Introduction
Malignant melanoma (melanoma) is the most aggressive
skin cancer which arises from pigment-producing cells, melanocytes,
or from dysplastic nevus cells. Despite various options basically
involving enlarged surgical excision with or without anticancer
drugs (chemotherapy) and various immune checkpoint inhibitors
(1), there is a high demand for the
development of new, more effective melanoma-targeted therapies.
The tumor microenvironment (TME) consists of tumor
cells and tumor stroma including cancer-associated fibroblasts
(CAFs), immune cells, and tumor endothelial cells (TECs). Cancer
cells, CAFs, and TECs secrete transforming growth factor-β (TGF-β),
a multifunctional cytokine, creating a favorable microenvironment
that promotes tumor progression (2).
There are three TGF-β isoforms that share structural similarity,
TGF-β1, TGF-β2, and TGF-β3, and signal through the same type I
(TβRI) and type II (TβRII) transmembrane receptors that are endowed
with serine/threonine kinase activity (3). The formation of a complex between TGF-β
and TβRII followed by binding of TβRI that results in activation of
TβRI and phosphorylation of downstream components Smad2/3, which is
followed by binding of Smad4 and translocation of the whole complex
to the nucleus and transcription of direct target genes,
plasminogen activator inhibitor-1 (PAI-1) (4) or transmembrane prostate androgen-induced
protein (TMEPAI) (5).
TGF-β signaling has been implicated in the
progression of various tumors by enhancing cell migration,
promoting the invasion of cancer cells, and suppressing immune
responses (6,7). The ability of TGF-β to induce
epithelial-mesenchymal transition (EMT), which endows cancer cells
of epithelial origin with the mesenchymal features, leading to loss
of cell-cell contact and enhanced motility has also been widely
studied (8). In the cells undergoing
EMT, decreased expression of epithelial cell markers, E-cadherin or
claudin-1 accompanied by increased expression of mesenchymal cell
markers, smooth muscle protein 22α (SM22α), α-smooth muscle actin
(αSMA), and fibronectin is observed (9). Although melanoma does not represent
epithelial tumors, a decrease in E-cadherin expression and
upregulated expression of EMT-related transcription factors, Snail,
Slug, Twist, and Zeb1 have been revealed to be correlated with the
enhanced invasion and acquisition of stem cell-like properties
(10,11).
Elevated expression levels of TGF-β have been
revealed to be associated with melanoma progression (12). Especially TGF-β2 can be detected in
early as well as in advanced melanomas. A positive association
between TGF-β2 expression level and tumor thickness (13) and high plasma levels of TGF-β2 have
been observed in melanomas at advanced stages. TGF-β signals have
been reported to stimulate melanoma cell dissemination from primary
tumors (14). Previous findings
indicated that activation of TGF-β signals in murine melanoma led
to the upregulation of PAI-1 expression that resulted in tumor
growth inhibition (15). Conversely,
other studies demonstrated that activation of TGF-β signals
facilitated progression of malignant melanoma (13,16) by
stimulating cell proliferation in vivo and metastasis
(17). In addition, TGF-β has also
been revealed to affect stromal, immune, or endothelial cells and
by this means exerts immunosuppressive and pro-angiogenic
activities (18).
Current attempts in the development of
melanoma-targeted therapies are based on the inhibition of proteins
involved in the mitogen-activated protein kinase (MAPK) signal
transduction pathway, B-Raf (BRAF) and MAPK kinase (1). In addition, the important role of
interleukin-13 signals in melanoma progression has also been
suggested (19). However, the
previously approved drugs vemurafenib, dabrafenib, and trametinib
which target the MAPK signaling pathway (20,21), may
potentiate the risk of developing other skin cancers or formation
of peripheral edema (22). As TGF-β
has been associated with progression of melanomas, there have also
been attempts to target TGF-β signals by using antisense
oligonucleotides (trabedersen; API12009) (23), monoclonal anti-TGF-β antibodies
(fresolimumab; GC1008) (24) or a low
molecular weight compound (vactosertib; TEW-7197) that inhibits
TβRI kinase activity (25). The
efficacy of these approaches is still under evaluation; however, at
present, various side effects such as cutaneous keratoacanthomas or
cardiac toxicity have been observed (26).
Recently there have been attempts to develop
chimeric proteins comprising the ligand-interacting ectodomains of
TGF-β receptors fused with Fc domain of human immunoglobulin (IgG).
The addition of the Fc region of IgG is known to prolong plasma
half-life of such chimeric Fc receptors (27–29) and to
affect their biological activities. As reviewed by Marotte and
Cimaz, etanercept, a human tumor necrosis factor (TNF) receptor p75
fused with the Fc domain of human IgG has been revealed to have a
prolonged half-life, resulting in an extended and more profound
biological effect than its native form (30). A TβRI-TβRII-Fc chimeric receptor
comprising extracellular domains of TGF-β type I and II receptors
fused with the Fc portion of human IgG, which could effectively
trap all TGF-β isoforms and inhibited EMT in oral squamous cell
carcinoma cells as well as primary tumor growth, was previously
designed by our research group (31).
As all TGF-β isoforms facilitate melanoma progression, it was
hypothesized that TβRI-TβRII-Fc chimeric receptor could potentially
interfere with tumor-inducing TGF-β signals in melanomas. Thus, in
the present study, using a melanoma syngeneic model and B16-F0 cell
line, the effect of soluble TβRI-TβRII-Fc chimeric receptors on the
induction of EMT and progression of melanoma were examined.
Materials and methods
Cell culture
The B16-F0 mouse melanoma cell line (cat. no.
JCRB0202) was purchased from Japanese Collection of Research
Bioresources (JCRB) Cell Bank and maintained in Eagle's Minimum
Essential Medium (EMEM; FUJIFILM Wako Pure Chemical Corporation)
supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich; Merck
KGaA), 100 U/ml penicillin and 100 µg/ml streptomycin (both from
Nacalai Tesque, Inc.) under mycoplasma-free conditions. Clone M3
(Cloudman S91) melanoma cell line was obtained from European
Collection of Authenticated Cell Cultures (ECACC) and cultured in
Ham's F10 medium (FUJIFILM Wako Pure Chemical Corporation)
supplemented with 15% FBS, 2 mM glutamine (Nacalai Tesque, Inc.),
100 U/ml penicillin and 100 µg/ml streptomycin. 293T and 293FT
cells were obtained from Invitrogen; Thermo Fisher Scientific, Inc.
HEK-Blue™ TGF-β cells were purchased from InvivoGen. 293T, 293FT
and HEK-Blue™ TGF-β cells were cultured in Dulbecco's modified
Eagle's medium (DMEM; Nacalai Tesque, Inc.) supplemented with 10%
FBS, 100 U/ml penicillin and 100 µg/ml streptomycin. The cultured
medium for 293FT cells was also supplemented with 1% non-essential
amino acid solution (Nacalai Tesque, Inc.). All cell lines were
cultured in a humidified incubator containing 5% CO2 at
37°C.
Reagents
TGF-β1 (PeproTech, Inc.), TGF-β2 (PeproTech, Inc.)
and TGF-β3 (R&D Systems, Inc.) were used at concentrations of 1
ng/ml or 3 ng/ml depending on the experiment. SB431542 (FUJIFILM
Wako Pure Chemical Corporation) was used at a concentration of 10
µM.
PrognoScan analysis
The correlation between the levels of expression of
genes encoding all human TGF-β isoforms, TGFB1, TGFB2 or TGFB3 and
overall survival of melanoma patients was performed using a public
database, PrognoScan (http://dna00.bio.kyutech.ac.jp/PrognoScan/) which
comprises multiple cancer microarray datasets (32). TGFB1, TGFB2 and TGFB3 were used as
queries. The analysis was conducted by minimum P-value approach,
which allowed grouping of patients into two groups based on the
expression levels of TGFB1, TGFB2 and TGFB3 at all possible cutoffs
(cutoff points providing the best minimum corrected P-value were
0.76 for TGFB1, 0.87 for TGFB2 and 0.66 for TGFB3 analyses,
respectively). The analysis results in the present study were based
on the evaluation of TGFB1, TGFB2 and TGFB3 expression levels and
survival of melanoma patients whose data was combined in dataset:
GSE19234 (33). The log-rank test was
used for statistical analysis.
Cell proliferation assay
The effect of expression of Fc chimeric receptors on
proliferation of B16 melanoma cells was evaluated by WST-1 assay.
The B16 cells (3.5×104) were seeded into a 12-well
culture plate and incubated overnight at 37°C, 5% CO2.
The following day the medium was refreshed, and the cells were
cultured for 72 h. The assay was conducted using the WST-1 reagent
(Dojindo Molecular Technologies, Inc.) according to the
manufacturer's protocol. The colorimetric changes in the substrate
were measured with a microplate reader (Model 680; Bio-Rad
Laboratories, Inc.) at 450 nm. To evaluate the effect of TGF-β
isoforms on the proliferation of B16 melanoma cells, B16 cells
(7.5×104 cells/well) were seeded into 6-well culture
plates and cultured overnight at 37°C in 5% CO2. The
following day, the medium was replaced with 1 ml of EMEM and cells
were treated with each TGF-β (3 ng/ml) isoform or SB431542 (10 µM)
for 72 h followed by direct cell counting with Bürker-Türk
hemocytometer (cat. no. 03-303-1; Erma, Inc.). The experiment was
performed in triplicate and repeated twice.
Production of Fc chimeric
receptors
Fc chimeric receptors were generated by transfection
of 293T cells with respective Fc chimeric receptor expression
vectors. The construction of expression vectors was performed as
previously described (31). Briefly,
to express Control-Fc protein, the Fc region of human IgG fused to
the interleukin-2 signal peptide was inserted into pENTR201 vector
(Invitrogen; Thermo Fisher Scientific, Inc.). For the expression of
TβRII-Fc chimeric receptor the extracellular domain (ECD) of
TβRII-Fc corresponding to the 184 amino acids (ECD1-184)
was fused with the Fc region of human IgG and inserted into
pENTR201 (Invitrogen; Thermo Fisher Scientific, Inc.). To express
TβRI-TβRII-Fc chimeric receptor the ECD of TβRI corresponding to
the 128 amino acids (ECD1-128) was fused with ECD of
TβRII-Fc lacking signal peptide (ECD23-184) followed by
the addition of the Fc region of human IgG and inserted into
pENTR201 (Invitrogen; Thermo Fisher Scientific, Inc.). The Gateway
Technology (Invitrogen; Thermo Fisher Scientific, Inc.) was used to
transfer Control-Fc, TβRII-Fc and TβRI-TβRII-Fc cDNAs into
pCSII-EF-RfA (a gift from Dr Hiroyuki Miyoshi, Keio University,
deceased) to generate lentiviral expression vectors;
pCSII-EF-RfA-Control-Fc, pCSII-EF-RfA-TβRII-Fc and
pCSII-EF-RfA-TβRI-TβRII-Fc. 293T cells (9.0×106) were
seeded into 10-cm tissue culture dishes and cultured overnight at
37°C and 5% CO2, followed by transfection with
pCSII-EF-RfA vectors (20 µg/dish) expressing each chimeric receptor
pCSII-EF-RfA-Control-Fc, pCSII-EF-RfA-TβRII-Fc and
pCSII-EF-RfA-TβRI-TβRII-Fc, using Lipofectamine 2000 Transfection
Reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. A total of 4 h post-transfection, the
medium was replaced with serum-free Opti-MEM (Gibco; Thermo Fisher
Scientific, Inc.) and the cells were incubated for 48 h to allow
accumulation of secreted Fc chimeric receptors in culture medium.
The accumulation of soluble chimeric receptors in the conditioned
media was evaluated by immunoblotting with rabbit polyclonal
anti-human IgG-Fc fragment (1:5,000; A80-105; Bethyl Laboratories,
Inc.) as described in the Immunoblot analysis section. The
concentration of each Fc chimeric receptor was assessed by
enzyme-linked immunosorbent assay (ELISA) with the Human IgG ELISA
Quantitation Set (E80-104; Bethyl Laboratories, Inc.). The
collected conditioned media were aliquoted and stored at −80°C
until use.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from B16 and Clone M3 cells.
The extraction was performed using Sepasol (R)-RNA I Super G
(Nacalai Tesque, Inc.) and reverse-transcribed to cDNA with
ReverTra Ace qPCR-RT Master Mix (Toyobo Life Science) according to
the manufacturer's protocol. Quantitative PCR analysis was
performed using SYBR Green (Roche Applied Science) on a Step One
Plus Real-Time PCR System (Applied Biosystems, Thermo Fisher
Scientific, Inc.) under the following cycling conditions: 95°C, 10
min, followed by 40 cycles at 95°C for 15 sec and 60°C for 30 sec
with a final incubation at 95°C for 5 sec. The relative standard
curve method was used to determine the relative expression of
target genes (34). All expression
data were normalized to the expression of β-actin. The genes and
corresponding primer sequences are listed in Table SI.
Protein extraction
B16, Clone M3 and 293T cells were lysed using
radioimmunoprecipitation assay buffer (RIPA Lysis Buffer System;
Santa Cruz Biotechnology, Inc.) in the presence of a protease
inhibitor (Sigma-Aldrich; Merck KGaA), followed by repeated freeze
and thawing. Cell lysates were cleared by centrifugation at 16,400
× g for 30 min at 4°C, and the supernatants were collected. The
protein concentration in obtained lysates was determined using a
BCA Protein Assay Kit (Thermo Fisher Scientific, Inc.).
Immunoblot analysis
Denatured cell lysates (30 µg of total proteins in
Figs. 5A and S5B; 20 µg of proteins in Figs. 3D, 4C,
S3B and S4B; or 10 µg of total protein in Fig. S1) were separated on 10.5% or 12%
SDS-PAGE gel depending on the experiment, followed by transfer onto
PVDF membranes (Merck KGaA). The membranes were then blocked with
3% bovine serum albumin (BSA; FUJIFILM Wako Pure Chemical
Corporation) for 30 min at room temperature and incubated with
appropriate primary antibodies diluted in 3% BSA (Nacalai Tesque,
Inc.): Rabbit monoclonal anti-αSMA (1:1,000; product no. 19245;
Cell Signaling Technology, Inc.), rabbit polyclonal
anti-TAGLN/Transgelin (SM22α; 1:2,000; product code ab14106;
Abcam), rabbit polyclonal anti-human IgG-Fc fragment (1:5,000), and
rabbit polyclonal anti-α-tubulin (1:10,000; product code ab4074;
Abcam) overnight at 4°C. The membranes were then incubated with
goat anti-rabbit IgG HRP-linked antibody (1:5,000; product no.
7074S; Cell Signaling Technology, Inc.) for 1 h at room
temperature. The target proteins were detected using an Enhanced
Chemiluminescence Kit (ECL detection reagent; Cytiva) and
visualized with a Fusion Solo S Imaging System (SOLO.6S.EDGE;
Vilber Lourmat).
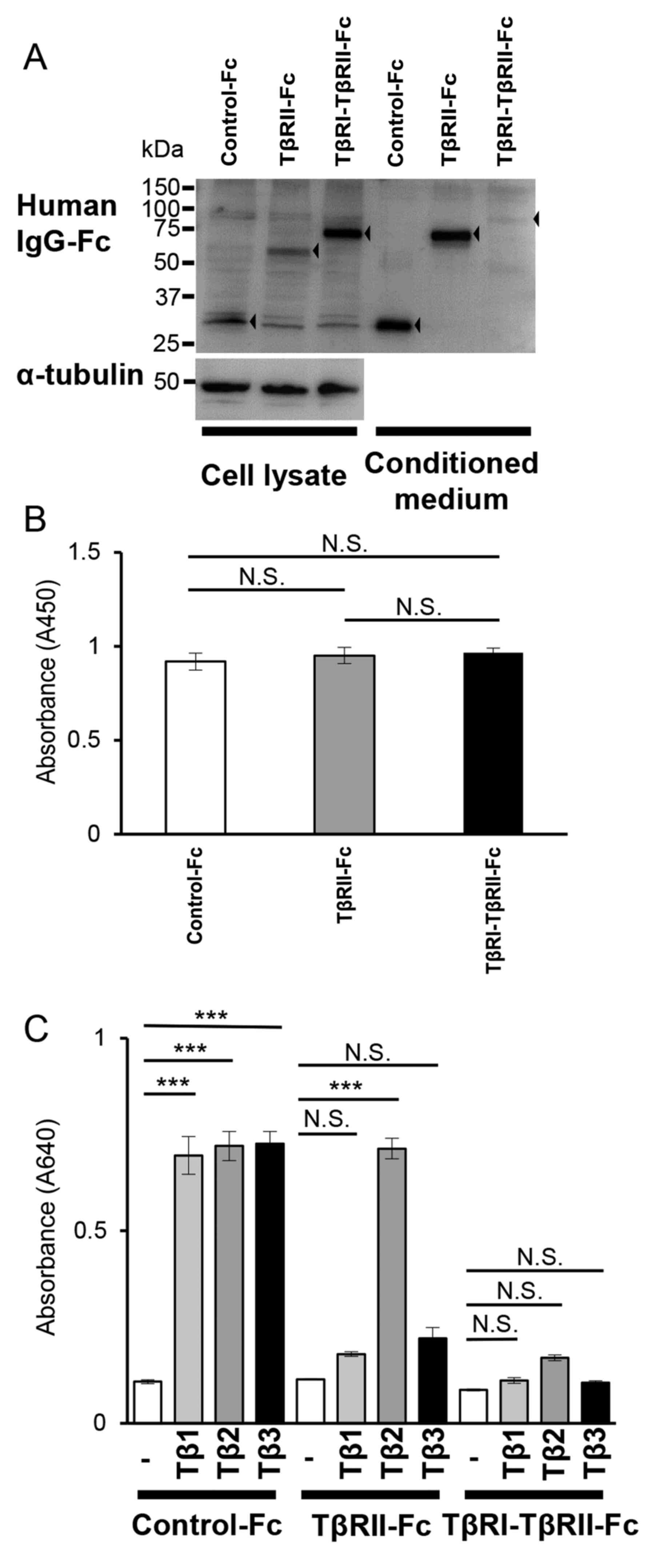 | Figure 5.TβRI-TβRII-Fc chimeric receptor
expressed and secreted by B16 cells traps all TGF-β isoforms. B16
cells were infected with lentivirus expressing each Fc chimeric
receptor (Control-Fc, TβRII-Fc, or TβRI-TβRII-Fc). (A) The Fc
chimeric receptors (black arrowhead; left, cell lysate; right,
conditioned medium) were visualized by immunoblotting analysis
using anti-human IgG-Fc antibody. α-tubulin was used as a loading
control for the cell lysate. (B) The WST-1 cell proliferation assay
of B16 cells expressing each Fc chimeric receptor. B16 cells were
seeded into a 12-well tissue culture plate and allowed to grow for
72 h followed by the WST-1 assay. Error bars, SD. (C) HEK-Blue
TGF-β reporter assay showing TGF-β-trapping ability of Fc chimeric
receptors secreted by B16 cells. The values represent TGF-β signal
activation corresponding to the colorimetric changes of the
Quanti-Blue substrate by SEAP at 640 nm. Each experiment was
performed in triplicate and repeated twice. Error bars, SD.
***P<0.001. TβRI, TGF-β type I receptor; TβRII, TGF-β type II
receptor; TGF-β, transforming growth factor-β; NS, not significant;
SEAP, secreted alkaline phosphatase; IgG, immunoglobulin G. |
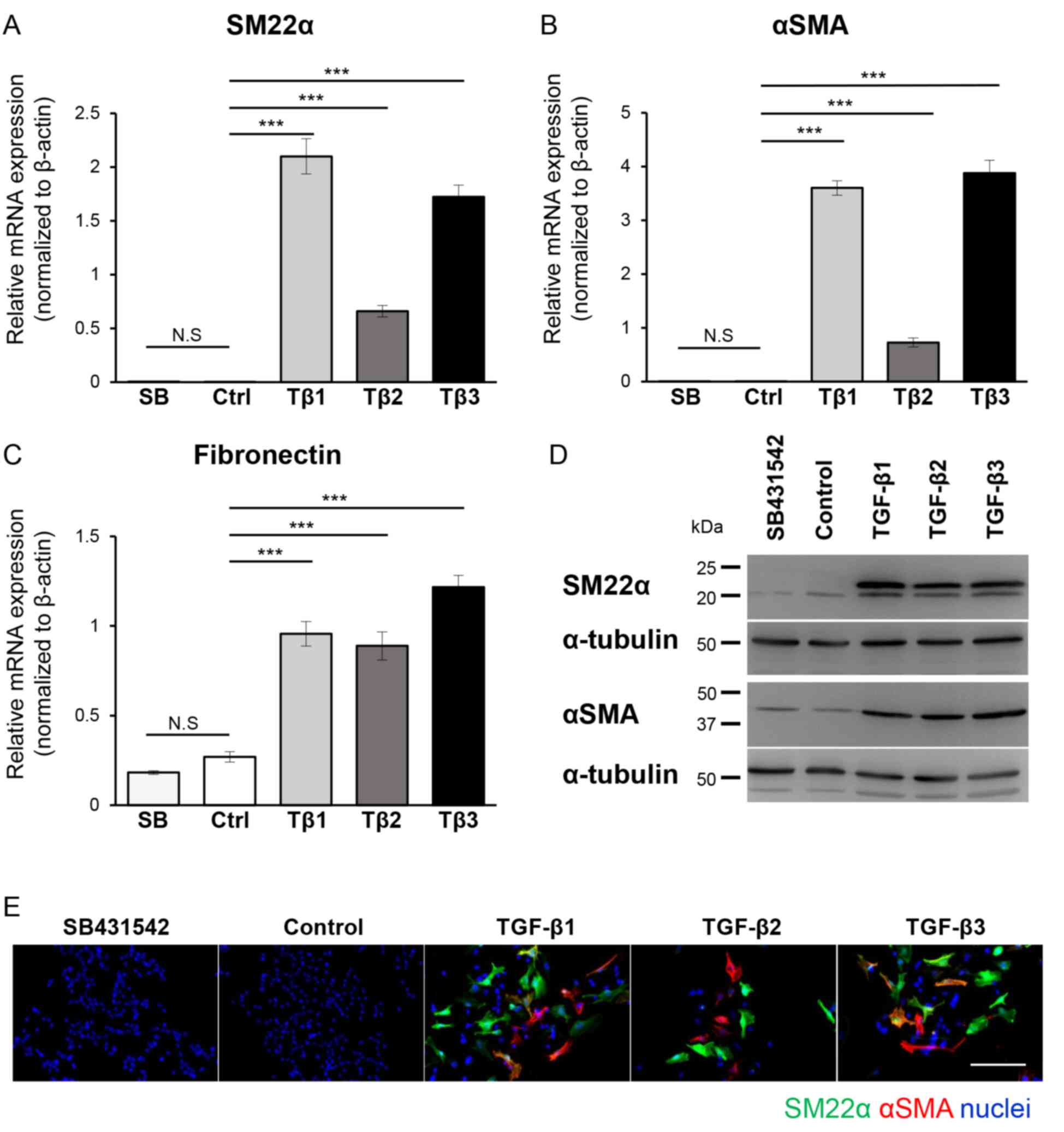 | Figure 3.EMT program in B16 melanoma cells is
induced by all TGF-β isoforms. B16 cells were cultured in the
absence (Ctrl or Control) or presence of TGF-β1 (Tβ1), TGF-β2
(Tβ2), or TGF-β3 (Tβ3) (3 ng/ml) or the TβRI kinase inhibitor,
SB431542 (SB; 10 µM) for 72 h, followed by (A-C) RT-qPCR, (D)
immunoblotting and (E) immunocytochemistry. Experiments were
performed in triplicate and repeated twice. (A-C) The expression of
mesenchymal markers (A) SM22α, (B) αSMA and (C) fibronectin were
evaluated by RT-qPCR analyses. All RT-qPCR data were normalized to
the β-actin expression. (D) The immunoblotting analysis with
antibodies specific to SM22α, αSMA and α-tubulin (loading control).
(E) Representative immunofluorescence images revealing staining of
SM22α (green), αSMA (red) and nuclei (blue). Scale bar, 100 µm.
Error bars, SD. ***P<0.001. EMT, epithelial-mesenchymal
transition; TGF-β, transforming growth factor-β; TβRI, TGF-β type I
receptor; RT-qPCR, reverse transcription-quantitative PCR; NS, not
significant; SM22α, smooth muscle protein 22α; αSMA, α-smooth
muscle actin. |
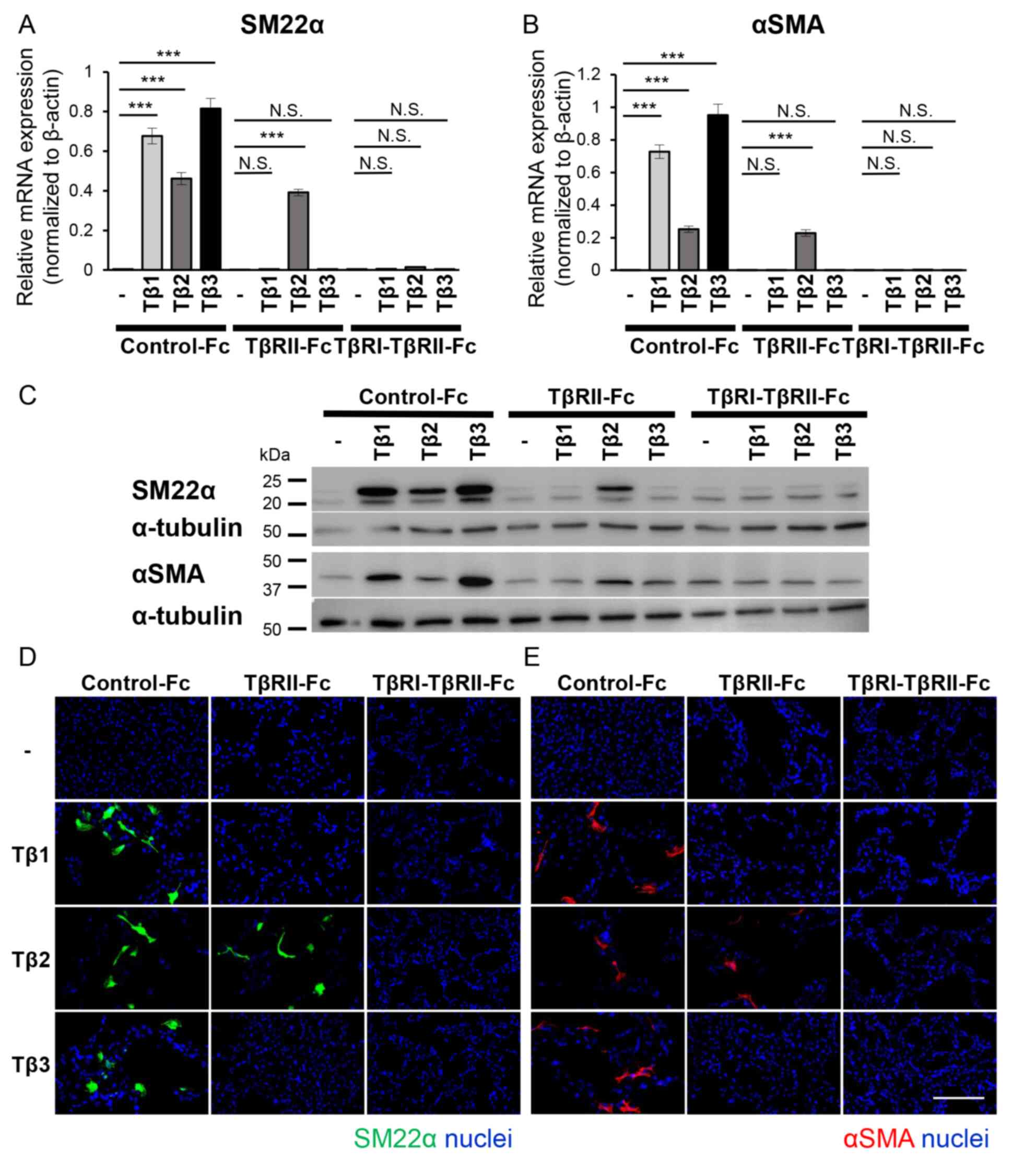 | Figure 4.TβRI-TβRII-Fc chimeric receptor
inhibits the TGF-β-induced EMT program in B16 cells. B16 cells were
incubated in the absence (−) or presence of TGF-β isoforms [TGF-β1
(Tβ1), TGF-β2 (Tβ2), or TGF-β3 (Tβ3)] (3 ng/ml) in combination with
conditioned media from 293T cells containing Fc chimeric receptors
(Control-Fc, TβRII-Fc, or TβRI-TβRII-Fc). The activation of the EMT
program, was estimated by (A and B) RT-qPCR analyses, (C)
immunoblotting and (D and E) immunocytochemistry. Each experiment
was performed in triplicate and repeated twice. (A and B) The
RT-qPCR analysis for the expression of mesenchymal markers (A)
SM22α and (B) αSMA. All data were normalized to the β-actin
expression. (C) The immunoblotting analysis of the expression
levels of SM22α, αSMA and α-tubulin (loading control). (D and E)
Immunofluorescence staining of (D) SM22α (green) and (E) αSMA
(red). The nuclei were stained with Hoechst (blue). Scale bar, 100
µm. Error bars, SD. ***P<0.001. TβRI, TGF-β type I receptor;
TβRII, TGF-β type II receptor; TGF-β, transforming growth factor-β;
EMT, epithelial-mesenchymal transition; RT-qPCR, reverse
transcription-quantitative PCR; SM22α, smooth muscle protein 22α;
αSMA, α-smooth muscle actin; NS, not significant. |
Immunocytochemistry
B16 and Clone M3 cells (3.5×104
cells/well) were seeded on cover glasses placed into 12-well tissue
culture plates and treated with TGF-β1, -β2, and -β3 in the
presence or absence of Fc chimeric receptors for 72 h, at 37°C in
5% CO2. The cells were then fixed with methanol/acetone
(1:1) for 20 sec on ice and blocked in PBS containing 1% BSA
(FUJIFILM Wako Pure Chemical Corporation) for 30 min at room
temperature and incubated with primary antibodies diluted in
Blocking One buffer: Rabbit polyclonal anti-TAGLN/Transgelin
(1:1,000), mouse monoclonal anti-actin, αSMA-Cy3™ (1:1,000; cat.
no. C6198-2ML; Sigma-Aldrich; Merck KGaA) overnight at 4°C. To
visualize SM22α and nuclei, samples were incubated for 1 h at room
temperature with a mixture of donkey polyclonal anti-rabbit IgG
(H+L) Alexa Fluor 488-conjugated secondary antibodies (1:1,000 in
Blocking One buffer; cat. no. A-21206; Thermo Fisher Scientific,
Inc.) and 500 ng/ml Hoechst33342 (Cell Signaling Technology, Inc.)
for nuclear staining. The samples were then embedded in
Fluoromount-G mounting medium (Cosmo Bio Co., Ltd.). Images were
captured under a fluorescence microscope (BZ-X710; Keyence
Corporation).
Treatment of B16 and Clone M3 cells
with Fc chimeric receptors
B16 or Clone M3 cells (3.5×104
cells/well) were seeded into tissue culture plates (12-well plate
or 6-well plate, depending on the experiment) and cultured
overnight at 37°C in 5% CO2. The next day, the medium
was replaced with 1 ml of serum-free Opti-MEM. The Fc chimeric
receptor/ligand complexes were generated by mixing the conditioned
medium from the 293T cells expressing Fc chimeric receptors
containing the equal amount of Fc chimeric receptors (600 ng of Fc
chimeric receptors in 500 µl of Opti-MEM) with TGF-β1, -β2, or -β3
at the concentration of 3 ng/ml. Samples were incubated for 2 h at
37°C to allow formation of the Fc chimeric receptor/ligand
complexes and added to the B16 or Clone M3 cells. The cells were
then incubated at 37°C and 5% CO2 with Fc chimeric
receptor/ligand complexes for 4 or 72 h (depending on the
experiment) and subjected to gene expression analysis by RT-qPCR,
immunocytochemistry, or immunoblotting. Cells treated with each
TGF-β ligand in serum-free Opti-MEM were used as controls for
upregulation of the TGF-β signal while samples treated with the
mixture of SB431542, a TβRI kinase inhibitor, and TGF-β in
serum-free Opti-MEM were used as controls for inhibition of the
TGF-β signal.
Lentivirus production and transduction
of B16 cells
The lentiviral particles were produced as previously
described (31). Briefly, the 293FT
(8.0×106) cells were co-transfected with 5.5 µg of
expression plasmids (pCSII-EF-RfA-Control-Fc,
pCSII-EF-RfA-TβRII-Fc, and pCSII-EF-RfA-TβRI-TβRII-Fc) and
packaging plasmids pCMV–VSV-G-RSV-Rev (3.25 µg; RIKEN BioResource
Center) and pCAG-HIVgp (3.25 µg; RIKEN BioResource Center) using
Lipofectamine 2000 Transfection Reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) in 5 ml Opti-MEM supplemented with 10% FBS. The
control lentiviral particles expressing green fluorescent protein
(GFP) were prepared by transfecting 293T cells with 5.5 µg of
pCSII-EF-RfA-GFP and packaging plasmids pCMV–VSV-G-RSV-Rev (3.25
µg) and pCAG-HIVgp (3.25 µg). A total of 24 h post-transfection,
the transfection medium was refreshed with 7.5 ml Opti-MEM, 10%
FBS, and the recombinant lentiviruses were harvested 48 h later.
The conditioned media containing viral particles were collected by
centrifugation at 4°C for 5 min at 1,700 × g, and incubated at 4°C
for 7 days on the rotary shaker with Lenti-X Concentrator (Takara
Bio, Inc.). The viral particles were then centrifuged at 4°C for 45
min, at 1,500 × g, and resuspended in 140 µl of Opti-MEM. The 70-µl
of concentrated lentiviral particles were used to infect B16
melanoma cells (3×105 cells/well in 12-well tissue
culture plates). Transduction efficiency was evaluated using
lentiviral particles expressing green GFP. The successful
expression was examined by immunoblotting using rabbit polyclonal
anti-human IgG-Fc antibody as described in the Immunoblot
analysis section. The second generation of transduced B16 cells
was used for further experiments.
Smad 2/3/4-responsive reporter assay
(HEK-Blue reporter assay)
The ability of Fc chimeric receptors, secreted by
transduced B16 cells, to trap TGF-β ligands was examined using the
HEK-Blue TGF-β reporter system. The B16 cells (1×106
cells) expressing each of the Fc chimeric receptors were seeded
into 10 cm tissue culture plates and incubated overnight at 37°C in
5% CO2. The following day, the medium was replaced with
5 ml of serum-free Opti-MEM and the cells were incubated for 48 h
to allow the accumulation of Fc chimeric receptors in the culture
supernatant. The conditioned media were collected and stored at
−80°C until use.
HEK-Blue TGF-β reporter cells (1.0×105)
were seeded into 96-well plates and incubated overnight at 37°C in
5% CO2. The following day, the medium was replaced with
200 µl of serum-free DMEM, and the cells were incubated for 3 h.
The B16 cell-derived conditioned medium containing Control-Fc,
TβRII-Fc, or TβRI-TβRII-Fc chimeric proteins, was mixed with TGF-β
ligands (1 ng/ml) and incubated for 2 h at 37°C to allow the
formation of Fc chimeric receptor/ligand complexes. Next, Fc
chimeric receptor/ligand complexes were added to the HEK-Blue TGF-β
reporter cells, followed by incubation for 24 h, at 37°C. The
activation of TGF-β/Smad signals was detected using QUANTI-Blue
substrate (InvivoGen) following incubation for 30 min at 37°C. The
colorimetric change of the substrate by the secreted alkaline
phosphatase (SEAP) was quantified at 640 nm using a microplate
reader (Bio-Rad Laboratories, Inc.).
Subcutaneous syngeneic tumor mouse
model
A total of 62 female C57/BL6 mice (5–6 weeks old;
average weight, 14–19 g) were purchased from Japan SLC, Inc. All
animal experimental protocols were approved (approval no.
R-02-017-1) by the Animal Experiment Committee of the Graduate
School of Dentistry, Osaka University (Osaka, Japan). The mice were
kept under a temperature of 23–24°C with 40–60% humidity and a 12-h
light/dark cycle. Mice were provided with access to food and water
ad libitum throughout the experiment. A total of 20 mice was
used for injection of B16-Control-Fc cells, 21 for B16-TβRII-Fc
cells, and 21 for B16-TβRI-TβRII-Fc cells. All animals underwent
general anesthesia with mixture of medetomidine (0.3 mg/kg; Nippon
Zenyaku Kogyo, Co., Ltd.) midazolam (4 mg/kg; Astellas Pharma,
Inc.) and butorphanol (5 mg/kg; Meiji Seika Kaisha, Ltd.) by
intraperitoneal administration (35,36). The
B16 cells (5.0×105) expressing the Fc chimeric
receptors, B16-Control-Fc, B16-TβRII-Fc, B16-TβRI-TβRII-Fc were
suspended in 50 µl serum-free EMEM and subcutaneously injected into
left flank region. Mice that did not develop any palpable tumor or
did not survive until the endpoint of the experiment were excluded
from the analysis. Tumor growth was monitored for 26 days. The
tumor volume was measured twice per week and estimated using the
following equation: Tumor volume (mm3) = [length (mm) ×
width (mm)2]/2. The size of developed tumors was
selected as the humane endpoint. Mice were sacrificed when the size
of the largest primary tumors started to exceed the 1,000
mm3. As large melanoma tumors often develop necrotic
changes that would likely affect the experimental outcome, a total
of 32 mice bearing primary tumors derived from Control-Fc (n=11),
TβRII-Fc (n=9), and TβRI-TβRII-Fc (n=11) were thus euthanized on
day 26 by intraperitoneal injection of the mixture of medetomidine
(3 mg/kg), midazolam (40 mg/kg) and butorphanol (50 mg/kg).
Statistical analysis
Statistical analysis was carried out by EZR software
(37). Results are presented as the
mean ± standard deviation (SD) or standard error (SE). Each
experiment was performed in triplicate and repeated twice.
Comparisons of quantitative data were conducted using one-way ANOVA
with post hoc Tukey's test or Mann-Whitney U test with post hoc
Bonferroni test depending on experiment. P<0.05 was considered
to indicate a statistically significant difference.
Results
TβRI-TβRII-Fc chimeric receptor
efficiently suppresses TGF-β signals in melanoma cells
TGF-β has been revealed to promote invasiveness and
progression of melanoma (13,16). Previous studies revealed that melanoma
cells expressed all three TGF-β isoforms (12) and that an elevated level of TGF-β in
melanoma patients was associated with metastatic outcomes (38). In addition, meta-analysis using a
public database, PrognoScan (http://dna00.bio.kyutech.ac.jp/PrognoScan/) (32) and dataset: GSE19234 (33), revealed that high expression levels of
TGF-β2, but not that of TGF-β1 or TGF-β3, were associated with
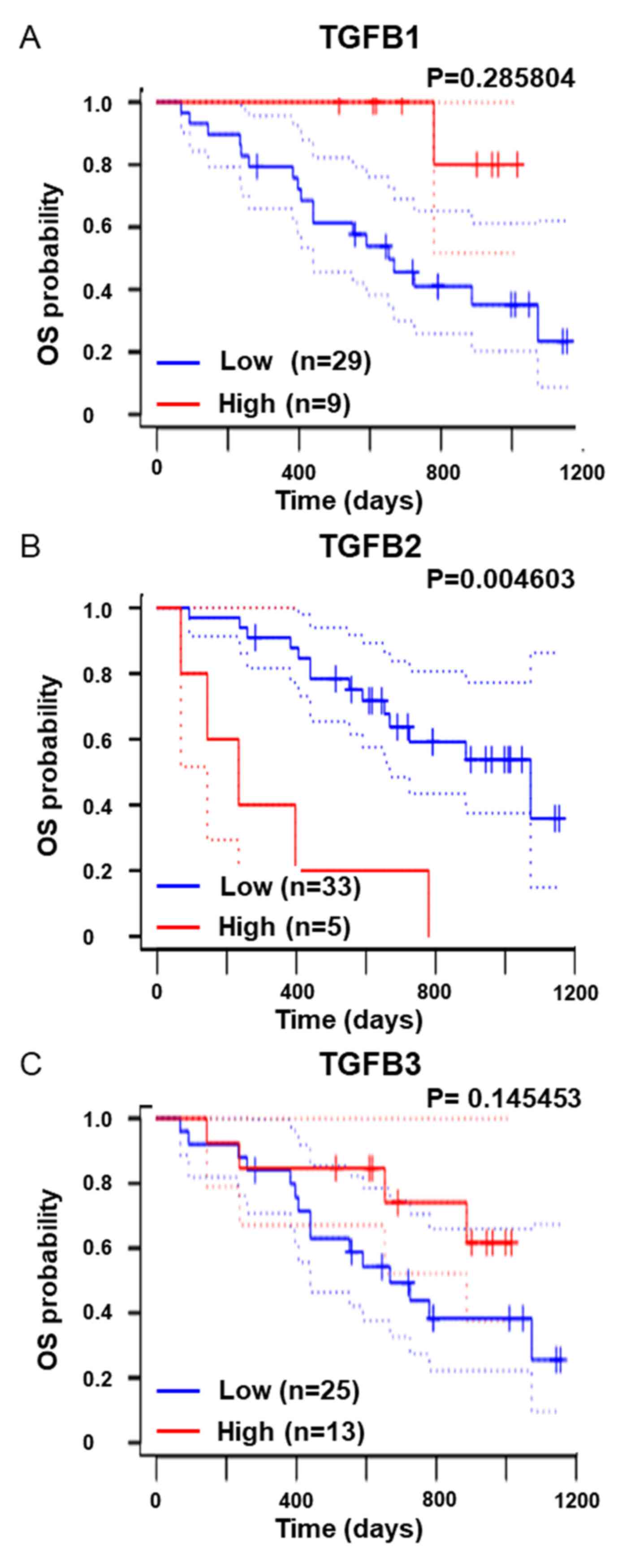
overall survival of melanoma patients (Fig. 1). Therefore, it was examined whether
TβRI-TβRII-Fc chimeric receptor could be applied in the melanoma
model. In our study, B16 melanoma cells were used, in which the EMT
program can be activated in response to TGF-β. 293T cells were
transfected with the vectors expressing Control-Fc, TβRII-Fc, or
TβRI-TβRII-Fc chimeric receptors and the accumulation of chimeric
proteins in the conditioned media was confirmed by immunoblotting
(Fig. S1). Such conditioned media
were then used to examine the effect of soluble chimeric receptors
on the activation of TGF-β signals. B16 cells were incubated in
conditioned media of 293T cells expressing Control-Fc, TβRII-Fc, or
TβRI-TβRII-Fc chimeric receptors in an absence or presence of
TGF-β1, -β2, or -β3, respectively, followed by an analysis of the
expression of genes directly responding to TGF-β, TMEPAI, and PAI-1
by RT-qPCR. Incubation of B16 melanoma cells with any of the TGF-β
isoforms upregulated the expression of both TMEPAI (Fig. 2A) and PAI-1 (Fig. 2B). As anticipated, SB431542, a TβRI
kinase inhibitor, reduced the expression of both direct target
genes to the background level (Fig.
2). Incubation of B16 melanoma cells with Control-Fc protein
did not reduce the expression of TMEPAI, and PAI-1 induced by
TGF-βs. Reduced expression of both genes in the presence of
TβRII-Fc chimeric receptor was only observed when cells were
treated with TGF-β1 and TGF-β3, but not by TGF-β2 (Fig. 2). Conversely, TβRI-TβRII-Fc chimeric
receptor significantly inhibited the expression levels of TMEPAI
and PAI-1 induced by all TGF-β isoforms indicating that
TβRI-TβRII-Fc chimeric receptor effectively interfered with TGF-β
signals also in the melanoma model. To generalize our findings, the
same set of experiments were performed with another melanoma cell
line, Clone M3. Clone M3 cells responded to all TGF-β isoforms as
indicated by upregulated expression of TMEPAI (Fig. S2A) and PAI-1 (Fig. S2B). In addition, incubation with
TβRI-TβRII-Fc decreased the expression of TMEPAI and PAI-1 induced
by all TGF-βs when compared with the expression of both genes
detected in the Clone M3 cells incubated with Control-Fc protein
indicating that TβRI-TβRII-Fc chimeric receptor suppressed TGF-β
signals in multiple types of melanoma cells.
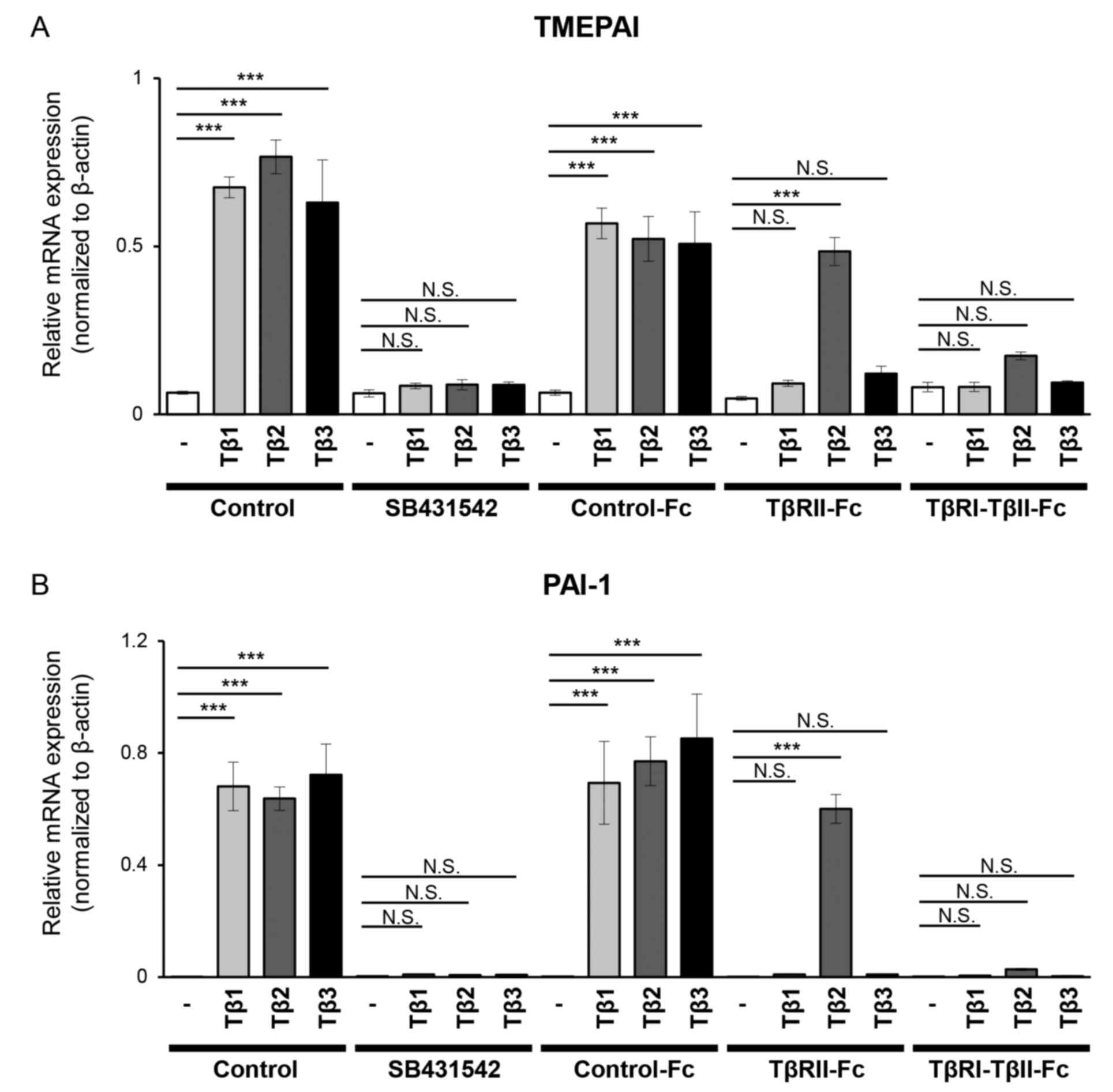 | Figure 2.TβRI-TβRII-Fc chimeric receptor
inhibits the expression of TGF-β direct target genes in B16
melanoma cells. B16 cells were incubated for 4 h in the absence (−)
or presence of TGF-β isoforms [(TGF-β1 (Tβ1), TGF-β2 (Tβ2), or
TGF-β3 (Tβ3) (3 ng/ml)] in combination with a vehicle (Control), 10
µM SB431542 or conditioned media from 293T cells containing Fc
chimeric receptors (Control-Fc, TβRII-Fc, or TβRI-TβRII-Fc). The
expression levels of (A) TMEPAI and (B) PAI-1 were determined
through reverse transcription-quantitative PCR analysis. Each
experiment was performed in triplicate and repeated twice. All data
are normalized to the expression of β-actin. Error bars, SD.
***P<0.001. TβRI, TGF-β type I receptor; TβRII, TGF-β type II
receptor; TGF-β, transforming growth factor-β; NS, not significant;
TMEPAI, transmembrane prostate androgen-induced protein; PAI-1,
plasminogen activator inhibitor-1. |
All TGF-β isoforms activate the EMT
program in melanoma cells
Melanoma cells can activate the EMT program in
response to TGF-β. Previous studies revealed that melanoma cells
treated with TGF-β upregulated the expression of mesenchymal
markers (14,17). Therefore, it was examined whether
similar changes can be observed in B16 melanoma cells. B16 melanoma
cells were cultured for 72 h in the absence or presence of each
TGF-β isoform or SB431542, and the expression of various
mesenchymal markers was determined using RT-qPCR. The treatment
with any of TGF-β isoform resulted in upregulated expression of all
mesenchymal markers; SM22α (Fig. 3A),
αSMA (Fig. 3B) and fibronectin
(Fig. 3C), while SB431542 did not
exhibit any effect (Fig. 3A-C). These
results were also confirmed at the protein level using
immunoblotting and immunocytochemical analysis. A significant
increase was observed in the band intensity corresponding to each
mesenchymal marker, SM22α and αSMA (Fig.
3D), as well as an increase in a fluorescent signal related to
the presence of SM22α-positive and αSMA-positive cells upon TGF-β
treatment (Fig. 3E), indicating that
B16 melanoma cells activated TGF-β-dependent EMT. The activation of
the EMT program was also confirmed using Clone M3 cells. Treatment
of the Clone M3 cells with any of the three TGF-β isoforms resulted
in upregulated expression of SM22α as revealed by RT-qPCR (Fig. S3A), immunoblotting (Fig. S3B), and immunocytochemical analysis
(Fig. S3C), supporting the findings
that treatment with TGF-β upregulated the expression of SM22α in
multiple types of melanoma cells.
EMT program, induced by all TGF-β
isoforms in melanoma cells, is inhibited in the presence of
TβRI-TβRII-Fc chimeric receptor
Our data indicated that the EMT program was
activated upon treatment with any of the three TGF-β isoforms. In
addition, as shown in Fig. 1B, high
TGFB2 expression was a poor prognostic factor in overall survival
in melanoma patients, indicating that inhibiting the EMT program
may have beneficial effects on melanoma treatment. Therefore, in
the following experiment, the effect of TβRI-TβRII-Fc chimeric
receptor on activation of the EMT program was examined in B16
melanoma cells. The B16 melanoma cells were treated without or with
TGF-β1, -β2 or -β3 in the presence of conditioned medium derived
from 293T cells expressing Control-Fc, TβRII-Fc, or TβRI-TβRII-Fc
chimeric receptors for 72 h, followed by RT-qPCR analysis for the
expression of mesenchymal markers, SM22α (Fig. 4A) and αSMA (Fig. 4B). The expression levels of both
mesenchymal markers were upregulated when B16 melanoma cells were
incubated in the conditioned medium containing Control-Fc protein.
A significant suppression of SM22α (Fig.
4A) and αSMA (Fig. 4B) expression
was observed in the cells stimulated with TGF-β1 or TGF-β3 in the
presence of TβRII-Fc or TβRI-TβRII-Fc chimeric receptors.
Conversely, the induction of the EMT program by TGF-β2 was
inhibited only in the presence of TβRI-TβRII-Fc chimeric receptor
(Fig. 4A and B), indicating that
TβRI-TβRII-Fc chimeric receptor could modulate the response to
TGF-β2 in the melanoma model. The aforementioned findings were also
confirmed at the protein level using immunoblotting (Fig. 4C) and immunocytochemical analyses
(Fig. 4D and E). In agreement with
the RT-qPCR results, changes in the intensity of bands
corresponding to upregulated expression of SM22α and αSMA proteins
were observed (Fig. 4C) in response
to all TGF-β isoforms, in the absence or presence of Fc chimeric
receptors, as well as the number of SM22α-positive and
αSMA-positive cells (Fig. 4D and E,
respectively). Consistent with the RT-qPCR results, effective
inhibition of the TGF-β2-induced EMT program was observed only in
the presence of TβRI-TβRII-Fc chimeric receptor while Control-Fc
and TβRII-Fc did not demonstrate such an effect (Fig. 4C-E). The suppressive effect of
TβRI-TβRII-Fc chimeric receptor on EMT-associated changes in SM22α
expression was also examined at both RNA and protein levels in
Clone M3 cells. As anticipated, the expression of SM22α induced by
any of the three TGF-β isoforms was inhibited only by TβRI-TβRII-Fc
chimeric receptor (Fig. S4),
indicating that TβRI-TβRII-Fc chimeric receptor could be
potentially used for targeting all TGF-β isoforms present in the
TME of melanoma tumors.
TβRI-TβRII-Fc chimeric receptor
inhibits melanoma tumor growth in vivo
As our in vitro data revealed effective
inhibition of the EMT program, examination of the effect of
TβRI-TβRII-Fc chimeric receptor on melanoma tumor growth in
vivo was performed. Therefore, B16 melanoma cells expressing
Control-Fc, TβRII-Fc, and TβRI-TβRII-Fc chimeric proteins were
established by infecting B16 melanoma cells with lentiviral vectors
(Fig. S5). As revealed in Fig. 5A, all Fc chimeric receptors were
expressed in B16 cells (Fig. 5A; cell
lysate) and secreted into the culture media (Fig. 5A; conditioned medium); however, the
amount of accumulated TβRI-TβRII-Fc chimeric receptor was lower
when compared with the secreted amount of Control-Fc or TβRII-Fc
chimeric receptors.
The anti-proliferative effect of TGF-β on normal
epithelial cells has been previously reported (39). Moreover, in the early stage of
melanoma progression, TGF-β has been revealed to inhibit cell
growth (15). In agreement with these
previous findings, B16 melanoma cells incubated in the presence of
TGF-βs for 72 h demonstrated decreased proliferation when compared
with the non-treated control cells, independently of the TGF-β
isoform used (Fig. S6). The
proliferation of B16 melanoma cells in the presence of SB431542, a
TβRI kinase inhibitor, did not differ from the proliferation of
control cells (Fig. S6). As B16
melanoma cells have been revealed to secrete active TGF-βs
(40), there was a possibility that
the expression of Fc chimeric receptors could alter their
extracellular environment and affect cell proliferation. Therefore,
the proliferation of B16 cells expressing each Fc chimeric receptor
was examined and it was revealed that there was not any difference
in the proliferation exhibited by B16 cells expressing TβRII-Fc and
TβRI-TβRII-Fc chimeric receptors when compared with B16 cells
expressing Control-Fc protein (Fig.
5B).
Next, the effect of TβRII-Fc and TβRI-TβRII-Fc
chimeric receptors accumulated in conditioned media of B16 cells on
TGF-β signaling was examined using HEK-Blue TGF-β reporter cells.
HEK-Blue cells were cultured in the conditioned medium of B16 cells
expressing Control-Fc, TβRII-Fc, or TβRI-TβRII-Fc chimeric
receptors in the absence or presence of TGF-β1, -β2, or -β3. As
anticipated, stimulation of HEK-Blue cells with any TGF-β isoform
in the presence of conditioned medium from B16 cells expressing
Control-Fc protein resulted in upregulation of TGF-β signals
(Fig. 5C). Conditioned medium derived
from B16 cells expressing TβRII-Fc chimeric receptor significantly
inhibited signals induced by TGF-β1 or TGF-β3 and had no effect on
signals induced by TGF-β2 (Fig. 5C).
Complete inhibition of TGF-β signals was observed only in the
presence of conditioned medium derived from B16 cells expressing
TβRI-TβRII-Fc chimeric receptor (Fig.
5C), indicating that TβRI-TβRII-Fc could trap all TGF-β
isoforms.
Finally, the effect of Fc chimeric receptors on
melanoma tumor growth was examined in vivo. The B16 cells
expressing Control-Fc, TβRII-Fc, or TβRI-TβRII-Fc chimeric
receptors were subcutaneously inoculated in the left flank of
C57/BL6 mice. The tumor growth and body weight were then monitored
for 26 days. Expression of TβRI-TβRII-Fc chimeric receptor
inhibited B16 melanoma tumor growth in vivo (Fig. 6A) when compared with Control-Fc.
Moreover, the size of tumors originating from B16 cells expressing
TβRI-TβRII-Fc chimeric receptor was significantly smaller than that
developed from B16 cells expressing Control-Fc protein (Fig. 6B) indicating that it could effectively
trap all TGF-β isoforms residing in the TME. Of note, no
significant difference was observed between Control-Fc and TβRII-Fc
or TβRII-Fc and TβRI-TβRII-Fc groups. In addition, no significant
differences in body weight were observed between the three
experimental groups (Fig. 6C).
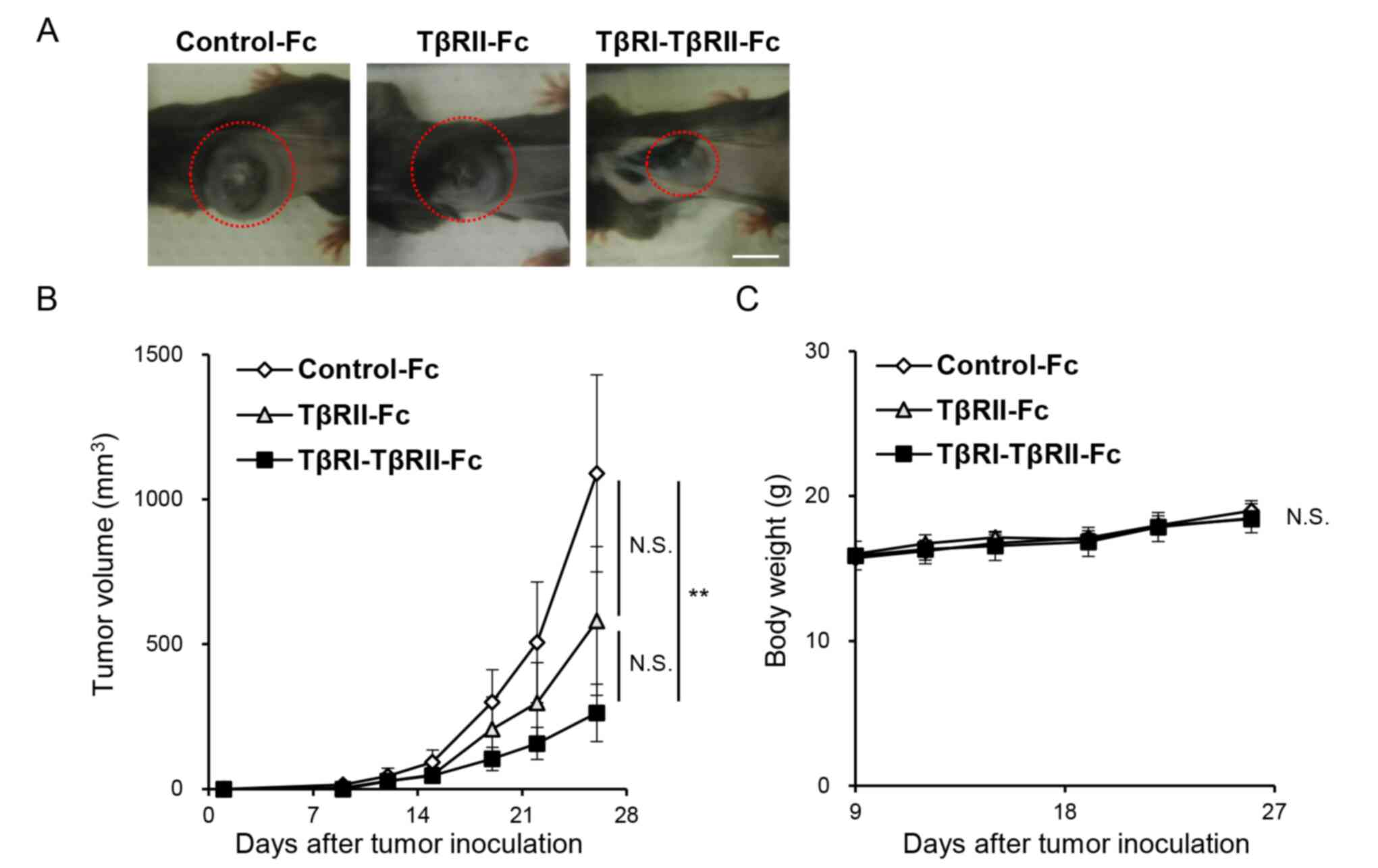 | Figure 6.TβRI-TβRII-Fc chimeric receptor
inhibits B16 melanoma tumor growth in vivo. B16 cells
expressing Control-Fc, TβRII-Fc, and TβRI-TβRII-Fc chimeric
receptors were subcutaneously inoculated into left flank region of
C57/BL6 mice. The experiment was repeated twice. (A) Representative
primary tumors formed by B16 melanoma cells expressing Control-Fc,
TβRII-Fc and TβRI-TβRII-Fc chimeric receptors on day 26 (marked by
red dashed circles). Scale bar, 10 mm. (B) Tumor growth was
monitored for 26 days. Control-Fc (n=11), TβRII-Fc (n=9), and
TβRI-TβRII-Fc (n=11). Error bars, SE. (C) The changes in body
weight of the mice inoculated with B16 cells expressing Control-Fc,
TβRII-Fc, and TβRI-TβRII-Fc chimeric receptors. Error bars, SE.
**P<0.01. TβRI, TGF-β type I receptor; TβRII, TGF-β type II
receptor; NS, not significant. |
Discussion
TGF-β ligands facilitate progression of various
types of cancer by affecting the components of the TME (6,7).
Therefore, targeting of TGF-β signals will have an outcome in the
development of effective agents. Recently, Fc chimeric receptors
bearing the extracellular domains of various receptors and the Fc
portion of IgG have attracted a significant amount of attention
(28). The presence of Fc can extend
the plasma half-life time of chimeric receptors and engage the
immune response, thus being a favorable choice to develop effective
agents (29). In the present study,
it was demonstrated that previously developed TβRI-TβRII-Fc
chimeric receptor could also trap all three TGF-β isoforms that
resulted in inhibition of the EMT program in B16 melanoma cells
in vitro. Our data also revealed that primary tumors
originated from B16 melanoma cells expressing TβRI-TβRII-Fc
chimeric receptor exhibited reduced growth in vivo, in a
subcutaneous murine xenograft model, indicating that TβRI-TβRII-Fc
chimeric receptor may represent a favorable strategy for the
development of a novel drug for melanoma treatment.
Our previous study with human oral cancer cells,
revealed that both TβRII-Fc and TβRI-TβRII-Fc chimeric receptors
could significantly suppress tumor formation originated from oral
cancer cells (31). However, in the
present study, only the tumors formed by TβRI-TβRII-Fc-expressing
cells appeared to be significantly smaller when compared with
tumors formed by B16 cells expressing Control-Fc protein. In our
study, TβRI-TβRII-Fc chimeric receptor was capable of interacting
with all three isoforms indicating that TβRI-TβRII-Fc chimeric
receptor could be used to control the level of TGF-β in the
melanoma TME. Since the melanoma cells expressing the chimeric
receptors in the present study were used, further experiments
employing the recombinant soluble TβRI-TβRII-Fc chimeric receptor
administered via blood system will shed light on the turnover of
TβRI-TβRII-Fc chimeric receptor and allow its validation.
The exact mechanism by which TβRI-TβRII-Fc chimeric
receptor inhibited melanoma tumor formation remains to be examined;
however our data with the oral cancer cell model revealed that
TβRI-TβRII-Fc chimeric receptor suppressed tumor formation by
affecting tumor angiogenesis (31).
As melanoma progression is tightly correlated with new vessel
formation (41), it is possible that
TβRI-TβRII-Fc chimeric receptor secreted by melanoma cells to the
TME affected the angiogenic response of endothelial cells and
resulted in reduced tumor size. The presence of TβRI-TβRII-Fc
chimeric receptor in the TME that results in a local decrease in
TGF-β level can also affect the formation of CAFs. Our group has
previously revealed that growth of primary tumors derived from A375
human melanoma was stimulated by CAFs originated from TECs treated
with TGF-β2, indicating that TGF-β2 conferred TECs with
myofibroblastic properties leading to the formation of
tumor-promoting CAFs (42).
Therefore, TβRI-TβRII-Fc chimeric receptor present in the TME would
likely trap TGF-β2, thus interfering with CAF formation and
affecting tumor growth. Conversely, various studies have revealed
the role of TGF-β in the regulation of immune responses, working
both as an immunosuppressor of macrophages and various types of
lymphocytes (43–45) or immune response inducer (7). Thus, depletion of TGF-β from the TME may
also result in altered antitumor immunity.
TGF-β2 along with bone morphogenetic protein 7
(BMP7) has been reported to be expressed at high levels by
proliferative and pro-invasive melanoma tumors (17). The aforementioned study indicated an
important role of TGF-β superfamily members in melanoma development
by regulating both melanoma invasion and proliferation. In
addition, the important role of TGF-β in the induction of the EMT
program in melanoma cells was also identified. A previous study has
also revealed that melanoma cells undergoing EMT activate
immunosuppressive regulatory T cells (Treg) (46). Kudo-Saito et al demonstrated
that overexpression of EMT-related transcription factor, Snail, in
mouse or human melanoma cells resulted in enhanced metastasis and
immunoresistance of formed tumors (46). Moreover, treatment of human melanoma
cells with TGF-β upregulated the expression of forkhead box P3
transcription factor (FOXP3), a marker of Treg. In our study,
TGF-β2 induced the EMT program in melanoma cells leading to the
myofibroblastic changes as revealed by upregulated αSMA expression.
This effect was inhibited by TβRI-TβRII-Fc chimeric receptor,
indicating that targeting TGF-β by the administration of
TβRI-TβRII-Fc chimeric receptor could be potentially used to
restore the immunocompetence in melanoma tumors.
Several chimeric receptors capable of inhibiting
TGF-β have been designed and successfully tested to demonstrate the
efficacy for the selective blockage of TGF-β family ligands in
pathological conditions (47,48). Particularly, TβRII-Fc chimeric
receptor has been applied in various studies (48,49).
However, as revealed by Yung et al, TβRII-Fc therapeutic
potential was isoform-selective, as it could trap only TGF-β1 and
TGF-β3, but not TGF-β2 (49). A
previous study has revealed that the elevated plasma expression
levels of TGF-β2 detected in melanoma patients were associated with
tumor progression, increased metastasis and poor prognosis
(33). In addition, other approaches
based on the small molecules targeting the kinase activity of TβRI
kinase (25) or short hairpin RNA
targeting TGF-β2 (23) revealed the
involvement of TGF-β signals in melanoma progression (50). Therefore, effective trapping of all
TGF-β isoforms may lead to improved clinical outcomes in treatment
of melanoma patients.
TGF-β regulates melanoma cell plasticity and
antitumor immunity by affecting the components of the TME (51). Therefore, targeting the TGF-β signals
will be beneficial for the development of effective antitumor
agents for melanoma. Considering the dual role of TGF-β and its
tumor-suppressive activities, the complete inhibition of TGF-β
signals may evoke tumorigenesis in normal epithelial cells or
result in unwanted side effects. From this point of view, by
adjusting the concentration of administered soluble Fc chimeric
receptors, it may be possible to maintain the concentration of
TGF-β at the level that exerts only tumor-suppressive effects
without unwanted pro-tumorigenic responses.
Supplementary Material
Supporting Data
Acknowledgements
The lentiviral vectors were kindly provided by Dr
Hiroyuki Miyoshi (Keio University, deceased). The authors would
like to thank the members at the Department of Biochemistry of
Tokyo Medical and Dental University for critical discussion.
Funding
The present study was supported by a research
program of the Japan Agency for Medical Research and Development
(AMED) (grant no. 20cm0106253h0002 to TW). The present study was
also supported in part by the Grant-in-Aid for Scientific Research
(C) (grant nos. 17K11828 and 20K10111 to KAPI) and Grant-in-Aid for
Early-Career Scientists (grant no. 19K19194 to TU) from the Japan
Society for the Promotion of Science (JSPS).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SK, KAPI, TU and TW conceived and designed the
experiments. SK, TU, KK, HT, AS and KT performed the experiments.
SK, KAPI, TU and TW analyzed and interpreted the data. TI, MK and
ST interpreted the data. SK, TU and KK performed the data
acquisition. SK, KAPI, TU and TW wrote the manuscript. SK, KAPI,
TU, TI, MK, ST and TW conducted the manuscript revision/review. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experimental protocols were approved
(approval no. R-02-017-1) by the Animal Experiment Committee of the
Graduate School of Dentistry, Osaka University (Osaka, Japan). The
molecular biology experimental procedures were approved (approval
no. G2019-026C3) by the Genetically Modified Organisms Safety
Committee of Tokyo Medical and Dental University (Tokyo,
Japan).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
BMP
|
bone morphogenetic protein
|
|
BRAF
|
B-Raf
|
|
BSA
|
bovine serum albumin
|
|
CAFs
|
cancer-associated fibroblasts
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
ECD
|
extracellular domain
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
|
EMEM
|
Eagle's minimum essential medium
|
|
EMT
|
epithelial-mesenchymal transition
|
|
FBS
|
fetal bovine serum
|
|
GFP
|
green fluorescent protein
|
|
IgG
|
immunoglobulin G
|
|
MAPK
|
mitogen-activated protein kinase
|
|
PAI-1
|
plasminogen activator inhibitor-1
|
|
RT-qPCR
|
reverse transcription-quantitative
PCR
|
|
SEAP
|
secreted alkaline phosphatase
|
|
αSMA
|
α-smooth muscle actin
|
|
SM22α
|
smooth muscle protein 22α
|
|
TECs
|
tumor endothelial cells
|
|
TGF-β
|
transforming growth factor-β
|
|
TβRI
|
TGF-β type I receptor
|
|
TβRII
|
TGF-β type II receptor
|
|
TME
|
tumor microenvironment
|
|
TMEPAI
|
transmembrane prostate
androgen-induced protein
|
|
Treg
|
regulatory T cells
|
References
|
1
|
Falcone I, Conciatori F, Bazzichetto C,
Ferretti G, Cognetti F, Ciuffreda L and Milella M: Tumor
microenvironment: Implications in melanoma resistance to targeted
therapy and immunotherapy. Cancers (Basel). 12:28702020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Miyazono K, Katsuno Y, Koinuma D, Ehata S
and Morikawa M: Intracellular and extracellular TGF-beta signaling
in cancer: Some recent topics. Front Med. 12:387–411. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Heldin CH and Moustakas A: Signaling
receptors for TGF-β family members. Cold Spring Harb Perspect Biol.
8:a0220532016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dennler S, Itoh S, Vivien D, ten Dijke P,
Huet S and Gauthier JM: Direct binding of Smad3 and Smad4 to
critical TGF beta-inducible elements in the promoter of human
plasminogen activator inhibitor-type 1 gene. EMBO J. 17:3091–3100.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Watanabe Y, Itoh S, Goto T, Ohnishi E,
Inamitsu M, Itoh F, Satoh K, Wiercinska E, Yang W, Shi L, et al:
TMEPAI, a transmembrane TGF-beta-inducible protein, sequesters Smad
proteins from active participation in TGF-beta signaling. Mol Cell.
37:123–134. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Morikawa M, Derynck R and Miyazono K:
TGF-beta and the TGF-β family: Context-dependent roles in cell and
tissue physiology. Cold Spring Harb Perspect Biol. 8:a0218732016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Batlle E and Massagué J: Transforming
growth factor-β signaling in immunity and cancer. Immunity.
50:924–940. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Katsuno Y, Lamouille S and Derynck R:
TGF-β signaling and epithelial-mesenchymal transition in cancer
progression. Curr Opin Oncol. 25:76–84. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moustakas A and Heldin CH: Mechanisms of
TGFβ-induced epithelial-mesenchymal transition. J Clin Med.
5:632016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Caramel J, Papadogeorgakis E, Hill L,
Browne GJ, Richard G, Wierinckx A, Saldanha G, Osborne J,
Hutchinson P, Tse G, et al: A switch in the expression of embryonic
EMT-inducers drives the development of malignant melanoma. Cancer
Cell. 24:466–480. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Heppt MV, Wang JX, Hristova DM, Wei Z, Li
L, Evans B, Beqiri M, Zaman S, Zhang J, Irmler M, et al:
MSX1-induced neural crest-like reprogramming promotes melanoma
progression. J Invest Dermatol. 138:141–149. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Javelaud D, Alexaki VI and Mauviel A:
Transforming growth factor-beta in cutaneous melanoma. Pigment Cell
Melanoma Res. 21:123–132. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Reed JA, McNutt NS, Prieto VG and Albino
AP: Expression of transforming growth factor-beta 2 in malignant
melanoma correlates with the depth of tumor invasion. Implications
for tumor progression. Am J Pathol. 145:97–104. 1994.PubMed/NCBI
|
|
14
|
Cantelli G, Orgaz JL, Rodriguez-Hernandez
I, Karagiannis P, Maiques O, Matias-Guiu X, Nestle FO, Marti RM,
Karagiannis SN and Sanz-Moreno V: TGF-β-induced transcription
sustains amoeboid melanoma migration and dissemination. Curr Biol.
25:2899–2914. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ramont L, Pasco S, Hornebeck W, Maquart FX
and Monboisse JC: Transforming growth factor-beta1 inhibits tumor
growth in a mouse melanoma model by down-regulating the plasminogen
activation system. Exp Cell Res. 291:1–10. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schlegel NC, von Planta A, Widmer DS,
Dummer R and Christofori G: PI3K signalling is required for a
TGFβ-induced epithelial-mesenchymal-like transition (EMT-like) in
human melanoma cells. Exp Dermatol. 24:22–28. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tuncer E, Calçada RR, Zingg D, Varum S,
Cheng P, Freiberger SN, Deng CX, Kleiter I, Levesque MP, Dummer R
and Sommer L: SMAD signaling promotes melanoma metastasis
independently of phenotype switching. J Clin Invest. 129:2702–2716.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Díaz-Valdés N, Basagoiti M, Dotor J,
Aranda F, Monreal I, Riezu-Boj JI, Borrás-Cuesta F, Sarobe P and
Feijoó E: Induction of monocyte chemoattractant protein-1 and
interleukin-10 by TGFbeta1 in melanoma enhances tumor infiltration
and immunosuppression. Cancer Res. 71:812–821. 2011. View Article : Google Scholar
|
|
19
|
Okamoto H, Yoshimatsu Y, Tomizawa T,
Kunita A, Takayama R, Morikawa T, Komura D, Takahashi K, Oshima T,
Sato M, et al: Interleukin-13 receptor α2 is a novel marker and
potential therapeutic target for human melanoma. Sci Rep.
9:12812019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chapman PB, Hauschild A, Robert C, Haanen
JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et
al: Improved survival with vemurafenib in melanoma with BRAF V600E
mutation. N Engl J Med. 364:2507–2516. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hauschild A, Grob JJ, Demidov LV, Jouary
T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr,
Kaempgen E, et al: Dabrafenib in BRAF-mutated metastatic melanoma:
A multicentre, open-label, phase 3 randomised controlled trial.
Lancet. 380:358–365. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Flaherty KT, Infante JR, Daud A, Gonzalez
R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N,
et al: Combined BRAF and MEK inhibition in melanoma with BRAF V600
mutations. N Engl J Med. 367:1694–1703. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schlingensiepen KH, Schlingensiepen R,
Steinbrecher A, Hau P, Bogdahn U, Fischer-Blass B and Jachimczak P:
Targeted tumor therapy with the TGF-beta 2 antisense compound AP
12009. Cytokine Growth Factor Rev. 17:129–139. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Morris JC, Tan AR, Olencki TE, Shapiro GI,
Dezube BJ, Reiss M, Hsu FJ, Berzofsky JA and Lawrence DP: Phase I
study of GC1008 (fresolimumab): A human anti-transforming growth
factor-beta (TGFβ) monoclonal antibody in patients with advanced
malignant melanoma or renal cell carcinoma. PLoS One. 9:e903532014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jin CH, Krishnaiah M, Sreenu D,
Subrahmanyam VB, Rao KS, Lee HJ, Park SJ, Park HJ, Lee K, Sheen YY
and Kim DK: Discovery of
N-((4-([1,2,4]triazolo[1,5-a]pyridin-6-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2-yl)methyl)-2-fluoroaniline
(EW-7197): A highly potent, selective, and orally bioavailable
inhibitor of TGF-β type I receptor kinase as cancer
immunotherapeutic/antifibrotic agent. J Med Chem. 57:4213–4238.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Colak S and Ten Dijke P: Targeting TGF-β
signaling in cancer. Trends Cancer. 3:56–71. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Roopenian DC and Akilesh S: FcRn: The
neonatal Fc receptor comes of age. Nat Rev Immunol. 7:715–725.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Czajkowsky DM, Hu J, Shao Z and Pleass RJ:
Fc-fusion proteins: New developments and future perspectives. EMBO
Mol Med. 4:1015–1028. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Duivelshof BL, Murisier A, Camperi J,
Fekete S, Beck A, Guillarme D and D'Atri V: Therapeutic Fc-fusion
proteins: Current analytical strategies. J Sep Sci. 44:35–62. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Marotte H and Cimaz R: Etanercept-TNF
receptor and IgG1 Fc fusion protein: Is it different from other TNF
blockers? Expert Opin Biol Ther. 14:569–572. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takahashi K, Akatsu Y, Podyma-Inoue KA,
Matsumoto T, Takahashi H, Yoshimatsu Y, Koinuma D, Shirouzu M,
Miyazono K and Watabe T: Targeting all transforming growth factor-β
isoforms with an Fc chimeric receptor impairs tumor growth and
angiogenesis of oral squamous cell cancer. J Biol Chem.
295:12559–12572. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mizuno H, Kitada K, Nakai K and Sarai A:
PrognoScan: A new database for meta-analysis of the prognostic
value of genes. BMC Med Genomics. 2:182009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bogunovic D, O'Neill DW, Belitskaya-Levy
I, Vacic V, Yu YL, Adams S, Darvishian F, Berman R, Shapiro R,
Pavlick AC, et al: Immune profile and mitotic index of metastatic
melanoma lesions enhance clinical staging in predicting patient
survival. Proc Natl Acad Sci USA. 106:20429–20434. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pfaffl MW: Relative quantification.
Real-time PCR. Dorak MT: 1st edition. Taylor & Francis; London:
2006, https://www.taylorfrancis.com/chapters/edit/10.4324/9780203967317-12/relative-quantification-michael-pfafflPubMed/NCBI
|
|
35
|
Kawai S, Takagi Y, Kaneko S and Kurosawa
T: Effect of three types of mixed anesthetic agents alternate to
ketamine in mice. Exp Anim. 60:481–487. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kirihara Y, Takechi M, Kurosaki K,
Kobayashi Y and Kurosawa T: Anesthetic effects of a mixture of
medetomidine, midazolam and butorphanol in two strains of mice. Exp
Anim. 62:173–180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kanda Y: Investigation of the freely
available easy-to-use software ‘EZR’ for medical statistics. Bone
Marrow Transplant. 48:452–458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Krasagakis K, Thölke D, Farthmann B,
Eberle J, Mansmann U and Orfanos CE: Elevated plasma levels of
transforming growth factor (TGF)-beta1 and TGF-beta2 in patients
with disseminated malignant melanoma. Br J Cancer. 77:1492–1494.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang Y, Alexander PB and Wang XF:
TGF-beta family signaling in the control of cell proliferation and
survival. Cold Spring Harb Perspect Biol. 9:a0221452017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Penafuerte C and Galipeau J: TGF beta
secreted by B16 melanoma antagonizes cancer gene immunotherapy
bystander effect. Cancer Immunol Immunother. 57:1197–1206. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Streit M and Detmar M: Angiogenesis,
lymphangiogenesis, and melanoma metastasis. Oncogene. 22:3172–3179.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Akatsu Y, Takahashi N, Yoshimatsu Y,
Kimuro S, Muramatsu T, Katsura A, Maishi N, Suzuki HI, Inazawa J,
Hida K, et al: Fibroblast growth factor signals regulate
transforming growth factor-β-induced endothelial-to-myofibroblast
transition of tumor endothelial cells via Elk1. Mol Oncol.
13:1706–1724. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gong D, Shi W, Yi SJ, Chen H, Groffen J
and Heisterkamp N: TGFβ signaling plays a critical role in
promoting alternative macrophage activation. BMC Immunol.
13:312012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen W and Ten Dijke P: Immunoregulation
by members of the TGFβ superfamily. Nat Rev Immunol. 16:723–740.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu M, Kuo F, Capistrano KJ, Kang D, Nixon
BG, Shi W, Chou C, Do MH, Stamatiades EG, Gao S, et al: TGF-β
suppresses type 2 immunity to cancer. Nature. 587:115–120. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kudo-Saito C, Shirako H, Takeuchi T and
Kawakami Y: Cancer metastasis is accelerated through
immunosuppression during Snail-induced EMT of cancer cells. Cancer
Cell. 15:195–206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Qin T, Barron L, Xia L, Huang H,
Villarreal MM, Zwaagstra J, Collins C, Yang J, Zwieb C, Kodali R,
et al: A novel highly potent trivalent TGF-β receptor trap inhibits
early-stage tumorigenesis and tumor cell invasion in murine
Pten-deficient prostate glands. Oncotarget. 7:86087–86102. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Muraoka RS, Dumont N, Ritter CA, Dugger
TC, Brantley DM, Chen J, Easterly E, Roebuck LR, Ryan S, Gotwals
PJ, et al: Blockade of TGF-beta inhibits mammary tumor cell
viability, migration, and metastases. J Clin Invest. 109:1551–1559.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yung LM, Nikolic I, Paskin-Flerlage SD,
Pearsall RS, Kumar R and Yu PB: A selective transforming growth
factor-β ligand trap attenuates pulmonary hypertension. Am J Respir
Crit Care Med. 194:1140–1151. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mohammad KS, Javelaud D, Fournier PG,
Niewolna M, McKenna CR, Peng XH, Duong V, Dunn LK, Mauviel A and
Guise TA: TGF-β-RI kinase inhibitor SD-208 reduces the development
and progression of melanoma bone metastases. Cancer Res.
71:175–184. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Javelaud D, Alexaki VI, Dennler S,
Mohammad KS, Guise TA and Mauviel A: The TGF-β/SMAD/GLI2 signaling
axis in cancer progression and metastasis. Cancer Res.
71:5606–5610. 2011. View Article : Google Scholar : PubMed/NCBI
|