Introduction
Primary liver cancers (PLCs), of which the most
predominant type is hepatocellular carcinoma, are the third leading
cause of cancer-related mortality worldwide after lung cancer
(1,2).
The morbidity of PLCs markedly varies between geographic regions
around the world, with the highest rates observed in sub-Saharan
Africa and Eastern Asia (3). The
major risk factors for PLC include chronic hepatitis B/C virus
infection, excessive alcohol consumption, non-alcoholic fatty liver
disease and aflatoxin exposure (3,4). Although
current treatments, including percutaneous local ablation, surgical
resection and liver transplantation, improve the survival of
patients with PLC, the mortality rate continues to increase due to
population growth and high recurrence rates (1,5,6). Therefore, further molecular studies on
PLCs are urgently required to identify novel targets for PLC
treatment.
Ferroptosis is an iron-dependent form of
non-apoptotic cell death first reported by Dixon et al
(7) in 2012. This type of cell death
is caused by lethal lipid peroxidation and its typical features,
such as the overproduction of lipid reactive oxygen species (ROS)
and the impairment of crista and outer mitochondrial membrane
(8), are different from other types
of regulated cell death. Solute carrier family 7 member 11
(SLC7A11) and solute carrier family 3 member 2 (SLC3A2) control the
import of extracellular cysteine and cystine, and regulate
glutathione (GSH) synthesis (9). GSH
depletion leads to the suppression of glutathione peroxidase
(GPX)4, a key glutathione peroxidase known to catalyze the
reduction of lipid ROS (9).
One of the major reasons for therapeutic resistance
in cancer was discovered to be the intrinsic or acquired resistance
of cancer cells to executioner-mediated apoptosis (10). Compared with non-cancer cells, cancer
cells often required increased iron to promote their proliferation.
Interestingly, this iron dependency also made cancer cells more
sensitive to ferroptosis (11).
Therefore, small molecules, such as erastin, sorafenib and
Ras-selective lethal small molecule-3/-5, which were found to be
capable of inducing iron-dependent accumulation of lipid ROS, are
currently being evaluated for their potential in cancer therapy
(12,13). The identification of drugs able to
induce ferroptosis may provide novel insights into the potential of
ferroptosis as a novel target for PLC treatment.
Artemisinin is a sesquiterpene trioxane lactone
originally extracted from Artemisia annua L., and its
derivates have been discovered to be effective anti-malarial agents
(14). To date, in addition to its
well-known anti-malarial application, artemisinins are currently
being evaluated for their potential in treating multiple cancer
types due to their established safety recorded in thousands of
patients with malaria (14). Ooko
et al (15) treated 60 cancer
cell lines with 11 artemisinin derivates, and analyzed the
expression profiles of 30 iron-related genes in these cell lines
via microarray hybridization. The results found that the
IC50 values of analyzed artemisinins were significantly
correlated with the expression of ≥20 iron-related genes. These
findings suggested the involvement of artemisinins in the induction
of ferroptosis in cancer cells. Artesunate is the most frequently
reported derivate of artemisinin, which has been found to trigger
ferroptosis in malignant cells (16,17).
Dihydroartemisinin (DHA), a semi-synthetic derivative of
artemisinin, has been demonstrated to exhibit antitumor activity in
PLC both in vitro and in vivo (18,19). The
role of DHA in inducing ferroptosis in cancer cells was first
reported in head and neck carcinoma cells by Lin et al
(20), and later in leukemia cells by
Du et al (21) and in glioma
cells by Chen et al (22).
However, to the best of our knowledge, there is currently no direct
evidence outlining the role of DHA in inducing ferroptosis in PLC.
Nonetheless, an earlier study from Wang et al (18) reported that DHA increased
intracellular ROS levels in LM3 liver cancer cells, suggesting that
DHA may trigger ferroptosis in PLC cells.
Cancer cells can thrive under hostile
microenvironmental conditions. Within tumor masses, endoplasmic
reticulum (ER) stress was reported to be induced by nutrient
deprivation, oxygen limitation and a high metabolic demand
(23). Malignant cells were
discovered to initiate the unfolded protein response (UPR) to cope
with the ER stress (24). The
ER-resident sensors, eukaryotic translation initiation factor 2 α
kinase 3 (eIF2), inositol-requiring transmembrane
kinase/endoribonuclease 1α (IRE1α) and activating transcription
factor (ATF)6, coordinate the UPR if misfolded proteins accumulate
and aggregate beyond a tolerable threshold (23,24). The
upregulation of ATF4, which is induced by ferroptosis inducers,
such as erastin and artesunate, is considered a compensatory
effect, as its knockdown was demonstrated to further augment
ferroptosis in cancer cells (16,25). In
glioma cells, the PERK/ATF4 signaling pathway was activated by DHA
and negatively regulated DHA-induced ferroptosis (22). However, to the best of our knowledge,
studies investigating the roles of the ATF6- and IRE1α-mediated UPR
branches in ferroptosis are scarce and how DHA affects these two
UPR mediators remains largely unknown.
In the present study, four PLC cell lines, Hep3B,
Huh7, PLC/PRF/5 and HepG2, were treated with different
concentrations of DHA in the presence or absence of ferroptosis
inhibitors or iron ions. To deactivate the UPRs, small interfering
RNA was used to knockdown the expression of ATF4, XBP1 or ATF6 in
PLC cells.
Materials and methods
Chemicals
Ferrostatin-1 and DHA were purchased from Shanghai
Aladdin Biochemical Technology Co., Ltd. and dissolved in DMSO to
final stock concentrations of 5 and 50 mM, respectively.
Deferoxamine mesylate salt (DFOM; MedChemExpress) and iron chloride
hexahydrate (Shanghai Aladdin Biochemical Technology Co., Ltd.)
were dissolved in distilled H2O to final stock
concentrations of 50 mM and 2 mg/l, respectively.
Cell lines and culture
PLC cell lines, Hep3B (p53 null), Huh7 (659 A>G
p53 mutant), PLC/PRF/5 (747 G>T p53 mutant) and HepG2 (p53
wild-type), were purchased from The Cell Bank of Type Culture
Collection of The Chinese Academy of Sciences. Each cell line
underwent short tandem repeat profiling for authentication. All
cell lines were free of mycoplasma. Hep3B, PLC/PRF/5 and HepG2
cells were maintained in Minimum Essential Medium Eagle (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% FBS (HyClone;
Cytiva), while Huh7 cells were cultured in DMEM (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS. Cells were
maintained in an HF-90 cell incubator (KG Medical Industries) in an
atmosphere with 5% CO2 at 37°C.
Cell transfection
Sequences of small interfering RNA (siRNA/si) ATF4,
si-X-box binding protein 1 (XBP1), si-ATF6 or si-Chac glutathione
specific γ-glutamylcyclotransferase 1 (CHAC1) are listed in
Table I. The coding region of CHAC1
gene was inserted into a pECMV-3×FLAG-N plasmid for overexpression
(Fenghui Biotechnology Co., Ltd.). PLC cells seeded in 6-well
plates were transfected with siRNA (100 pM) or pECMV-3×FLAG-N
plasmid (vector containing the CHAC1 gene) (2 µg) mixed with 6 µl
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) at 37°C for 24 h, then treated with DHA for an
additional 24 h.
 | Table I.siRNA sequences. |
Table I.
siRNA sequences.
| siRNA | Sequence,
5′-3′ |
|---|
| siATF4-1 |
|
|
Forward |
GUCCUCCACUCCAGAUCAUTT |
|
Reverse |
AUGAUCUGGAGUGGAGGACTT |
| siATF4-2 |
|
|
Forward |
UGGAUAUCACUGAAGGAGATT |
|
Reverse |
UCUCCUUCAGUGAUAUCCATT |
| siXBP1-1 |
|
|
Forward |
GCAAGUGGUAGAUUUAGAATT |
|
Reverse |
UUCUAAAUCUACCACUUGCTT |
| siXBP1-2 |
|
|
Forward |
CCUAAAGUUCUGCUUCUGUTT |
|
Reverse |
ACAGAAGCAGAACUUUAGGTT |
| siATF6-1 |
|
|
Forward |
GAAAUGUCGGUUCAGAUAUTT |
|
Reverse |
AUAUCUGAACCGACAUUUCTT |
| siATF6-2 |
|
|
Forward |
GAGCCACUGAAGGAAGAUATT |
|
Reverse |
UAUCUUCCUUCAGUGGCUCTT |
| siCHAC1-1 |
|
|
Forward |
GACGCUCCUUGAAGAUCAUTT |
|
Reverse |
AUGAUCUUCAAGGAGCGUCTT |
| siCHAC1-2 |
|
|
Forward |
GCCACAACCUUGAAUACUUTT |
|
Reverse |
AAGUAUUCAAGGUUGUGGCTT |
| si*CHAC1 |
|
|
Forward |
UGGGAAGCUCAUCACUACATT |
|
Reverse |
UGUAGUGAUGAGCUUCCCATT |
| Negative
control |
|
|
Forward |
UUCUCCGAACGUGUCACGUTT |
|
Reverse |
ACGUGACACGUUCGGAGAATT |
Cell Counting Kit (CCK)-8 assay
PLC cells were treated with different concentrations
of DHA, then the cell viability was determined using a CCK-8 assay
(Sigma-Aldrich; Merck KGaA). Briefly, PLC cells were plated into
96-well plates at a density of 3×103 cells/well and
cultured for 24 h. Following the incubation, the cells were treated
with 2, 5, 7.5, 10, 20, 30, 40 or 50 µM DHA for 24 h. Cells were
subsequently incubated with 10 µl CCK-8 for 1 h and the absorbance
of each well was measured at a wavelength of 450 nm using a
microplate reader (BioTek Instruments, Inc.). For some experiments,
DFOM (10 µM for Huh7 cells and 50 µM for the other cell lines),
ferrostatin-1 (5 µM for HepG2 cells and 1 µM for the other cell
lines) or exogenous iron ions (10 µg/ml iron chloride hexahydrate)
were used to treat PLC cells in the presence of DHA.
Establishment of a PLC cell xenograft
tumor mouse model
A total of 32 male BALB/c nude mice (age, 6–8 weeks;
weight, 18–20 g) were obtained from HFK Bioscience Co. Ltd. and
housed in a specific pathogen-free facility. The mice were randomly
divided into four groups (n=8/group) and PLC cells were
subcutaneously injected into the nude mice. The tumor volumes were
determined using the following formula: 0.5 × tumor length × (tumor
width)2. When the xenografted tumors had grown to 80–100
mm3, half of the mice in each group were administered
with 100 mg/kg DHA for 5 days/week by gavage. After 21 days, all
mice were anesthetized with isoflurane (2.5%; Yuyan) and sacrificed
by cervical dislocation immediately (26,27). The
present study was approved by the Ethics Committee of Zhengzhou
University (Zhengzhou, China).
Hematoxylin and eosin (H&E)
Tumor tissues were collected, fixed with 4%
paraformaldehyde (Xilong Chemicals) overnight at 4°C, embedded in
paraffin and sliced into 5-µm thick sections. The sections were
subsequently deparaffinized and rehydrated, then incubated with
H&E staining agents (Beijing Solarbio Science & Technology
Co., Ltd.), according to the manufacturer's protocol.
Measurement of ROS levels
Total (Jiancheng Bioengineering Institute) and lipid
ROS levels (Invitrogen; Thermo Fisher Scientific, Inc.) were
determined using ROS assay kits, according to the manufacturers'
protocols, which were based on detecting 2′,7′-dichlorofluorescin
(DCF) diacetate (cat. no. E004) and C11-BODIPY® 581/591
fluorescence (cat. no. D3861), respectively. The DCF and C11-BODIPY
fluorescence intensities were analyzed using a M200 PRO automatic
microplate reader (Tecan Group, Ltd.) or a NovoCyte flow cytometer
(Agilent Technologies, Inc.), respectively. Raw data from the flow
cytometer was analyzed using NovoExpress version 1.4.1 (Agilent
Technologies, Inc.).
Measurement of iron
concentrations
Intracellular ferrous iron levels were analyzed
using an iron assay kit (cat. no. TC1015; Leagene Biotech),
according to the manufacturer's protocol. The output was measured
on a M200 PRO automatic microplate reader at a wavelength of 562
nm.
Measurement of malondialdehyde (MDA)
and glutathione (GSH) levels, and glutathione peroxide (GSH-PX)
activity
Kits (all from Jiancheng Bioengineering Institute)
were used to determine MDA (cat. no. A003-1) and GSH (cat. no.
A061-1) levels, and GSH-PX (cat. no. A005) activity, according to
the manufacturers' protocols.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from PLC cells treated with
DHA for 1, 6, 12 or 24 h with a RNAsimple kit (Tiangen Biotech Co.,
Ltd.). RNAs were reverse transcribed to cDNA using a TIANSeq M-MLV
kit (Tiangen Biotech Co., Ltd.) according to the manufacturer's
protocol. Briefly, RNAs were incubated in a LifeExpress Classic PCR
machine (Hangzhou Bioer Co., Ltd.) at 25°C for 10 min, 42°C for 50
min, and then at 80°C for 10 min to stop the reaction. The splicing
of XBP1 mRNA (NM_005080 and NM_001079539) was assessed using qPCR
using one pair of primers: Forward, 5′-AAACTTTTGCTAGAAAATCAGC-3′
and reverse, 5′-CAATACCGCCAGAATCCA-3′, which spanned the splice
site. The thermocycling conditions were: 95°C for 5 min; followed
by 36 of 95°C for 10 sec, 52°C for 20 sec, 72°C for 30 sec and 25°C
for 5 min. This enabled two fragments (252 and 226 bp) to be
detected. The PCR products were subsequently analyzed using a 1.5%
agarose gel.
Western blotting
Total protein was extracted from the whole cell
lysates using RIPA lysis buffer (Beijing Solarbio Science &
Technology Co., Ltd.) supplemented with 1% PMSF. Nuclear proteins
were isolated using a Nuclear and Cytoplasmic Protein Extraction
kit, according to the manufacturer's protocol (Beyotime Institute
of Biotechnology). Protein concentration was determined using a BCA
assay kit (Beijing Solarbio Science & Technology Co., Ltd.)
and, 10–20 µg protein sample was separated via SDS-PAGE (8–15%).
The resolved proteins were subsequently transferred to PVDF
membranes (MilliporeSigma) and blocked with 5% non-fat milk for 1 h
at room temperature. The membranes were then incubated with primary
antibodies (listed in Table II) at
4°C overnight. Following primary antibody incubation, the membranes
were incubated with secondary antibodies at 37°C for 1 h. Protein
bands were visualized using ECL reagents (Beijing Solarbio Science
& Technology Co., Ltd.). GAPDH was used as the loading control
for whole cell lysates, and Histone H3 was used as the nuclear
loading control.
| Table II.Antibodies used for western blot
analysis. |
Table II.
Antibodies used for western blot
analysis.
| Primary
antibodies | Supplier | Cat. no. | Dilution |
|---|
| ATF4 | Cell Signaling
Technologies, Inc. | 11815 | 1:1,000 |
| SLC7A11 |
| 12691 | 1:1,000 |
| ATF6 | ABclonal Biotech
Co., Ltd. | A0202 | 1:1,000 |
| p-eIF2α |
| AP0692 | 1:100 |
| eIF2α |
| A0764 | 1:500 |
| GPX4 | Abcam | AB125066 | 1:3,000 |
| SLC3A2 |
| AB108300 | 1:5,000 |
| CHAC1 |
| AB76386 | 1:1,000 |
| p-IRE1α | Affinity
Biosciences | DF8322 | 1:1,000 |
| IRE1α |
| DF7709 | 1:1,000 |
| PERK |
| AF5304 | 1:1,000 |
| p-PERK | Thermo Fisher
Scientific, Inc. | PA5-40294 | 1:500 |
| GAPDH | ProteinTech, Group,
Inc. | 60004-1-Ig | 1:10,000 |
| Histone H3 | Bioss
Antibodies | bs-0349R | 1:1,000 |
| IgG-horseradish
peroxidase | Beijing Solarbio
Science & Technology Co., Ltd. | SE131 | 1:3,000 |
|
|
| SE134 | 1:3,000 |
pGL3 dual luciferase reporter
assay
The potential binding sites for ATF4, XBP1 and ATF6
on the CHAC1 gene promoter were predicted using JASPAR (jaspar.genereg.net) and PROMO (alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3).
The promoter (−2,000 to +30 bp) of the CHAC1 gene was inserted into
a pGL3 luciferase reporter and the promoter activity was determined
by analyzing the firefly/Renilla luciferase ratio, according
to the manufacturer's protocol (Promega Corporation).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism version 8.0 (GraphPad Software, Inc.) and data are presented
as the mean ± SD. Statistical differences between groups were
determined using a one-way or two-way ANOVA followed by a
Bonferroni's multiple comparison test. P<0.05 was considered to
indicate a statistically significant difference.
Results
DHA induced-ferroptosis in PLC cells
is irrelevant of p53 status
Considering the involvement of p53 in ferroptosis
(25), four PLC cell lines with
different p53 statuses, Hep3B (p53 null), Huh7 and PLC/PRF/5 (both
p53 mutant) and HepG2 (p53 wild-type), were treated with increasing
concentrations of DHA for 24 h. The data revealed that DHA
inhibited the survival of all the PLC cell lines, irrelevant of
their p53 status (Fig. 1A).
Subsequently, two different ferroptosis inhibitors, DFOM (an iron
chelator) and ferrostatin-1 (a lipid peroxidation inhibitor) were
used to treat PLC cells in the presence of DHA. The results
revealed that the cytotoxic effects of DHA on PLC cells were
attenuated by both ferroptosis inhibitors (Fig. 1A). Conversely, the addition of
exogenous iron ions further augmented the effects of DHA (Fig. 1A).
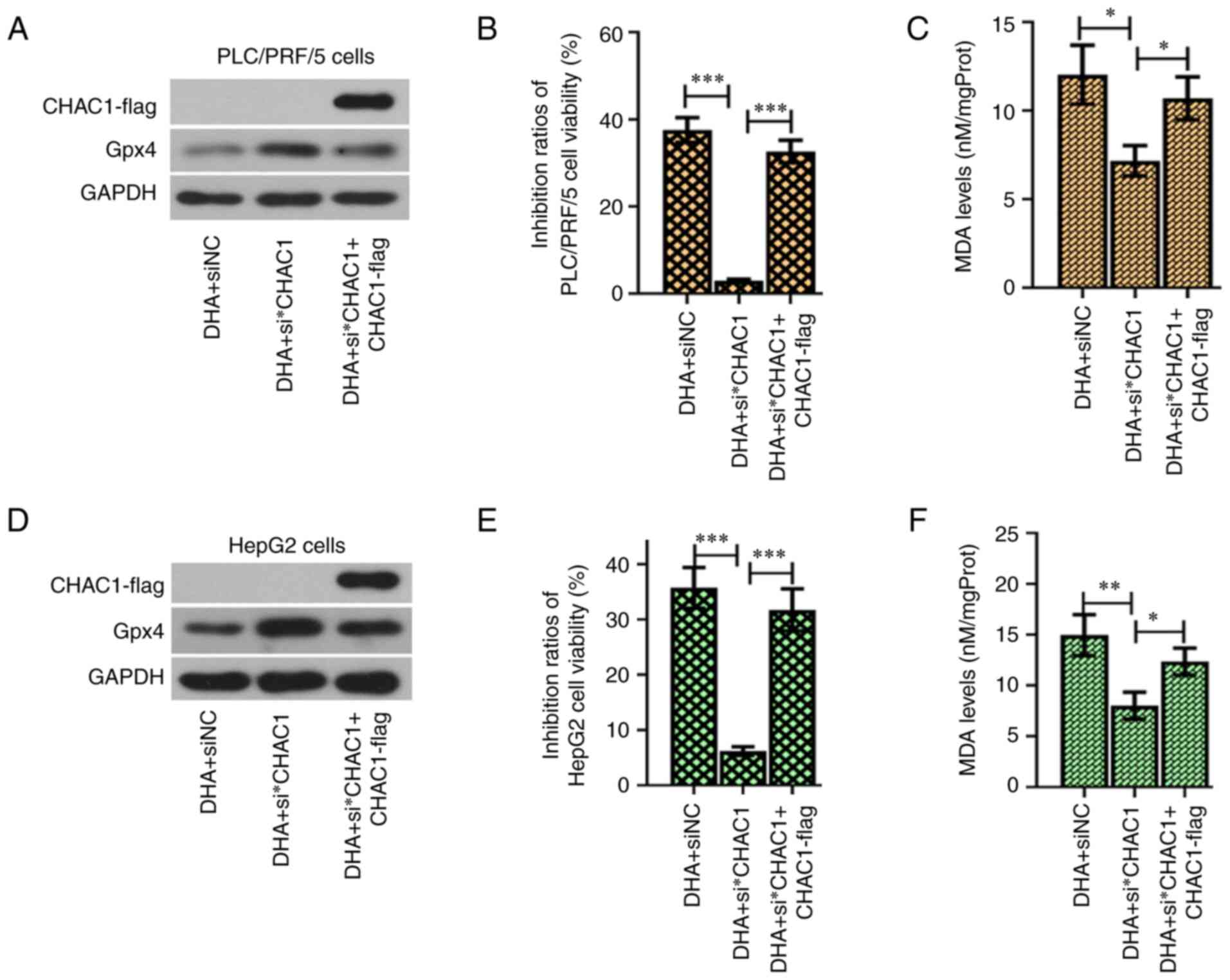
 | Figure 1.DHA induces ferroptosis in PLC cells
in vitro. (A) Hep3B (p53 null), Huh7 and PLC/PRF/5 (both p53
mutant) and HepG2 (p53 wild-type) cells were treated with
increasing concentrations of DHA for 24 h. Two different
ferroptosis inhibitors, DFOM (an iron chelator; 10 µM for Huh7
cells and 50 µM for the other cell lines) and ferrostatin-1 (a
lipid peroxidation inhibitor; 5 µM for HepG2 and 1 µM for the other
cell lines) were used to treat PLC cells in the presence of DHA.
Iron chloride hexahydrate (10 µg/ml) was used to increase the iron
concentration. The vitality of PLC cells were determined using Cell
Counting Kit-8 assays. (B) IC50 values were calculated.
(C) Based on the IC50 values, HepG2, Huh7, Hep3B and
PLC/PRF/5 were treated with 40, 35, 30 and 25 µM DHA, respectively.
After incubation with DHA for 1, 6, 12 or 24 h, (C) total ROS, (D)
lipid ROS generation. (E) MDA levels, (F) iron concentrations, (G)
GSH/GSSG ratios (H) and GSH-PX activity, as well as (I) the protein
expression levels of GPX4, SLC7A11, SLC3A2 and CHAC1 in PLC cells
were determined. Data are presented as the mean ± standard
deviation of three repeats. *P<0.05, **P<0.01, ***P<0.001
vs. control (0 h). DHA, dihydroartemisinin; PLC, primary liver
cancer; DFOM, deferoxamine mesylate salt; ROS, reactive oxygen
species; MDA, malondialdehyde; GSSG, oxidized glutathione; GSH-PX,
glutathione-peroxidase; GPX4, glutathione peroxidase 4; SLC, solute
carrier family; CHAC, ChaC glutathione specific
γ-glutamylcyclotransferase; DCF, dichlorofluorescin; M, % of
control. |
The IC50 value of DHA was 29.4±1.7 µM for
Hep3B cells, 32.1±4.5 µM for Huh7 cells, 22.4±3.2 µM for PLC/PRF/5
cells and 40.2±2.1 µM for HepG2 cells (Fig. 1B). These findings suggested that HepG2
cells were the most resistant to DHA, followed by Huh7 and Hep3B
cells, while PLC/PRF/5 cells were the least resistant. According to
the IC50 values, HepG2, Huh7, Hep3B and PLC/PRF/5 cells
were further treated with 40, 35, 30 or 25 µM DHA,
respectively.
Following incubation with DHA for 1, 6, 12 or 24 h,
PLC cells were harvested to analyze the total and lipid ROS
contents (Fig. 1C and D), MDA levels
(Fig. 1E) and iron concentrations
(Fig. 1F). The results revealed that
the ROS, MDA and iron levels were increased in PLC cells exposed to
DHA. These data suggested that DHA may act as a ferroptosis inducer
in PLC cells in vitro.
Blocking GSH synthesis is known to facilitate the
accumulation of toxic ROS (7).
Therefore, the content of GSH in DHA-treated PLC cells was analyzed
using a commercial kit. Compared with the blank cells, the
GSH/glutathione disulfide (oxidized form of GSH) ratio was
decreased in cells exposed to DHA (Fig.
1G). The expression levels of GPX4, SLC7A11 and SLC3A2, and the
activity of GSH-PX were also inhibited by DHA treatment (Fig. 1H and I). Conversely, the expression
levels of CHAC1 were upregulated in response to DHA treatment
(Fig. 1I). These data suggested that
DHA may act as a negative regulator of GSH synthesis in PLC
cells.
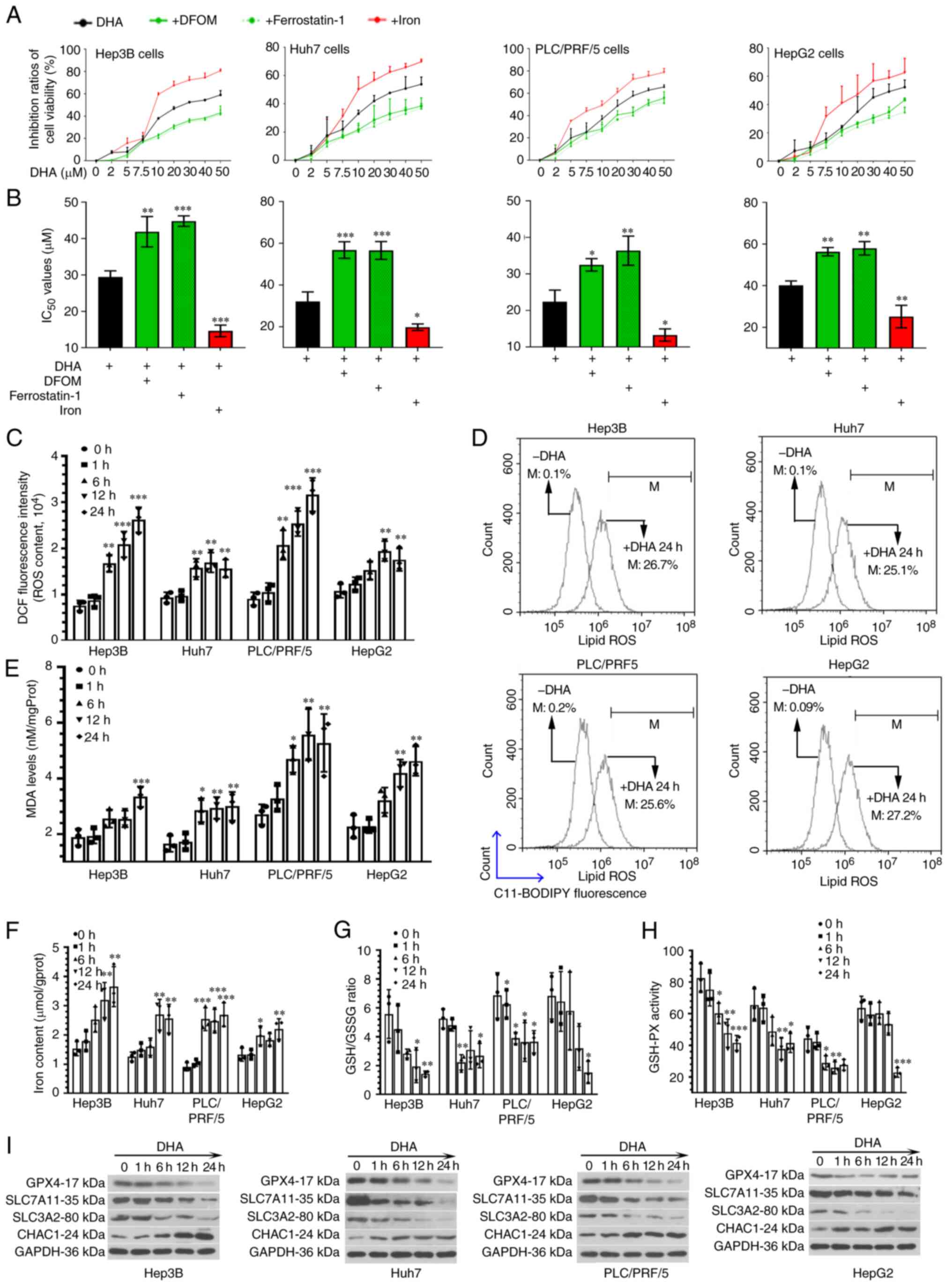
DHA limits PLC xenograft tumor growth
in vivo
The effects of DHA on PLC growth in vivo were
further investigated by subcutaneously injecting PLC cells into
immune-deficient mice; no weight loss or health problems were
observed in the mice. DHA was then administered to the mice upon
the tumor volumes reaching 80–100 mm3. The tumor volumes
were recorded every 3 days and 3 weeks later, the xenografted
tumors were collected. As shown in Fig.
2A-C, DHA limited, but did not completely inhibit, the
formation of PLC xenograft tumors. Furthermore, DHA increased ROS
accumulation (Fig. 2D) and decreased
GSH-PX activity (Fig. 2E) in the
tumor masses. These data, together with the aforementioned results
from the in vitro experiments, indicated that DHA may induce
ferroptosis both in vitro and in vivo.
 | Figure 2.DHA reduces PLC tumor growth in nude
mice. (A and B) PLC cells were subcutaneously injected into immune
deficient mice. DHA (100 mg/kg/day, 5 days/week) was administered
to mice when tumor volumes reached 80–100 mm3. The tumor
volumes were recorded every 3 days, and (C) 3 weeks later, the
xenografted tumors were collected for hematoxylin and eosin
staining (scale bar, 100 µm). (D) ROS and (E) GSH-PX activities in
tumor tissues were determined. Data are presented as the mean ±
standard deviation of four repeats. *P<0.05, **P<0.01,
***P<0.001 vs. control (0 h). DHA, dihydroartemisinin; PLC,
primary liver cancer; ROS, reactive oxygen species; GSH-PX,
glutathione-peroxidase. |
DHA activates the three branches of
the UPR in PLC cells
As DHA induced ferroptosis in all the analyzed PLC
cells both in vivo and in vitro, alterations in
UPR-associated molecules were next investigated in PLC/PRF/5 and
HepG2 cells. The data demonstrated that all three branches of the
UPR signaling pathways were activated by DHA, which was evidenced
through the upregulated expression levels of phosphorylated PERK,
eukaryotic translation initiation factor 2A (eIF2α), IRE1α, ATF4,
nuclear ATF6 and spliced XBP1 (Fig.
3A-D). Notably, DHA was found to activate PERK/eIF2α/ATF4
signaling earlier in PLC/PRF/5 cells than in HepG2 cells (Fig. 3A-D). To determine the role of the
activated UPR in DHA-mediated ferroptosis, ATF4, XBP1 and ATF6 were
silenced with their respective siRNAs (Fig. 4A and E). The transfection efficiencies
of siRNAs were first determined in PLC cells via western blotting
or RT-PCR analysis (Fig. S1).
Following the knockdown of all three UPR sensors, PLC cell
viability increased (Fig. 4B and F),
and total (Fig. 4C and G) and lipid
ROS (Fig. 4D and H) content was
decreased. The expression levels of GPX4, SLC7A11, SCL3A2 and CHAC1
were restored to different degrees in cells following the knockdown
of the three UPR transcription factors (Fig. 5A-F). To the best of our knowledge,
these data indicated, for the first time, that DHA may activate the
UPR as an anti-survival mechanism in PLC cells.
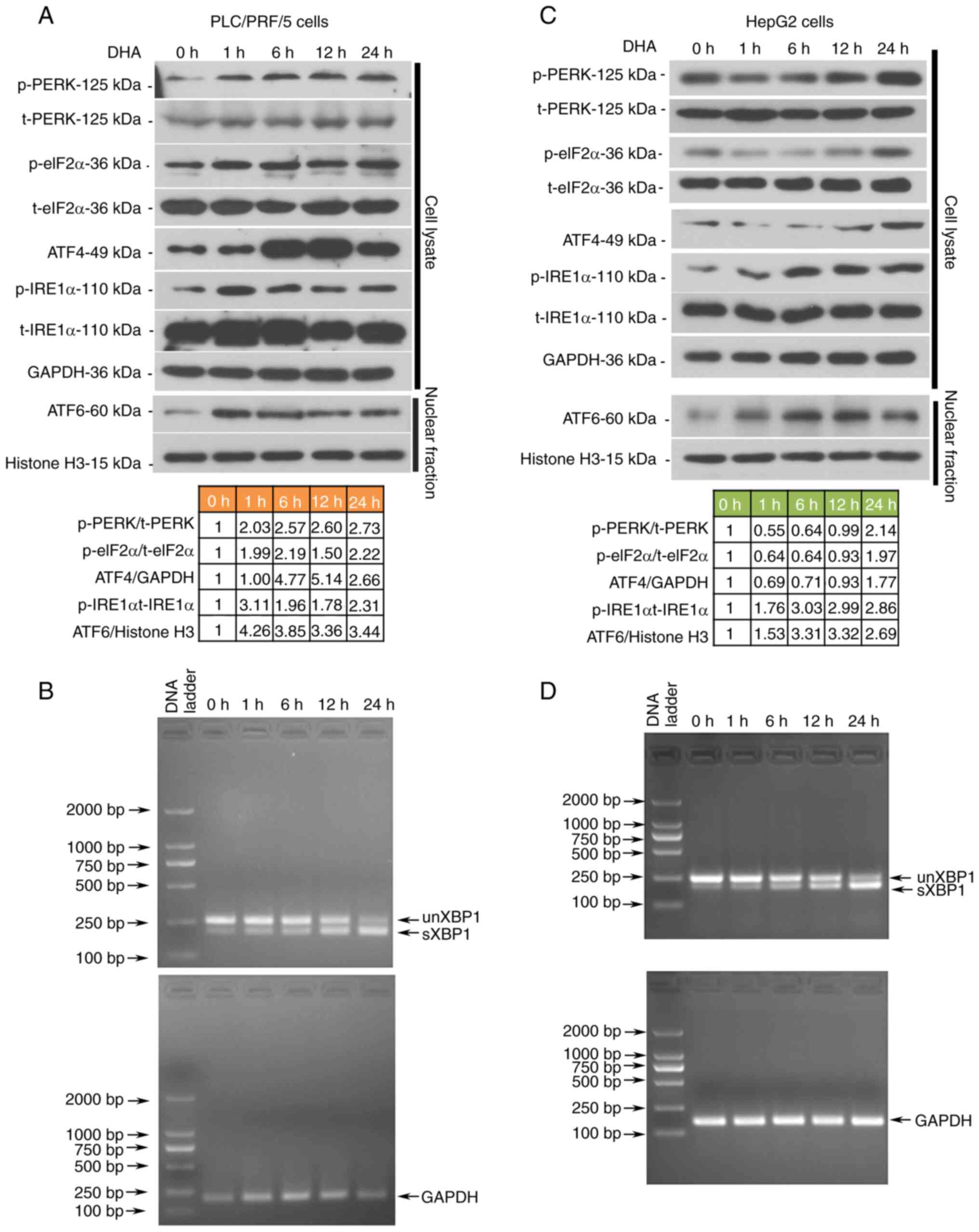 | Figure 3.DHA activates the UPRs in PLC cells
in vitro. HepG2 and PLC/PRF/5 cells were treated with 40 and
25 µM DHA, respectively, for 1, 6, 12 or 24 h. (A and C) The
protein expression levels of UPR-associated molecules were assessed
by western blotting. Relative protein expression levels of
indicated molecules are listed below the blots. (B and D) Total
RNAs were isolated to analyze the formation of sXBP1 in PLC cells.
DHA, dihydroartemisinin; PLC, primary liver cancer; UPR, unfolded
protein response; p-, phosphorylated; t-, total; PERK, protein
kinase R-like ER kinase; eIF2α, eukaryotic initiation factor 2α;
IRE1α, inositol-requiring transmembrane kinase/endoribonuclease 1α;
ATF, activating transcription factor. |
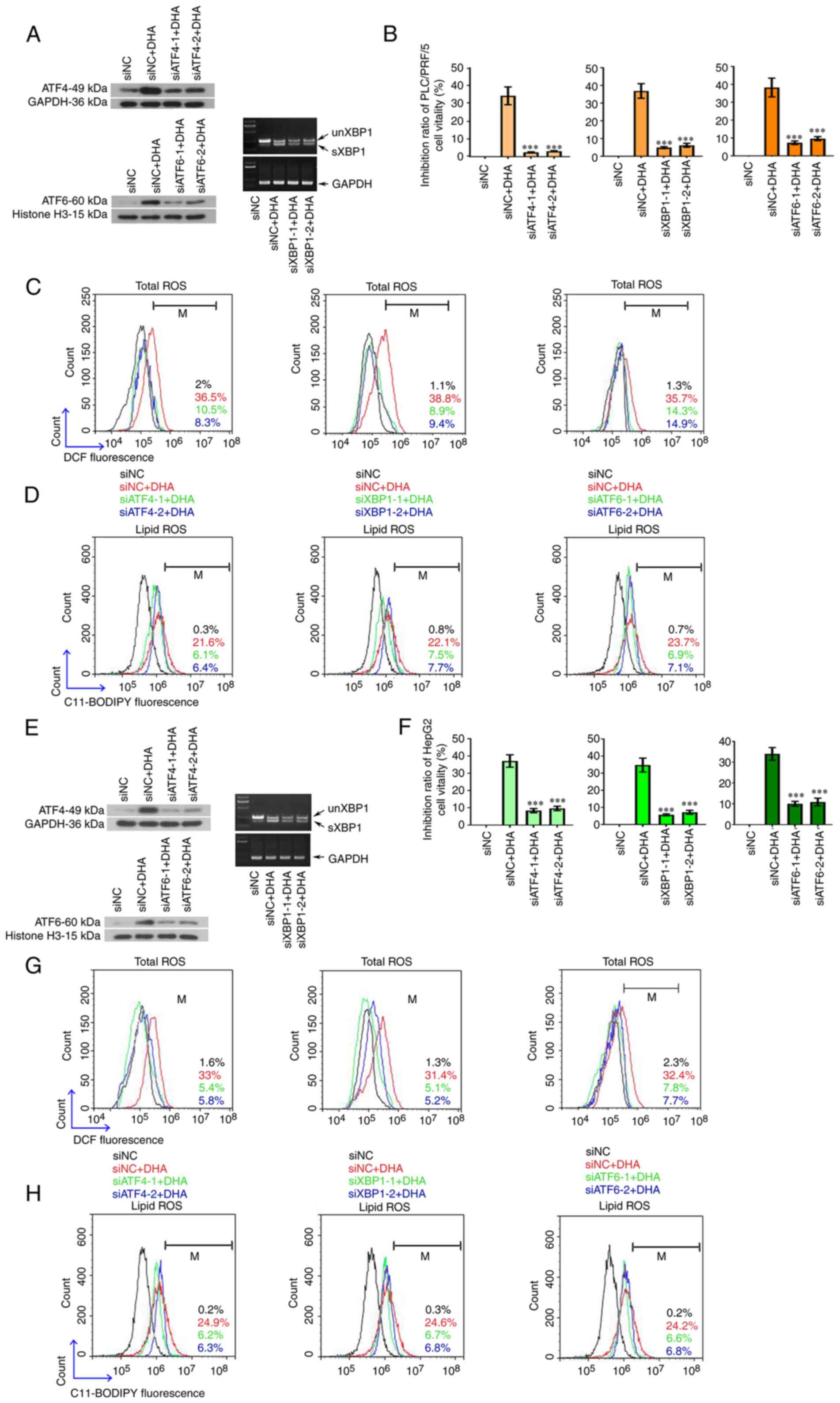 | Figure 4.The cytotoxic effects of DHA are
attenuated following suppression of the UPR. (A and E) Specific
siRNAs were synthesized to target ATF4, XBP1 and ATF6 in PLC cells.
A total of 24 h post siRNA transfection, HepG2 and PLC/PRF/5 cells
were treated with 40 or 25 µM DHA, respectively, for 24 h. (B and
F) Cell viability was determined using a Cell Counting Kit-8, and
(C and G) intracellular total ROS and (D and H) lipid ROS levels
were determined by flow cytometry. Data are presented as the mean ±
standard deviation of three repeats. ***P<0.001 vs. DHA + siNC.
DHA, dihydroartemisinin; UPR, unfolded protein response; siRNA,
small interfering RNA; ATF, activating transcription factor; XBP1,
X box-binding protein 1; sXBP1, spliced form of XBP1; unXBP1,
unspliced form of XBP1; PLC, primary liver cancer; ROS, reactive
oxygen species; NC, negative control; M, % of control. |
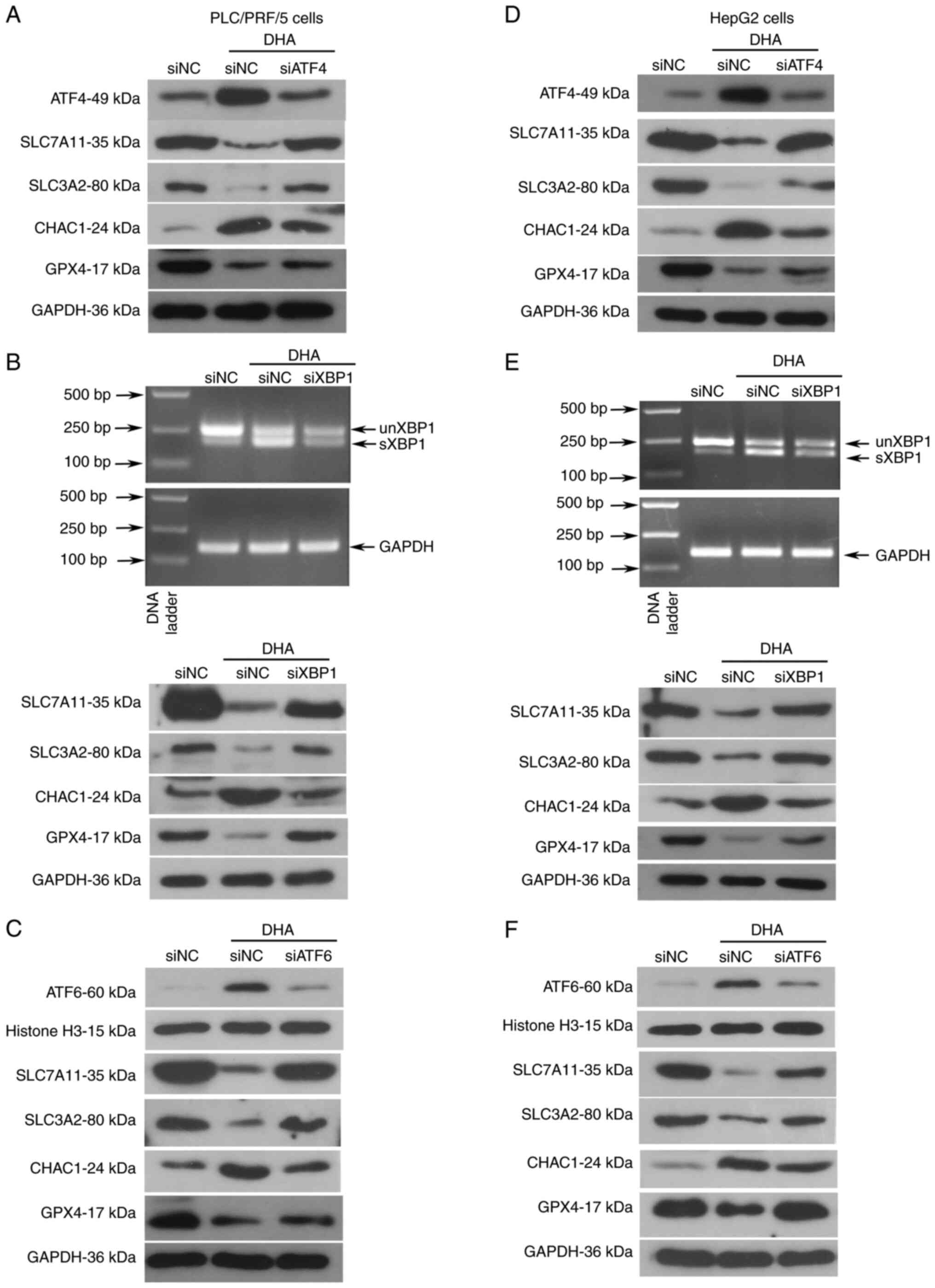 | Figure 5.Knockdown of UPR proteins partly
restores the expression of molecules associated with ferroptosis in
PLC cells in the presence of DHA. (A-F) Specific siRNAs were
synthesized to target ATF4, XBP1 and ATF6 in PLC cells. A total of
24 h post-siRNA transfection, HepG2 and PLC/PRF/5 were treated with
40 or 25 µM DHA, respectively, for 24 h. Cellular proteins were
extracted to analyze the protein expression levels of proteins
associated with the UPR via western blot analysis. (B and E) Total
RNAs were isolated to analyze the mRNA expression levels sXBP1 in
PLC cells. siRNA, small interfering RNA; ATF, activating
transcription factor; XBP1, X box-binding protein 1; sXBP1, spliced
form of XBP1; unXBP1, unspliced form of XBP1; PLC, primary liver
cancer; UPR, unfolded protein response; NC, negative control; GPX4,
glutathione peroxidase 4; SLC, solute carrier family; CHAC, ChaC
glutathione specific γ-glutamylcyclotransferase. |
DHA promotes the transcription of
CHAC1 by activating the UPRs in PLC cells
Multiple binding sites for the transcription factors
were found on the CHAC1 promoter (Fig.
6A). Since DHA could effectively upregulate the expression of
ATF4, XBP1 and ATF6, it was hypothesized that this agent may
promote CHAC1 transcription. The results from the pGL3 dual
luciferase reporter assay validated that DHA enhanced the promoter
activity of the CHAC1 gene (Fig. 6B and
C, left panel), and that this effect was suppressed following
knockdown of ATF4, XBP1 or ATF6 (Fig. 6B
and C, right panel). To further validate the role of CHAC1 in
DHA-induced ferroptosis, siRNA was used to knockdown CHAC1
expression (Fig. 6D and E). The
results revealed that DHA-induced ferroptosis was suppressed by
siCHAC1 (Fig. 6F-I). CHAC1 encoding
fragment with a lack of the 3′UTR was inserted into an expression
vector, which resulted in a lack of effect of the siRNA targeting
CHAC1 3′UTR (si*CHAC1) on its expression. Additionally, it was
further found that overexpression of CHAC1 sensitized PLC cells to
DHA (Fig. 7).
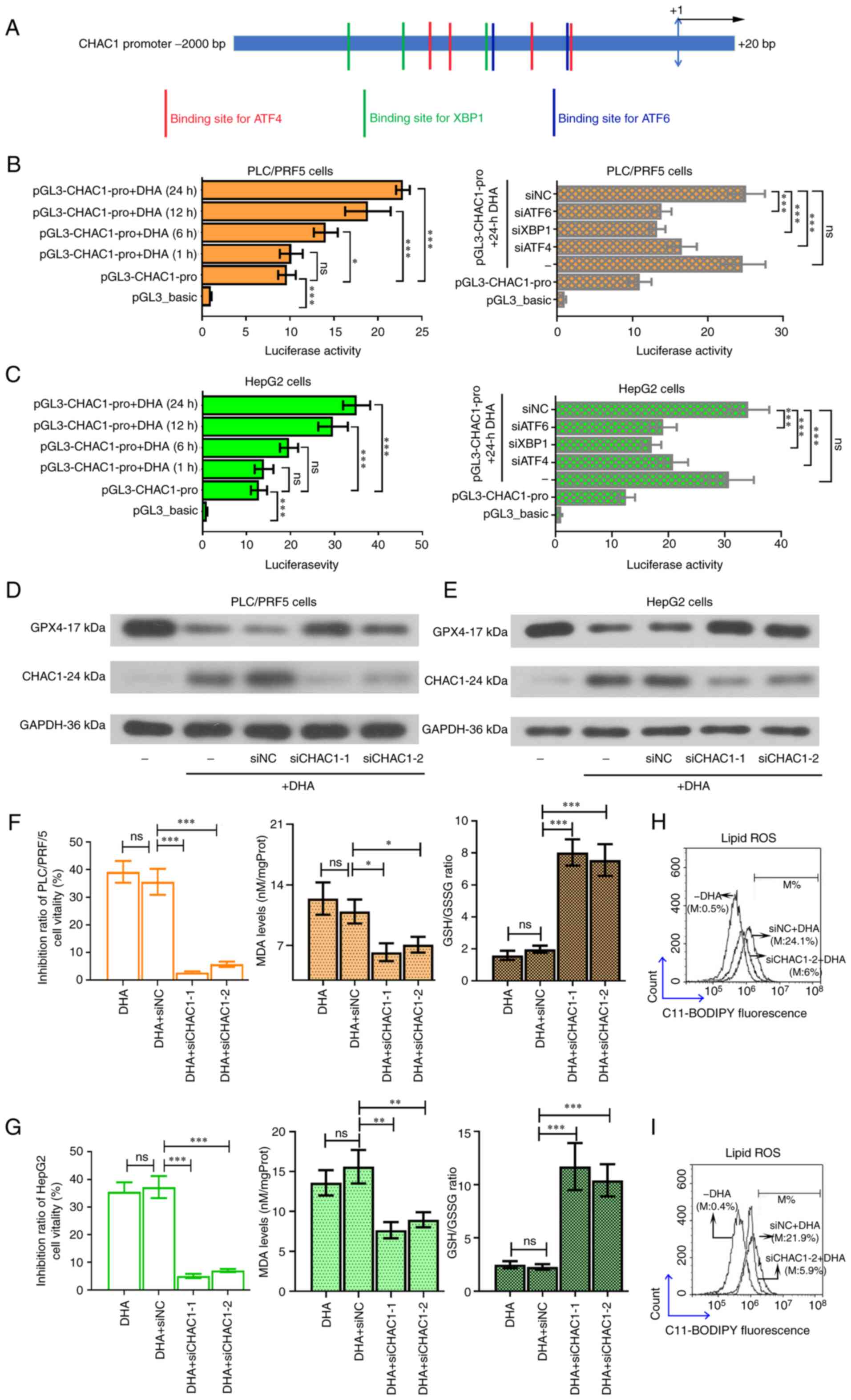 | Figure 6.DHA enhances the promoter activity of
the CHAC1 gene. (A) Potential binding sites for ATF4, XBP1 and ATF6
on the CHAC1 gene promoter. (B and C) pGL3 luciferase reporter
assay was performed to determine the promoter activity of CHAC1. To
knockdown CHAC1 expression, 2 siRNAs were synthesized to target
CHAC1 in PLC cells. A total of 24 h post siRNA transfection, HepG2
and PLC/PRF/5 cells were treated with 40 or 25 µM DHA, respectively
for 24 h. (D and E) Protein expression levels of GPX4 and CHAC1
were determined by western blotting. (F and G) Cell viability, MDA
levels and GSH/GSSG ratios, as well as (H and I) intracellular
lipid ROS levels were determined. Data are presented as the mean ±
standard deviation of three repeats. *P<0.05, **P<0.01,
***P<0.001 vs. control. DHA, dihydroartemisinin; PLC, primary
liver cancer; CHAC1, ChaC glutathione specific
γ-glutamylcyclotransferase 1; ATF, activating transcription factor;
XBP1, X box-binding protein 1; siRNA, small interfering RNA; GPX4,
glutathione peroxidase 4; ROS, reactive oxygen species; MDA,
malondialdehyde; GSSG, oxidized glutathione; siRNA, small
interfering RNA; NC, negative control; M, % of control. |
Discussion
The induction of ferroptosis is currently being
investigated as an alternative approach to eradicate
apoptosis-resistant cancer cells (11). Several artemisinin derivates, such as
artesunate, artemether and arteether, have been demonstrated to
kill cancer cells by inducing ferroptosis (15). In addition, previous studies have
revealed that DHA acts as a ferroptosis inducer in cancer cells
(20–22). Consistent with the findings of these
previous studies, the present study demonstrated that DHA killed
PLC cells by inducing ferroptosis.
The classic method to determine whether a drug can
induce ferroptosis is to co-treat cancer cells with ferroptosis
blockers or iron ions (12,13). The antitumor effects of ferroptosis
inducers, such erastin and sorafenib, were found to be attenuated
by iron chelators (such as DFOM) and inhibitors of lipid
peroxidation inhibitors (such as ferrostatin-1), but augmented
following the addition of exogenous iron ions (7,28). In the
current study, based on the IC50 results, it was
suggested that PLC cells were more resistant to DHA-induced
cytotoxicity when ferroptosis was suppressed by DFOM or
ferrostatin-1. In contrast, the addition of iron chloride
hexahydrate sensitized PLC cells to DHA. Of note, treatment with
DFOM or ferrostatin-1 did not completely abolish the antitumor
effects of DHA, suggesting that triggering ferroptosis may not be
the only method through which DHA can induce cytotoxicity in PLC
cells. Although DHA has been shown to induce ferroptosis in other
types of cancer cells (20–22), the present study was the first to
demonstrate such effects in PLC cells, to the best of our
knowledge.
p53 is a key tumor suppressor gene, and mutations in
the gene or its loss of function is known to promote tumorigenesis
(29). SLC7A11 and SLC3A2 control the
import of extracellular cysteine and cystine, and regulate GSH
synthesis (9,30). Notably, both wild-type and
acetylation-defective mutant p53 were discovered to downregulate
SLC7A11 expression, thereby inhibiting GSH synthesis and
sensitizing tumor cells to ferroptosis (31). In light of the key role of p53 in
SLC7A11-associated ferroptosis, the present study aimed to
investigate the cytotoxicity of DHA in four PLC cell lines with
different p53 statuses. The current data demonstrated that DHA
could effectively induce ferroptosis in the analyzed PLC cells,
even in the p53 null cells (Hep3B cells). It is worth noting that
DHA also exhibited antitumor activity against PLC by activating
caspase 3, a key apoptosis regulator, regardless of the p53 status
(32). This finding, together with
the results of the present study, suggested that DHA may exert its
antitumor effects in a p53-independent manner. Thus, DHA may
represent an attractive drug for treating p53-mutant or -deficient
cancers.
GPX4 was first identified by Ursini et al
(33) in 1982 and it has since been
shown to reduce reactive phosphatidylcholine hydroperoxides and
suppress lipid peroxidation (8). GSH
depletion was found to downregulate GPX4 expression (34). In cancer cells, ferroptosis was shown
to be induced by GPX4 inactivation (11). To further explore how DHA induces
ferroptosis in PLC cells, GSH content and the expression and
activity of GPX4 were analyzed. The present data revealed that DHA
reduced GSH synthesis and downregulated GPX4 expression in PLC
cells. The decreased activity of GPX4 observed in PLC cells exposed
to DHA may result from the downregulated expression of GPX4. While
a study by Lin et al (20)
supported the findings of the present study, a recent study from
Chen et al (22) did not
report consistent results. The latter study demonstrated that DHA
induced ferroptosis in glioma cells and upregulated GPX4 expression
in U251 and U373 cancer cells as a compensatory mechanism (22). At present, a reasonable explanation
for such paradoxical findings has not been found. Nonetheless,
these findings suggested that the effects of DHA on regulating GPX4
expression in different solid tumor types may be inconsistent, and
further investigations into the underlying mechanisms are
warranted.
The present study also sought to determine whether
the DHA-activated UPR played a role in ferroptosis. The UPR is
coordinated by three branches, namely, the PERK/eIF2α/ATF4,
IRE1α/XBP1 and ATF6 branches (23,24). In
response to ER stress, PERK is self-phosphorylated to induce the
phosphorylation of eIF2α, thereby promoting ATF4 translation
(35). Once phosphorylated, IRE1α
splices XBP1 into the short form (35). ATF6 translocates into the cell nucleus
to function as a transcription factor after cleavage from the ER
(36). ATF4 was suggested to protect
against ferroptosis due to its ability to activate the
transcription of SLC7A11 (25). Our
previous study also demonstrated that ATF4 acted as a pro-survival
factor in erastin-induced ferroptosis in PLC cells (37). Therefore, it was hypothesized that
blocking the PERK/eIF2α/ATF4 signaling pathway may further augment
the antitumor effects of DHA in PLC cells. However, by determining
the cell viability and ROS contents, the results found that the
knockdown of ATF4 attenuated the cytotoxic effects of DHA. Notably,
DHA-induced ferroptosis was attenuated following the knockdown of
XBP1 or ATF6. Unlike the majority of previous studies revealing the
anti-ferroptotic roles of these UPR proteins (22,25), the
findings of the present study suggested that the UPR proteins acted
as pro-ferroptosis molecules in DHA-treated PLC cells.
Due to the observed role of ATF4 in inducing SLC7A11
expression (25), little previous
evidence supports the pro-ferroptotic role of ATF4 observed in the
current study. Nonetheless, an earlier study evaluating the effects
of artesunate, another classic ferroptosis inducer, in Burkitt's
lymphoma cells indirectly supported the findings of the present
study (16). A previous study showed
that ATF4 activated the transcription of CHAC1 to augment
artesunate-induced ferroptosis in DAUDI and CA-46 cells (16). CHAC1 was found to function as a
γ-glutamyl cyclotransferase to induce GSH degradation (38). These findings suggested that ATF4
upregulation may also lead to GSH degradation. It is possible that
DHA, like artesunate, activates mechanisms important for GSH
degradation, which are mediated by UPR proteins, rather than GSH
synthesis. In the present study, the results of the dual luciferase
reporter assay demonstrated that DHA enhanced the promoter activity
of the CHAC1 gene. While the effects of ATF4 on CHAC1 expression
have been reported, the current study was the first to demonstrate
that XBP1 and ATF6 also upregulated CHAC1 expression, to the best
of our knowledge.
There are several limitations to the present study.
First, only the expression of the key UPR regulators, ATF4, XBP1
and ATF6, were knocked down in PLC cells. Since the endogenous
expression levels of these factors are already abundant in PLC
cells stimulated with DHA, in the present study, knockdown of their
expression was deemed more appropriate. Activation of UPR branches
by different stimuli leads to different results in HCC cells, both
pro-apoptotic (39) and
anti-apoptotic (40) results have
been reported before. Simple overexpression of ATF4, XBP1 or ATF6
in HCC cells without DHA may demonstrate their own contribution to
the viability of HCC cells. However, such effects may not be
related to DHA. Nonetheless, to further elucidate the roles of the
activated UPR in DHA-induced ferroptosis, further studies should
investigate the effects of the overexpression of these regulators
in PLC cells. Second, several binding sites for XBP1 and ATF6 were
identified on the CHAC1 promoter. Although the aim of the present
study was to demonstrate how DHA affected the promoter activity of
CHAC1, the specific locations that these UPR sensors bind to
remains to be determined.
In conclusion, the findings of the present study
suggested that DHA may effectively induce ferroptosis and activate
the UPRs in PLC cells. The knockdown of key regulators of the UPRs
suppressed the toxic effects of DHA on PLC cells, which was
demonstrated through the downregulation of CHAC1 expression, a key
gene responsible for GSH degradation. DHA also promoted the
transcription of CHAC1. These findings may provide novel insights
into the antitumor effects of DHA, suggesting that the DHA-induced
UPR promotes cell death in PLC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated and/or analyzed during the
present study are included in the published article.
Authors' contributions
ZW and TB designed the study, performed the
experiments and analyzed the data. ZW drafted the manuscript. ML,
YL and ZQ, performed the experiments and assisted with data
analysis. LY and BL performed the experiments and revised the
manuscript. All authors have read and approved the final
manuscript. ZW and TB confirmed the authenticity of the data.
Ethics approval and consent to
participate
All institutional and national guidelines for the
care and use of laboratory animals were followed. The present study
was approved by the Ethics Committee of Zhengzhou University
(Zhengzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DHA
|
dihydroartemisinin
|
|
ROS
|
reactive oxygen species
|
|
MDA
|
malondialdehyde
|
|
GSH
|
glutathione
|
|
GSSG
|
oxidized glutathione
|
|
GPX4
|
glutathione peroxidase 4
|
|
PERK
|
protein kinase R-like ER kinase
|
|
eIF2
|
eukaryotic initiation factor 2
|
|
ATF4
|
activating transcription factor 4
|
|
ATF6
|
activating transcription factor 6
|
|
IRE1α
|
inositol-requiring transmembrane
kinase/endoribonuclease 1α
|
|
XBP1
|
X box-binding protein 1
|
|
CHAC1
|
ChaC glutathione specific
γ-glutamylcyclotransferase 1
|
|
SLC7A11
|
solute carrier family 7 member 11
|
|
SLC3A2
|
SLC family 3 member 2
|
References
|
1
|
Yang JD, Hainaut P, Gores GJ, Amadou A,
Plymoth A and Roberts LR: A global view of hepatocellular
carcinoma: Trends, risk, prevention and management. Nat Rev
Gastroenterol Hepatol. 16:589–604. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tang A, Hallouch O, Chernyak V, Kamaya A
and Sirlin CB: Epidemiology of hepatocellular carcinoma: Target
population for surveillance and diagnosis. Abdom Radiol (NY).
43:13–25. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
El-Serag HB: Epidemiology of viral
hepatitis and hepatocellular carcinoma. Gastroenterology.
142:1264–1273 e1261. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tabrizian P, Jibara G, Shrager B, Schwartz
M and Roayaie S: Recurrence of hepatocellular cancer after
resection: Patterns, treatments, and prognosis. Ann Surg.
261:947–955. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shiina S, Sato K, Tateishi R, Shimizu M,
Ohama H, Hatanaka T, Takawa M, Nagamatsu H and Imai Y: Percutaneous
ablation for hepatocellular carcinoma: Comparison of various
ablation techniques and surgery. Can J Gastroenterol Hepatol.
2018:47561472018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hirschhorn T and Stockwell BR: The
development of the concept of ferroptosis. Free Radic Biol Med.
133:130–143. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cao JY and Dixon SJ: Mechanisms of
ferroptosis. Cell Mol Life Sci. 73:2195–2209. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Okada H and Mak TW: Pathways of apoptotic
and non-apoptotic death in tumour cells. Nat Rev Cancer. 4:592–603.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hassannia B, Vandenabeele P and Vanden
Berghe T: Targeting ferroptosis to iron out cancer. Cancer Cell.
35:830–849. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kagan VE, Mao G, Qu F, Angeli JP, Doll S,
Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al: Oxidized
arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem
Biol. 13:81–90. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ho WE, Peh HY, Chan TK and Wong WS:
Artemisinins: Pharmacological actions beyond anti-malarial.
Pharmacol Ther. 142:126–139. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ooko E, Saeed ME, Kadioglu O, Sarvi S,
Colak M, Elmasaoudi K, Janah R, Greten HJ and Efferth T:
Artemisinin derivatives induce iron-dependent cell death
(ferroptosis) in tumor cells. Phytomedicine. 22:1045–1054. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang N, Zeng GZ, Yin JL and Bian ZX:
Artesunate activates the ATF4-CHOP-CHAC1 pathway and affects
ferroptosis in Burkitt's Lymphoma. Biochem Biophys Res Commun.
519:533–539. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Eling N, Reuter L, Hazin J, Hamacher-Brady
A and Brady NR: Identification of artesunate as a specific
activator of ferroptosis in pancreatic cancer cells. Oncoscience.
2:517–532. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang D, Meng G, Zheng M, Zhang Y, Chen A,
Wu J and Wei J: The Glutaminase-1 inhibitor 968 enhances
dihydroartemisinin-mediated antitumor efficacy in hepatocellular
carcinoma cells. PLoS One. 11:e01664232016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang CZ, Zhang H, Yun J, Chen GG and Lai
PB: Dihydroartemisinin exhibits antitumor activity toward
hepatocellular carcinoma in vitro and in vivo. Biochem Pharmacol.
83:1278–1289. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lin R, Zhang Z, Chen L, Zhou Y, Zou P,
Feng C, Wang L and Liang G: Dihydroartemisinin (DHA) induces
ferroptosis and causes cell cycle arrest in head and neck carcinoma
cells. Cancer Lett. 381:165–175. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X,
Ren X, An Y, Wu Y, Sun W, et al: DHA inhibits proliferation and
induces ferroptosis of leukemia cells through autophagy dependent
degradation of ferritin. Free Radic Biol Med. 131:356–369. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen Y, Mi Y, Zhang X, Ma Q, Song Y, Zhang
L, Wang D, Xing J, Hou B, Li H, et al: Dihydroartemisinin-induced
unfolded protein response feedback attenuates ferroptosis via
PERK/ATF4/HSPA5 pathway in glioma cells. J Exp Clin Cancer Res.
38:4022019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cubillos-Ruiz JR, Bettigole SE and
Glimcher LH: Tumorigenic and immunosuppressive effects of
endoplasmic reticulum stress in cancer. Cell. 168:692–706. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang M and Kaufman RJ: The impact of the
endoplasmic reticulum protein-folding environment on cancer
development. Nat Rev Cancer. 14:581–597. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen D, Fan Z, Rauh M, Buchfelder M,
Eyupoglu IY and Savaskan N: ATF4 promotes angiogenesis and neuronal
cell death and confers ferroptosis in a xCT-dependent manner.
Oncogene. 36:5593–5608. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fang C, Dai CY, Mei Z, Jiang MJ, Gu DN,
Huang Q and Tian L: microRNA-193a stimulates pancreatic cancer cell
repopulation and metastasis through modulating
TGF-beta2/TGF-betaRIII signalings. J Exp Clin Cancer Res.
37:252018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tadvalkar G, Pal-Ghosh S, Pajoohesh-Ganji
A and Stepp MA: The impact of euthanasia and enucleation on mouse
corneal epithelial axon density and nerve terminal morphology. Ocul
Surf. 18:821–828. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Luo M, Wu L, Zhang K, Wang H, Zhang T,
Gutierrez L, O'Connell D, Zhang P, Li Y, Gao T, et al: miR-137
regulates ferroptosis by targeting glutamine transporter SLC1A5 in
melanoma. Cell Death Differ. 25:1457–1472. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Amaral JD, Castro RE, Steer CJ and
Rodrigues CM: p53 and the regulation of hepatocyte apoptosis:
Implications for disease pathogenesis. Trends Mol Med. 15:531–541.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Banjac A, Perisic T, Sato H, Seiler A,
Bannai S, Weiss N, Kölle P, Tschoep K, Issels RD, Daniel PT, et al:
The cystine/cysteine cycle: A redox cycle regulating susceptibility
versus resistance to cell death. Oncogene. 27:1618–1628. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jiang L, Kon N, Li T, Wang SJ, Su T,
Hibshoosh H, Baer R and Gu W: Ferroptosis as a p53-mediated
activity during tumour suppression. Nature. 520:57–62. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hou J, Wang D, Zhang R and Wang H:
Experimental therapy of hepatoma with artemisinin and its
derivatives: In vitro and in vivo activity, chemosensitization, and
mechanisms of action. Clin Cancer Res. 14:5519–5530. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ursini F, Maiorino M, Valente M, Ferri L
and Gregolin C: Purification from pig liver of a protein which
protects liposomes and biomembranes from peroxidative degradation
and exhibits glutathione peroxidase activity on phosphatidylcholine
hydroperoxides. Biochim Biophys Acta. 710:197–211. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dixon SJ, Patel DN, Welsch M, Skouta R,
Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS
and Stockwell BR: Pharmacological inhibition of cystine-glutamate
exchange induces endoplasmic reticulum stress and ferroptosis.
Elife. 3:e025232014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Walter F, Schmid J, Dussmann H, Concannon
CG and Prehn JH: Imaging of single cell responses to ER stress
indicates that the relative dynamics of IRE1/XBP1 and PERK/ATF4
signalling rather than a switch between signalling branches
determine cell survival. Cell Death Differ. 22:1502–1516. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bai T, Liang R, Zhu R, Wang W, Zhou L and
Sun Y: MicroRNA-214-3p enhances erastin-induced ferroptosis by
targeting ATF4 in hepatoma cells. J Cell Physiol. 235:5637–5648.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kumar A, Tikoo S, Maity S, Sengupta S,
Sengupta S, Kaur A and Bachhawat AK: Mammalian proapoptotic factor
ChaC1 and its homologues function as gamma-glutamyl
cyclotransferases acting specifically on glutathione. EMBO Rep.
13:1095–1101. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ma MKF, Lau EYT, Leung DHW, Lo J, Ho NPY,
Cheng LKW, Ma S, Lin CH, Copland JA, Ding J, et al: Stearoyl-CoA
desaturase regulates sorafenib resistance via modulation of ER
stress-induced differentiation. J Hepatol. 67:979–990. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nakagawa H, Umemura A, Taniguchi K,
Font-Burgada J, Dhar D, Ogata H, Zhong Z, Valasek MA, Seki E,
Hidalgo J, et al: ER stress cooperates with hypernutrition to
trigger TNF-dependent spontaneous HCC development. Cancer Cell.
26:331–343. 2014. View Article : Google Scholar : PubMed/NCBI
|