Introduction
The association between thrombosis and cancer is
well established; 25% of patients with cancer suffer from venous
thromboembolic complications, the second most common cause of
cancer-related death (1). Low
molecular weight heparins (LMWHs) are currently used for
antithrombotic treatment (2,3); antithrombin limits the coagulation
process, while its anticoagulant activity is stimulated by heparin
(4). Tinzaparin is a
LMWH-anticoagulant used for the treatment of deep vein thrombosis
(5), and is recommended for optimum
antithrombotic interventions in the secondary prophylaxis of cancer
patients (6) with renal failure
(7) and brain tumors (8). Several studies (9–17),
reviewed for experimental models (18), have shown the anticancer properties of
tinzaparin However, in local tumor growth, tinzaparin failed to
impede cellular proliferation in an in vitro model of human
breast cancer cells (11).
Furthermore, it has been reported that tinzaparin has no effect on
primary tumor growth in the B16F10 metastasis model, and that
non-anticoagulant heparin inhibits metastasis, but not primary
tumor growth (19).
Tumor growth and metastatic potential are
angiogenesis-dependent (20), and
VEGF is a major angiogenic growth factor involved in human tumors
and angiogenic diseases (21). In
addition to the potent inhibition of angiogenesis (15), tinzaparin also has an important effect
on tumorigenesis and metastasis. Specifically, in the B16
melanoma-injectable model of metastasis, treatment of mice with
tinzaparin for 4 h prior to injection of melanoma cells, or daily
administration for 14 days, was shown to reduce lung tumor
formation by 89 and 96%, respectively (14). Furthermore, tinzaparin has been
reported to inhibit the extracellular vesicle-induced migration of
carcinoma cells (22), as well as
pancreatic tumor growth and metastasis (23). Finally, tinzaparin also has effects at
the nuclear level, modulating (among other diverse processes) the
expression of genes that regulate transcription and chromatin
modification in human A2780cis ovarian cancer cells; notably,
tinzaparin treatment caused marked transcriptional reprogramming of
3,776 tinzaparin-regulated genes, and antagonized
cisplatin-resistance (17).
Epithelial-mesenchymal transition (EMT) is an
important process involved in cancer progression, influencing
cellular invasion and metastasis. During EMT, epithelial cells lose
polarity and cell-to-cell contact, which is accompanied by
cytoskeleton remodeling, and the acquisition of migratory
properties, as well as a mesenchymal-like profile of gene
expression (24,25). In addition to the induction of
angiogenesis, VEGF also induces EMT in human pancreas carcinoma
cells (26). Strong evidence suggests
a link between EMT and cancer stem cells (CSCs) (27,28), and
human breast tumors characterized by a small population of CSCs are
also termed as tumor-initiating cells (29). Furthermore, a number of EMT-related
properties of CSCs have been shown to be associated with the
establishment of breast cancer metastasis (27).
Retrotransposons are DNA sequences that constitute
~45% of the human and mouse genome, and play a crucial role in
nuclear organization, structure and evolution (30). Categorized as long terminal repeat
(LTR) and non-LTR subtypes, retrotransposons are mobilized in the
genome by the intracellular process of retrotransposition. LTR
retrotransposition requires a retrotransposon RNA-intermediate,
which upon reverse transcription to cDNA, is integrated into a new
genomic site (31).
Retrotransposition is a potent mutagenic phenomenon, as new
retrotransposon copy integrations can inactivate or deregulate the
expression of nearby genes. Currently, 124 independent
retrotransposon insertions have been correlated with 65 human
diseases, including cancer (32).
Retrotransposition is a rare phenomenon that occurs at a low
frequency of up to 10−6 events/cell in each cell
generation (33). Low-rate
retrotransposition events mainly occur during oogenesis and
embryogenesis (34,35), while induced or uncontrolled
retrotransposon mobility occasionally result in the onset of
genetic diseases or tumorigenesis (35).
VL30 elements (VL30s) are a family of endogenous
retrovirus-like LTR-retrotransposons, present in the mouse and rat
genomes. Their internal sequences bear multiple stop codons, and
with a lack of protein coding capacity (36), VL30s are non-infectious. VL30
transcripts form a complex with the poly-pyrimidine tract binding
protein-associated splicing factor (PSF), and upregulate gene
expression and proto-oncogene transcription, as well as affecting
embryo-genesis and steroidogenesis (37). Furthermore, the PSF/VL30 RNA complex
promotes cellular proliferation and oncogenesis (37). VL30 LTRs bear a large number of common
and unique transcription factor binding sites, which may justify
the versatile and tissue-specific expression of VL30 RNA, as well
as its upregulation by various or pleiotropic stimuli, respectively
(38) (for example; histone
phosphorylation, acetylation and DNA demethylation) (39). VL30 transcription is also induced by
the Simian virus 40 large T antigen (40) and heavy metals (41,42), as
well as steroid hormones, 5′-azacytidine, C2-ceramide and a mouse
dominant-negative p53 gene (43).
Transcriptional induction by various inducers is a prominent
feature of VL30s, classified as early response genes (36). In reference to the mutagenic or
deleterious effects of retrotransposition, induction of VL30
retrotransposition has been associated with cytotoxicity (44) and cell death (45). Notably, our previous study revealed
VL30 retrotransposition induced by either arsenic (42) or H2O2 (44) is effectively reduced by the
anti-oxidant N-acetylcysteine (NAC).
Previously, VL30 retrotransposition in mouse HC11
epithelial mammary cells (with stem-like properties) was reported
in association with induced EMT, CSC generation and tumor growth
(46). Since tinzaparin modulates the
expression of a large number of genes (17), and LTR-retrotransposition is
accomplished through a retrotransposon RNA-intermediate, the aim of
the present study was to establish whether tinzaparin also affected
VL30 retrotransposition. Applying a previous model of HC11/VL30
retrotransposition (40), the current
study indicates novel activities of tinzaparin as an anti-oxidant,
anti-VEGF and anti-retroviral agent for the inhibition of VL30
retrotransposition. Furthermore, tinzaparin was shown to cause both
disaggregation and proliferative inhibition of VL30
retrotransposition-induced mammosphere/tumor-initiating CSCs,
suggesting an inhibition of VL30 retrotransposition-induced
tumorigenesis.
Materials and methods
Cell lines, cloning and
treatments
HC11 cells are immortalized mouse mammary epithelial
cells (47) originating from
mid-pregnancy BALB/c mice, with stem-like or progenitor cell
properties (48). HC11 cells were
cultured in RPMI 1640 medium containing 10% FBS, 2 mM L-glutamine,
5 mg/ml insulin, 10 ng/ml EGF (47),
100 units penicillin and 100 µg/ml streptomycin (all Thermo Fisher
Scientific, Inc.) at 37°C in an incubator supplemented with 5%
CO2. A total of 2.5×105 HC11 cells were
transfected at room temperature with 2.5 µg of our previously
constructed pNVL-3*/EGFP-INT DNA (40) using the PolyFect®
transfection reagent (Qiagen, Inc.), and then cultured for 16 h
under cell culture conditions. Hygromycin B-resistant cell clones
were isolated following application of 100 µg/ml hygromycin B for
18 days. Isolated clone cells were further cultured for 20 days in
cell culture treated flasks (Corning, Inc.), and used for
subsequent experimentation. Notably this plasmid harbors a
recombinant VL30 element tagged with an EGFP gene-based
retrotransposition indicator cassette, and the expression of EGFP
protein occurs solely after a retrotransposition event. Thus, VL30
retrotransposition-positive cells can be enumerated by flow
cytometry (40). Our previously
isolated Pcl.10 cell clone derived from NIH3T3 mouse embryo
fibroblasts transfected with pNVL-3*/EGFP-INT (40), as aformentioned. Pcl.10 cells were
cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented
with 10% FBS, 2 mM glutamine and antibiotics (100 units penicillin
and 100 µg/ml streptomycin, and maintained as aforementioned. Serum
starvation was performed in serum-free medium for 24 h at 37°C.
Recovery of 30% confluency HC11 cells treated with 1- or 2 IU
tinzaparin pre-incubation for 3 h, and/or 40 pg/ml glucose oxidase
(GO) for 24 h, was performed by washing the cells twice with RPMI
1640 medium at room temperature. Tinzaparin sodium (syringe 10,000
anti-factor Xa IU/ml) was obtained from LEO Pharma A/S, and
appropriate dilutions were made with culture medium. VEGF treatment
at 3–12 ng/ml was performed in both HC11 and Pcl.10 cells with
mouse recombinant VEGF A (ImmunoTools GmbH) 24 h after serum
starvation. Tinzaparin and/or VEGF treatment was performed at
37°C.
Paraformaldehyde fixation and
observation of EGFP/VL30 retrotransposition-positive cells
HC11 cl.11 assay cells cultured at the desired
confluence on sterilized glass coverslips were fixed with 3.7%
paraformaldehyde for 30 min on ice. Then coverslip-samples mounted
onto microscope slides were observed and photographed under normal
or UV light (Nikon Eclipse E800 Fluorescent Microscope).
Measurement of VL30 retrotransposition
frequency and cellular proliferation
All tested hygromycin B-resistant HC11 clone cells
were trypsinized, washed with PBS, centrifuged at 1,600 x g for 3
min at room temperature, and resuspended in PBS. Samples of 15,000
clone or non-transfected cells (as the negative control) were used
to determine EGFP-positivity by flow cytometry, as previously
described (40). The fluorescence
intensity thresholds were evaluated using non-transfected cells,
and sample fluorescence ≥99.60% was considered as negative, and
0.4% as false positive. Above the false-positive threshold, samples
were considered as EGFP- or VL30 retrotransposition-positive. The
continuous cell proliferation rate (cell index) was measured using
a microelectronic biosensor system (xCELLigence®
real-time cell analysis DP) in E-plates using 3,500 seeded cells in
a volume of 60 µl RPMI (supplemented with 10% FBS) for up to 80 h.
Cellular proliferation was assessed by seeding 60,000 cells per
well (~10% confluent) into a 6-well plate. Cells from three wells
were trypsinized, stained with trypan blue and counted using a
Neubauer hemocytometer every 24 h for a period of 5 days (44).
Soft agar-foci formation assay
The foci formation assay was based on a previously
described protocol (49). Briefly,
6-well plates bearing a 0.66% nobble agar-base layer (prepared in
10% FBS/RPMI medium supplemented with 100 units penicillin and 100
µg/ml streptomycin), a 0.33% upper- and 0.33% feeder layer, were
used. The upper layer was prepared after mixing nobble agar with
trypsinized cells from either control or test cell groups, in the
presence or absence of 2 IU/ml tinzaparin, respectively. Dishes
were incubated at 37°C in a 5% CO2 cell culture
incubator, and supplemented with 200 µl medium every two days to
avoid desiccation. The examination of dishes for cell foci was
formation performed with an optical microscope.
Mammosphere preparation from
tumorigenic HC11 cl.19 cells
For mammosphere preparation, VL30
retrotransposition- tumorigenic HC11 cl.19 cells cultured in normal
culture plates were trypsinized and seeded into non-adherent plates
at 30% density. Following 20 days of culture, the cells were
replenished with RPMI-1640 medium every 2 days, and the resulting
anchorage-independent/floating mammospheres in the culture medium
were collected using a pipette for further use.
DNA lysate preparation and PCR
analysis
A total of 1.5×106 HC11 cl.19 cells were
trypsinized, washed with PBS and centrifuged at 1,600 x g for 3 min
at room temperature. Cell pellets were resuspended in 800 µl PCR
wash buffer [10 mM Tris-HCl (pH 8.4), 50 mM KCl, 1.5 mM
MgCl2 and 0.001% (w/v) gelatin], and centrifuged once
more. The pellets were then gently resuspended in 100 µl PCR lysis
buffer [10 mM Tris-HCl pH 8.4, 50 mM KCl, 1.5 mM MgCl2,
0.001% gelatin, 0.1% (v/v) Triton X-100, 0.45% (v/v) Nonidet P-40
and 0.45% (v/v) Tween-20], heated at 80°C for 10 min, cooled at
room temperature for 15 min and then treated with 4 µl proteinase K
(10 mg/ml) overnight at 55°C. Following heat inactivation at 95°C
for 15 min, the samples were centrifuged at 1,600 x g for 3 min at
room temperature, and the supernatant, representing an extracted
DNA lysate, was collected. A volume of 3–5 µl DNA lysate was used
for PCR analysis. PCR reactions were performed using 2.5 units Taq
polymerase (Invitrogen; Thermo Fisher Scientific, Inc.) in a final
volume of 50 µl, with specific EGFP primers: GFP968 forward,
5′-GCACCATCTTCTTCAAGGACGAC-3, and GFP1013 reverse,
5′-TCTTTGCTCAGGGCGGACTG-3′ (50)
using conditions previously reported (40).
Semi-quantitative reverse
transcription (RT)-PCR analysis
Quantitation of endogenous reverse transcriptase
(enRT) and VL30 transcripts was performed by RT-PCR. Total RNA was
extracted from untreated cells, cells treated with VEGF alone or
VEGF/tinzaparin using a RNeasy Mini Kit (Qiagen, Inc.).
Subsequently, 100 ng cDNA was prepared from 1 µg total RNA. A set
of degenerate primers designed to target the enRT conserved domains
(4 and 5 amino acid sequences identified in the amino-terminal
coding regions of most known enRT polymerases including that of the
Moloney murine leukemia virus) (51),
as well as a set of previously reported VL30-specific primers:
Forward, 5′-CCTTTGTTGCCCAGGTAAGTC-3′ and reverse,
5′-CACTGTAGCCAGTTGTGACCAG-3′ (52).
mRNA expression of the mouse β-actin gene was quantitated using the
following primers: β-actin forward, 5′-TTGCTGACAGGATGCAGAAG-3′ and
reverse, 5′-ACATCTGCTGGAAGGTGGAC-3′. The enRT thermocycling
conditions were as follows: 94°C for 4 min; 27 cycles of 94°C for
30 sec, 50°C for 45 sec, and 72°C for 2 min; and 72°C for 2 min.
The VL30 thermocycling conditions were: 94°C for 4 min; 30 cycles
of 94°C for 30 sec, 57°C for 30 sec and 72°C for 2 min; and 72°C
for 2 min. Finally, the β-actin conditions were: 94°C for 4 min; 27
cycles of 94°C for 30 sec, 57°C for 30 sec, and 72°C for 1.5 min;
and 72°C for 2 min. All PCR products were fractionated using 1.2%
(w/v) agarose gels, stained with ethidium bromide (EtBr), and
visualized under ultraviolet light. enRTs and VL30 RNA expression
values were normalized to that of β-actin, and densitometric
analysis was performed using Fiji (ImageJ2) software (National
institutes of Health).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism version 5.0 (GraphPad Software, Inc.). Comparisons between
multiple groups of retrotransposition frequency values were
determined by one-way ANOVA followed by the Tukey's post hoc-test.
A paired Student's t-test was used to assess statistically
significant differences between two groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
Isolation and characterization of VL30
retrotransposition-positive HC11 clones
In a previous study, VL30
retrotransposition-positive HC11 cell clones were generated,
exhibiting an induced EMT phenotype, CSC properties and solid tumor
production in syngeneic Balb/c mice (46), and with a retrotransposition frequency
of ~2-5.5%. Notably, as HC11 cells possess stem-like properties,
the occurrence of retrotransposition events could be explained by
their hypomethylation status (46),
as has been suggested for human stem cells (53). To examine the effect of tinzaparin
and/or VEGF treatment on VL30 retrotransposition, new clones were
isolated with a retrotransposition frequency of 7–15%, allowing a
more accurate measurement of retrotransposition changes, especially
in the case of inhibition.
Following transfection with pNVL-3*/EGFP-INT, 20
hygromycin B-resistant HC11 clones were isolated. Using
non-transfected HC11 cells as the control, the isolated clones were
flow cytometrically examined for retrotransposition positivity, as
exemplified for clone 11 (Figs. S1A
and S2). Among all clones examined,
a set of seven clones (cl.) exhibited the following percentages of
retrotransposition frequency: cl.12, 7%; cl.15, 8%; cl.6 and cl.9,
~9%; cl.11, 11.5%; cl.5, 12%; and cl.17, 15.2%, falling within the
range of 7–15.2% (Fig. S1B). PCR
analysis confirmed retrotransposition-positivity at the genomic
level in four representative clones, through the diagnostic
intron-less 342 bp band [indicative of a retrotransposition event
(40) (Fig. S1C)]. Furthermore, microscopic
observation revealed two common VL30 retrotransposition-derived
features, as shown for representative cl.11 (Fig. S3): i) An expected VL30
retrotransposition-induced EMT phenotype (46) observed at low confluence (Fig. S3B); and ii) cytoplasmic EGFP
fluorescence (Fig. S3D), confirming
the occurrence of VL30 retrotransposition events (40), associated with genomic instability
manifested by multinucleated cells bearing enlarged cytoplasm, in
cell clusters formed at high confluence (Fig. S3C). Thus, isolated clones with a
retrotransposition value of 7–15.2% may be used for studying
factors that modulate the frequency of VL30 retrotransposition.
Tinzaparin inhibits VEGF-induced VL30
retrotransposition in HC11 cells
To address the potential role of VEGF and/or
tinzaparin treatment on VL30 retrotransposition, HC11 cl.17 cells
were initially cultured in serum-free medium for 24 h to exclude
the presence of serum associated-VEGF. Following serum starvation
for 24 h, (a condition used in all subsequent treatments), the
retrotransposition frequency of HC11 cl.17 cells was increased from
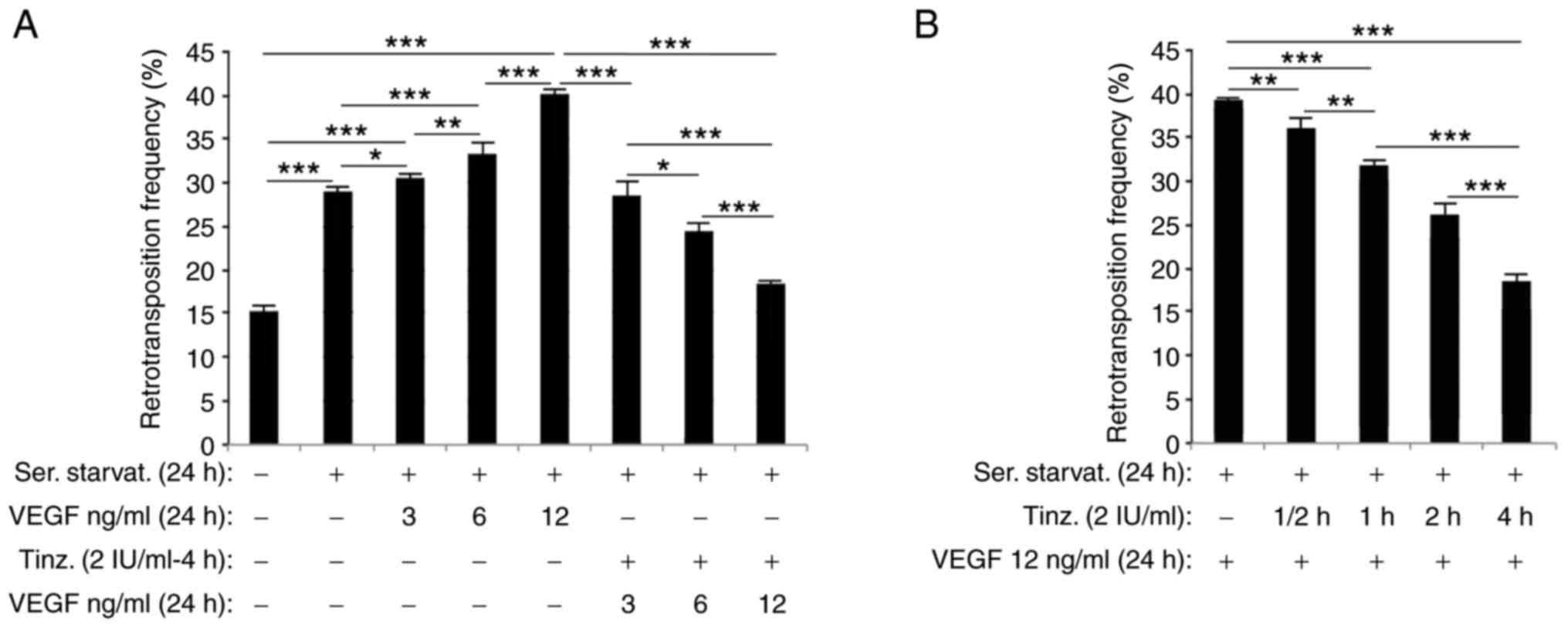
15.2 to 29% (Fig. 1A). Then, HC11
cl.17 cells were serum-starved in the absence or presence of 3, 6
or 12 ng/ml VEGF for 24 h and their retrotransposition was compared
with serum-starved cells without VEGF as the control, VEGF
treatment increased retrotransposition in a dose-dependent manner
in all cases (Fig. 1A). The level of
retrotransposition under serum starvation (29%) increased in the
presence of increasing VEGF concentrations (3, 6 and 12 ng/ml) to
30.4, 33.4 and 40.3% corresponding to a net VEGF-dependent
induction of 1.4, 4.4 and 11.3%, respectively.
To investigate the action of tinzaparin on
VEGF-induced VL30 retrotransposition, serum-starved cells were
initially pre-incubated with 2 IU tinzaparin/ml for 4 h, and then
treated, in the presence of tinzaparin, with 3, 6 or 12 ng/ml VEGF
for 24 h. Compared with those treated with VEGF alone, the
retrotransposition frequencies of tinzaparin/VEGF-treated samples
were significantly reduced. Specifically, at 3, 6 and 12 ng/ml
VEGF, tinzaparin reduced retrotransposition frequency from 30.4 to
28.7%, 33.4 to 24.6%, and 40.3 to 18.3%, respectively,
corresponding to a net reduction in retrotransposition frequency of
1.7, 8.8 and 22% (Fig. 1A).
Next, the effects of tinzaparin pre-incubation time
on the inhibition of retrotransposition were assessed using
treatment times <4 h. HC11 cl.17 cells were pre-incubated with 2
IU tinzaparin/ml for 0.5, 1, 2 or 4 h, and then treated with 12
ng/ml VEGF for 24 h. Using the 39.42% retrotransposition frequency
of tinzaparin-untreated cells as the control, tinzaparin reduced
VEGF-induced retrotransposition frequency in a time-dependent
manner (Fig. 1B). At 0.5, 1, 2 and 4
h, respective retrotransposition frequencies of 36.1, 32.2, 26.3
and 18.7% were recorded, corresponding to a net inhibition of 8.37,
18.27, 33.24 and 52.53%.
In preliminary experiments, a higher concentration
of VEGF (i.e. 15 compared with 12 ng/ml) induced an even greater
retrotransposition frequency than the 40.3% presented in Fig. 1 (data not shown). It was concluded
that: i) 40.3% induced retrotransposition was suitable enough to
reliably document the effects of tinzaparin with VEGF treatment;
and ii) such a high value (40.3%) was adequate to more accurately
assess the effectiveness of tinzaparin, especially at low
concentrations (such as the 1 or 2 IU used), and to retain HC11
cell functionality at this level of VEGF-derived
retrotransposition, as a higher VEGF concentration may induce many
more mutations [since 1% induced retrotransposition equates to an
additional ‘retrotransposition burden’ of 1000-fold
genome-mutations/cell (33)].
Collectively, these data revealed that tinzaparin was effective in
simultaneously reducing serum-starved/VEGF-induced VL30
retrotransposition.
Tinzaparin inhibits VEGF-induced VL30
retrotransposition in NIH3T3 cells
The aforementioned data prompted the investigation
of VEGF and/or VEGF/tinzaparin treatment on cell types other than
epithelial HC11 mammary progenitors. A previously generated VL30
retrotransposition-positive clone (Pcl.10), derived from mouse
NIH3T3 fibroblast cells, was used. Notably, Pcl.10 is a hygromycin
B-resistant NIH3T3 fibroblast clone, isolated as HC11 clones, but
VL30 retrotransposition events are elicited solely following a
stimulus such as the SV40 large T antigen (40), heavy-metal vanadium (VOSO4)
(41) or arsenic (42). Serum starvation for 24 h induced VL30
retrotransposition frequency by 2.4%, compared with non-serum
starved Pcl.10 cells. Furthermore, treatment with 12 ng/ml further
increased retrotransposition frequency to 7.44% (Fig. S4), corresponding to a net increase of
5.04%. Notably, VEGF concentrations up to 50 ng/ml did not increase
retrotransposition frequency further (data not shown). Then, Pcl.10
cells were pre-incubated with 2 IU tinzaparin/ml for 0.5, 1, 2 or 4
h, and then treated with 12 ng/ml VEGF for 24 h. Of note, 7.44%
VEGF-induced retrotransposition frequency (used as control), was
inhibited in a time-dependent manner to 6.5, 4.75, 3.8 and 2.45%,
respectively (Fig. S4). The use of
Pcl.10 cells showed that the reduction of VEGF-induced
retrotransposition by tinzaparin is not limited to mouse mammary
epithelial stem-like HC11 cells.
Tinzaparin inhibits oxidative
stress-induced VL30 retrotransposition in HC11 cells
Serum starvation has been reported to induce the
production of reactive oxygen species (ROS) in prostate cancer cell
lines (54), and our previous study
showed that H2O2-derived oxidative stress
induces VL30 retrotransposition of NIH3T3 fibroblast Pcl.10 clone
cells to a high level (44).
Therefore, the effect of oxidative stress on HC11
retrotransposition, and whether it is affected by tinzaparin
treatment, was investigated in the present study. VL30
retrotransposition signals activation of a caspase-independent and
p53-dependent death pathway (45),
while HC11 cells, harboring a mutated p53 gene (55), alleviated cell death allowing the
measurement of high retrotransposition frequencies.
In the present study, HC11 cl.17 cells (Fig. S1B) were initially treated with 5–40
pg/ml GO for 24 h, and retrotransposition frequency was measured
after a 48 h recovery in normal medium, using non-treated cells as
the control. At 10–40 pg/ml GO, the 15.2% nominal
retrotransposition frequency of HC11 cl.17 cells was further
increased in a concentration-dependent manner, up to ~82.5% at 40
pg/ml (Fig. 2A). Next, considering
that 40 pg/ml GO strongly induced VL30 retrotransposition, HC11
cl.17 cells were pre-incubated with 1 or 2 IU/ml tinzaparin for 3
h, then treated with 40 pg/ml GO for 24 h. Pre-incubation with 1
IU- or 2 IU/ml tinzaparin reduced the ~82.5% GO-induced
retrotransposition frequency to respective values of 69 and 57%,
corresponding to a 13.5 or 25.5% level of inhibition (Fig. 2B). The data indicate that tinzaparin,
reducing GO-induced retrotransposition, acts as an anti-oxidant
agent.
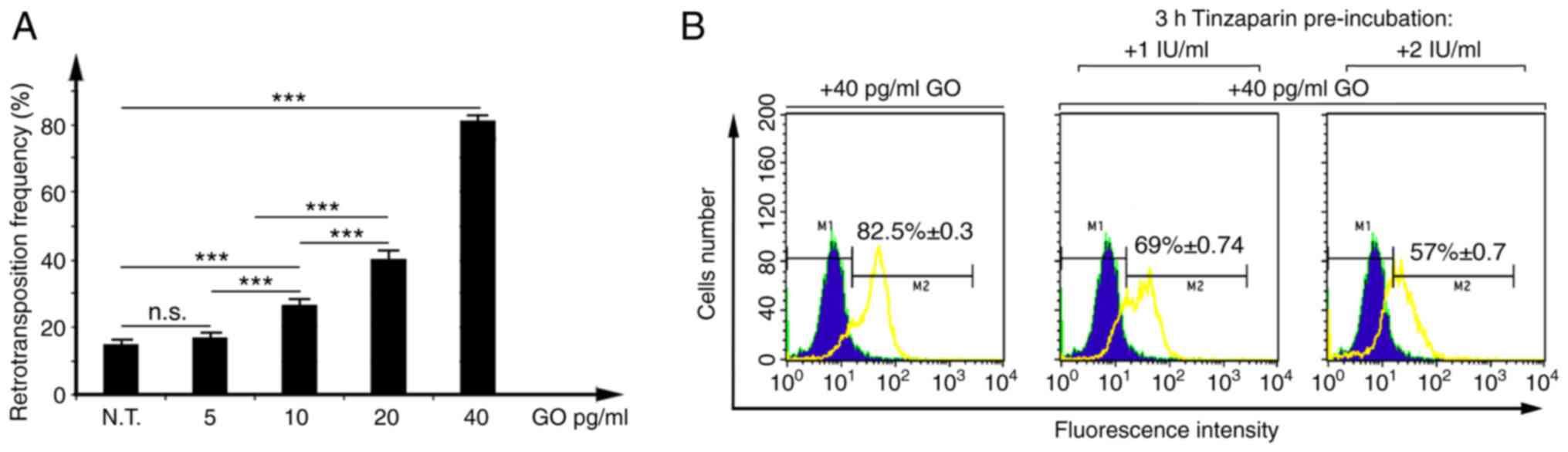 | Figure 2.Tinzaparin inhibits the GO-induced
VL30 retrotransposition frequency of HC11 cl.17 cells. (A) HC11
cl.17 cells, with a 15.2±0.3% nominal retrotransposition frequency,
were treated with 5–40 pg/ml GO for 24 h, and retrotransposition
frequency was measured by flow cytometry 48 h post-treatment
recovery, using N.T. HC11 cl.17 cells as the control. Data are
presented as the mean ± SD, with duplicate samples from three
independent experiments. ***P<0.001; one-way ANOVA followed by
Tukey's test. (B) HC11 cl.17 cells were either treated with 40
pg/ml GO for 24 h, or pre-incubated with 1- or 2 IU tinzaparin for
3 h and then treated with 40 pg/ml GO for 24 h. N.T. cells or
treated cultures were assessed for EGFP-positivity by flow
cytometry. Overlaid green circumscribed-solid blue and yellow
histograms represent fluorescence of control and
tinzaparin/GO-treated HC11 cl.17 cells, respectively. M1 and M2
gates correspond to arbitrarily set fluorescence intensity
thresholds, up to 99.60% considered as negative and 0.4% false
positive. The indicated percentage values with standard error ± SE
shown inside the histogram panels represent net retrotransposition
frequencies, subtracting a 0.4% self-fluorescence percentage (false
positive at M2). Percentage of retrotransposition values shown on
the left, middle and right panels, respectively, are the mean ± SE
of duplicate samples from three independent experiments. GO,
glucose oxidase; N.T., non-treated; n.s., not significant. |
Tinzaparin inhibits CSC features of
VL30 retrotransposition- positive HC11 cells
Our previous study reported that VL30
retrotransposition-positive HC11 cells acquire tumorigenic features
such as a high rate of proliferation, induced-EMT phenotype and
anchorage-independent mammosphere formation (46). These findings prompted the
investigation of tinzaparin on the features of VL30
retrotransposition-positive HC11 cells.
Using both the previously well-characterized HC11
cl.19 for its retrotransposition-induced CSCs and mammosphere
formation properties (46), and HC11
cl.15 from the present study, the effect of tinzaparin on HC11 cell
proliferation rate was initially examined. Trypsinized cells from
these clones were subjected to real-time cell analysis in the
absence or presence of 2 IU/ml tinzaparin for up to 80 h, using
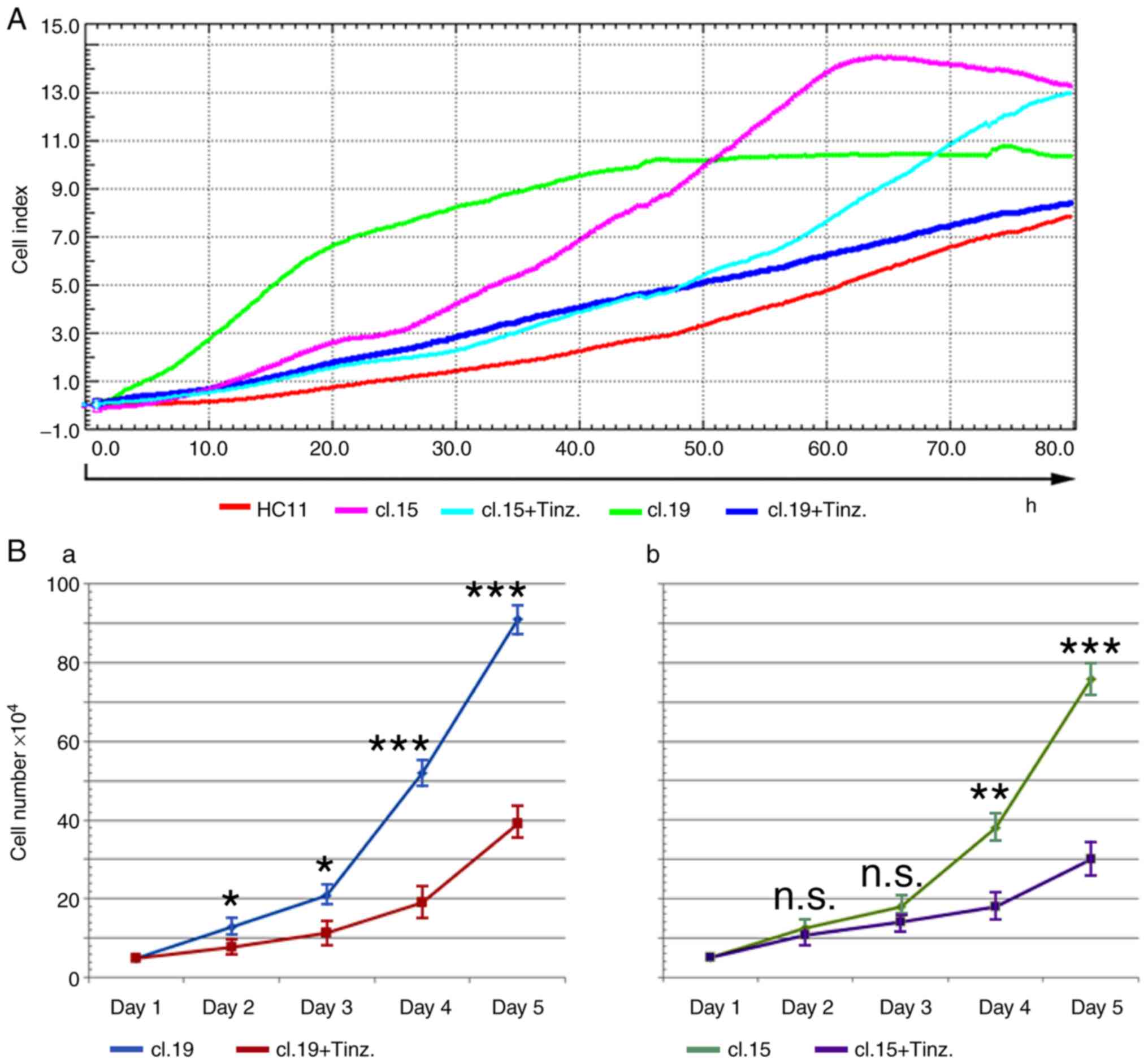
normal HC11 cells as the control. In the absence of tinzaparin, the
continuous proliferation rate of both retrotransposition-positive
clones was markedly higher than that of the control cells, but was
inhibited to varying degrees in the presence of tinzaparin
(Fig. 3A). However, the
tinzaparin-induced reduction of proliferation was retained up to 60
h, as previously observed (46), most
probably due to the continuous cell proliferation in the absence of
culture media replenishment. To quantify the inhibitory effect of
tinzaparin on cell proliferation at longer time-points, cells from
each clone were cultured in the absence or presence of 2 IU/ml
tinzaparin, and counted daily for 5 days. Tinzaparin inhibited the
proliferation of both clones in a time-dependent manner, reaching
up to ~55 and ~60% for clones 19 and 15, respectively, at day 5
(Fig. 3Ba and b).
VL30 retrotransposition-positive HC11 clones produce
CSCs forming anchorage-independent mammospheres (46); therefore their ability to produce cell
foci in semi-solid media, and whether this is affected by
tinzaparin, was assessed. Trypsinized cells from either normal HC11
or HC11 cl.19 cultures were maintained for three weeks in soft-agar
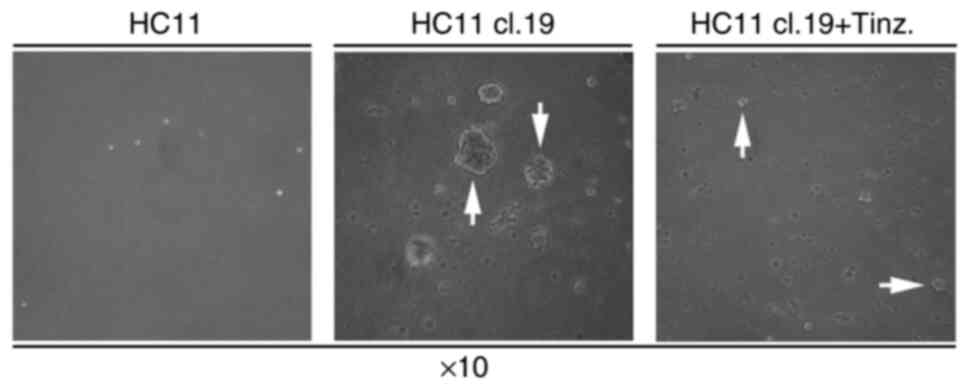
plates containing normal RPMI medium. While normal HC11 cells were
not capable of forming foci (Fig. 4,
left panel), the respective HC11 cl.19 cells produced large
expanding foci (Fig. 4, middle
panel). Furthermore, in the presence of 2 IU/ml tinzaparin, the
resultant foci were smaller in size (Fig.
4, right panel).
Next, the effect of tinzaparin on mammosphere
formation was investigated. HC11 cl.19 cells were seeded into
normal culture dishes and their growth was monitored. 10 days after
reaching full confluence, multinucleated cells bearing cytoplasmic
vacuoles were observed, as well as cell clusters with emerging
mammospheres (Fig. 5Aa, arrows). In
parallel, examining growth in non-adherent culture dishes for 20
days, the formation of large spheroid cell masses, or mammospheres
floating in the culture medium, was apparent (Fig. 5Ab). Next, selected mammospheres
transferred into fresh dishes were further cultured in the presence
of 2 IU/ml tinzaparin for two or three weeks. After two weeks of
tinzaparin administration, substantially reduced mammosphere-size,
accompanied by a small number of either disaggregated floating or
single mammosphere cells attached to the culture dish, were
observed (Fig. 5Ba and b). Notably,
after three weeks, the effect of tinzaparin was augmented, as even
smaller-sized mammospheres were observed, and the vast majority of
single disaggregated mammosphere cells were attached to the dish,
bearing a clear mesenchymal phenotype (Fig. 5Bc and d).
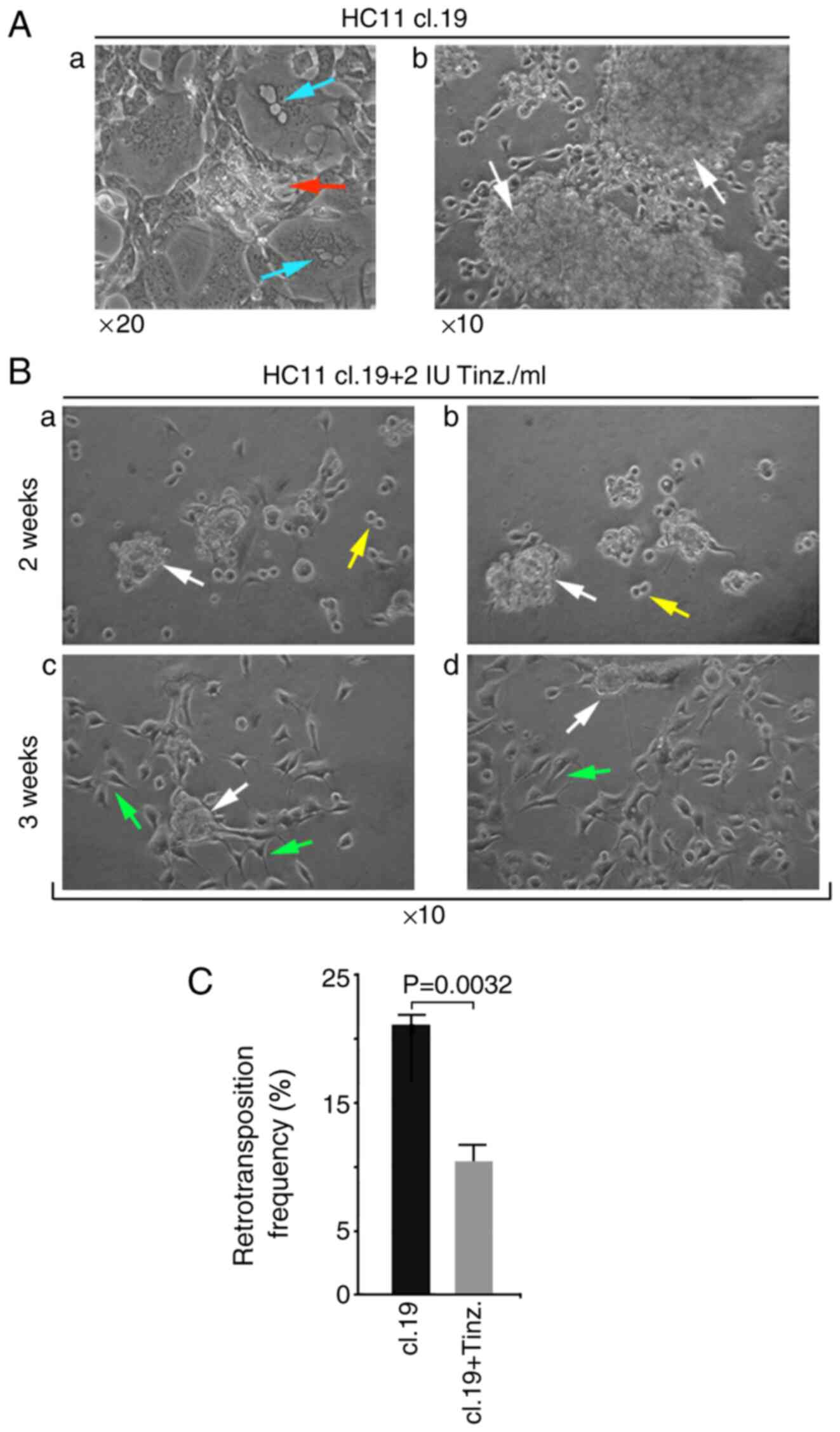 | Figure 5.Tinz treatment elicits disaggregation
of growing tumorigenic VL30 retrotransposition-positive HC11 cl.19
mammospheres, and is associated with retrotransposition inhibition.
(A) Confluent HC11 cl.19 cells were cultured for an additional 10
days in normal culture dishes (Aa) or for 20 days in non-adherent
surface dishes (Ab). Red arrow in panel (Aa) shows outgrowth of a
single early mammosphere, while turquoise arrows show
multinucleated cells bearing cytoplasmic vacuoles. White arrows in
panel (Ab) indicate growing mammospheres floating in the culture
medium. Magnification, ×20 and ×10 in panels (Aa) and (Ab),
respectively. (B) Samples of pipette-collected floating
mammospheres were further cultured in non-adherent surface dishes
in the presence of 2 IU/ml tinzaparin either for two (panels Ba
& Bb) or three weeks (panels Bc & Bd). White arrows in
panels Ba, Bb, Bc and Bd indicate small-sized disaggregated
mammospheres, while yellow arrows in Ba & Bb indicate single
cells, and green arrows in Bc & Bd, dish-attached
mesenchymal-like cells. Magnification, ×10. (C) Samples of cells
from either non-treated HC11 cl.19 cultures or disaggregated HC11
cl.19-mammosphere cells treated with 2 IU tinzaparin for 3 weeks,
were flow cytometrically assessed for EGFP-positivity cells.
Relative growth media of (B) and (C) assays, in the presence or
absence of tinzaparin, were replenished every 3 days. Columns
represent the mean value of retrotransposition frequencies ± SD, of
duplicate samples from three independent experiments. Paired sample
Student's t-test. Tinz., tinzaparin. |
Since tinzaparin resulted in mammosphere
disaggregation, the potential link between tinzaparin and the
modulated retrotransposition of disaggregated cells was
investigated. Compared with untreated HC11 cl.19 cells, the
retrotrans-position frequency of disaggregated HC11 cl.19
mammosphere-derived mesenchymal-like cells was assessed after
treatment with 2 IU tinzaparin for 3 weeks. The retrotransposition
frequency was reduced by 49.7% compared with the control, namely
from their nominal HC11 cl.19 retrotransposition value of 21.1, to
10.5% (Fig. 5C). Collectively, these
data link the inhibitory effect of tinzapapin on VL30
retrotransposion frequency with the inhibition of cellular
proliferation, reduction of cell foci size, and the disaggregation
of mammospheres of tumorigenic VL30 retrotransposition-positive
HC11 cells.
Tinzaparin inhibits the RNA expression
of VL30s and endogenous reverse transcriptase retroviral genes
The retrotransposition of autonomous
LTR-retrotransposons requires a retrotransposon RNA-intermediate,
and the activity of a functionally self-encoded reverse
transcriptase (31). To investigate
the effect of tinzaparin on the inhibition of VL30
retrotransposition in HC11 cells, semi-quantitative RT-PCR analysis
was used to quantity transcription of the retrotransposon
RNA-intermediate and/or various enRT gene transcripts.
cDNAs were prepared from normal HC11 cl.19 cells
used as control, as well as HC11 cl.19 cells treated with either
tinzaparin or VEGF alone, or pre-incubated with tinzaparin and then
treated with VEGF for 24 h. Fractionation of PCR products revealed
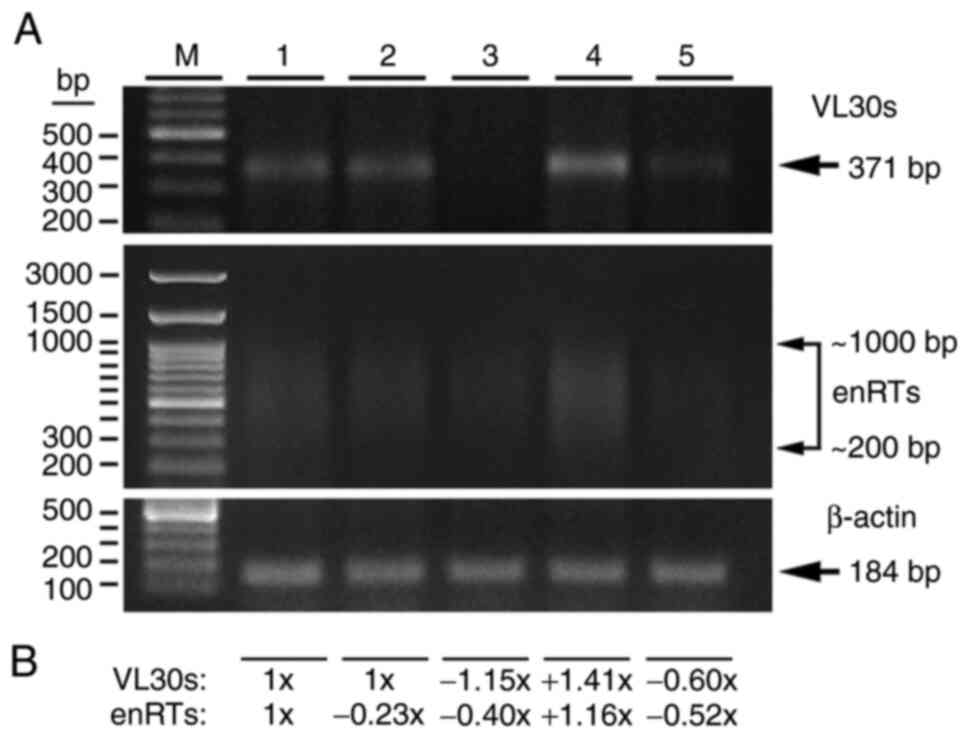
a primary 371 bp band of transcriptionally active VL30s, as well as
a smear of enRTs transcripts representing transcriptional active
enRT genes, mostly in the range of 1,000-200 bp (Fig. 6A). While 2 IU/ml tinzaparin for 2 h
had almost no effect on VL30 RNA expression, the relative
expression of enRTs was inhibited ~0.23-fold in comparison to the
control (arbitrarily considered as 1-fold). Notably, at 4 h of
tinzaparin treatment, the RNA expression of VL30s was inhibited
~1.15-fold, while that of enRTs was further inhibited 0.40-fold.
Regarding cell treatment, 12 ng/ml VEGF alone induced RNA
expression of VL30s and enRTs of ~1.41- and 1.16-fold. However, 4 h
pre-incubation with 2 IU/ml tinzaparin, followed by 12 ng/ml VEGF,
inhibited both relative RNA expression of VL30s and enRTs at the
level of 0.60- and 0.52-fold, respectively, compared with control
levels (Fig. 6A and B). Given that a
4 h tinzaparin pre-incubation simultaneously inhibited the
VEGF-induced RNA expression of both VL30s and enRTs, the data
indicate that tinzaparin acts rapidly as an anti-retroviral drug
inhibiting the necessary VEGF-induced transcripts required for a
retrotransposition event.
Discussion
The principal finding of the present study was the
inhibitory effect of the angiogenic inhibitor tinzaparin (15) on VL30 retrotransposition, induced
either by VEGF or oxidative stress, in mouse cells. VEGF-induced
retrotransposition was documented using two different cell types:
HC11 epithelial mammary stem-like cells and NIH3T3 fibroblasts.
Serum starvation of HC11 cl.17 cells (Fig. S1B) increased their nominal
retrotransposition value (15.2%) to 29% (Fig. 1A). In addition, this frequency (in the
absence of serum-containing VEGF) was further increased by VEGF
treatment in a dose-dependent manner (Fig. 1A). A second line of evidence,
clarifying and confirming the effect of VEGF on induction of VL30
retrotransposition, was obtained with NIH3T3 Pcl.10 cells (without
pre-existing retrotransposition), where inducible VL30
retrotransposition is achieved solely after an external stimulus
(35–37). In particular, while serum-starvation
of Pcl.10 cells elicited 2.4% retrotransposition, this was further
increased to 7.44% with 12 ng/ml VEGF, attributed to both
serum-starvation and VEGF (Fig. S4).
A third finding was that VEGF treatment of non-serum starved HC11
cl.19 cells significantly augmented VL30 and enRT RNA expression
(Fig. 6, lane 4), required for the
retrotransposition event. Thus, we hypothesize that VEGF is an
effective inducer of VL30 retrotransposition in both breast
epithelial stem-like cells and mouse fibroblasts, and that its
action is intensified by serum starvation. Of note, VEGF is
expressed in a wide variety of tumors, including human breast
carcinoma (56), and transformed
serum-starved v-H-ras or v-raf NIH3T3 cell lines express increased
levels of VEGF mRNA and protein, compared with untransformed cells
(57). These data imply that
endogenous VEGF expression in HC11 cl.17 cells, similar to
tumorigenic HC11 cl.19 cells (39),
is involved either in their nominal, serum starvation- and
exogenous VEGF-induced retrotransposition. By contrast, only serum
starvation may be linked with endogenous VEGF expression- and serum
starvation/VEGF-induced retrotransposition in NIH3T3 Pcl.10 cells
(Fig. S4). To the best of our
knowledge, the present study was the first such investigation in
HC11 retrotransposition-positive cells and serum-starved NIH3T3
cells, thus the respective level of endogenous VEGF expression
remains for detailed future investigation.
Regarding oxidative stress, concentration-dependent
GO/H2O2-induced retrotransposition was
observed in HC11 cl.17 cells, compared with the nominal
retrotransposition value (Fig. 2A).
This supports a previous study showing strong
H2O2-induced VL30 retrotransposition in
Pcl.10 fibroblasts (44).
Furthermore, considering HC11 cl.17 cells as tumorigenic (as the
respective HC11 cl.19 cells) (39),
and that tumor cells produce ROS (46), this explains the nominal
retrotransposition of HC11 cl.17 cells. Accordingly, the strong
GO/H2O2-induced retrotransposition was
apparently due to the action of exogenously added
H2O2, resulting in an increase in
intracellular ROS in HC11 cl.17 cells. In agreement with these
findings, the present study revealed that the sum of serum
starvation and 12 ng/ml VEGF-induced retrotransposition in HC11
cl.17 (Fig. 1A) and Pcl.10 cells
(Fig. S4) was higher than their
respective serum starvation-treatment alone. It is known that
serum-starved cells produce ROS, particularly
H2O2 (54,58),
therefore this may underline the contribution of serum starvation
to the VEGF-induced retrotransposition of HC11 cl.17 and Pcl.10
cells. Finally, in reference to the action of VEGF and
intracellular H2O2, VEGF-treated HC11 cl.19
cells exhibited an induced VL30 and enRT RNA expression (Fig. 6, lane 4) ~1.41- and 1.16-fold,
respectively, exceeding those by intracellular-produced ROS
(Fig. 6, lane 1). Thus, this
justifies a higher VEGF-induced retrotransposition frequency
compared with that induced by serum-starvation alone in HC11 cl.17
and Pcl.10 cells (Figs. 1A and
S3), respectively. In addition, it
is worth noting that oxidative stress and VEGF are linked, as
intracellular oxidants and extracellular H2O2
correlate with the concomitant upregulation of VEGF transcription
(59), that highlights oxidative
stress as a primary stimulus of VL30 retrotransposition. Hence, we
hypothesize that H2O2 per se is a
strong inducer of VL30 retrotransposition in epithelial stem-like
HC11 cells, and consequently, the augmented retrotransposition
under serum starvation is explained by the cooperation of
intracellular H2O2 with VEGF activity.
Serum-starved HC11 cl.17 cells pre-incubated with
tinzaparin exhibited significantly reduced retrotransposition at
all concentrations of VEGF tested. This was most evident with 12
ng/ml VEGF (inducing 40.3% retrotransposition), which was reduced
to 18.3% by tinzaparin (Fig. 1A).
Furthermore, the tinzaparin effect was also time dependent
(Fig. 1B). Apparently, the similar
18.3- and 18.7% lowest values, even lower than the 29% serum
starvation-induced retrotransposition scored in two independent
experiments (Fig. 1A and B),
corresponding to a ~53% mean reduction value, document the strength
of the tinzaparin effect. In addition, two more lines of evidence
support this tinzaparin effect. First, tinzaparin pre-incubation of
serum starved 12 ng/ml VEGF treated Pcl.10 cells reduced their
retrotransposition in a time-dependent manner (Fig. S4). Second, a 3 h tinzaparin (1 or 2
IU) pre-incubation of HC11 cl.17 cells reduced their expected
(unusually high) GO-induced retrotransposition (Fig. 2B). Overall, given that: i) Tinzaparin
pre-incubation strongly reduces VEGF- and/or oxidative
stress-induced VL30 retrotransposition in epithelial HC11 and
NIH3T3 fibroblasts; ii) a single 2 IU/ml dose elicits a strong
reduction of VEGF-induced retrotransposition; iii) its drastic
effect emerges rapidly, at a 3- to 4 h time point; and iv) it acts
in a dose- and time-dependent manner; the current data highlight
tinzaparin as a strong inhibitor of VL30 retrotransposition.
Based on the mechanism of LTR-retrotransposition,
requiring both a homologous retrotransposon-intermediate transcript
and RT activity (31), the action of
tinzaparin was addressed at the transcription level. In particular,
the RNA expression of both VL30s and enRTs in untreated HC11 cl.19
cells (Fig. 6, lane 1) primarily
explains their nominal retrotransposition (due to
intracellular-oxidative stress), given that these cells are
tumorigenic (46) and produce ROS
(60). Notably, the inhibitory effect
of tinzaparin was documented: i) A direct tinzaparin treatment with
2 IU/ml for 4 h inhibited the RNA expression of VL30 elements and
enRT below control levels (Fig. 6,
lane 3); ii) their respective induced RNA expression with VEGF
alone (Fig. 6, lane 4) was also
inhibited upon pre-incubation with tinzaparin (Fig. 6, lane 5). Interestingly; and iii) 2 h
tinzaparin pre-incubation inhibited only the RNA expression of
enRTs (Fig. 6, lane 2), which mirrors
its rapid effect on transcribed enRTs compared with VL30s. These
data are in line with oxidative stress- and/or VEGF-induced
retrotransposition inhibited by tinzaparin treatment. Furthermore,
direct tinzaparin application: inhibited: i) The intracellular
ROS-dependent VL30 and enRT RNA expression (Fig. 6, lane 3), and retrotransposition of
disaggregated HC11 cl.19 cell mammospheres (Fig. 5C); ii) the induced retrotransposition
(Figs. 1A and S4), attributed to the sum of intracellular-
and serum starvation-ROS (54); and
iii) strong retrotransposition induction by extracellular
GO/H2O2 (Fig.
2B). In reference to tinzaparin pre-incubation, it also
inhibited both the VEGF-induced RNA expression of VL30s and enRTs
(Fig. 6, lane 5), and VEGF-induced
retrotransposition of HC11 cl. 17 (Fig.
1) and Pcl.10 cells (Fig. S4).
These findings support that tinzaparin is an efficient inhibitor of
VL30 retrotransposition, independently induced by either oxidative
stress or VEGF, acting at the transcriptional level through
inhibition of enRT and VL30 RNA expression.
The precise molecular pathways associated with the
modulation of VL30 retrotransposition by oxidative stress, VEGF
and/or tinzaparin, are unknown. Our previous studies have shown
that VL30 retrotransposition is induced either by
vanadium-generated H2O2 (41), arsenic-generated
ROS/H2O2 (42)
or H2O2 alone (44). Furthermore,
H2O2-induced retrotransposition was reported
to be inhibited (as in the present study) by the anti-oxidant NAC
(42,44), or by the non-toxic/non-nucleoside
specific reverse transcriptase inhibitors efavirenz or etravirine
(44). As tinzaparin inhibited the
RNA expression of VL30 and enRT genes, we hypothesize that
tinzaparin acts similarly, both as an anti-oxidant and
anti-retroviral drug.
Recombinant NVL-3/9 VL30 retrotransposon bears two
activator protein-1 (AP-1) and an NF-kB transcription factor
binding motifs at the U3 region of its LTR (38). An intermediate amount of ROS activates
both NF-kB and AP-1 transcription factors (61), while VEGF also induces the expression
of AP-1 family proteins (62).
Therefore, NF-kB and AP-1 pathways may support the serum
starvation- and/or VEGF or ROS/H2O2-induced
VL30 RNA expression required for the occurrence of a
retrotransposition event. Regarding an active reverse
transcriptase, an endogenous VL30-specific reverse transcriptase is
yet to be identified, since as noncoding RNAs, VL30s are
non-autonomous retrotransposons (36,38). Our
previous studies have shown that ectopic expression of a
MoMLV-reverse transcriptase gene (40), acting in trans-complementation,
induces VL30 retrotransposition that is further augmented by
H2O2 (41).
Given that MoMLV primers were included in the degenerate
primers-set in the present study (to detect enRT RNA expression),
this suggests that respective endogenous MoMLV-RT expression may be
implicated in induced retrotransposition. While there are no data
on induced MoMLV-RT enzyme activity (to the best of our knowledge),
a possible mechanism may be similar to that of telomerase,
activated by phosphorylation through the protein phosphatase
2-subunit A, that is implicated in its negative regulation
(63). It is, thus, reasonable to
speculate that MoMLV activity was activated through the
phosphatases known inhibition of H2O2
(64). Accordingly, this may endorse
the inhibitory effect of tinzaparin on oxidative stress-induced
VL30 retrotransposition preventing MoMLV-RT enzyme activation.
The present data support the inhibitory effect of
tinzaparin on the proliferation of retrotransposition-positive HC11
cells, since tinzaparin-cultured HC11 cl.19 or cl.15 cells showed
time-dependent proliferation inhibition (Fig. 3A) up to ~55 and ~60%, respectively, at
5 days of culture (Fig. 3Ba and b).
In addition, the effect of tinzaparin on cell foci and mammosphere
formation was also shown. As HC11 cl.19 cells are tumorigenic CSCs
(46), and as CSCs are referred to as
tumor-initiating cells (29), the
formation of cell foci either in semi-solid media (Fig. 4) or mammospheres in non-adherent
surface dishes (Fig. 5Ab) was
expected. Regarding cell foci formation, tinzaparin treatment led
to cellular disaggregation with a reduced size of ~3–6 cells
(Fig. 4). This effect was more
evident in preformed mammospheres, as their treatment produced
single mesenchymal-like cells (Fig. 5Bc
and d) accompanied by a significant inhibition of
retrotransposition (Fig. 5C). This
shows an association between tinzaparin-inhibited
retrotransposition and mammosphere disaggregation, without
affecting the EMT-induced phenotype (46) of HC11 cl.19 cells. It is thought that
a low intracellular level of ROS is tightly controlled to promote
proliferation and survival of stem and progenitor cells (65), while enRTs, active in germ cells,
embryo and tumor tissues, are considered mediators of cellular
differentiation and proliferation (66). These findings may explain the
inhibited proliferation and associated retrotransposition caused by
the anti-oxidant and/or anti-retroviral effects of tinzaparin.
The exact molecular mechanisms underlying the dual
action of tinzaparin on mammosphere-disaggregation, and
proliferative inhibition of disaggregated cells, is yet to be
elucidated. It appears that the cell surface heparan sulfate
proteoglycans (HSPGs) are implicated in the mammosphere formation
of HC11 cl.19 cells (Fig. 5Ab), as
they most commonly bind to secondary sites on cell adhesion
molecules, and increase the strength of intercellular adhesions and
cell-cell stability (67). However,
heparin potentially inhibits cell-cell interactions by binding to
cellular adhesion molecules (68).
Hence, we hypothesize that tinzaparin interferes with the
intercellular adhesion process, resulting in mammosphere
disaggregation (Fig. 5Bc and d), and
reduces foci size (Fig. 4). In
addition, the proliferation of disaggregated cells can be inhibited
either by soluble HSPGs, as shown in pancreatic cancer cells
(69), or by soluble HSPGs or
heparin, shown in neuroblastoma cells (70). Accordingly, the present study supports
that, in addition to its anti-oxidant/anti-retroviral effects,
tinzaparin (as a low molecular weight heparin) stimulates the
production of soluble HSPGs, which in turn inhibit the
proliferation of disaggregated tumorigenic HC11 cl.19 cells, a
matter that warrants further investigation.
It is noteworthy that the natural
retrotransposition frequency for a defective retrotransposon, such
as VL30, is estimated to be up to 10−6 events per
cell/generation (33). By contrast,
unusually high retrotransposition frequencies were noted in the
present study. For example; 40% by serum-starvation/VEGF treatment
(Fig. 1A), corresponding to a
400,000-fold induced retrotransposition. Apparently, such
accumulation of new genomic integrated retrotransposon copies can
cause numerous mutations, underlining the detrimental effects of
VL30 retrotransposition. Therefore, the present data, showing that
tinzaparin inhibits retrotransposition, underlines the prophylactic
property of tinzaparin against accumulated VL30 retrotransposition
mutations. Finally, human breast cancer mammosphere-forming cells
display resistance to chemotherapeutic drugs (71,72). In
this sense, our mouse HC11-VL30 retrotransposition model could be
used as a preliminary in vitro assay to evaluate the
efficacy of novel chemotherapeutic drugs.
To the best of our knowledge, the present study
associates, for the first time, the action of tinzaparin with the
inhibition of VL30 retrotransposition, being a causative factor of
tumorigenesis in HC11 mammary epithelial stem-like cells. The data,
extending the properties of tinzaparin as an anticoagulant agent
and angiogenesis inhibitor, reveal three additional
tinzaparin-interconnected actions: i) Tinzaparin acts as an
anti-oxidant and anti-retroviral drug against VL30
retrotransposition induced by oxidative-stress and/or VEGF,
providing prophylaxis against new VL30 retrotransposition-derived
genomic mutations; ii) tinzaparin exerts anti-proliferative action
on CSCs; and iii) tinzaparin is effective in disaggregating
mammosphere-formation to intercept tumor growth. As tinzaparin
activity is associated with transcriptional reprogramming of a
plethora of genes (17), the VL30
retrotransposition model may potentially be used in elucidating its
impact on diverse biological processes in HC11 cells.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was financially supported by two
funds (grant no. 80899 and 81423) from the LEO Pharmaceuticals
(Athens, Greece).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
SM performed the laboratory experiments. GM
directed the study, evaluated the flow cytometric data and prepared
the original manuscript. GV and DN performed the flow cytometric
analysis. ST and FG assisted with the cell culture experiments. TT
conceived, designed and supervised the experiments, and wrote the
manuscript. SM and TT confirm the authenticity of all the raw data.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests, or any other personal connections with, or are employed
by, LEO Pharmaceuticals.
References
|
1
|
Khorana AA and Connolly GC: Assessing risk
of venous thromboembolism in the patient with cancer. J Clin Oncol.
27:4839–4847. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mandalà M, Falanga A and Roila F; ESMO
Guidelines Working Group, : Management of venous thromboembolism
(VTE) in cancer patients: ESMO clinical practice guidelines. Ann
Oncol. 22 (Supp 6):vi85–vi92. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lyman GH, Bohlke K, Khorana AA, Kuderer
NM, Lee AY, Arcelus JI, Balaban EP, Clarke JM, Flowers CR, Francis
CW, et al: Venous thromboembolism prophylaxis and treatment in
patients with cancer: American society of clinical oncology
clinical practice guideline update 2014. J Clin Oncol. 33:654–656.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dahlbäck B: Blood coagulation. Lancet.
355:1627–1632. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wong NN: Tinzaparin. Heart Dis. 4:331–340.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Horton J: Venous thrombotic events in
cancer: The bottom line. Cancer Control. 12 (Suppl 1):S31–S37.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Scotté F, Rey JB and Launay-Vacher V:
Thrombosis, cancer and renal insufficiency: Low molecular weight
heparin at the crossroads. Support Care Cancer. 20:3033–3042. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Perry SL, Bohlin C, Reardon DA, Desjardins
A, Friedman AH, Friedman HS and Vredenburgh JJ: Tinzaparin
prophylaxis against venous thromboembolic complications in brain
tumor patients. J Neurooncol. 95:129–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stevenson JL, Choi SH and Varki A:
Differential metastasis inhibition by clinically relevant levels of
heparins-correlation with selectin inhibition, not antithrombotic
activity. Clin Cancer Res. 11:7003–7011. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schlesinger M, Roblek M, Ortmann K, Naggi
A, Torri G, Borsig L and Bendas G: The role of VLA-4 binding for
experimental melanoma metastasis and its inhibition by heparin.
Thromb Res. 133:855–862. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Harvey JR, Mellor P, Eldaly H, Lennard TW,
Kirby JA and Ali S: Inhibition of CXCR4-mediated breast cancer
metastasis: A potential role for heparinoids? Clin Cancer Res.
13:1562–1570. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Alyahya R, Sudha T, Racz M, Stain SC and
Mousa SA: Anti-metastasis efficacy and safety of non-anticoagulant
heparin derivative versus low molecular weight heparin in surgical
pancreatic cancer models. Int J Oncol. 46:1225–1231. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bauer AT, Suckau J, Frank K, Desch A,
Goertz L, Wagner AH, Hecker M, Goerge T, Umansky L, Beckhove P, et
al: von Willebrand factor fibers promote cancer-associated platelet
aggregation in malignant melanoma of mice and humans. Blood.
125:3153–3163. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Amirkhosravi A, Mousa SA, Amaya M and
Francis JL: Antimetastatic effect of tinzaparin, a
low-molecular-weight heparin. J Thromb Haemost. 1:1972–1976. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mousa SA and Mohamed S: Anti-angiogenic
mechanisms and efficacy of the low molecular weight heparin,
tinzaparin: Anti-cancer efficacy. Oncol Rep. 12:683–688.
2004.PubMed/NCBI
|
|
16
|
Mousa SA and Mohamed S: Inhibition of
endothelial cell tube formation by the low molecular weight
heparin, tinzaparin, is mediated by tissue factor pathway
inhibitor. Thromb Haemost. 92:627–633. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pfankuchen DB, Stölting DP, Schlesinger M,
Royer HD and Bendas G: Low molecular weight heparin tinzaparin
antagonizes cisplatin resistance of ovarian cancer cells. Biochem
Pharmacol. 97:147–157. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dimakakos EP, Vathiotis I and Syrigos K:
The role of tinzaparin in oncology. Clin Appl Thromb Hemost.
24:697–707. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kragh M, Binderup L, Vig Hjarnaa PJ, Bramm
E, Johansen KB and Frimundt Petersen C: Non-anti-coagulant heparin
inhibits metastasis but not primary tumor growth. Oncol Rep.
14:99–104. 2005.PubMed/NCBI
|
|
20
|
Folkman J: Angiogenesis in cancer,
vascular, rheumatoid and other disease. Nat Med. 1:27–31. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Carmeliet P and Jain RK: Angiogenesis in
cancer and other diseases. Nature. 407:249–257. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gamperl H, Plattfaut C, Freund A, Quecke
T, Theophil F and Gieseler F: Extracellular vesicles from malignant
effusions induce tumor cell migration: Inhibitory effect of LMWH
tinzaparin. Cell Biol Int. 40:1050–1061. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sudha T, Yalcin M, Lin HY, Elmetwally AM,
Nazeer T, Arumugam T, Phillips P and Mousa SA: Suppression of
pancreatic cancer by sulfated non-anticoagulant low molecular
weight heparin. Cancer Lett. 350:25–33. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guarino M, Rubino B and Ballabio G: The
role of epithelial-mesenchymal transition in cancer pathology.
Pathology. 39:305–318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hugo H, Ackland ML, Blick T, Lawrence MG,
Clements JA, Williams ED and Thompson EW: Epithelial-mesenchymal
and mesenchymal-epithelial transitions in carcinoma progression. J
Cell Physiol. 213:374–383. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang AD, Camp ER, Fan F, Shen L, Gray MJ,
Liu W, Somcio R, Bauer TW, Wu Y, Hicklin DJ and Ellis LM: Vascular
endothelial growth factor receptor-1 activation mediates epithelial
to mesenchymal transition in human pancreatic carcinoma cells.
Cancer Res. 66:46–51. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hayashida T, Jinno H, Kitagawa Y and
Kitajima M: Cooperation of cancer stem cell properties and
epithelial-mesenchymal transition in the establishment of breast
cancer metastasis. J Oncol. 2011:5914272011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Al-Hajj M, Wicha MS, Benito-Hernandez A,
Morrison SJ and Clarke MF: Prospective identification of
tumorigenic breast cancer cells. Proc Natl Acad Sci USA.
100:3983–3988. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cordaux R and Batzer MA: The impact of
retrotransposons on human genome evolution. Nat Rev Genet.
10:691–703. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Boeke JD, Garfinkel DJ, Styles CA and Fink
GR: Ty elements transpose through an RNA intermediate. Cell.
40:491–500. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hancks DC and Kazazian HH Jr: Roles for
retrotransposon insertions in human disease. Mob DNA. 7:92016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Heidmann T, Heidmann O and Nicolas JF: An
indicator gene to demonstrate intracellular transposition of
defective retroviruses. Proc Natl Acad Sci USA. 85:2219–2223. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Georgiou I, Noutsopoulos D, Dimitriadou E,
Markopoulos G, Apergi A, Lazaros L, Vaxevanoglou T, Pantos K,
Syrrou M and Tzavaras T: Retrotransposon RNA expression and
evidence for retrotransposition events in human oocytes. Hum Mol
Genet. 18:1221–1228. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Goodier JL and Kazazian HH Jr:
Retrotransposons revisited: The restraint and rehabilitation of
parasites. Cell. 135:23–35. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
French NS and Norton JD: Structure and
functional properties of mouse VL30 retrotransposons. Biochim
Biophys Acta. 1352:33–47. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Garen A and Song X: Regulatory roles of
tumor-suppressor proteins and noncoding RNA in cancer and normal
cell functions. Int J Cancer. 122:1687–1689. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Markopoulos G, Noutsopoulos D, Mantziou S,
Gerogiannis D, Thrasyvoulou S, Vartholomatos G, Kolettas E and
Tzavaras T: Genomic analysis of mouse VL30 retrotransposons. Mob
DNA. 7:102016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Brunmeir R, Lagger S, Simboeck E, Sawicka
A, Egger G, Hagelkruys A, Zhang Y, Matthias P, Miller WJ and Seiser
C: Epigenetic regulation of a murine retrotransposon by a dual
histone modification mark. PLoS Genet. 6:e10009272010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Noutsopoulos D, Vartholomatos G, Kolaitis
N and Tzavaras T: SV40 large T antigen up-regulates the
retrotransposition frequency of viral-like 30 elements. J Mol Biol.
361:450–461. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Noutsopoulos D, Markopoulos G, Koliou M,
Dova L, Vartholomatos G, Kolettas E and Tzavaras T: Vanadium
induces VL30 retrotransposition at an unusually high level: A
possible carcinogenesis mechanism. J Mol Biol. 374:80–90. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Markopoulos G, Noutsopoulos D, Mantziou S,
Vartholomatos G, Monokrousos N, Angelidis C and Tzavaras T: Arsenic
induces VL30 retrotransposition: The involvement of oxidative
stress and heat-shock protein 70. Toxicol Sci. 134:312–322. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tzavaras T, Eftaxia S, Tavoulari S, Hatzi
P and Angelidis C: Factors influencing the expression of endogenous
reverse transcriptases and viral-like 30 elements in mouse NIH3T3
cells. Int J Oncol. 23:1237–1243. 2003.PubMed/NCBI
|
|
44
|
Konisti S, Mantziou S, Markopoulos G,
Thrasyvoulou S, Vartholomatos G, Sainis I, Kolettas E, Noutsopoulos
D and Tzavaras T: H2O2 signals via iron induction of VL30
retrotransposition correlated with cytotoxicity. Free Radic Biol
Med. 52:2072–2081. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Noutsopoulos D, Markopoulos G,
Vartholomatos G, Kolettas E, Kolaitis N and Tzavaras T: VL30
retrotransposition signals activation of a caspase-independent and
p53-dependent death pathway associated with mitochondrial and
lysosomal damage. Cell Res. 20:553–562. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Thrasyvoulou S, Vartholomatos G,
Markopoulos G, Noutsopoulos D, Mantziou S, Gkartziou F, Papageorgis
P, Charchanti A, Kouklis P, Constantinou AI and Tzavaras T: VL30
retrotransposition is associated with induced EMT, CSC generation
and tumorigenesis in HC11 mouse mammary stem-ike epithelial cells.
Oncol Rep. 44:126–138. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ball RK, Friis RR, Schoenenberger CA,
Doppler W and Groner B: Prolactin regulation of beta-casein gene
expression and of a cytosolic 120-kd protein in a cloned mouse
mammary epithelial cell line. EMBO J. 7:2089–2095. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Williams C, Helguero L, Edvardsson K,
Haldosén LA and Gustafsson JA: Gene expression in murine mammary
epithelial stem cell-like cells shows similarities to human breast
cancer gene expression. Breast Cancer Res. 11:R262009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Borowicz S, Van Scoyk M, Avasarala S,
Karuppusamy Rathinam MK, Tauler J, Bikkavilli RK and Winn RA: The
soft agar colony formation assay. J Vis Exp. e519982014.PubMed/NCBI
|
|
50
|
Ostertag EM, Prak ET, DeBerardinis RJ,
Moran JV and Kazazian HH Jr: Determination of L1 retrotransposition
kinetics in cultured cells. Nucleic Acids Res. 28:1418–1423. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Xiong Y and Eickbush TH: Origin and
evolution of retroelements based upon their reverse transcriptase
sequences. EMBO J. 9:3353–3362. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Puschendorf M, Stein P, Oakeley EJ,
Schultz RM, Peters AH and Svoboda P: Abundant transcripts from
retrotransposons are unstable in fully grown mouse oocytes. Biochem
Biophys Res Commun. 347:36–43. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bloushtain-Qimron N, Yao J, Snyder EL,
Shipitsin M, Campbell LL, Mani SA, Hu M, Chen H, Ustyansky V,
Antosiewicz JE, et al: Cell type-specific DNA methylation patterns
in the human breast. Proc Natl Acad Sci USA. 105:14076–14081. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
White EZ, Pennant NM, Carter JR, Hawsawi
O, Odero-Marah V and Hinton CV: Serum deprivation initiates
adaptation and survival to oxidative stress in prostate cancer
cells. Sci Rep. 10:125052020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Merlo GR, Venesio T, Taverna D, Marte BM,
Callahan R and Hynes NE: Growth suppression of normal mammary
epithelial cells by wild-type p53. Oncogene. 9:443–453.
1994.PubMed/NCBI
|
|
56
|
Ferrara N and Davis-Smyth T: The biology
of vascular endothelial growth factor. Endocr Rev. 18:4–25. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Grugel S, Finkenzeller G, Weindel K,
Barleon B and Marmé D: Both v-Ha-Ras and v-Raf stimulate expression
of the vascular endothelial growth factor in NIH 3T3 cells. J Biol
Chem. 270:25915–25919. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Scherz-Shouval R, Shvets E, Fass E, Shorer
H, Gil L and Elazar Z: Reactive oxygen species are essential for
autophagy and specifically regulate the activity of Atg4. EMBO J.
26:1749–1760. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wu Y, Meitzler JL, Antony S, Juhasz A, Lu
J, Jiang G, Liu H, Hollingshead M, Haines DC, Butcher D, et al:
Dual oxidase 2 and pancreatic adenocarcinoma: IFN-γ-mediated dual
oxidase 2 overexpression results in H2O2-induced, ERK-associated
up-regulation of HIF-1α and VEGF-A. Oncotarget. 7:68412–68433.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Szatrowski TP and Nathan CF: Production of
large amounts of hydrogen peroxide by human tumor cells. Cancer
Res. 51:794–798. 1991.PubMed/NCBI
|
|
61
|
Gloire G, Legrand-Poels S and Piette J:
NF-kappaB activation by reactive oxygen species: Fifteen years
later. Biochem Pharmacol. 72:1493–1505. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jia J, Ye T, Cui P, Hua Q, Zeng H and Zhao
D: AP-1 transcription factor mediates VEGF-induced endothelial cell
migration and proliferation. Microvasc Res. 105:103–108. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Liu JP: Studies of the molecular
mechanisms in the regulation of telomerase activity. FASEB J.
13:2091–2104. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Reth M: Hydrogen peroxide as second
messenger in lymphocyte activation. Nat Immunol. 3:1129–1134. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kobayashi CI and Suda T: Regulation of
reactive oxygen species in stem cells and cancer stem cells. J Cell
Physiol. 227:421–430. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Spadafora C: Endogenous reverse
transcriptase: A mediator of cell proliferation and
differentiation. Cytogenet Genome Res. 105:346–350. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bernfield M, Götte M, Park PW, Reizes O,
Fitzgerald ML, Lincecum J and Zako M: Functions of cell surface
heparan sulfate proteoglycans. Annu Rev Biochem. 68:729–777. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Bendas G and Borsig L: Cancer cell
adhesion and metastasis: Selectins, integrins, and the inhibitory
potential of heparins. Int J Cell Biol. 2012:6767312012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Knelson EH, Nee JC and Blobe GC: Heparan
sulfate signaling in cancer. Trends Biochem Sci. 39:277–288. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Knelson EH, Gaviglio AL, Nee JC, Starr MD,
Nixon AB, Marcus SG and Blobe GC: Stromal heparan sulfate
differentiates neuroblasts to suppress neuroblastoma growth. J Clin
Invest. 124:3016–3031. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Creighton CJ, Li X, Landis M, Dixon JM,
Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A,
Herschkowitz JI, et al: Residual breast cancers after conventional
therapy display mesenchymal as well as tumor-initiating features.
Proc Natl Acad Sci USA. 106:13820–13825. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ji P, Zhang Y, Wang SJ, Ge HL, Zhao GP, Xu
YC and Wang Y: CD44hiCD24lo mammosphere-forming cells from primary
breast cancer display resistance to multiple chemotherapeutic
drugs. Oncol Rep. 35:3293–3302. 2016. View Article : Google Scholar : PubMed/NCBI
|