Introduction
Cancers arise as a result of genetic changes in the
genome such that cancer genomes contain inherited and sporadic
nucleotide sequence changes. In addition to hereditary
predisposition, direct mutational events and/or epigenetic
modifications initiate changes in signalling pathways that can
result in cellular transformation (1). At a molecular level, cancer development
is a multistep process with alterations in the expression of three
gene classes; namely oncogenes, tumour suppressor genes and
stability genes (2,3). Overexpression of oncogenes and loss of
tumour suppressor gene functions result in constitutive activation
of pathways under conditions in which the wild-type gene is not
active (4,5). By contrast, mutations in stability genes
increase genetic instability, resulting in inefficient DNA repair
mechanisms and cell cycle control (6,7).
The DNA sequence changes may include insertions and
deletions (InDels), gene duplication, genomic rearrangements, loss
of heterozygosity in somatic cells or inherited mutations (8). Given that InDels are more likely to be
driver gene mutations, a substantial number of InDels in the coding
DNA sequence is not surprising, since InDels are often deleterious
as evidenced by their frequent association with human disease
(9,10). InDels have also been shown to be
components of gene and pseudogene evolution that are important in
the long-term evolution of genome size (11). Thus, the abundance of InDels, which
have a comparatively greater influence than single nucleotide
polymorphisms (SNPs), is consistent with the abnormal function and
devastating characteristics of cancer (10).
Our previous Whole Genome Sequencing (WGS) study in
patients with oesophageal squamous cell carcinoma (OSCC) revealed
large scale deletions of the BCL2 associated transcription factor 1
(BCLAF1) gene (12).
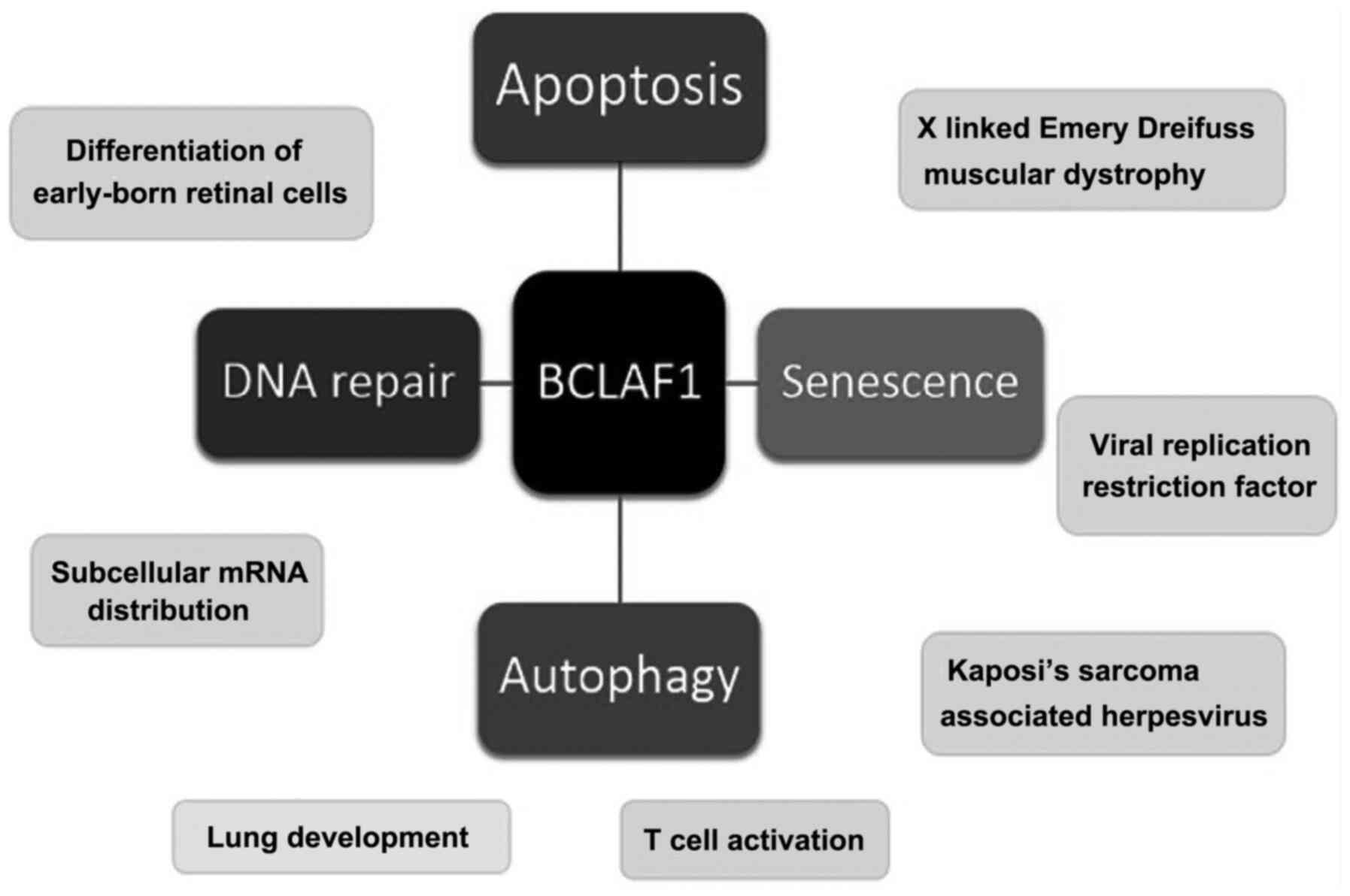
BCLAF1 encodes a multifunctional protein with four splice
variants that perform a wide range of seemingly unrelated functions
(13). Some of these functions
include tumour suppression (through promoting apoptosis,
senescence, DNA repair and autophagy), embryonic development, T
cell maturation and mRNA processing, as illustrated in Fig. 1.
BCLAF1 was originally identified in a yeast
protein screen for E1B 19K interacting factors. E1B 19K is a
homolog of anti-apoptotic BCL-2 family members and inhibits cell
death signal transduction cascades activated by adenoviral proteins
(13,14). Overexpression of BCLAF1 induces
apoptosis, suggesting that BCLAF1 is a pro-apoptotic factor
with potential tumour suppressor activity (13). However, the role of BCLAF1 as a
tumour suppressor gene that promotes apoptosis and autophagy is
debatable due to conflicting lines of evidence (15,16).
Despite this, BCLAF1 mutations and loss of gene expression
have been implicated in a range of cancers, including oesophageal
cancer, non-Hodgkin's lymphoma, Burkitt's lymphoma, colon cancer,
bladder cancer and rectal cancer (17). Understanding of the genomic
abnormalities in OSCC is very limited, hence the need to identify
genomic abnormalities underlying OSCC and to elucidate the
molecular basis of the disease in order to guide the development of
effective targeted therapies (18,19).
Although, BCLAF1 deletions have been observed
in a number of tumour types, very little is known concerning
BCLAF1 expression patterns in human cancers and the effects
of its deficiency on gene expression and tumourigenesis (13,20). The
present study aimed to identify structural variants (SVs),
primarily large InDels, and the downstream molecular mechanisms
involved in cellular transformation following the loss of
BCLAF1 gene expression in a cultured cell line model.
Materials and methods
Sample collection
Patients were recruited at Groote Schuur Hospital,
University of Cape Town (Cape Town, South Africa) in the
gastrointestinal clinic between January 2010 and December 2021.
Written informed consent was obtained from all patients before
recruitment into the study. Patient ages ranged between 40 and 80
years, and the ratio of female to male patients was 60:40. Tumours
were identified by endoscopic examination and biopsy after staining
with acetic iodide solution. Normal biopsies were taken at least 10
cm from the tumour. Patients who had previously received any form
of chemo- or radiotherapy were excluded from the study (21). Ethics approval was obtained from the
University of Cape Town/Groote Schuur Hospital Human Research
Ethics Committee (Cape Town, South Africa; approval no.
HEC040/2005), and all methods were performed in accordance with the
Declaration of Helsinki.
DNA extraction from tissue
biopsies
Frozen tissue was thawed on ice and placed into a
sterile 1.5 ml microcentrifuge tube containing 0.5 ml QIAzol Lysis
Reagent™ (Qiagen, Inc.). The tip of a disposable probe was then
placed in the tube and homogenised at full speed until the tissue
lysate was uniformly homogeneous (1–5 min). An additional 0.5 ml
QIAzol Lysis Reagent was added and mixed by vortexing for 30 sec,
after which the samples were incubated for 5 min at room
temperature (20–25°C) to permit the complete dissociation of
nucleoprotein complexes. A total of 0.2 ml chloroform per 1 ml
QIAzol Lysis Reagent was then added to each tube, capped and shaken
vigorously by hand for 15 sec and incubated at room temperature for
2–3 min. The samples were centrifuged at 12,000 × g for 15 min at
4°C to separate the mixture into three phases: A colourless upper
aqueous phase containing RNA, a white middle interphase containing
DNA and a lower organic phenol phase containing protein. The upper
aqueous phase was transferred to a new tube and stored at 4°C for
later RNA extraction. A total of 0.3 ml 100% ethanol was then added
to the interphase and mixed by inversion followed by incubation on
ice for 30 min. The samples were then centrifuged at 14,000 × g for
5 min at 4°C to pellet the DNA, the ethanol was discarded and the
pellet was rinsed in 1 ml 75% ethanol. The samples were centrifuged
at 14,000 × g for 5 min at 4°C, and the supernatant was carefully
removed and the DNA pellet was air dried for 5–15 min. The DNA
pellet was dissolved in 200 µl 10 mM Tris/1 mM EDTA and quantitated
on a NanoDrop 2000/2000c Spectrophotometer (NanoDrop Technologies;
Thermo Fisher Scientific, Inc.). Samples were stored at −20°C until
required.
Variant calling
Four OSCC DNA biopsy samples were submitted to the
Centre for Proteomic and Genomic Research (Cape Town, South Africa)
for paired-end sequencing on the Illumina HiSeq2000 (Illumina,
Inc.) using TruSeq SBS chemistry v3. or WGS. Quality control (QC)
measures were carried out to validate the integrity of the samples
for WGS with the Quant-iT™ PicoGreen™ dsDNA kit (cat. no. P7589;
Invitrogen; Thermo Fisher Scientific, Inc.). Paired-end sequencing
was performed on the Illumina HiSeq2000, with 2×100 bp reads. Reads
were aligned to the NCBI37 Homo sapiens reference genome using
ELAND and CASAVA version 1.8.2 software (22). Since matched normal samples were not
included, variants with global allele frequency >10% in either
the Exome Variant Server or 1000 Genomes Project were filtered out
to reduce background noise. SVs reported from the alignment were
collated with gene loci, (upstream, downstream and overlapping)
using the Variant Effect Predictor of Ensembl (23). The samples were genotyped on an
Illumina HumanOmni2.5–8 array (Illumina, Inc.) and the affected
genes were subsequently cross-checked against the Genetic
Association Database for disease and cancer associations (24). The deleted genes common to all the
four tumour samples were described in HUGO Gene Nomenclature
Committee (HGNC) (25) and Medical
Subject Headings (MeSH) gene and disease terms, respectively
(Courtesy of the U.S. National Library of Medicine).
WGS
DNA isolated from tumour biopsies obtained from
patients with oesophageal squamous cell carcinoma (OSCC) were sent
to the Centre for Proteomic and Genomic Research (CPGR), Cape Town,
South Africa for WGS. The samples were taken from the Black and
Mixed Ancestry population groups since these are the population
groups most affected by OSCC in South Africa. The Quant-iT™
PicoGreen™ dsDNA kit (cat. no. P7589; Invitrogen; Thermo Fisher
Scientific, Inc.) was used to determine DNA integrity and 10 nM DNA
sample was used for sequencing. Paired-end sequencing was performed
on the Illumina HiSeq2000, with 2×100 bp reads. PCR-free paired end
libraries were manually generated from 500 ng 1 µg genomic DNA
using a modified version of TruSeq DNA v2 Sample Preparation kits
(cat. no. FC-121-2001; Illumina, Inc.). Fragmentation was performed
using Covaris LE220 (Covaris, Inc.). After fragmentation and end
repair, libraries were manually size-selected using bead-based size
selection targeting 300 bp inserts (Agencourt AMPure XP A63881;
Thermo Fisher Scientific, Inc.). Following size selection,
libraries were A-tailed, and ligated before proceeding to library
QC. Final Libraries were quality checked on a Caliper GX
(PerkinElmer, Inc.) and quantified by reverse
transcription-quantitative (RT-q)PCR on the Roche LightCycler 480
(Roche Diagnostics). Samples were clustered on three lanes with the
Illumina v3 flowcells (Illumina, Inc.) using the Illumina cBot
(Illumina, Inc.) with the TruSeq Paired End Cluster Kit (v3).
Flowcells were sequenced as 100 bp-end reads on the Illumina
HiSeq2000 using TruSeq SBS chemistry v3. DNA sequences were
compared to the human reference genome (NCBI37), to perform a
preliminary screen for InDels.
Maintenance of cells in culture
CT1 cells were obtained from Dr Namba and cultured
as previously described (26) were
cultured in complete medium [Dulbecco's modified Eagle's medium
(DMEM; Sigma-Aldrich; Merck KGaA) supplemented with 10% heat
inactivated foetal bovine serum (FBS; Sigma-Aldrich; Merck KGaA)
and 100 U/ml penicillin and 100 mg/ml streptomycin] at 37°C in a
humidified incubator with 5% CO2. Every 2 days, the
medium was changed and the cells were sub-cultured when they
reached 90% confluency, by detaching the cells with 0.05%
trypsin-EDTA. Once detached, the trypsin-EDTA was inactivated by
the addition of 5 ml complete DMEM. Cells were collected by
centrifugation at 3,000 × g at 4°C for 5 min, suspended in complete
DMEM and re-plated at a ratio of 1:3. Cells were regularly checked
for mycoplasma contamination as previously described (27).
Small interfering (si)RNA
transfection
Transient transfection with siRNA was used to knock
down the expression of BCLAF1. Briefly, 2×105
cells were plated in a six-well dish (35 mm per well) with complete
medium and incubated overnight at 37°C, in a humidified 5%
CO2 incubator. The following day, the cells were washed
once with 2 ml DMEM, then transfected using TransFectin™ Lipid
Reagent (Bio-Rad Laboratories, Inc.), according to the
manufacturer's instructions. Two transfection mixtures were
prepared separately for each siRNA; the first mixture contained 1.5
µl (75 pmoles) siRNA in 50 µl DMEM and the second mixture had 10 µl
TransFectin Lipid Reagent in 45 µl DMEM. After combining the
mixtures and incubating them at room temperature for 45 min, the
combined mixture was pipetted drop-wise into each well. The plates
were incubated at 37°C in a humidified atmosphere of 5%
CO2 for 5–7 h, after which, 1.750 ml DMEM containing 10%
FBS, 50 U/ml penicillin and 50 µg/ml streptomycin was added and the
cells incubated for an additional 48 h. Untransfected cells and a
non-targeting siRNA were used as controls. Each experiment was
conducted in triplicate. The siRNA duplex sequences are listed in
Table SI.
RNA preparation and RT-qPCR
Total RNA was extracted as previously described
using TRIzol® reagent (Thermo Fisher Scientific, Inc.)
(27). cDNA was prepared from 2 µg
total RNA using the ImProm-II™ Reverse Transcription System
(Promega Corporation), according to the manufacturers'
instructions. RT-qPCR assays were performed using SYBR®
FAST qPCR kit (Roche Diagnostics) in a reaction volume of 20 µl.
All amplifications were performed as follows: Initial denaturation
at 95°C for 5 min, followed by 40 cycles at 95°C for 30 sec, 60°C
for 20 sec and 72°C for 5 sec. Analysed genes were amplified in
triplicate using the LightCycler® 480II (Roche
Diagnostics) and normalized to GAPDH expression in the same sample
using the 2−ΔΔCq method (28). A list of the primers used and their
annealing temperatures are shown in Table SII.
Western blot analysis
Control cells and cells transfected with siRNA were
lysed in RIPA buffer [10 mM Tris-HCl pH 7.6, 10 mM NaCl, 3 mM MgCl2
and 1% (v/v) Nonidet P-40 containing 50 µg/ml each of pepstatin,
leupeptin and aprotinin]. Total protein concentration was
determined using the Bradford assay (Bio-Rad Laboratories, Inc.).
Total cell lysates (50 µg protein) were separated by
electrophoresis on 12% SDS-PAGE gels under reducing conditions (50
mM β-mercaptoethanol). After electrophoresis, proteins were
transferred to nitrocellulose membranes and blocked in 5% fat-free
milk in Tris-buffered saline (TBS) containing 0.1% Tween-20 with
shaking for 1 h at room temperature. The membranes were incubated
overnight at 4°C with one of the following primary antibodies
(1:1,000; all purchased from Abcam): Rabbit polyclonal anti-BCL2
associated transcription factor 1 (BTF; cat. no. ab107177), rabbit
polyclonal anti-caspase-3 (phospho S150; cat. no. ab59425), rabbit
polyclonal anti-Caspase-9 (cat. no. ab209495), rabbit monoclonal
anti-BAX (cat. no. ab182733), rabbit polyclonal anti-Bcl-2 (cat.
no. ab115807), rabbit polyclonal anti-Exonuclease 1 (EXO1; cat. no.
ab106303), rabbit polyclonal anti-γ H2AX (phospho S139; cat. no.
ab11174), rabbit polyclonal anti-p53 (cat. no. ab17990) and rabbit
polyclonal anti-p21 (cat. no. ab219811). After several washes in
TBS with 0.1% Tween-20 buffer, the nitrocellulose membranes were
incubated by shaking at room temperature for 1 h with the required
horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary
antibody (1:3,000; cat. no.1706515; Bio-Rad Laboratories, Inc.) and
detected using LumiGLO® substrate (KPL, Inc.).
Soft agar assay
The anchorage dependence and colony formation
ability of the cells was assessed using a soft agar assay. Cells
(2×105) were transfected with 75 ρmoles siRNA. Following
transfection, cells were washed with 2 ml PBS and trypsinised with
500 µl 0.5% trypsin-EDTA. Then, 500 µl DMEM was added to inactivate
trypsin. The cells were centrifuged at 3,000 × g at 4°C for 5 min
and suspended in 1 ml DMEM. A base agar was prepared by mixing 1%
agarose gel at 40°C with 2X DMEM (20% FBS and 2% P/S). Following
which, 1 ml base agar was added to each well of a 6-well plate and
gently swirled to cover the surface. The agar was allowed to set
for 30 min at room temperature. Top agar was prepared by dissolving
0.7% agarose in a microwave, then cooling it to 40°C. A suspension
of 2,500 cells in 0.1 ml was mixed with 3 ml 0.7% agar and 3 ml 2X
DMEM. Then, 1 ml cell suspension was pipetted onto the base agar in
triplicate. The cells were incubated at 37°C in a humidified 5%
CO2 incubator for 21 days. The cells were fed twice a
week with 1 ml complete medium. After 21 days, 0.5 ml 0.005%
crystal violet (Sigma-Aldrich; Merck KGaA) was added for 1 h and
pink stained colonies were counted in 15 different fields using an
Olympus CKY41 light microscope (Olympus Corporation) at ×100
magnification. ImageJ (version 1.53i) software (National Institutes
of Health) was used for semi-quantification of the number of
colonies >150 µm in diameter.
Statistical analysis
The data are presented as the mean ± SEM. All
experiments were performed in triplicate unless stated otherwise.
Statistical analysis of the data was performed using GraphPad Prism
5 (GraphPad Software, Inc.). A two tailed unpaired Student's t-test
was performed. P<0.05 was considered to indicate a statistically
significant difference.
Results
SVs in OSCC
WGS data generated 4.27 billion reads of 100 bp
covering at least 95% of the human reference genome. QC indicators,
such as the duplication rate, coverage and mean depth of coverage,
indicated that the experiments were successful in acquiring
high-quality sequence reads (Table
SIII). The minimum depth of coverage was 33.7, and the library
preparation was successful across all samples without being
compromised by PCR amplification bias after having obtained a 98%
unique read proportion (Non-N reference) to the human reference
genome (NCBI37).
The samples were genotyped on an Illumina
HumanOmni2.5–8 array, and had a high quality score with a
percentage base calling of >Q30 indicating a smaller probability
of error. Fig. S1A shows that sample
B381 had 89.0% of filtered and aligned reads with a mean depth of
coverage of 40.7, while Fig. S1B
shows that sample B450 had 90.7% of filtered and aligned reads with
a mean depth of coverage of 35.0. Sample M456 had 89.3% of filtered
and aligned reads with a mean depth of coverage of 33.6 (Fig. S1C). Whereas, sample M478 had 88.3% of
filtered and aligned reads with a mean depth of coverage of 34.2
(Fig. S1D). Thus, the sequencing
phase of this experiment achieved high quality reads that allowed
for greater confidence in the variant calling procedure and
results.
Alignment and variant calling
Illumina's proprietary alignment algorithm ELAND and
CASAVA 1.8.2 software were used to align the reads to the NCBI37
human reference genome (22). As
matched normal samples were not included, variants with global
allele frequency >10% in either the Exome Variant Server or 1000
Genomes Project were filtered out to reduce background noise
(29,30). SVs reported from the alignment were
collated with gene loci (upstream, downstream and overlapping)
using the variant effect predictor of Ensembl (23). The affected genes were subsequently
cross-checked against the Genetic Association Database for disease
and cancer associations (24). A
large number of SNP effects from all four samples were observed
(Table I), having 98% mean array
agreement between the samples and the reference genome NCBI37 of
which 38% were associated with Genes.
 | Table I.Summary of SNP consequences across
all four tumour samples. |
Table I.
Summary of SNP consequences across
all four tumour samples.
|
| Tumour samples |
|---|
|
|
|
|---|
| Variant
effects | B381 | B450 | B456 | M478 |
|---|
| Array agreement,
% | 98.5 | 99.2 | 97.6 | 99.2 |
| SNP total, n | 4,919,446 | 5,334,173 | 4,185,808 | 5,018,129 |
| Genes, % | 38.9 | 38.9 | 38.8 | 39.1 |
| Exons, % | 1.6 | 1.6 | 1.6 | 1.6 |
| Ti/Tv, n | 2.1 | 2.1 | 2.1 | 2.1 |
| Novel, n | 591,283 | 617,474 | 476,394 | 656,181 |
| Ti/Tv novel, n | 1.5 | 1.6 | 1.4 | 1.7 |
| Synonymous, n | 14,266 | 15,310 | 11,750 | 14,562 |
| Missense, n | 13,349 | 14,556 | 11,243 | 13,660 |
| Nonsense, n | 140 | 144 | 118 | 135 |
| InDel total, n | 1,308,522 | 1,358,218 | 1,153,901 | 1,303,928 |
| Frameshift InDel,
n | 417 | 431 | 378 | 446 |
| Splice
regiona, n | 1,819 | 1,892 | 1,450 | 1,857 |
The transition/transversion (Ti/Tv) ratio has been
used in multiple studies for assessing the specificity of SNP
calls, with an empirical Ti/Tv ratio of ~2.1 for genome-wide
variants (31–33). Ti/Tv ratio is computed as the number
of transition SNPs divided by the number of transversion SNPs
(34). Typically, the Ti/Tv ratio is
lower in novel SNPs than in known SNPs because of residual false
positives and a relative deficit of transitions due to sequencing
context bias (31). All the samples
in the present study had a Ti/Tv ratio of 2.1, thus further
validating the accuracy of SNP calling. The SNP sequence variation
in the chromosome showed 90% call accuracy to the reference genome
(NCBI37) across all the four samples, which was replicated with the
autosomal, mitochondrial and the X-specific sites (data not
shown).
The most prevalent SVs were insertions, deletions
and copy number variation. Very few insertions were observed in all
the four samples compared to deletions and copy number variations,
which were common (data not shown). This study concentrated on
identifying large SVs (>300 bp) and the role that they could
play in OSCC. Since no common insertions were identified in all
four samples, the evaluation of insertions was not explored further
in this study.
Effects associated with deletions
A total of 697 deletions were observed in all four
samples of which 349 within genes and 127 were associated with
various cancers (Table II). Only 12
genes were shown to have deletions in the coding region or were
associated with OSCC (Table III).
Among the twelve genes; alcohol dehydrogenase 4 (ADH4),
EGFR, muts homolog 3 (MSH3) and BCLAF1 have
previously been associated with cancer (35,36), but
only BCLAF1 was identified in the present study as a
possible noteworthy SV owing to its novel association with
OSCC.
| Table II.Summary of deletions common to all
four tumour samples that shows the breakdown of the total number of
deletions described in the samples. |
Table II.
Summary of deletions common to all
four tumour samples that shows the breakdown of the total number of
deletions described in the samples.
| Deletions | Common in all four
samples, n |
|---|
| Number of common
deletions | 697 |
| Deletions
associated with genes | 349 |
| Deletions
associated with cancer | 127 |
| Table III.Description of gene deletions common
to all four tumour samples. |
Table III.
Description of gene deletions common
to all four tumour samples.
| Genes (HGNC) | Location | Variant
consequence | MeSH disease
terms | Percentage protein
coding overlap, % |
|---|
| NXPE1 | Chr.11 | Non-coding exon
variant | Ulcerative colitis;
inflammatory bowel disease | 0.66 |
| DLEU1 | Chr.13 | Non-coding exon
variant | Chronic lymphocytic
leukaemia | 0.32 |
| TMBIM4 | Chr.12 | 3′UTR variant | Venezuelan
haemorrhagic fever; Bolivian haemorrhagic fever | 0.73 |
| ADH4 | Chr.4 | Upstream gene
variant | Adenocarcinoma;
oesophageal neoplasm; Parkinson's disease | 0.12 |
| HYDIN | Chr.16 | Coding sequence
variant | Primary ciliary
dyskinesia 5; Hydin-related primary ciliary dyskinesia | 0.49 |
| KCNJ12 | Chr.17 | Frameshift variant
3′UTR | Hypokalemic
periodic paralysis | 3.53 |
| BCLAF1 | Chr.6 | Coding sequence
variant | Non-Hodgkin's
lymphoma; colorectal cancer | 53.20 |
| CACNA1B | Chr.9 | Coding sequence
variant | Pulmonary
aspergilloma | 0.34 |
| EGFR | Chr.7 | Intron variant | Oesophageal
neoplasms; colonic neoplasms; cervical squamous cell carcinoma | 0.24 |
| FAM118A | Chr.22 | Coding sequence
variant | Cardiovascular
diseases; Diabetes Mellitus, Type 2 | 1.70 |
| OR6K5P | Chr.1 | Non-coding exon
variant | Neuronitis | 25.35 |
| MSH3 | Chr.5 | Intron variant | Adenocarcinoma;
oesophageal neoplasm; colonic neoplasms | 0.31 |
The BCLAF1 deletion had previously been shown
to be associated with Non-Hodgkin's lymphoma and colorectal cancer
(13,17). In the current study, the BCLAF1
deletion spanned 53.20% of the protein coding region as opposed to
the other SVs and was proposed to have significant implications for
the progression of OSCC.
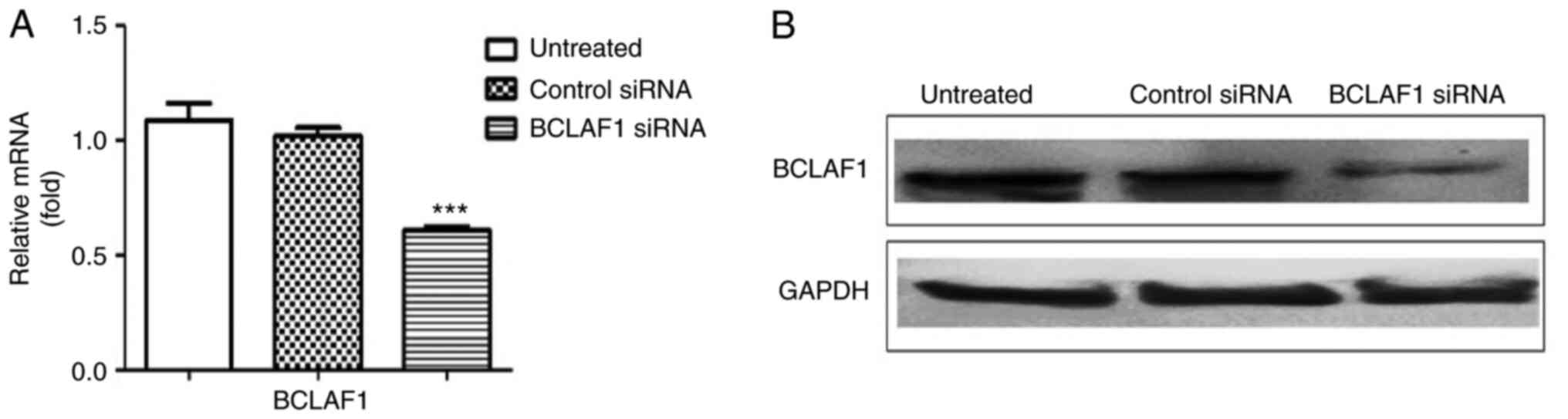
siRNA-mediated BCLAF1 silencing
siRNA-mediated knockdown of BCLAF1 was
determined at both the protein and mRNA levels. A decreased
expression of BCLAF1 expression at both the mRNA and protein
levels was observed after transfection with BCLAF1-siRNA
(Figs. 2A and B and S2A).
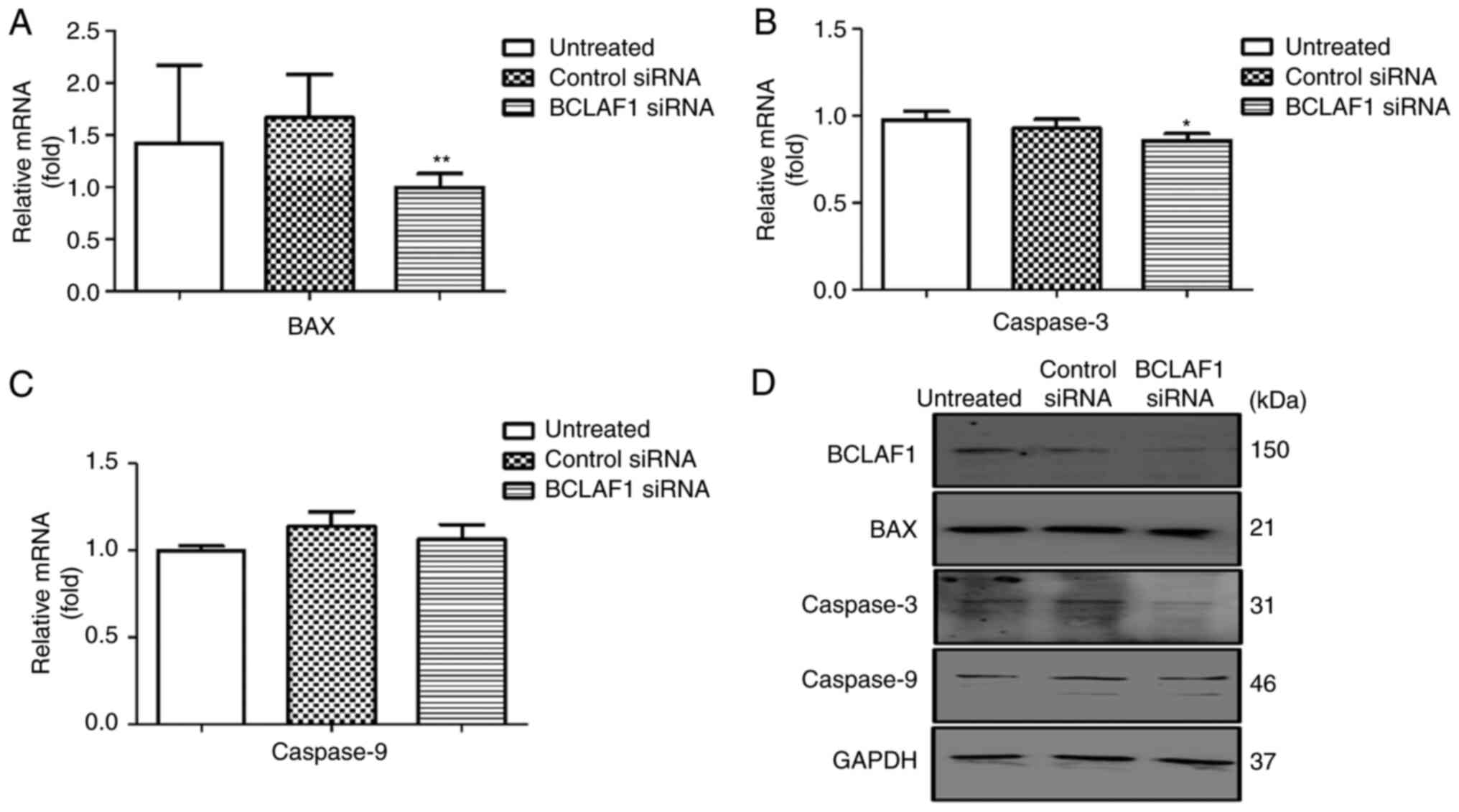
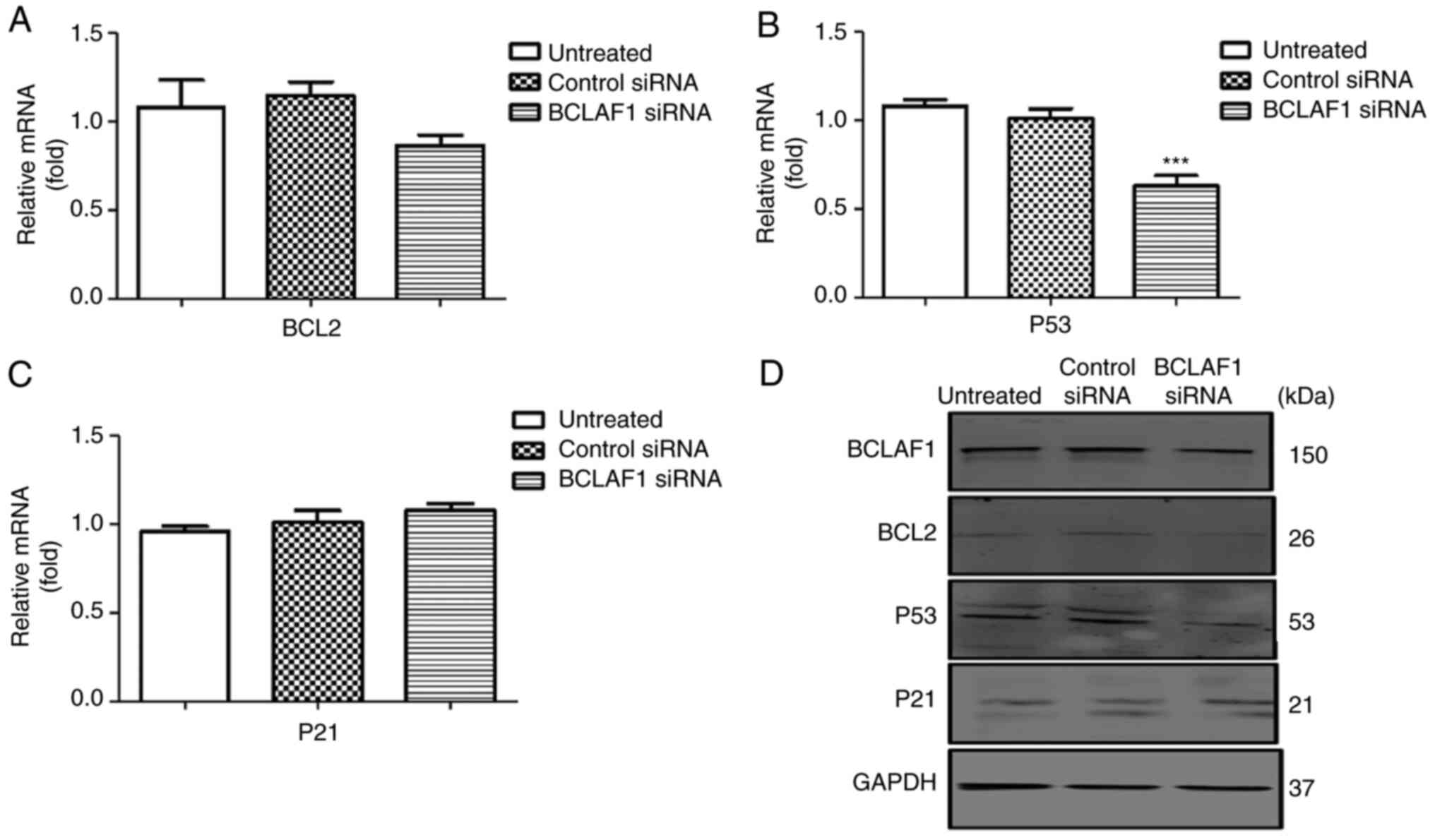
Effect of BCLAF1 silencing on cellular
gene expression
To assess whether the expression of genes downstream
of BCLAF1 was altered by the targeted disruption of
BCLAF1 expression, RNA and protein extracts from cells
transfected with either non-specific scrambled control siRNA,
BCLAF1 siRNA and untreated cells were collected 24 h
post-transfection. RT-qPCR and western blot analyses were performed
to determine the effects of BCLAF1 deficiency on
pro-apoptotic, anti-apoptotic, DNA damage response (DDR) genes and
cell cycle regulators. BCLAF1 knockdown resulted in a
significant decrease in the expression of the apoptotic genes, such
as BAX and Caspase-3, gene expression (Figs. 3A-D and S2B-D and Table
SIV). Analysis of both the protein and mRNA levels showed no
significant effect on the expression of the anti-apoptotic gene
BCL2, but a significant effect was observed on the expression of
cell cycle regulators such as P53 (Figs.
4A-D and S2E and H and Table SIV). Depletion of BCLAF1 also
resulted in reduced expression levels of EXO1, ATRIP and BACH1 mRNA
and the slight attenuation of EXO1 and H2AX protein levels
(Figs. 5A-F and S2F and G and Table SIV).
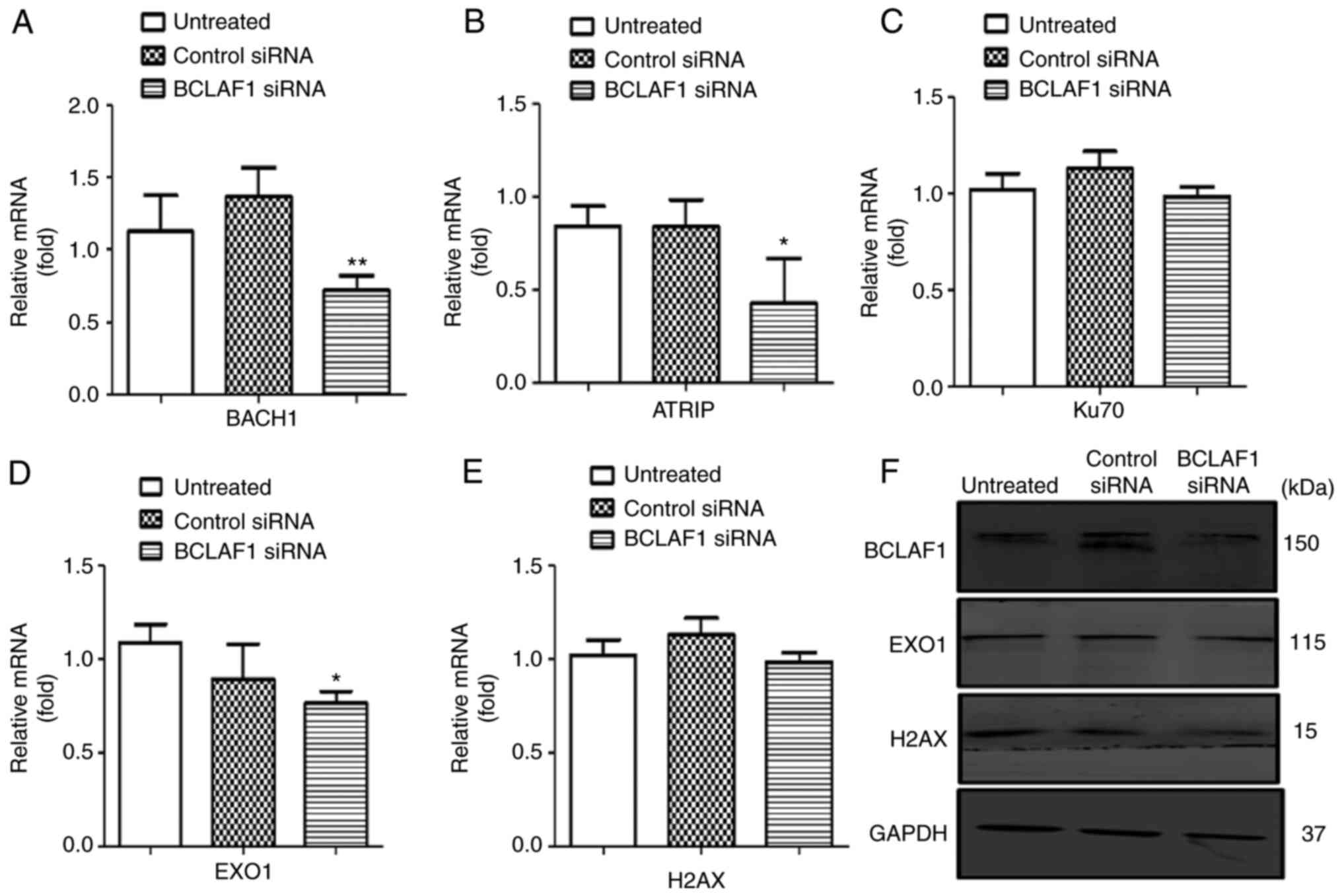 | Figure 5.Effect of BCLAF1 knockdown on
double strand DNA repair gene expression. (A-E) Reverse
transcription-quantitative PCR analysis of the DDR genes BACH1,
ATRIP, Ku70, H2AX and EXO1. (F) Representative western blot
analysis of DDR proteins, including BCLAF1, EXO1 and H2AX in
BCLAF1 siRNA-treated cells. Separate amplification of GAPDH
served as a housekeeping gene to control for loading. Each
experiment was performed in triplicate. *P<0.05, **P<0.01 vs.
control siRNA. DDR, DNA damage response; BCLAF1, BCL2 associated
transcription factor 1; siRNA, small interfering RNA; BACH1,
transcription regulator protein BACH1; ATRIP, ATR-interacting
protein; EXO1, exonuclease 1; Ku70, 70 kDa subunit of Ku
antigen. |
Effect of BCLAF1 silencing on
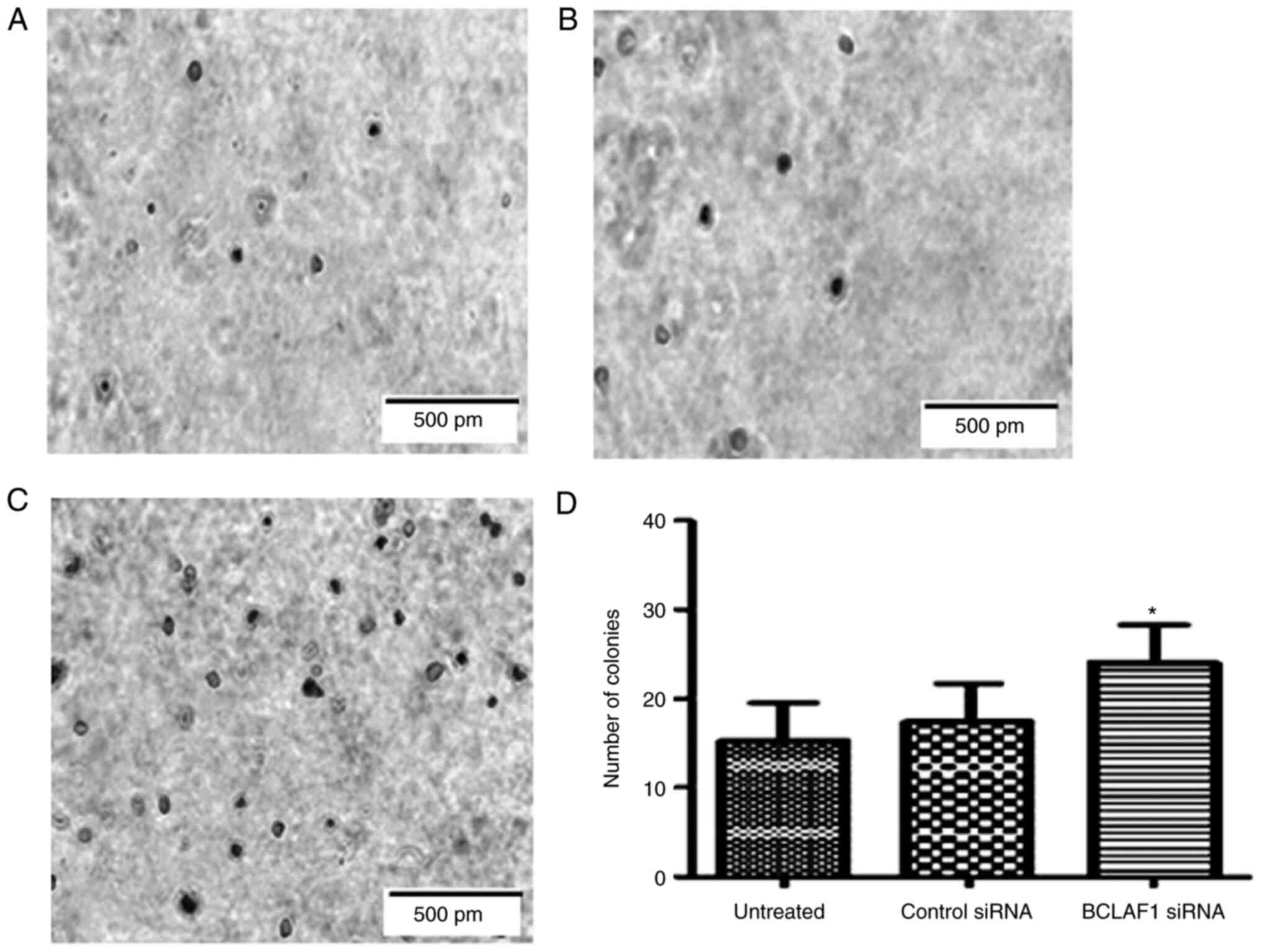
anchorage-dependent growth
To determine the effect of BCLAF1 on
anchorage-dependent growth and cellular transformation, soft agar
colony forming assays were performed. Normal cells are anchorage
dependent and undergo apoptosis in the absence of a binding
substrate. Transformed cells on the other hand, proliferate to form
colonies (27). After incubation for
21 days, colonies were stained with 0.005% crystal violet,
visualised and counted. There was a notable increase in size and
number of colonies following BCLAF1 silencing compared with
the controls (Fig. 6A-D).
Discussion
Cancer-derived cell lines are generally used as
models in the study of cancer, but there is no perfect cell line
model due to a number of well-known limitations (37–42).
Although cancer research is commonly conducted in epithelial cells,
tumours are heterogeneous and the tumour microenvironment aids
carcinogenesis (43). Due to the
ability of immortalized cells to surpass replicative barriers in
culture, they constitute a robust model system that can be used to
study human cell transformation (43,44). CT1
fibroblasts were used as a model cell line in the present study to
validate the effects of BCLAF1 knockdown on cellular
transformation. The CT1 cells used in this study did not contain
any p53 mutations and were thus suitable for studying the
relationship between cell immortalization and changes in p53 gene
expression (26,45) as opposed to human cell lines that are
immortalized by treatment with oncogenic DNA viruses, such as SV40,
papilloma and adenoviruses, since these viruses bind to and
inactivate the proteins, such as p53 and Rb (46,47).
In the present study, four OSCC samples were
subjected to WGS to identify large SVs (InDels) that may be
associated with OSCC. A total of 4.27 billion reads of 300 bp
fragments were generated covering 95% of the human reference genome
(NCBI37) at a mean read depth of 33.7. This allowed for the
reliable calling of SNPs and SVs across the human reference genome
(NCBI37) and agreed with previous studies that also showed that an
average read depth that exceeded 30X was the de facto
standard for achieving reliable variant calling across 95% of the
reference genome (48,49).
The Ti/Tv ratio is also a critical metric for
assessing the specificity of SNP calling (41) and has been used as a quality control
parameter for assessing the overall SNP quality in multiple studies
with the expected Ti/Tv ratios in WGS being 2.1, generally
indicating higher accuracy in variant calling (31,33). The
present results showed a Ti/Tv ratio of 2.1 across all four
samples, further confirming the reliability of these SNPs and SV
calling.
SVs in the human genome play an important role in
the genetics of complex diseases (1,48).
Although the analysis of the present study reduced the number of
variants of specific interest from the pool of SVs to a select few,
large numbers of potentially functional alterations that could
contribute to OSCC susceptibility were not investigated. From the
12 potential genes that were common in all four samples, only three
genes have been previously shown to be associated with OSCC;
ADH4, EGFR, MSH3 (17,34,35,50–52)
and one novel gene BCLAF1 that has not been reported in OSCC
to date.
A reduction in ADH4 expression in poorly
differentiated OSCC contributes to the maintenance of the tumour
state by inhibiting the retinoic acid signalling pathway (53), while mutations in EGFR have
also been shown to occur in primary OSCC (54,55). Other
studies by Vogelsang et al (36,56) also
showed that MSH3 polymorphisms, together with exposure to
tobacco smoking increased the risk of oesophageal cancer. Although
these genes (ADH4, EGFR and MSH3) had previously been
associated with OSCC, we did not analyse them further for
functional significance in the current study.
The BCLAF1 deletion on the other hand, is a
notable SV due to its novel association with OSCC. Although the
exact molecular role of BCLAF1 remains to be elucidated,
BCLAF1 has been shown to have functional connections and
mutation patterns consistent with the known hallmarks of cancer
(20,57) suggesting BCLAF1 as a
contributor to the progression of OSCC. Although there are
currently no extensive studies depicting the role of BCLAF1
deletion in OSCC, previous studies have shown a role for BCLAF1
deletion in a number of cancers other than OSCC. Ungerbäck et
al (51) identified BCLAF1
deletion as among the tumour suppressor candidate genes that may be
responsible for or a contributing factor to colorectal cancer.
Other cancers also linked with the loss of BCLAF1 include
hepatocellular carcinoma (58),
leukaemia (59), lung cancer
(60), bladder cancer (15) and colon cancer (20,61).
BCLAF1 has also been shown to directly
interact with and activate P53 gene transcription and that
silencing of BCLAF1 reduces P53-dependent apoptosis,
thus implicating P53 as a downstream target of BCLAF1
(62). This is also supported by
other studies that identified P53 as among the significantly
mutated genes in OSCC (63,64). DDR pathways protect cells from
extensive DNA damage and cellular transformation (51,57,65). The
network of DDR pathways consists of homologous recombination,
mismatch repair, non-homologous end joining and double strand break
repair (66). Since there is
redundancy in these repair mechanisms and the genes are expressed
at low levels in the absence of external DNA damaging agents
(5,67), the expression levels of genes that are
essential to these processes, including H2AX, BACH1, Ku70, EXO1 and
ATRIP, were investigated.
BCLAF1 silencing reduced ATRIP expression in
the present study, this was expected due to decreased
BCLAF1/BRCA1 complex formation (65). BCLAF1 silencing had no
significant impact on EXO1 expression, but then transcription
factors such as MYC and E2F1 also positively regulate the EXO1
promoter (68). Additionally, the
RAS/PI3K/AKT signalling pathways induce EXO1 gene expression
(69). The activation of these
pathways in the transformed cells could compensate for the
decreased BCLAF1 levels. Thus, the BCLAF1 knockdown
was insufficient to cause a significant change in EXO1
expression.
The siRNA-mediated silencing of BCLAF1 also
altered the expression of important DNA damage response regulators,
such as ATRIP, H2AX and EXO1. In the event of replicative stress,
cells activate several genes to halt cell cycle progression and
proliferation. P53 and P21 are potent inhibitors of G1/S phase
progression through their interaction with CDK2 and CDK4/6
(70,71). In the G1 phase cell cycle arrest, P53
triggers transcription of P21 (cip1/waf1) a key component in cell
cycle regulation (72). The present
study confirmed that silencing BCLAF1 significantly
attenuated P53 expression, which was in agreement with the results
on BCLAF1 deficiency and downregulation of Caspase-3 and BAX
protein levels.
Caspases regulate the intrinsic and extrinsic
apoptosis pathways, resulting in DNA fragmentation, cytoskeletal
breakdown and protein degradation (73,74). This
current study showed that the expression of Caspase 3 was decreased
after knockdown of BCLAF1 expression. This is likely
mediated via the resulting increase in anti-apoptotic Ku70-Bax
complexes that causes inhibition of Caspase-3 expression and
activation (16). Thus, apoptosis
resistance and cell survival are enhanced in siBCLAF1
treated cells. The BCL-2 family of genes are essential in apoptosis
regulation and are either pro-apoptotic (Bax, Bad, Bak) or
anti-apoptotic (BCL-2, BCL-XL) (75).
The specific underlying mechanisms regarding the induction of
apoptosis by BCLAF1 repression in transformed cells is still
largely unknown. Although the transformed cells undergo apoptotic
cell death following siBCLAF1 treatment, they still formed
colonies on soft agar.
Although the present study showed that the
abrogation of BCLAF1 expression in CT1 fibroblasts resulted in the
disruption of several signalling pathways, the fact that a
fibroblast cell line rather than an oesophageal cell line for the
functional analysis is a limitation in this study.
The current study demonstrated that BCLAF1 deletion
may be an important determinant in the progression of OSCC. To the
best of our knowledge this is the first study linking BCLAF1
deletion in OSCC. It is possible that the link between BCLAF1
deletion and the progression of OSCC is via the downregulation of
P53 and the deregulation of cellular processes involved in
carcinogenesis. This hypothesis is also supported by studies that
showed the role of BCLAF1 as a tumour suppressor and to promote the
stability of genes required for DNA repair and maintenance of
genomic stability (13,65).
To conclude, this study contributes towards our
understanding of the underlying mechanisms involved in genomic
alterations. It also provides evidence that abrogation of
BCLAF1 expression results in the dysregulation of several
cancer signalling pathways leading to cellular transformation. This
in turn presents an opportunity for the design of novel therapeutic
protocols based on the genomic traits of tumours.
Supplementary Material
Supporting Data
Acknowledgements
The present study forms part of the PhD Thesis ‘The
Role of Viral Sequences in Genetic Aberrations and Malignant
Transformation’ by LM submitted to the Faculty of Health Sciences,
University of Cape Town, September 2014.
Funding
This study was supported by funds from the National
Department of Health and the Medical Research Council (UK) with
funds from the UK Government's Newton Fund and Glaxo Smith Klein
(grant no. 046). This work was also supported by the National
Research Foundation and the University of Cape Town.
Availability of data and materials
All data generated or analysed during the current
study are available in the NLM NCBI dbVar repository (accession no.
nstd195; http://www.ncbi.nlm.nih.gov/dbvar/studies/nstd195/).
Authors' contributions
LMM and MIP conceived and designed the experiments.
LMM, VC and HS performed the experiments. LM, VC, HS and MIP
analysed the data. MIP contributed reagents, materials and analysis
tools. LMM and MIP wrote the manuscript. LMM and MIP confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Ethics approval was obtained from the University of
Cape Town/Groote Schuur Hospital Human Research Ethics Committee
(Cape Town, South Africa; approval no. HEC040/2005), and all
methods were performed in accordance with the Declaration of
Helsinki. Written informed consent was obtained from all patients
before recruitment into the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ADH4
|
alcohol dehydrogenase 4
|
|
BCLAF1
|
BCL2 associated transcription factor
1
|
|
DDR
|
DNA damage response
|
|
DMEM
|
Dulbecco's modified Eagle's
medium
|
|
DSB
|
double strand break
|
|
InDels
|
insertions and deletions
|
|
MSH3
|
muts homolog 3
|
|
OSCC
|
oesophageal squamous cell
carcinoma
|
|
SNP
|
single nucleotide polymorphisms
|
|
WGS
|
Whole Genome Sequencing
|
References
|
1
|
Feuk L, Carson AR and Scherer SW:
Structural variation in the human genome. Nat Rev Genet. 7:85–97.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vogelstein B and Kinzler KW: Cancer genes
and the pathways they control. Nat Med. 10:789–799. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fearon ER and Vogelstein B: A genetic
model for colorectal tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Knudson AG: Cancer genetics. Am J Med
Genet. 111:96–102. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hoeijmakers JH: Genome maintenance
mechanisms for preventing cancer. Nature. 411:366–374. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li GM: Mechanisms and functions of DNA
mismatch repair. Cell Res. 18:85–98. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Motoyama N and Naka K: DNA damage tumor
suppressor genes and genomic instability. Curr Opin Genet Dev.
14:11–16. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Popova T, Manié E, Stoppa-Lyonnet D,
Rigaill G, Barillot E and Stern MH: Genome Alteration Print (GAP):
A tool to visualize and mine complex cancer genomic profiles
obtained by SNP arrays. Genome Biol. 10:R1282009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kondrashov AS and Rogozin IB: Context of
deletions and insertions in human coding sequences. Hum Mutat.
23:177–185. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang H, Zhong Y, Peng C, Chen JQ and Tian
D: Important role of indels in somatic mutations of human cancer
genes. BMC Med Genet. 11:1282010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gregory TR: Insertion-deletion biases and
the evolution of genome size. Gene. 324:15–34. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mwapagha LM: The role of viral sequences
in genetic aberrations and malignant transformation. University of
Cape Town, 2014. http://hdl.handle.net/11427/12870
|
|
13
|
Kasof GM, Goyal L and White E: Btf, a
novel death-promoting transcriptional repressor that interacts with
Bcl-2-related proteins. Mol Cell Biol. 19:4390–4404. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fulda S, Gorman AM, Hori O and Samali A:
Cellular stress responses: Cell survival and cell death. Int J Cell
Biol. 2010:2140742010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shen B, Tan M, Mu X, Qin Y, Zhang F, Liu Y
and Fan Y: Upregulated SMYD3 promotes bladder cancer progression by
targeting BCLAF1 and activating autophagy. Tumor Biol.
37:7371–7381. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee YY, Yu YB, Gunawardena HP, Xie L and
Chen X: BCLAF1 is a radiation-induced H2AX-interacting partner
involved in γH2AX-mediated regulation of apoptosis and DNA repair.
Cell Death Dis. 3:e3592012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lawrence MS, Stojanov P, Mermel CH,
Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander
ES and Getz G: Discovery and saturation analysis of cancer genes
across 21 tumour types. Nature. 505:495–501. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Beroukhim R, Mermel CH, Porter D, Wei G,
Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J,
Urashima M, et al: The landscape of somatic copy-number alteration
across human cancers. Nature. 463:899–905. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hu N, Clifford RJ, Yang HH, Wang C,
Goldstein AM, Ding T, Taylor PR and Lee MP: Genome wide analysis of
DNA copy number neutral loss of heterozygosity (CNNLOH) and its
relation to gene expression in esophageal squamous cell carcinoma.
BMC Genomics. 11:5762010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou X, Li X, Cheng Y, Wu W, Xie Z, Xi Q,
Han J, Wu G, Fang J and Feng Y: BCLAF1 and its splicing regulator
SRSF10 regulate the tumorigenic potential of colon cancer cells.
Nat Commun. 5:45812014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Matejcic M, Mathew CG and Parker MI: The
relationship between environmental exposure and genetic
architecture of the 2q33 Locus with esophageal cancer in South
Africa. Front Genet. 10:4062019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kidd JM, Graves T, Newman TL, Fulton R,
Hayden HS, Malig M, Kallicki J, Kaul R, Wilson RK and Eichler EE: A
human genome structural variation sequencing resource reveals
insights into mutational mechanisms. Cell. 143:837–847. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
McLaren W, Pritchard B, Rios D, Chen Y,
Flicek P and Cunningham F: Deriving the consequences of genomic
variants with the Ensembl API and SNP effect predictor.
Bioinformatics. 26:2069–2070. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Becker KG, Barnes KC, Bright TJ and Wang
SA: The genetic association database. Nat Genet. 36:431–432. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tweedie S, Braschi B, Gray K, Jones TEM,
Seal RL, Yates B and Bruford EA: Genenames. org: The HGNC and VGNC
resources in 2021. Nucleic Acids Res. 49:D939–D946. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Namba M, Nishitani K and Kimoto T:
Characteristics of WI-38 cells (WI-38 CT-1) transformed by
treatment with Co-60 gamma rays. Gann. 71:300–307. 1980.PubMed/NCBI
|
|
27
|
Mwapagha LM, Tiffin N and Parker MI:
Delineation of the HPV11E6 and HPV18E6 pathways in initiating
cellular transformation. Front Oncol. 7:2582017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bailey JA, Yavor AM, Massa HF, Trask BJ
and Eichler EE: Segmental duplications: Organization and impact
within the current human genome project assembly. Genome Res.
11:1005–1017. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bailey JA, Gu Z, Clark RA, Reinert K,
Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW and Eichler EE:
Recent segmental duplications in the human genome. Science.
297:1003–1007. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
DePristo MA, Banks E, Poplin R, Garimella
KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA,
Hanna M, et al: A framework for variation discovery and genotyping
using next-generation DNA sequencing data. Nat Genet. 43:491–498.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li H and Durbin R: Fast and accurate
long-read alignment with Burrows-Wheeler transform. Bioinformatics.
26:589–595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu Q, Guo Y, Li J, Long J, Zhang B and
Shyr Y: Steps to ensure accuracy in genotype and SNP calling from
Illumina sequencing data. BMC Genomics. 13 (Suppl 8):S82012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Guo Y, Zhao S, Sheng Q, Ye F, Li J,
Lehmann B, Pietenpol J, Samuels DC and Shyr Y: Multi-perspective
quality control of Illumina exome sequencing data using QC3.
Genomics. 103:323–328. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kato H, Arao T, Matsumoto K, Fujita Y,
Kimura H, Hayashi H, Nishiki K, Iwama M, Shiraishi O, Yasuda A, et
al: Gene amplification of EGFR, HER2, FGFR2 and MET in esophageal
squamous cell carcinoma. Int J Oncol. 42:1151–1158. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vogelsang M, Paccez JD, Schäfer G, Dzobo
K, Zerbini LF and Parker MI: Aberrant methylation of the MSH3
promoter and distal enhancer in esophageal cancer patients exposed
to first-hand tobacco smoke. J Cancer Res Clin Oncol.
140:1825–1833. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sharma SV, Haber DA and Settleman J: Cell
line-based platforms to evaluate the therapeutic efficacy of
candidate anticancer agents. Nat Rev Cancer. 10:241–253. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mirabelli P, Coppola L and Salvatore M:
Cancer cell lines are useful model systems for medical research.
Cancers (Basel). 11:10982019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Borrell B: How accurate are cancer cell
lines? Nature. 463:8582010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Weinstein JN: Cell lines battle cancer.
Nature. 483:544–545. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gillet JP, Varma S and Gottesman MM: The
clinical relevance of cancer cell lines. J Natl Cancer Inst.
105:452–458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bhowmick NA, Neilson EG and Moses HL:
Stromal fibroblasts in cancer initiation and progression. Nature.
432:332–337. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Boehm JS and Hahn WC: Immortalized cells
as experimental models to study cancer. Cytotechnology. 45:47–59.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Namba M, Nishitani K and Kimoto T: Effects
of theophylline on the cell growth of normal and malignant human
cells transformed in culture. Gann. 71:621–627. 1980.PubMed/NCBI
|
|
46
|
Itjima M, Mihara K, Kondo T, Tsuji T,
Ishioka C and Namba M: Mutation in p53 and de-regulation of
p53-related gene expression in three human cell lines immortalized
with 4-nitroquinoline 1-oxide or 60Co gamma rays. Int J Cancer.
66:698–702. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rhim JS, Yoo JH, Park JH, Thraves P,
Salehi Z and Dritschilo A: Evidence for the multistep nature of in
vitro human epithelial cell carcinogenesis. Cancer Res. 50 (Suppl
17):5653S–5657S. 1990.PubMed/NCBI
|
|
48
|
Yoon S, Xuan Z, Makarov V, Ye K and Sebat
J: Sensitive and accurate detection of copy number variants using
read depth of coverage. Genome Res. 19:1586–1592. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sims D, Sudbery I, Ilott NE, Heger A and
Ponting CP: Sequencing depth and coverage: Key considerations in
genomic analyses. Nat Rev Genet. 15:121–132. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Iafrate AJ, Feuk L, Rivera MN, Listewnik
ML, Donahoe PK, Qi Y, Scherer SW and Lee C: Detection of
large-scale variation in the human genome. Nat Genet. 36:949–951.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ungerbäck J, Belenki D, Jawad ul-Hassan A,
Fredrikson M, Fransén K, Elander N, Verma D and Söderkvist P:
Genetic variation and alterations of genes involved in
NFκB/TNFAIP3-and NLRP3-inflammasome signaling affect susceptibility
and outcome of colorectal cancer. Carcinogenesis. 33:2126–2134.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kuwano H, Kato H, Miyazaki T, Fukuchi M,
Masuda N, Nakajima M, Fukai Y, Sohda M, Kimura H and Faried A:
Genetic alterations in esophageal cancer. Surg Today. 35:7–18.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Han CL, Liao CS, Wu CW, Hwong CL, Lee AR
and Yin SJ: Contribution to first-pass metabolism of ethanol and
inhibition by ethanol for retinol oxidation in human alcohol
dehydrogenase family. Eur J Biochem. 254:25–31. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hanawa M, Suzuki S, Dobashi Y, Yamane T,
Kono K, Enomoto N and Ooi A: EGFR protein overexpression and gene
amplification in squamous cell carcinomas of the esophagus. Int J
Cancer. 118:1173–1180. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Guo M, Liu S, Herman JG, Zhuang H and Lu
F: Brief communication Gefitinib-sensitizing mutation in esophageal
carcinoma cell line Kyse450. Cancer Biol Ther. 5:152–155. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Vogelsang M, Wang Y, Veber N, Mwapagha LM
and Parker MI: The cumulative effects of polymorphisms in the DNA
mismatch repair genes and tobacco smoking in oesophageal cancer
risk. PLoS One. 7:e369622012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Shao AW, Sun H, Geng Y, Peng Q, Wang P,
Chen J, Xiong T, Cao R and Tang J: Bclaf1 is an important NF-κB
signaling transducer and C/EBPβ regulator in DNA damage-induced
senescence. Cell Death Differ. 23:865–875. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wen Y, Zhou X, Lu M, He M, Tian Y, Liu L,
Wang M, Tan W, Deng Y, Yang X, et al: Bclaf1 promotes angiogenesis
by regulating HIF-1α transcription in hepatocellular carcinoma.
Oncogene. 38:1845–1859. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Dell'Aversana C, Giorgio C, D'amato L,
Lania G, Matarese F, Saeed S, Di Costanzo A, Belsito Petrizzi V,
Ingenito C, Martens JHA, et al: miR-194-5p/BCLAF1 deregulation in
AML tumorigenesis. Leukemia. 31:2315–2325. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jiang T, Liu B, Wu D and Zhang F: BCLAF1
induces cisplatin resistance in lung cancer cells. Oncol Lett.
20:2272020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wang H, Liang L, Fang J-Y and Xu J:
Somatic gene copy number alterations in colorectal cancer: New
quest for cancer drivers and biomarkers. Oncogene. 35:2011–2019.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Liu H, Lu ZG, Miki Y and Yoshida K:
Protein kinase C delta induces transcription of the TP53 tumor
suppressor gene by controlling death-promoting factor Btf in the
apoptotic response to DNA damage. Mol Cell Biol. 27:8480–8491.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lin DC, Hao JJ, Nagata Y, Xu L, Shang L,
Meng X, Sato Y, Okuno Y, Varela AM, Ding LW, et al: Genomic and
molecular characterization of esophageal squamous cell carcinoma.
Nat Genet. 46:467–473. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Song Y, Li L, Ou Y, Gao Z, Li E, Li X,
Zhang W, Wang J, Xu L, Zhou Y, et al: Identification of genomic
alterations in oesophageal squamous cell cancer. Nature. 509:91–95.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Savage KI, Gorski JJ, Barros EM, Irwin GW,
Manti L, Powell AJ, Pellagatti A, Lukashchuk N, McCance DJ,
McCluggage WG, et al: Identification of a BRCA1-mRNA splicing
complex required for efficient DNA repair and maintenance of
genomic stability. Mol Cell. 54:445–459. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Dietlein F, Thelen L and Reinhardt HC:
Cancer-specific defects in DNA repair pathways as targets for
personalized therapeutic approaches. Trends Genet. 30:326–339.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Friedberg EC: DNA damage and repair.
Nature. 421:436–440. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Leung J, Ehmann G, Giangrande P and Nevins
J: A role for Myc in facilitating transcription activation by E2F1.
Oncogene. 27:4172–4179. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Muthuswami M, Ramesh V, Banerjee S, Viveka
Thangaraj S, Periasamy J, Bhaskar Rao D, Barnabas GD, Raghavan S
and Ganesan K: Breast tumors with elevated expression of 1q
candidate genes confer poor clinical outcome and sensitivity to
Ras/PI3K inhibition. PLoS One. 8:e775532013. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: The cell cycle: A review of regulation, deregulation
and therapeutic targets in cancer. Cell Prolif. 36:131–149. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Bertoli C, Skotheim JM and De Bruin RA:
Control of cell cycle transcription during G1 and S phases. Nat Rev
Mol Cell Biol. 14:518–828. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kruse JP and Gu W: Modes of p53
regulation. Cell. 137:609–622. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Riedl SJ and Shi Y: Molecular mechanisms
of caspase regulation during apoptosis. Nat Rev Mol Cell Biol.
5:897–904. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Thomadaki H and Scorilas A: BCL2 family of
apoptosis-related genes: Functions and clinical implications in
cancer. Crit Rev Clin Lab Sci. 43:1–67. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Cory S and Adams JM: The Bcl2 family:
Regulators of the cellular life-or-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|