Introduction
Glioblastoma multiforme (GBM) is the most prevalent
malignant tumor in the adult central nervous system, with a median
survival time of only 12–14 months following initial diagnosis, and
a 5-year survival rate of only 5.5% (1,2). It is
characterized by an extremely poor prognosis, high recurrence rate
and short median survival time following initial diagnosis.
According to pathological histology, primary brain tumors have been
classified by the World Health Organization (WHO) into four grades
(grades I–IV), with GBM (grade IV) as the most malignant glioma
(3). To date, the main treatments of
glioma are surgical resection, radiotherapy and chemotherapy;
however, obtaining an optimal effect is challenging (4). Therefore, an improved understanding of
the underlying mechanisms of the occurrence and progression of
glioma are essential for the development of diagnostic markers and
novel effective treatments.
Angiomotin (AMOT) belongs to the motin family of
angiostatin-binding proteins (5). The
motin family, also known as AMOTs, consists of three members: AMOT,
AMOT-like 1 (AMOTL1) and AMOTL2 (5).
Among these, AMOTL1 and AMOTL2 belong to human protein sequences
and have similar structure. Members of the motin family are a type
of adaptor proteins mainly distributed in the cytomembrane,
cytoplasm or nucleus, and have a higher expression in the
endothelial cells of capillaries and in larger vessels of the
placenta (6). During the formation of
new blood vessels, angiostatin can inhibit the tube formation and
migration of endothelial cells toward growth factors, while members
of the motin family can regulate this effect to maintain normal
physiological functions (7). Although
there is a high similarity between the members of AMOTs, their
various functions remain mostly unknown (8). Functionally, AMOTs have been revealed to
directly regulate the initiation and progression of cancer through
various pathways. For example, AMOTs can interact with core protein
Yes-associated protein (YAP)1 in the Hippo signaling pathway, which
leads to its inhibition, and bind to AKT pleckstrin homology domain
to block membrane localization of AKT and negatively regulate the
AKT signaling pathway (9,10). At present, AMOTL1 has been primarily
reported as an oncogene in cervical and breast cancer, while it
could suppress tumorigenesis in GBM (6).
The Wnt signal transduction cascade is a major
regulator of development throughout the animal kingdom and a key
signaling pathway mediating development and stemness (11). In adult mammals, the Wnt/β-catenin
signaling pathway is also a key driver of most types of tissue stem
cells. Recently, with the progress of sequencing technology and the
deepening of the understanding of structural characterization of
cancer genomes, certain Wnt signaling pathway components have been
revealed to play key roles in various growth-related pathologies
and cancers (12). Wnt/β-catenin
signaling regulates tumorigenesis and progression mainly through
the transcriptional regulation of its downstream genes, including
cyclin D1 (13), Myc (14), MMP7 (15), Snail (16) and Sox9 (17), thus resulting in cancer cell
proliferation, migration, invasion and cancer stem cell property
maintenance (18,19). Therefore, delineation of the
mechanisms of Wnt/β-catenin signaling in cancer may provide novel
insights into the development of targeted cancer therapies.
Currently, there are few studies on the mechanism of
action of AMOTL2 in glioma, however, our study found abnormal
expression of AMOTL2 in glioma and its ability to combine with
β-catenin. In order to further understand the mechanism, in the
present study, a series of experiments including western blotting,
qPCR, and immunofluorescence (IF) staining were conducted to
explore the effect of AMOTL2 on glioma and the relationship between
AMOTL2 and β-catenin.
Materials and methods
Public data collection
Gene expression and survival analysis data were
obtained from the Chinese Glioma Genome Atlas (http://www.cgga.org.cn; dataset ID: mRNAseq_693)
(20,21) and The Cancer Genome Atlas (TCGA;
http://cancergenome.nih.gov/) databases.
Gene Ontology (GO) (22) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) (23) analyses were conducted by DAVID
(https://david.ncifcrf.gov/gene2gene.jsp) (version 6.8)
(24) using data downloaded from the
CGGA database. The GO and KEGG analysis of genes were drawn using
the ggplot2 package of R software (https://cran.r-project.org/web/packages/ggplot2/index.html).
Patients and specimens
The GBM specimens were collected from the Tianjin
Medical University General Hospital (Tianjin, China). The inclusion
criteria was a pathologically confirmed diagnosis of GBM. The age
range of the patients was from 30 to 60 years and the sex ratio was
1:1. Informed consent for the use of these tissues and data in the
present study was obtained from the patients, their immediate
family members or their guardians. The study was approved by the
Ethics Committee of Tianjin Medical University General Hospital
(Tianjin, China).
Immunohistochemistry (IHC)
Glioma tissues of clinical patients were fixed with
4% formaldehyde for 24 h at room temperature, paraffin-embedded at
54°C for 4 h and cut into 4-µm paraffin sections. Following heating
at 60°C for 2 h, the paraffin sections were deparaffinized by
xylene and rehydrated in graded ethanol. Following antigen
extraction with 0.1% sodium citrate buffer (pH 6.0) at 95°C, the
tissue sections were quenched of endogenous peroxidase activity
with 3% H2O2·dH2O and blocked with
5% goat serum at room temperature for 30 min. Next, the tissue
sections were incubated with antibodies against AMOTL2 (1:200; cat.
no. 36101; Signalway Antibody LLC) overnight at 4°C and then
incubated with HRP-labeled goat anti-rabbit antibody (product no.
ZB-2010; ZSGB-BIO, Inc.) at room temperature for 15 min. Following
three washes by PBS for 5 min/wash, the slides were stained by
diaminobenzidine (cat. no. DA1010; Beijing Solarbio Science &
Technology Co., Ltd.) for 3 min at room temperature and
counterstained by hematoxylin (cat. no. B3671; APeXBIO Technology
LLC) for 10 min at room temperature. Finally, graded ethanol was
used to dehydrate these tissues, which were then sealed using
neutral balsam and visualized using a fluorescence microscope (cat.
no. IX81; Olympus Corporation; magnification, ×20).
Cell lines
Human GBM cell lines U251 and glioblastoma of
unknown origin U87MG were purchased from American Type Culture
Collection. Cells were cultured at 37°C in a humidified 5%
CO2 atmosphere with Dulbecco's Modified Eagle's Medium
(DMEM; Thermo Fisher Scientific, Inc.) containing 10% FBS (Thermo
Fisher Scientific, Inc.). These cell lines underwent mycoplasma
tests and were authenticated by Short Tandem Repeat (Beijing
Microread Genetics Co., Ltd).
Lentiviral vectors with AMOTL2 short
hairpin (sh)RNA and overexpressed plasmids of AMOTL2
The lentiviral vectors with AMOTL2 and control shRNA
were purchased from Shanghai GeneChem Co., Ltd. A 2nd generation
system was used and the interim cell line used was 293T (Shanghai
GeneChem Co., Ltd.). The packaging vector: Envelope was 3:1 and the
duration of transfection was 48–72 h. The 4-µg lentivirus was
transfected into the target cells (the MOI was ~1-3) when the cell
density was 70–80%. After maintaining for 12–16 h, the medium with
lentivirus was replaced to fresh DMEM containing 10% FBS. After
infection, cells were selected using a 2 µg/ml puromycin solution
(Thermo Fisher Scientific, Inc.) and maintained using a 1 µg/ml
puromycin solution to create stable infection cell lines to conduct
follow-up experiments. Subsequent experiments were performed 72 h
after transfection.
AMOTL2 plasmid was created using Shanghai GeneChem
Co., Ltd., and the overexpressed plasmids of AMOTL2 were purchased
from Addgene, Inc. The 723 ng/µl plasmid was transfected into
target cells using Lipofectamine 3000 (Thermo Fisher Scientific,
Inc.) at room temperature for 72 h and subsequent experiments were
conducted.
Western blotting
Cell samples were lysed in RIPA lysis and extraction
buffer (cat. no. 89901; Thermo Fisher Scientific, Inc.) containing
protease inhibitor (cat. no. 36978; Thermo Fisher Scientific,
Inc.). After evaluating the concentration using a BCA protein assay
kit, protein samples were denatured by heating at 100°C and mixed
with loading buffer. The protein samples were separated on 10%
SDS-PAGE gels at 10 µg per lane and electrophoretically transferred
to a PVDF membrane (EMD Millipore). Following blocking with 5%
skimmed milk for 1 h at 37°C, incubation with primary antibodies
overnight at 4°C and incubation with secondary antibodies for 1 h
at room temperature, the membrane was exposed through G:BOX
(Syngene Europe) using immobilon western chemiluminescent HRP
substrate (cat. no. WBKLS0500; EMD Millipore) to achieve the target
proteins. The primary antibodies used in this study were as
follows: Anti-AMOTL2 rabbit monoclonal antibody (1:1,000; product
no. 43130; Cell Signaling Technology, Inc.), anti-β-catenin rabbit
monoclonal antibody (1:5,000; product code ab32572; Abcam),
anti-cyclin D1 rabbit monoclonal antibody (1:1,000; product no.
2922; Cell Signaling Technology, Inc.), anti-c-Myc rabbit
monoclonal antibody (1:1,000; product no. 18583; Cell Signaling
Technology, Inc.), anti-GAPDH mouse monoclonal antibody (1:2,000;
cat. no. 40493; Signalway Antibody LLC), anti-histone H3 Rabbit
monoclonal antibody (1:1,000; product no. 4499; Cell Signaling
Technology, Inc.). The secondary antibodies used in this study were
as follows: Goat anti-rabbit IgG H&L (HRP) (1:5,000; product
code ab6721) and goat anti-mouse IgG H&L (HRP) (1:5,000;
product code ab6789; both from Abcam).
Reverse transcription-quantitative
(RT-q)PCR assay
Total RNA from cells was extracted by TRIzol (cat.
no. 15596018; Thermo Fisher Scientific, Inc.) and
reverse-transcribed using GoTaq® Reverse Transcription
system (cat. no. A3500; Promega Corporation), according to the
manufacturer's instructions. Moreover, 2X SYBR Green qPCR Master
mix (low ROX) was purchased from Bimake, and the reaction
conditions used were according to the manufacturer's instructions
(denaturation: 95°C for 15 sec; annealing/extension: 60°C for 30–60
sec; 40 cycles). Comparative quantification was performed using the
2−ΔΔCq method with GAPDH as the endogenous control
(25). All primers were synthesized
by Beijing Tianyi Huiyuan Bioscience & Technology Inc. The
primer sequences used were as follows: GAPDH forward,
5′-GGAGCGAGATCCCTCCAAAAT-3′ and reverse,
5′-GGCTGTTGTCATACTTCTCATGG-3′; AMOTL2 forward,
5′-TGGAGAAGACCATGCGGAAC-3′ and reverse,
5′-CTTCTCTTGCTCCTGCTGCT-3′.
Colony formation assay
Cells were generated by seeding 500 cells/well in
6-well plates and subsequently incubating for 10–14 days at 37°C.
Cells were fixed with 4% paraformaldehyde for 10 min at room
temperature and stained with 0.5% crystal violet blue for 10 min at
room temperature. The cell colonies were observed and images were
captured by camera and colonies with >10 cells were counted
using a fluorescence microscope (cat. no. IX81; Olympus
Corporation; magnification, ×10).
Cell Counting Kit-8 (CCK-8) cell
viability assay
A CCK-8 assay was conducted using a CCK-8 kit (cat.
no. PA5-84814; Thermo Fisher Scientific, Inc.) at 1–5/6 days. GBM
cells were seeded at 3×103 cells/well in 96-well plates
and cultured for 24 h at 37°C. The CCK-8 mixture was configured and
added to each well, and the cells were incubated for 1 h at 37°C.
Finally, the optical density value was measured using a microplate
reader (BioTek Instruments, Inc.) at 450 nm.
Wound-healing assay
Glioma cells were seeded at 4×105
cells/well in 6-well plates and incubated for 12 h. A total of 4
vertical straight scratches were drawn in each well using a 10-µl
sterile pipette tip. Plates were washed using PBS and incubated
with serum-free medium. Images of the scratches were captured at 0,
12 and 24 h by a fluorescence microscope (cat. no. IX81; Olympus
Corporation; magnification, ×4) to evaluate the rate of wound
healing. Gap width analysis was performed with ImageJ 1.43 software
(National Institutes of Health) and the maximum confluence
percentage reached 93%.
Transwell migration and
Matrigel® invasion assays
A total of 2×104 cells were seeded in the
upper chambers of 24-well plates (pore size: 3 µm; Corning, Inc.),
incubated with 200 µl DMEM without FBS in the upper chambers, and
500 µl DMEM with 10% FBS was added at the bottom of the 24-well
plates. Following incubation for 24 h at 37°C, cells were fixed
with 4% paraformaldehyde for 10 min and stained with 0.5% crystal
violet blue for 5 min at room temperature. Images of the Transwell
were captured by a fluorescence microscope (cat. no. IX81; Olympus
Corporation; magnification, ×10). For the Matrigel®
invasion assay, 5×104 cells were added to the bottom
membrane of the upper chamber, which was coated with 100 µl
Matrigel® (1:5 dilution with DMEM without FBS) for 1 h
at 37°C, and the aforementioned steps as in the migration assay
were conducted.
Co-immunoprecipitation (Co-IP)
assay
Cells were lysed in 400 µl IP lysis buffer (cat. no.
87787; Thermo Fisher Scientific, Inc.), supplemented with complete
protease inhibitor cocktail (CAS no. 329-98-6; Beijing Solarbio
Science & Technology Co., Ltd.). The lysates were incubated
with 40 µl Protein A Agarose (cat. no. AS046; ABclonal Biotech Co.,
Ltd.) and appropriate antibodies were placed in a rotating
incubator overnight at 4°C. Then, the cells were centrifuged at
5,939 × g at 4°C for 5 min, to remove the supernatant and the
precipitation was washed three times with PBS. After boiling with
5X SDS-PAGE loading buffer (cat. no. Top2225; Biotopped Life
Sciences, Inc.) for 10 min at 100°C, western blotting was conducted
to further analyze the immunoprecipitated samples. The antibodies
used in Co-IP assay in this study were as follows: Anti-AMOTL2
rabbit monoclonal antibody (1:200; product no. 43130; Cell
Signaling Technology, Inc.), anti-β-catenin rabbit monoclonal
antibody (1:200; product code ab32572; Abcam).
Immunofluorescence (IF) staining
Glioma cells were seeded on cover slides in a
24-well plate chamber with 500 µl medium containing 10% FBS
incubated for 24 h at 37°C. Cells were then fixed with 4%
paraformaldehyde for 30 min, permeabilized by PBS with 0.1% Triton
X-100 (PBST) for 5 min, and blocked with 1% BSA in PBST at room
temperature. Immunostaining was conducted using the primary
antibodies against β-catenin (1:50; cat. no. ab32572; Abcam)
overnight at 4°C and goat anti-rabbit secondary antibodies
conjugated with Alexa Fluor® 594 (1:200; cat. no.
A11037; Thermo Fisher Scientific, Inc.) for 1 h at room
temperature. Images were captured using a confocal microscope
(×20).
β-catenin pathway inhibitor
treatment
β-Catenin pathway inhibitor (XAV-939; cat. no.
S1180) was purchased from Selleck Chemicals. The AMOTL2-knockdown
U87MG and U251 cells were treated with 20 µM of XAV939 for 24
h.
Statistical analysis
All quantitative data are expressed as the mean ±
SD. Statistical analysis was performed with GraphPad Prism 8
software (GraphPad Software, Inc.). Unpaired Student's t-test was
used to determine the statistical significance of the data between
two experimental groups, while one-way ANOVA was used to analyze
the significance of multiple group comparisons. Tukey's post hoc
test was performed after ANOVA. All statistical analyses were
two-sided, and different cut-off values, (P<0.05, P<0.01,
P<0.001), were considered to indicate statistically significant
differences. All experiments were repeated at least in
triplicate.
Results
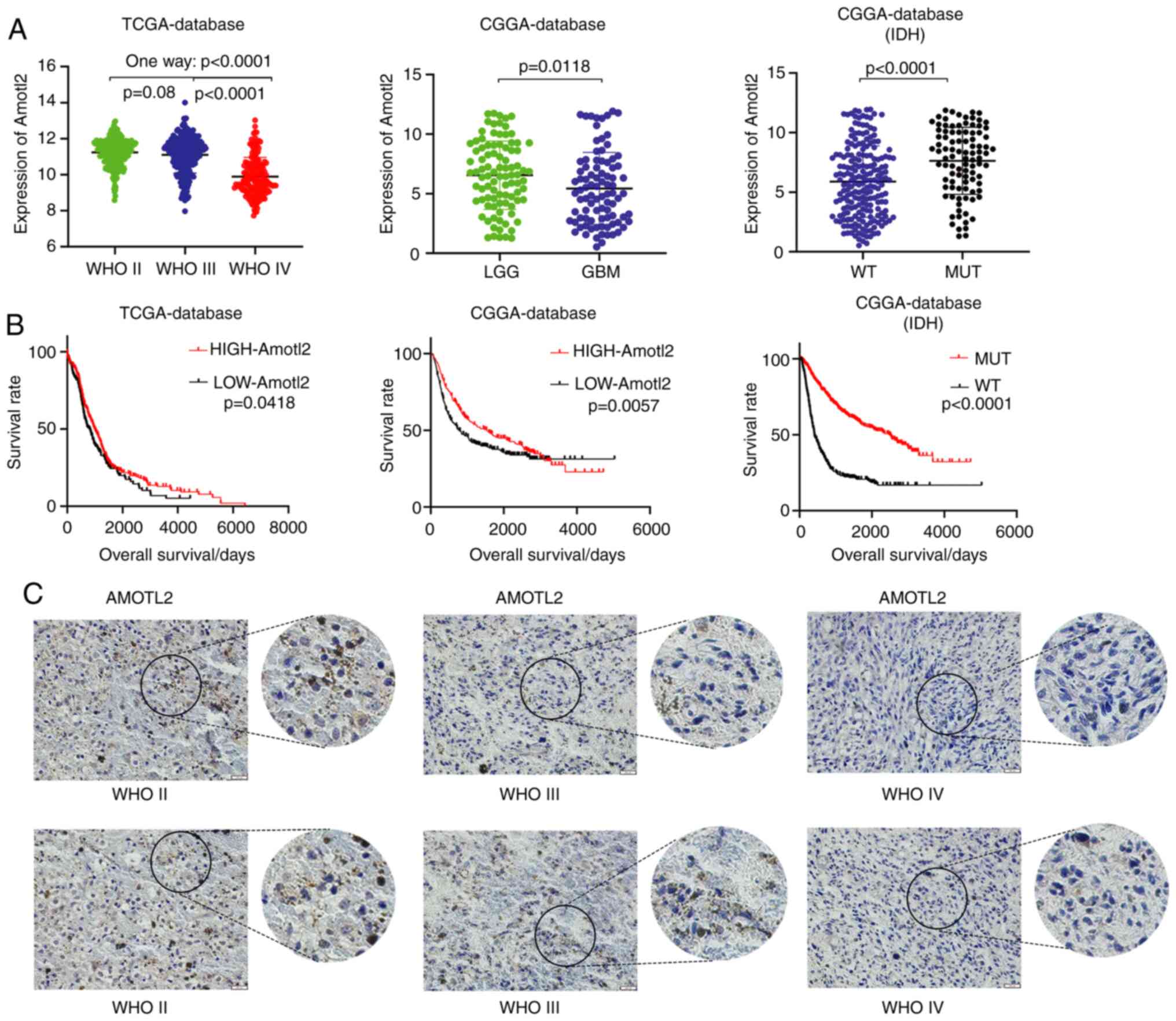
AMOTL2 expression is decreased in GBM,
as compared with low-grade glioma
To assess the differences in AMOTL2 expression
between different glioma grades, data on the expression of AMOTL2
in glioma from TCGA and CGGA databases were analyzed. Data from
TCGA database revealed that AMOTL2 expression was significantly
decreased in WHO IV grade glioma, as compared with WHO II and III
grade glioma (Fig. 1A). Similarly,
data from the CGGA database indicated that AMOTL2 expression was
significantly decreased in GBM, as compared with low-grade glioma
(LGG; Fig. 1A). Notably, the overall
survival analysis of these data indicated that glioma patients with
a high AMOTL2 expression had a high survival rate (Fig. 1B). Furthermore, data from the CGGA
database on isocitrate dehydrogenase (IDH) indicated that the
AMOTL2 expression level of the IDH mutant (MUT) was significantly
higher than that of the IDH wild-type (WT) (Fig. 1A). Consistently, the results of
survival analysis revealed that patients with IDH MUT had a longer
survival rate than patients with IDH WT (Fig. 1B). Next, IHC of clinical specimens
from patients with glioma was performed to further examine the
expression level of AMOTL2. The results indicated that the
expression of AMOTL2 was decreased as the malignancy grades
increased (Fig. 1C). In conclusion,
it was revealed that AMOTL2 was clearly decreased in GBM, and may
act as an inhibitor of glioma to prolong the survival time of
patients with glioma.
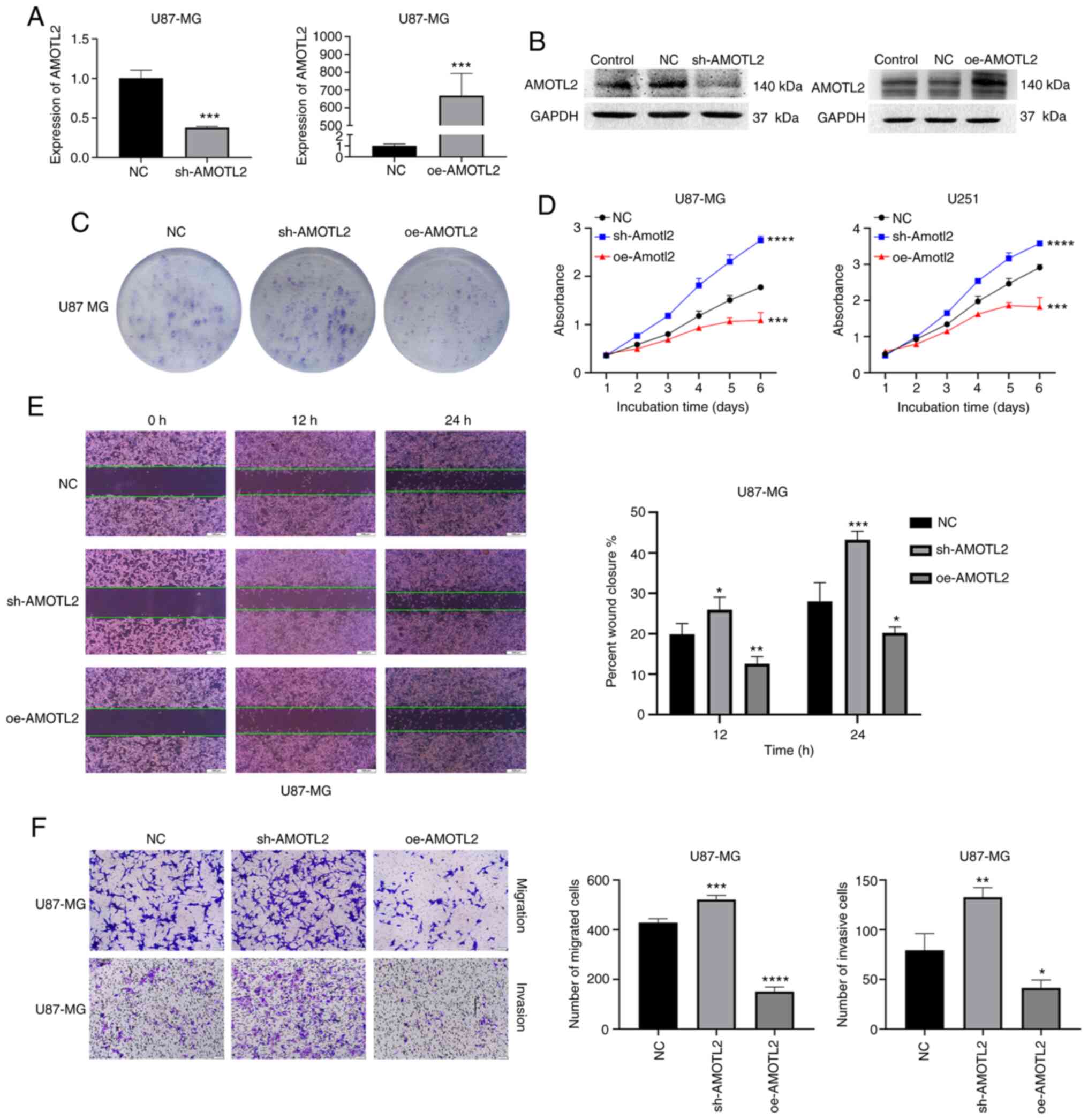
AMOTL2 inhibits the proliferation,
migration and invasion of glioma cells
To identify the effect of the differential
expression of AMOTL2 in glioma progression, the classic glioma cell
lines U87MG and U251 were selected for subsequent tests. First,
lentiviral vectors with AMOTL2 shRNA and overexpressed plasmids of
AMOTL2 were used to transfect the U87MG and U251 cells to induce
AMOTL2-knockdown or -overexpression. Western blotting and RT-qPCR
were used to examine the efficiency of the infection of shRNAs and
plasmids in U87MG and U251 cell lines (Figs. 2A and B, and S1A and B). Colony formation assays revealed
that the downregulation of AMOTL2 significantly promoted colony
formation, and AMOTL2-overexpression inhibited colony formation
both in U87MG and U251 cells (Figs.
2C and S1C). Consistently, as
revealed in Fig. 2D, the result of
CCK-8 assays also demonstrated that AMOTL2 overexpression could
inhibit the proliferation of U87MG and U251 cells.
It is well-known that the migrating and invasive
abilities of tumor cells cause tumor metastasis and poor prognosis.
In the present study, following the incubation of cells for 24 h in
6-well plates, 4 vertical straight scratches were drawn. Next, the
wound-healing rate was detected at 0, 12 and 24 h to examine the
migrating ability of cells, which was revealed to be significantly
inhibited by AMOTL2 overexpression and increased following AMOTL2
silencing (Figs. 2E and S1F). Next, a Transwell assay with or
without Matrigel® was conducted to further detect the
migrating and invasive abilities of glioma cells. The results
revealed that the knockdown of AMOTL2 clearly promoted the
migration and invasion of U87MG and U251 cells. On the contrary,
the overexpression of AMOTL2 significantly reduced the cell
migratory and invasive abilities of the two cell lines (Figs. 2F, and S1D
and E). In conclusion, AMOTL2 was revealed to inhibit
proliferation, migration and invasion in glioma cells.
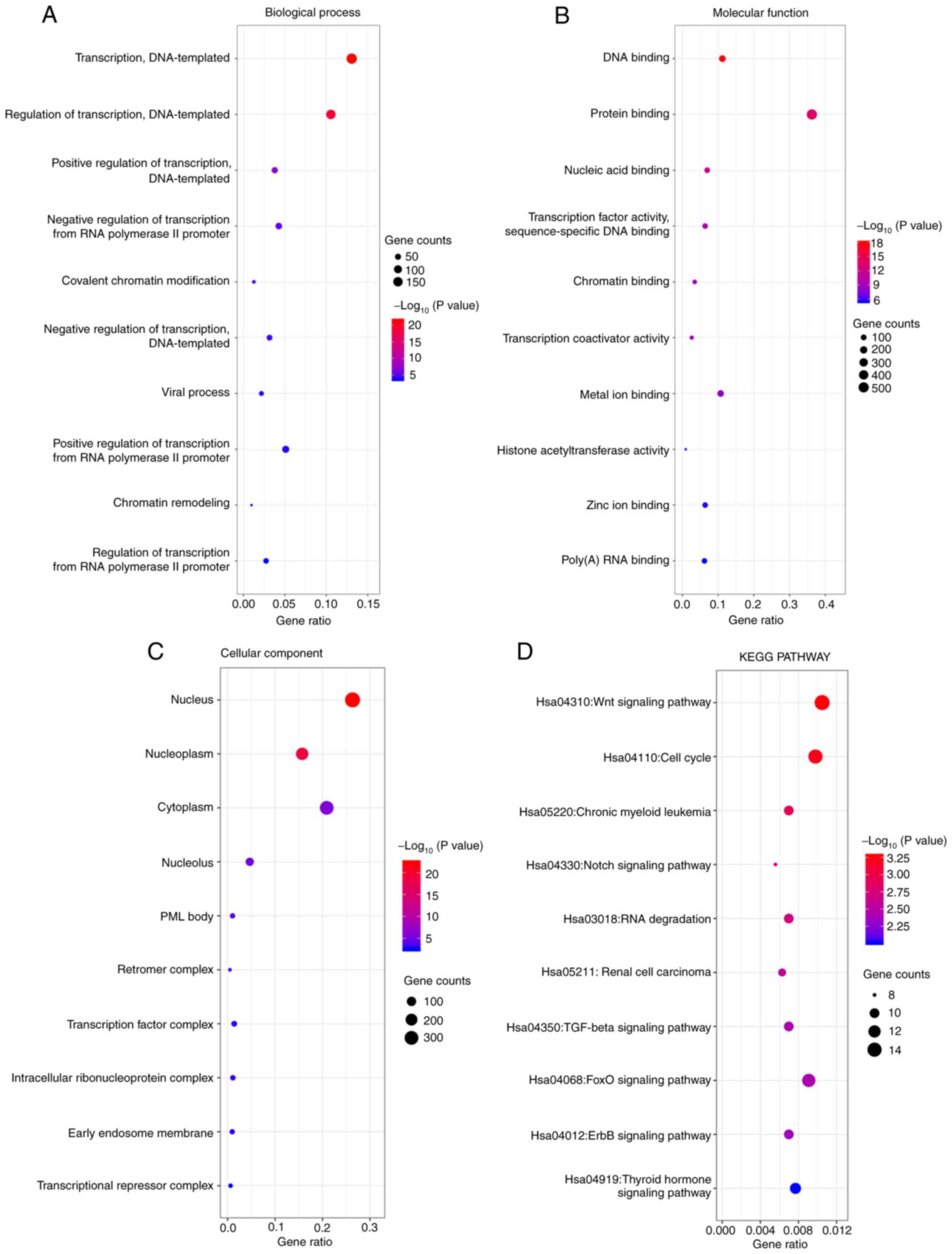
KEGG and GO enrichment analyses of
AMOTL2
To further explore the mechanism of AMOTL2,
functional and pathway enrichment analyses were performed using
data from the CGGA database by ‘ggplot2’ R language package. The GO
analysis results revealed that changes in AMOTL2 biological
processes (BP) were significantly enriched in ‘DNA-templated
transcription’ and ‘DNA-templated regulation of transcription’
(Fig. 3A). Changes in molecular
function (MF) were mainly enriched in ‘protein and DNA binding’
(Fig. 3B). The results of cellular
component (CC) analysis revealed that AMOTL2 was primarily enriched
in the ‘nucleus’, ‘cytoplasm’ and ‘nucleoplasm’ (Fig. 3C). Furthermore, the results of KEGG
pathway analysis revealed that the changes of AMOTL2 were mainly
enriched in the ‘Wnt’, ‘cell cycle’ and ‘FoxO signaling pathways’
(Fig. 3D).
Downregulation of AMOTL2 promotes
β-catenin nuclear localization and activates the Wnt/β-catenin
signaling pathway
Based on the results of GO and KEGG analysis,
whether AMOTL2 regulates the Wnt/β-catenin signaling pathway via
protein binding was next examined. Co-IP assay between AMOTL2 and
β-catenin was performed, and the results revealed that AMOTL2 and
β-catenin proteins could mutually combine (Fig. 4A). It was consistent with our
hypothesis that AMOTL2 probably plays its role in tumor suppression
by directly binding to β-catenin. Subsequently, western blotting
demonstrated that AMOTL2-knockdown could increase the expression of
certain downstream genes of the Wnt/β-catenin signaling pathway,
such as c-Myc and cyclin D1, to activate Wnt signaling; however, no
change was observed in the expression of β-catenin (Fig. 4B). This indicated that the function of
AMOTL2 in the regulation of Wnt pathway genes activity occurs other
than via direct action on the β-catenin destruction complex. It has
been reported that, in addition to the β-catenin destruction
complex, the activation of the Wnt pathway was also regulated by
the distribution of β-catenin in the nucleus and cytoplasm
(26). Therefore, to further identify
the mechanism of AMOTL2 in the Wnt signaling pathway, the nuclear
and cytoplasmic localization of β-catenin in control and
AMOTL2-silenced cells were next examined.
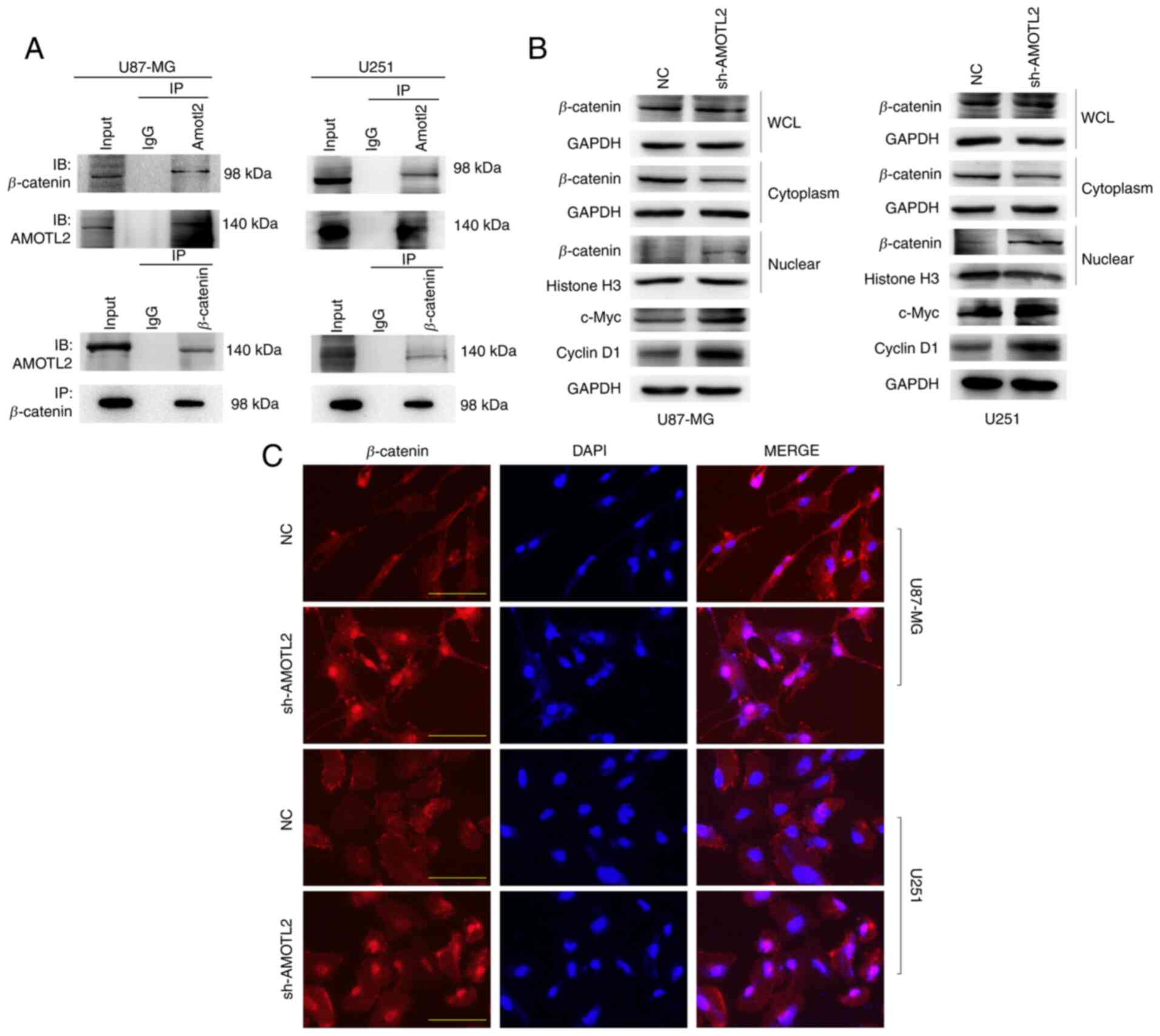 | Figure 4.AMOTL2-knockdown activates the Wnt
signaling pathway by promoting β-catenin nuclear translocation. (A)
Extracts of U87MG and U251 cells were lysed in IP lysis buffer,
incubated with AMOTL2 antibody or control IgG, immunoblotted with
β-catenin antibody and AMOTL2 antibody (upper images). Reciprocal
IP was performed using β-catenin antibody or control IgG, and
immunoblotted with AMOTL2 antibody and β-catenin antibody (lower
images). (B) Western blotting examined the expression of β-catenin
in the whole cell lysate, cytoplasm and nucleus, as well as that of
c-Myc and Cyclin D1. (C) Double immunofluorescence staining for
β-catenin (red) and nuclear DAPI (blue) was conducted in sh-AMOTL2
or NC U87MG and U251 cells (scale bar, 50 µm). AMOTL2,
angiomotin-like 2; IP, immunoprecipitation; NC, negative control;
sh-, short hairpin; WCL, whole cell lysate. |
First, western blotting of nuclear and cytoplasmic
proteins in AMOTL2-silenced and control cells was performed. AMOTL2
was revealed to significantly increase the nuclear distribution and
decrease the cytoplasmic distribution of β-catenin proteins
(Fig. 4B). Next, immunofluorescence
assays were performed to further illustrate the effect of AMOTL2 on
β-catenin protein localization. AMOTL2 downregulation was revealed
to clearly promote the nuclear transfer of β-catenin protein, both
in U87MG and U251 cells (Fig.
4C).
In conclusion, these results indicated that AMOTL2
could prevent β-catenin from translocating to the nucleus by
directly binding to the β-catenin protein.
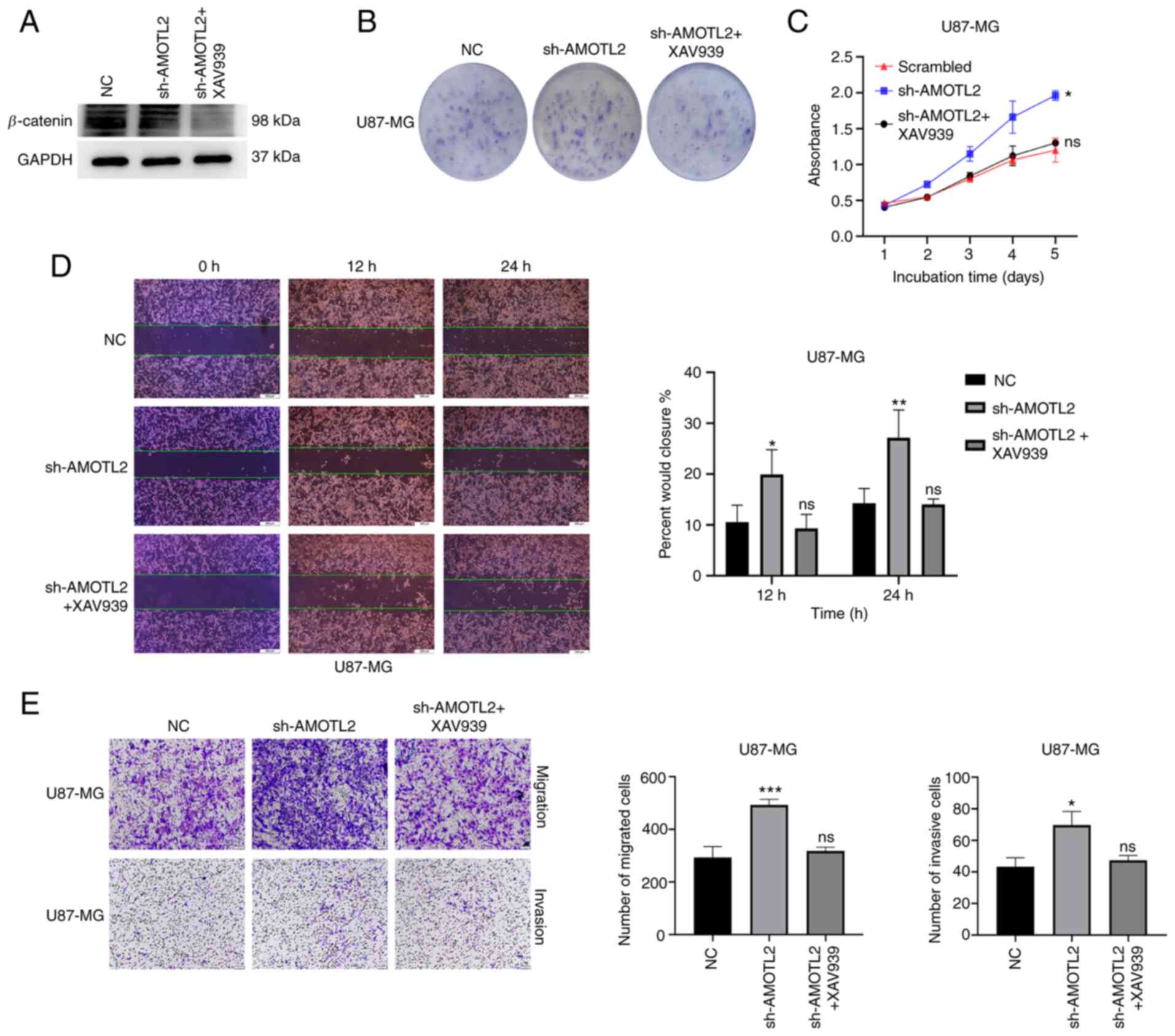
Wnt/β-catenin pathway inhibitor can
reverse the AMOTL2-knockdown-induced proliferation, migration and
invasion of glioma
It is unclear whether the regulatory function of
AMOTL2 over proliferation, migration and invasion of glioma is
realized through the Wnt/β-catenin signaling pathway. Therefore,
the AMOTL2-knockdown U87MG and U251 cells were treated with
Wnt/β-catenin pathway inhibitor XAV939 with 20 µM for 24 h. The
efficiency of the Wnt/β-catenin pathway inhibition was identified
by western blotting, and the results showed that the treatment
group exhibited a clearly lower β-catenin protein expression, as
compared with the control (Figs. 5A
and S2A). Colony formation assays
revealed that the AMOTL2-silencing-induced increased colony
formation ability decreased following XAV939 treatment (Figs. 5B and S2B). Notably, it was revealed by CCK-8
assay that XAV939 reduced the proliferation of AMOTL2-knockdown
cells (Figs. 5C and S2C). These results indicated that the Wnt
pathway inhibitor XAV939 could reverse the AMOTL2-silencing-induced
increase in the proliferative ability of glioma.
Subsequently, rescue assays of migration and
invasion were conducted. It was shown by the wound healing assay
results that XAV939 could suppress the increased wound-healing rate
of AMOTL2-silenced cells (Figs. 5D
and S2D). The Transwell assays with
or without Matrigel® indicated that the
AMOTL2-downregulation-induced increase of migration and invasion of
glioma were significantly reduced following XAV939 treatment
(Figs. 5E and S2E). These results indicated that Wnt
pathway inhibitor XAV939 could reverse the AMOTL2-silencing-induced
increase in the migratory and invasive abilities of glioma.
In conclusion, these results revealed that the
Wnt/β-catenin signaling pathway inhibitor XAV939 could reverse the
AMOTL2-knockdown-induced proliferation, migration and invasion of
glioma. Collectively, AMOTL2 could regulate the proliferation,
migration and invasion of glioma through the Wnt/β-catenin
signaling pathway.
Discussion
AMOTL2 is a member of the motin family of
angiostatin-binding proteins. During the formation of new blood
vessels, angiostatin can inhibit the tube formation and migration
of endothelial cells toward growth factors, while AMOTL2 can
regulate this effect to maintain normal physiological functions
(7). Previous studies have reported
that AMOTL2 is closely associated with the occurrence and
progression of cancer. However, these studies mainly focused on
breast, lung, pancreatic, liver and cervical cancer (10,27–29).
Research on the relationship between AMOTL2 and glioma is limited.
Similarly, research that focused on the mechanism of AMOTL2 in
cancer, on the regulation of YAP, AKT protein and Hippo signaling
pathways was also limited (10,30,31). In
the present study, a novel mechanism through which AMOTL2 regulates
the development of glioma was reported.
Expression and survival analyses of glioma were
performed using data from TGGA and CGGA databases. The results
indicated that the expression of AMOTL2 was reduced in GBM, as
compared with low-grade glioma, and the patients with a high AMOTL2
expression had a longer survival. IHC of clinical glioma specimens
yielded the same result. The data from the phenotype experiment
demonstrated that AMOTL2 inhibited the tumor proliferation,
migration and invasion of glioma. To further explore the mechanism
of AMOTL2 in glioma, GO and KEGG analyses were conducted using data
from the CGGA. The results revealed that AMOTL2 performed its
function by ‘protein binding’, and the pathway AMOTL2 was
significantly enriched in was the Wnt/β-catenin signaling
pathway.
The Wnt/β-catenin signaling pathway is one of the
classic pathways that can affect several physiological processes,
such as cell proliferation, differentiation, organogenesis, tissue
regeneration and tumorigenesis (12,32–35).
Several studies have revealed that the aberrant regulation of the
Wnt/β-catenin signaling pathway occurs frequently in glioma
(36,37). Therefore, several studies were
performed to examine the potential role of the Wnt signaling
pathway as a novel therapeutic target for glioma. In this context,
it appears using aberrant Wnt/β-catenin signaling as a therapeutic
target is a lucrative approach to combat cancer (38). Treating cancer through the suppression
of the Wnt signaling is an emerging therapeutic method, and several
inhibitors of the Wnt pathway, such as PORCN inhibitors, Wnt ligand
antagonists and FZD antagonists/monoclonal antibodies (39–41) have
been used in various cancer-related clinical trials. However, due
to the positive regulatory function of Wnt/β-catenin signaling over
biological processes, including the maintenance of stem cell pool
stability and regeneration of tissues and organs (42), the interdiction of Wnt signaling may
cause irreversible damage to physiological homeostasis and
regenerative ability (43).
Therefore, to further use these molecules in the clinical setting,
an understanding of their potential mechanism needs to be obtained
through additional preclinical and clinical work.
To demonstrate whether AMOTL2 can directly interact
with β-catenin to regulate the Wnt pathway, Co-IP assay and western
blotting were performed. It was revealed that AMOTL2 could bind to
the β-catenin protein and the knockdown of AMOTL2 could activate
downstream genes of the Wnt pathway, such as c-Myc and cyclin D1.
Nevertheless, the expression level of β-catenin protein was not
altered when AMOTL2 was silenced.
It was reported that, in the canonical Wnt pathway,
when the Wnt signal was in an inactivated state, the β-catenin
destruction complex, which contains the scaffold protein Axin, APC
and the kinases GSK3β and casein kinase, would be activated,
leading to β-catenin protein phosphorylatation by the destruction
complex, and degradation by proteasomes (12). Conversely, when Wnt signaling is
activated, β-catenin proteins tend to be stable and are constantly
accumulated, finally translocating into the nucleus. In the
nucleus, β-catenin forms an active complex with T-cell factor and
lymphoid enhancer factor proteins by displacing the TLE/Groucho
complexes and recruiting histone modifying co-activators, such as
CBP/p300, BRG1, BCL9 and Pygo (44).
This transcriptional switch leads to changes in multiple cellular
processes (45,46).
Therefore, it was hypothesized that the role of
AMOTL2 in regulating Wnt pathway genes was not directly played
through the β-catenin destruction complex, but rather by
controlling its nuclear translocation. To verify this hypothesis,
nuclear and cytoplasmic proteins of the control and knockdown
groups, respectively, were extracted to conduct western blotting.
Next, immunofluorescence staining was performed to locate the
distribution of β-catenin in cells. The results revealed that the
downregulation of AMOTL2 promoted the nuclear translocation of
β-catenin. These results indicated that AMOTL2 could keep β-catenin
from translocating into the nucleus by directly binding to
β-catenin protein.
To further demonstrate that AMOTL2 regulates the
proliferative, migrating and invasive abilities of glioma cells
through the Wnt/β-catenin signaling pathway, the Wnt/β-catenin
pathway inhibitor XAV939 was used to perform the phenotype rescue
assays. The results indicated that the Wnt/β-catenin pathway
inhibitor XAV939 could reverse the AMOTL2-silencing-induced
proliferation, migration and invasion of glioma. It was reported
that AMOTL2 could regulate the development of glioma cells through
the Wnt/β-catenin pathway.
In conclusion, the present study revealed that
AMOTL2 could regulate the proliferation, migration and invasion of
glioma cells by directly binding to the β-catenin protein to
control the nuclear translocation of β-catenin. Therefore, AMOTL2
may serve as a novel therapeutic target in glioma treatment.
Furthermore, based on the association between AMOTL2 and β-catenin,
AMOTL2 may contribute to the study of targeting therapeutics of Wnt
signaling. In view of the several side effects of Wnt signaling
inhibitors, further research should be performed on the interaction
between AMOTL2 and β-catenin. In addition, there are some
limitations in the clinical transformation of this study. The
present study only demonstrated the effect of AMOLT2 on glioma
through the Wnt/β-catenin pathway at the cellular level, but lacks
verification at the animal level. Moreover, the application of
AMOTL2 as an inhibitor of the Wnt pathway is only at a preliminary
stage, and more basic and clinical validation is required.
Therefore, additional studies are necessary to understand the
potential mechanism and clinical value of AMOTL2.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural
Science Foundation of China (grant nos. 81172405 and 81572490), the
Tianjin Science and Technology Committee (grant no. 18JCZDJC98600)
and the Science and Technology fund of Tianjin Binhai New Area
Health and Family Planning Commission (grant no. 2018BWKZ003).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
XC and YL performed the experiments and compiled the
manuscript. GG contributed to the design of the study and analyzed
data. YZ and YS conducted experiments and analyzed data. LG, RL and
YN acquired and interpreted the data. XY, JD and XJ collected
clinical specimens and analyzed the data. QH contributed to the
conception and design of the present study and revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All clinical specimen tissues were reviewed and
approved by the Ethics Committee of Tianjin Medical University
General Hospital (Tianjin, China), and informed consents were
signed by the patients according to the Declaration of Helsinki.
(approval no. IRB2021-WZ-006).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Anton K, Baehring JM and Mayer T:
Glioblastoma multiforme: Overview of current treatment and future
perspectives. Hematol Oncol Clin North Am. 26:825–853. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ostrom QT, Gittleman H, Liao P,
Vecchione-Koval T, Wolinsky Y, Kruchko C and Barnholtz-Sloan JS:
CBTRUS Statistical Report: Primary brain and other central nervous
system tumors diagnosed in the United States in 2010–2014. Neuro
Oncol. 19 (Suppl 5):v1–v88. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 World Health Organization
classification of tumors of the central nervous system: A summary.
Acta Neuropathol. 131:803–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Omuro A and DeAngelis LM: Glioblastoma and
other malignant gliomas: A clinical review. JAMA. 310:1842–1850.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lv M, Shen Y and Yang J, Li S, Wang B,
Chen Z, Li P, Liu P and Yang J: Angiomotin family members:
Oncogenes or tumor suppressors? Int J Biol Sci. 13:772–781. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang T, Zhou Y, Zhang J, Cheng ASL, Yu J,
To KF and Kang W: The physiological role of Motin family and its
dysregulation in tumorigenesis. J Transl Med. 16:982018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lv M, Li S, Luo C, Zhang X, Shen Y, Sui
YX, Wang F, Wang X and Yang J, Liu P and Yang J: Angiomotin
promotes renal epithelial and carcinoma cell proliferation by
retaining the nuclear YAP. Oncotarget. 7:12393–12403. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Moreau J, Lord M, Boucher M, Belleau P and
Fernandes MJG: Protein diversity is generated within the motin
family of proteins by alternative pre-mRNA splicing. Gene.
350:137–148. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim M, Kim M, Park S-J, Lee C and Lim DS:
Role of angiomotin-like 2 mono-ubiquitination on YAP inhibition.
EMBO Rep. 17:64–78. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Han H, Yang B and Wang W: Angiomotin-like
2 interacts with and negatively regulates AKT. Oncogene.
36:4662–4669. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nusse R and Clevers H: Wnt/β-catenin
signaling, disease, and emerging therapeutic modalities. Cell.
169:985–999. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhan T, Rindtorff N and Boutros M: Wnt
signaling in cancer. Oncogene. 36:1461–1473. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vallée A and Lecarpentier Y: Crosstalk
between peroxisome proliferator-activated receptor gamma and the
canonical WNT/β-catenin pathway in chronic inflammation and
oxidative stress during carcinogenesis. Front Immunol. 9:7452018.
View Article : Google Scholar
|
|
14
|
He TC, Sparks AB, Rago C, Hermeking H,
Zawel L, da Costa LT, Morin PJ, Vogelstein B and Kinzler KW:
Identification of c-MYC as a target of the APC pathway. Science.
281:1509–1512. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brabletz T, Jung A, Dag S, Hlubek F and
Kirchner T: Beta-catenin regulates the expression of the matrix
metalloproteinase-7 in human colorectal cancer. Am J Pathol.
155:1033–1038. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
ten Berge D, Koole W, Fuerer C, Fish M,
Eroglu E and Nusse R: Wnt signaling mediates self-organization and
axis formation in embryoid bodies. Cell Stem Cell. 3:508–518. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Blache P, van de Wetering M, Duluc I,
Domon C, Berta P, Freund JN, Clevers H and Jay P: SOX9 is an
intestine crypt transcription factor, is regulated by the Wnt
pathway, and represses the CDX2 and MUC2 genes. J Cell Biol.
166:37–47. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Clevers H and Nusse R: Wnt/β-catenin
signaling and disease. Cell. 149:1192–1205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Espada J, Calvo MB, Díaz-Prado S and
Medina V: Wnt signalling and cancer stem cells. Clin Transl Oncol.
11:411–427. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Y, Qian T, You G, Peng X, Chen C, You
Y, Yao K, Wu C, Ma J, Sha Z, et al: Localizing seizure-susceptible
brain regions associated with low-grade gliomas using voxel-based
lesion-symptom mapping. Neuro Oncol. 17:282–288. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu X, Li Y, Qian Z, Sun Z, Xu K, Wang K,
Liu S, Fan X, Li S, Zhang Z, et al: A radiomic signature as a
non-invasive predictor of progression-free survival in patients
with lower-grade gliomas. Neuroimage Clin. 20:1070–1077. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The Gene
Ontology Consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kanehisa M: The KEGG database. Novartis
Found Symp. 247:91–103, 119-128, 244–252. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang DW, Sherman BT, Tan Q, Collins JR,
Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC and Lempicki
RA: The DAVID Gene functional classification tool: A novel
biological module-centric algorithm to functionally analyze large
gene lists. Genome Biol. 8:R1832007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Krieghoff E, Behrens J and Mayr B:
Nucleo-cytoplasmic distribution of beta-catenin is regulated by
retention. J Cell Sci. 119:1453–1463. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jiang WG, Watkins G, Douglas-Jones A,
Holmgren L and Mansel RE: Angiomotin and angiomotin like proteins,
their expression and correlation with angiogenesis and clinical
outcome in human breast cancer. BMC Cancer. 6:162006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cui R, Jiang N, Zhang M, Du S, Ou H, Ge R,
Ma D and Zhang J: AMOTL2 inhibits JUN Thr239 dephosphorylation by
binding PPP2R2A to suppress the proliferation in non-small cell
lung cancer cells. Biochim Biophys Acta Mol Cell Res.
1868:1188582020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guo Z, Wang X, Yang Y, Chen W, Zhang K,
Teng B, Huang C, Zhao Q and Qiu Z: Hypoxic tumor-derived exosomal
long noncoding RNA UCA1 promotes angiogenesis via miR-96-5p/AMOTL2
in pancreatic cancer. Mol Ther Nucleic Acids. 22:179–195. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hong W: Angiomotin'g YAP into the nucleus
for cell proliferation and cancer development. Sci Signal.
6:pe272013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhao B, Li L, Lu Q, Wang LH, Liu CY, Lei Q
and Guan KL: Angiomotin is a novel Hippo pathway component that
inhibits YAP oncoprotein. Genes Dev. 25:51–63. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Acebron SP, Karaulanov E, Berger BS, Huang
YL and Niehrs C: Mitotic Wnt signaling promotes protein
stabilization and regulates cell size. Mol Cell. 54:663–674. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Atlasi Y, Noori R, Gaspar C, Franken P,
Sacchetti A, Rafati H, Mahmoudi T, Decraene C, Calin GA, Merrill BJ
and Fodde R: Wnt signaling regulates the lineage differentiation
potential of mouse embryonic stem cells through Tcf3
down-regulation. PLoS Genet. 9:e10034242013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Clevers H, Loh KM and Nusse R: Stem cell
signaling. An integral program for tissue renewal and regeneration:
Wnt signaling and stem cell control. Science. 346:12480122014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Green JL, Inoue T and Sternberg PW:
Opposing Wnt pathways orient cell polarity during organogenesis.
Cell. 134:646–656. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kristensen BW, Priesterbach-Ackley LP,
Petersen JK and Wesseling P: Molecular pathology of tumors of the
central nervous system. Ann Oncol. 30:1265–1278. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
He L, Zhou H, Zeng Z, Yao H, Jiang W and
Qu H: Wnt/β-catenin signaling cascade: A promising target for
glioma therapy. J Cell Physiol. 234:2217–2228. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jung YS and Park JI: Wnt signaling in
cancer: Therapeutic targeting of Wnt signaling beyond β-catenin and
the destruction complex. Exp Mol Med. 52:183–191. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Madan B, Ke Z, Harmston N, Ho SY, Frois
AO, Alam J, Jeyaraj DA, Pendharkar V, Ghosh K, Virshup IH, et al:
Wnt addiction of genetically defined cancers reversed by PORCN
inhibition. Oncogene. 35:2197–2207. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Giraudet AL, Cassier PA, Iwao-Fukukawa C,
Garin G, Badel JN, Kryza D, Chabaud S, Gilles-Afchain L, Clapisson
G, Desuzinges C, et al: A first-in-human study investigating
biodistribution, safety and recommended dose of a new radiolabeled
MAb targeting FZD10 in metastatic synovial sarcoma patients. BMC
Cancer. 18:6462018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jimeno A, Gordon M, Chugh R, Messersmith
W, Mendelson D, Dupont J, Stagg R, Kapoun AM, Xu L, Uttamsingh S,
et al: A first-in-human phase I study of the anticancer stem cell
agent ipafricept (OMP-54F28), a decoy receptor for Wnt ligands, in
patients with advanced solid tumors. Clin Cancer Res. 23:7490–7497.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Patel S, Alam A, Pant R and Chattopadhyay
S: Wnt signaling and its significance within the tumor
microenvironment: Novel therapeutic insights. Front Immunol.
10:28722019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Galluzzi L, Spranger S, Fuchs E and
López-Soto A: WNT signaling in cancer immunosurveillance. Trends
Cell Biol. 29:44–65. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lien WH and Fuchs E: Wnt some lose some:
Transcriptional governance of stem cells by Wnt/β-catenin
signaling. Genes Dev. 28:1517–1532. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Clevers H: Wnt/beta-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
MacDonald BT, Tamai K and He X:
Wnt/beta-catenin signaling: Components, mechanisms, and diseases.
Dev Cell. 17:9–26. 2009. View Article : Google Scholar : PubMed/NCBI
|