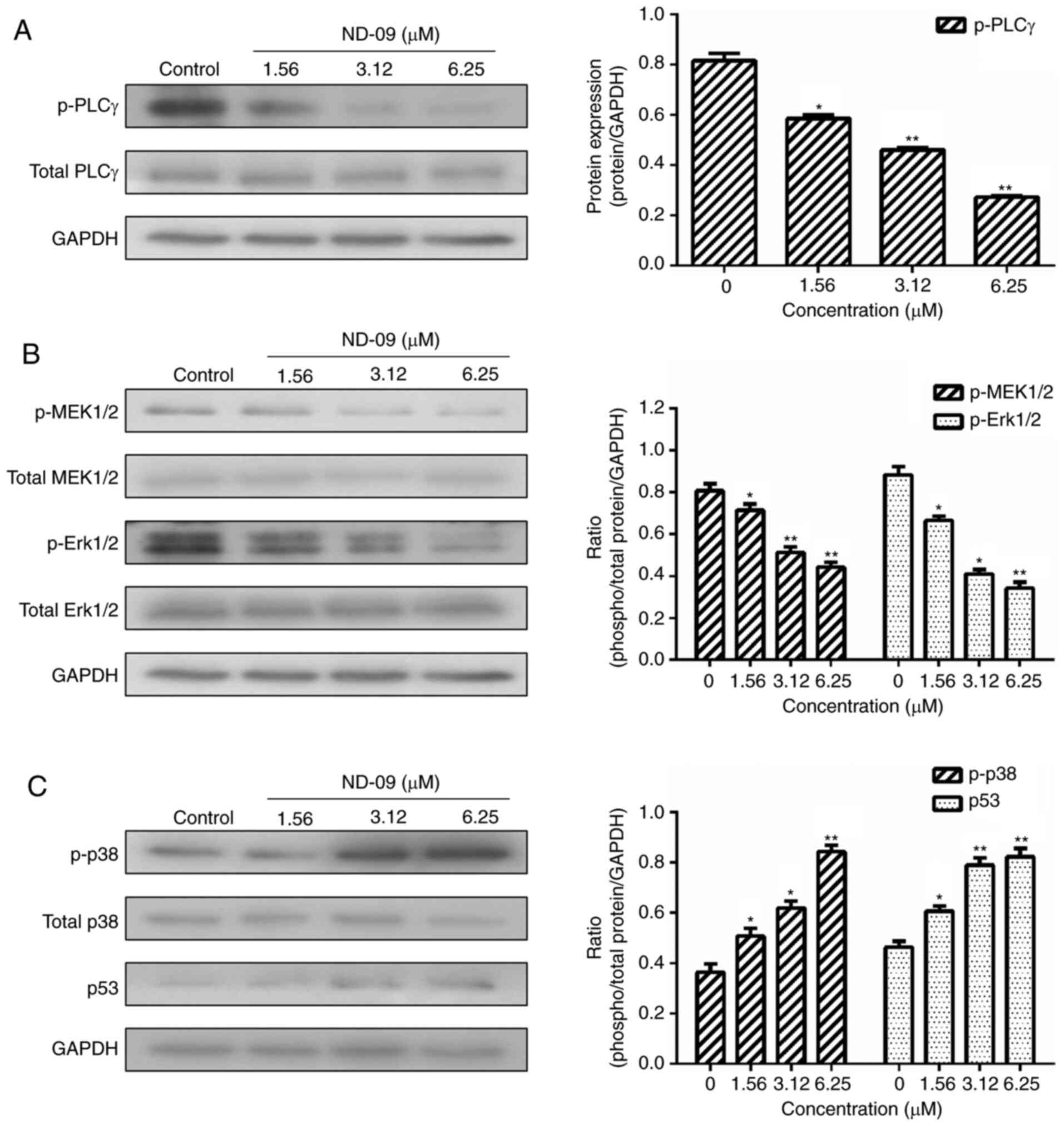
ND-09 regulates the BCR-ABL downstream
signaling pathway
As shown in Fig. 5,
ND-09 was found to significantly inhibit the phosphorylation of
JAK/STAT signaling pathway members (JAK2, JAK3, STAT3, and STAT5).
Furthermore, ND-09 treatment led to an increase in the amount of
PTEN and a decrease in the amount of PI3K subunits (110α, 110β,
p-p85/p55 and p85), as well as decreased phosphorylation of
downstream molecules (AKT and mTOR) in K562 cells (Fig. 6). ND-09 upregulated p-p38 and p53
levels and significantly inhibited the phosphorylation of PLC-γ,
which in turn inhibited MEK1/2 and ERK1/2 phosphorylation (Fig. 7).
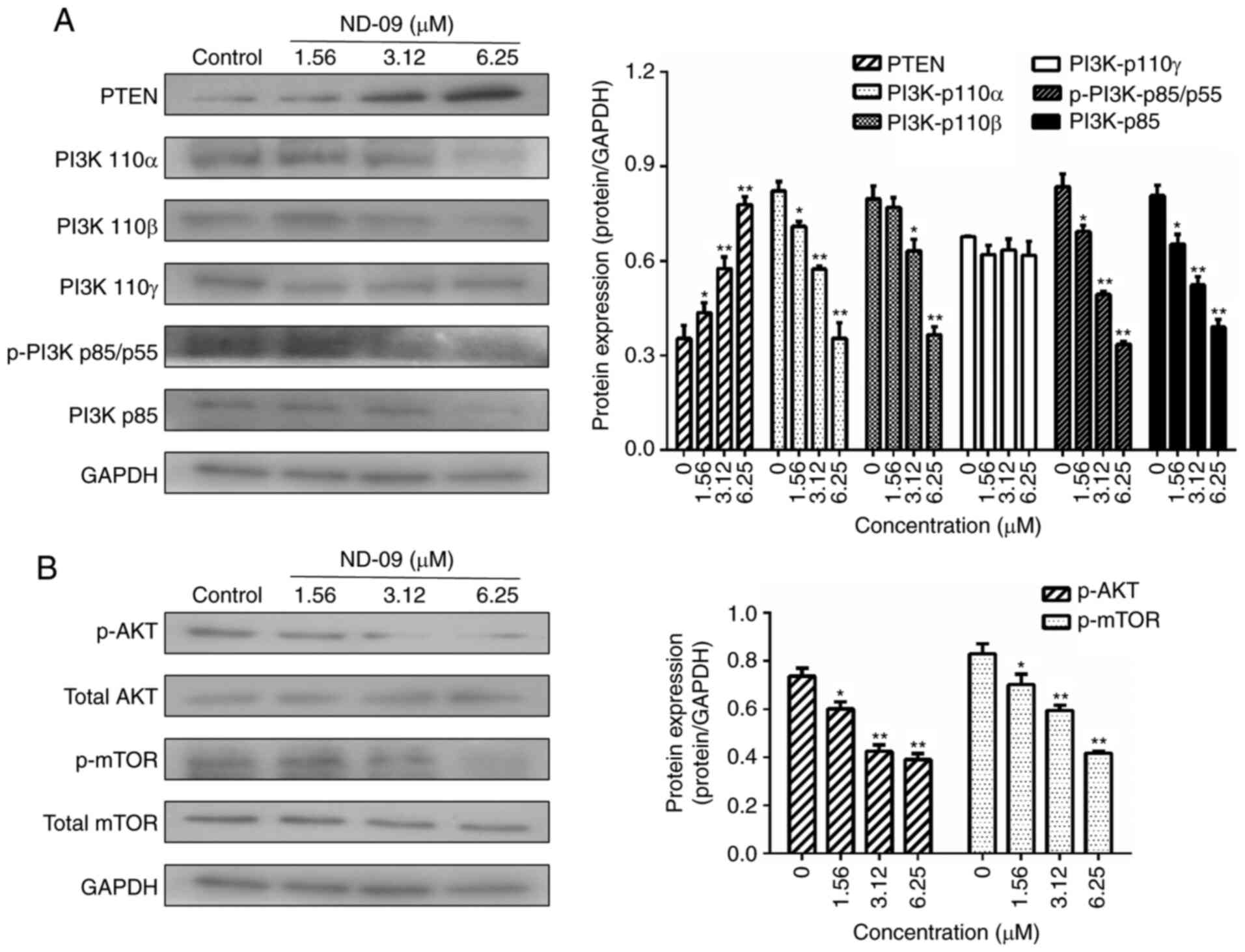 | Figure 6.Effects of ND-09 on PI3K/AKT
signaling protein levels. (A) Protein levels of PTEN, PI3K-p110α,
PI3K-p110β, PI3K-p110γ, p-PI3K p85/p55, and PI3K-p85 in K562 cells
treated with ND-09 were evaluated by western blot analysis. (B)
Protein levels of AKT, p-AKT, mTOR, and p-mTOR in K562 cells
treated with ND-09 were evaluated by western blot analysis. Results
were quantified by densitometric analysis of the bands and were
normalized to GAPDH (internal control). Samples were derived from
the same experiment, and blots were processed in parallel. Values
represent the average of three independent experiments. Data are
presented as mean ± SEM (n=3). *P<0.05, **P<0.01 compared to
the untreated control group. |
Discussion
CML, a myeloproliferative malignancy driven by the
constitutively active BCR-ABL1 tyrosine kinase, is currently
treated with TKIs that have proven to be clinically effective
(17). However, while TKI treatment
was initially successful, the emerging development of TKI
resistance presents a considerable challenge (18,19). In
this study, we developed ND-09, which exhibited potent anticancer
activity against CML by targeting BCR-ABL.
K562 cells, which express high levels of
BCR-ABL, were most sensitive to ND-09, suggesting that ND-09
potentially inhibits cell proliferation via targeting
BCR-ABL (Fig. 1). Furthermore,
ND-09 inhibited the growth of K562 cells in a dose- and
time-dependent manner. We proceeded to verify whether
BCR-ABL is the target of ND-09. The results revealed that
combining ND-09 with BCR-ABL siRNA knockdown led to an
enhanced inhibitory effect on cell proliferation, and this growth
effect of BCR-ABL siRNA could be fully rescued by
transfection with BCR-ABL (Fig.
4A-C). Taken together, ND-09 appears to inhibit K562 cell
proliferation via targeting BCR-ABL.
As ND-09 targets BCR-ABL to inhibit cell
proliferation, MD was used to simulate the binding properties of
ND-09 and BCR-ABL. Notably, ND-09 fit well within BCR-ABL and
occupied the BCR-ABL ATP-binding pocket (Fig. 4E). As a consequence, ND-09 could alter
BCR-ABL kinase activity and inhibit the phosphorylation of BCR-ABL
and ABL (Fig. 4F and G). Our data
strongly support the hypothesis that ND-09 is a BCR-ABL-specific
inhibitor.
Apoptosis is a major mechanism of programmed cell
death. Findings have shown that BCR-ABL exerts anti-apoptotic
effects that play an important role in the development of CML
(20). Furthermore, the intrinsic
mitochondrial pathway is one of the two main apoptotic pathways
(21). In the current study, ND-09
was found to induce apoptosis in K562 cells, an effect that was
relatively higher in K562 cells than in JURKAT and HUT78 cells
(Fig. 2A-C). More importantly,
combined ND-09 and BCR-ABL siRNA treatment led to an
enhanced induction of apoptosis in K562 cells (Fig. 4D), indicating that ND-09 could induce
cell apoptosis by targeting BCR-ABL. ND-09 treatment significantly
reduced Δψm in K562 cells (Fig. 2D and
E). The mitochondrial apoptotic pathway is mainly regulated by
the Bcl-2 protein family, which includes the anti-apoptotic members
Bad, Bak, and Bax, and pro-apoptotic members Mcl-1 and Bcl-2.
Findings have shown that p53 could activate the expression of
proapoptotic Bcl-2 proteins, thus triggering apoptosis (12). In the present study, we found that
ND-09 could significantly upregulate p53 protein levels (Fig. 7C). Accordingly, ND-09 increased the
protein levels of Bad, Bax, and Bak, and downregulated Mcl-1 and
Bcl-2 protein levels (Fig. 2F).
Previous findings have shown that the BCR-ABL
oncoprotein plays an important role in regulating the cell cycle of
cancer cells (22). Furthermore,
silencing BCR-ABL causes CML cell cycle arrest in the G0/G1 phase
(23,24). Our data revealed that ND-09 treatment
arrested K562 cell cycle in the G0/G1 phase (Fig. 3A). As CDK/cyclin proteins play a
critical role in cell cycle regulation, we examined whether ND-09
regulated CDK/cyclin protein levels. Results revealed that ND-09
treatment resulted in a decrease in the protein levels of cyclin D1
and cyclin E in K562 cells (Fig.
3D).
As BCR-ABL is a key factor for K562 cell growth,
major downstream molecules of the BCR-ABL pathway were evaluated.
BCR-ABL activates a number of signaling pathways (JAK/STAT,
PI3K/AKT, and MAPK), promotes the proliferation of hematopoietic
stem cells, and prevents cell apoptosis (25). Inhibition of JAK/STAT and PI3K/AKT
signals can inhibit CML cell growth, indicating JAK/STAT and
PI3K/AKT are both involved in BCR-ABL-driven leukemias (26). BCR-ABL reportedly induces
Grb2-mediated MAPK activation, which regulates cell proliferation,
cell cycle progression, cell survival, differentiation, and induces
leukemogenesis (27). Our data
revealed that ND-09 could downregulate the phosphorylation of
JAK/STAT signaling members, including JAK2, JAK3, STAT3, and STAT5
(Fig. 5). In addition, ND-09
treatment led to an increase in the amount of PTEN. The main PI3K
subunit proteins (110α, 110β, p-p85/p55, and p85) and
phosphorylated AKT and mTOR were significantly decreased (Fig. 6). Furthermore, ND-09 treatment led to
the decreased phosphorylation of PLC-γ and subsequently inhibited
the phosphorylation of MEK1/2 and Erk1/2, while upregulating the
amount of p-p38 and p53 in K562 cells (Fig. 7). The aforementioned results suggest
that ND-09 regulated cell proliferation through modulation of
JAK/STAT, PI3K/AKT, and MAPK/ERK signaling.
In conclusion, our study has established ND-09 as a
selective inhibitor of BCR-ABL. The findings suggest that BCR-ABL
pathway downregulation by ND-09 drives growth arrest in CML.
Acknowledgements
Not applicable.
Funding
This study was supported by the Natural Science
Basic Research Program of Shaanxi Province (grant no. 2018JQ8019),
the Fundamental Research Funds for the Central Universities (grant
no. xzy012019078).
Availability of data and materials
The authors declare that all the data supporting the
results in this study are available within the article.
Authors' contributions
WNM was responsible for the conceptualization,
collection of material, and writing and reviewing the manuscript.
YHL was responsible for data collection, curation and analysis, as
well as writing the original draft. MZ contributed to the curation
and interpretation of data. PPL contributed to the design and data
analysis, and writing of the manuscript. XYP was responsible for
the synthesis of compound ND-09. All authors read and approved the
study.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CML
|
chronic myeloid leukemia
|
|
BCR
|
breakpoint cluster region
|
|
ABL
|
Abelson murine leukemia
|
|
MAPK
|
mitogen-activated protein kinase
|
|
JAK/STAT
|
Janus-activated kinase/signal
transducer and activator of transcription
|
|
PI3K/AKT
|
phosphatidylinositide 3-kinase/protein
kinase B
|
|
TKIs
|
tyrosine kinase inhibitors
|
|
IMDM
|
Iscove's modified Dulbecco's
medium
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2.5-diphenyl-2H-tetrazolium
bromide
|
|
FBS
|
fetal bovine serum
|
|
PI
|
propidium iodide
|
References
|
1
|
Chen Y, Wang TT, Du J, Li YC, Wang X, Zhou
Y, Yu XX, Fan WM, Zhu QJ, Tong XM and Wang Y: The critical role of
PTEN/PI3K/AKT signaling pathway in shikonin-induced apoptosis and
proliferation inhibition of chronic myeloid leukemia. Cell Physiol
Biochem. 47:981–993. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kaleem B, Shahab S, Ahmed N and Shamsi TS:
Chronic myeloid leukemia-prognostic value of mutations. Asian Pac J
Cancer Prev. 16:7415–7423. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yu CJ, Gorantla SP, Müller-Rudorf A,
Muller TA, Kreutmair S, Albers C, Jakob L, Lippert LJ, Yue ZY,
Engelhardt M, et al: Phosphorylation of BECLIN-1 by BCR-ABL
suppresses autophagy in chronic myeloid leukemia. Haematologica.
105:1285–1293. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yin XL, Zhou MR, Fu Y, Yang L, Xu M, Sun
T, Wang XM, Huang T and Chen CY: Histone demethylase RBP2 mediates
the blast crisis of chronic myeloid leukemia through an
RBP2/PTEN/BCR-ABL cascade. Cell Signal. 63:1093602019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Valencia-Serna J, Kucharski C, Chen M,
Remant KC, Jiang XY, Brandwein J and Uludag H: siRNA-mediated
BCR-ABL silencing in primary chronic myeloid leukemia cells using
lipopolymers. J Control Release. 310:141–154. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Burslem GM, Schultz AR, Bondeson DP, Eide
CA, Stevens SL, Druker BJ and Crews CM: Targeting BCR-ABL1 in
chronic myeloid leukemia by PROTAC-mediated targeted protein
degradation. Cancer Res. 79:4744–4753. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bugler J, Kinstrie R, Scott MT and Vetrie
D: Epigenetic reprogramming and emerging epigenetic therapies in
CML. Front Cell Dev Biol. 7:1362019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
De Rosa V, Monti M, Terlizzi C, Fonti R,
Del Vecchio S and Iommelli F: Coordinate modulation of glycolytic
enzymes and OXPHOS by imatinib in BCR-ABL driven chronic
myelogenous leukemia cells. Int J Mol Sci. 20:31342019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang XF, Yang JL, Guo GJ, Feng RY, Chen K,
Liao Y, Zhang LF, Sun LP, Huang SL and Chen JL: Novel lncRNA-IUR
suppresses Bcr-Abl-induced tumorigenesis through regulation of
STAT5-CD71 pathway. Mol Cancer. 18:842019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhu ZW, Yang C, Wen LL, Liu L, Zuo XB,
Zhou FS, Gao JP, Zheng XD, Shi YJ, Zhu CH, et al: Bach2 regulates
aberrant activation of B cell in systemic lupus erythematosus and
can be negatively regulated by BCR-ABL/PI3K. Exp Cell Res.
365:138–144. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kumar H, Chattopadhyay S, Das N, Shree S,
Patel D, Mohapatra J, Gurjar A, Kushwaha S, Singh AK, Dubey S, et
al: Leprosy drug clofazimine activates peroxisome
proliferator-activated receptor-γ and synergizes with imatinib to
inhibit chronic myeloid leukemia cells. Haematologica. 105:971–986.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Carter BZ, Mak PY, Mu H, Wang XM, Tao WJ,
Mak DH, Dettman EJ, Cardone M, Zernovak O, Seki T and Andreeff M:
Combined inhibition of MDM2 and BCR-ABL1 tyrosine kinase targets
chronic myeloid leukemia stem/progenitor cells in a murine model.
Haematologica. 105:1274–1284. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Miura M: Therapeutic drug monitoring of
imatinib, nilotinib, and dasatinib for patients with chronic
myeloid leukemia. Biol Pharm Bull. 38:645–654. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huguet F, Cayuela JM, Cambier N,
Carpentier N, Tindel M, Violet I, Zunic P, Lascaux A, Etienne G and
AdheRMC Investigators: Nilotinib efficacy, safety, adherence and
impact on quality of life in newly diagnosed patients with chronic
myeloid leukaemia in chronic phase: A prospective observational
study in daily clinical practice. Br J Haematol. 187:615–626. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Minson AG, Cummins K, Fox L, Costello B,
Yeung D, Cleary R, Forsyth C, Tatarczuch M, Burbury K, Motorna O,
et al: The natural history of vascular and other complications in
patients treated with nilotinib for chronic myeloid leukemia. Blood
Adv. 3:1084–1091. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pan X, Wang F, Zhang Y, Gao HP, Hu ZG,
Wang SC and Zhang J: Design, synthesis and biological activities of
Nilotinib derivates as antitumor agents. Bioorg Med Chem.
21:2527–2534. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Braun TP, Eide CA and Druker BJ: Response
and resistance to BCR-ABL1-targeted therapies. Cancer Cell.
37:530–542. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Clapper E, Wang S, Raninga PV, Di Trapani
G and Tonissen KF: Cross-talk between Bcr-abl and the thioredoxin
system in chronic myeloid leukaemia: Implications for CML
treatment. Antioxidants (Basel). 9:2072020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Maiti A, Franquiz MJ, Ravandi F, Cortes
JE, Jabbour EJ, Sasaki K, Marx K, Daver NG, Kadia TM, Konopleva MY,
et al: Venetoclax and BCR-ABL tyrosine kinase inhibitor
combinations: Outcome in patients with philadelphia
chromosome-positive advanced myeloid leukemias. Acta Haematol.
14:1–7. 2020.
|
|
20
|
Sun H, Kapuria V, Peterson LF, Fang DX,
Bornmann WG, Bartholomeusz G, Talpaz M and Donato NJ: Bcr-Abl
ubiquitination and Usp9× inhibition block kinase signaling and
promote CML cell apoptosis. Blood. 117:3151–3162. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhu M, Yang L, Shi XP, Gong ZY, Yu RZ,
Zhang DD, Zhang YM and Ma WN: TPD7 inhibits the growth of cutaneous
T cell lymphoma H9 cell through regulating IL-2R signalling
pathway. J Cell Mol Med. 24:984–995. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Steelman LS, Pohnert SC, Shelton JG,
Franklin RA, Bertrand FE and McCubrey JA: JAK/STAT, Raf/MEK/ERK,
PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis.
Leukemia. 18:189–218. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rangatia J and Bonnet D: Transient or
long-term silencing of BCR-ABL alone induces cell cycle and
proliferation arrest, apoptosis and differentiation. Leukemia.
20:68–76. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fathi E, Farahzadi R, Valipour B and
Sanaat Z: Cytokines secreted from bone marrow derived mesenchymal
stem cells promote apoptosis and change cell cycle distribution of
K562 cell line as clinical agent in cell transplantation. PLoS One.
14:e02156782019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen CW, Lee YL, Liou JP, Liu YH, Liu CW,
Chen TY and Huang HM: A novel tubulin polymerization inhibitor,
MPT0B206, downregulates Bcr-Abl expression and induces apoptosis in
imatinib-sensitive and imatinib-resistant CML cells. Apoptosis.
21:1008–1018. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Moriyama K and Hori T: BCR-ABL induces
tyrosine phosphorylation of YAP leading to expression of Survivin
and Cyclin D1 in chronic myeloid leukemia cells. Int J Hematol.
110:591–598. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ma L, Xu Z, Wang J, Zhu ZC, Lin GB, Jiang
LJ, Lu XZ and Zou C: Matrine inhibits BCR/ABL mediated ERK/MAPK
pathway in human leukemia cells. Oncotarget. 8:108880–108889. 2017.
View Article : Google Scholar : PubMed/NCBI
|