Introduction
In order to improve effectiveness and minimize the
adverse effects of cancer treatment, more specific targeted agents
have been identified (1). These
novel agents have changed the course of cancer treatment and are
capable of improving patient outcomes. One of the most promising
classes of targeted antineoplastic agents is the poly (ADP-ribose)
polymerase (PARP) inhibitors (2).
PARP inhibitors may selectively eliminate those cells that have
lost the homologous recombination repair pathway (3). The antitumor activity of PARP
inhibitors involves inhibition of PARP enzymatic activity and an
increase in the formation of PARP-DNA complexes, resulting in DNA
damage, apoptosis and cell death, particularly in cancer cells
(4). PARP inhibitors, including
olaparib, niraparib, rucaparib, talazoparib and veliparib, are
administered orally, which has an advantage in terms of
flexibility, convenience and quality of life compared with
traditional chemotherapy (5).
However, as oral PARP inhibitors are extensively used, patients
with cancer are at increased risk for drug-drug interactions
(DDIs). As a consequence, the pharmacokinetics (PK) of PARP
inhibitors may display high inter-individual variability in
patients with cancer and a subsequently increased risk for serious
toxicity or therapeutic failure (6–8).
DDIs are a major and growing clinical health
problem, and could lead to unwanted toxicities or therapeutic
failure. DDIs could be divided into pharmacodynamic (PD) and PK
interactions (9). PD-based DDIs
occur when medications cause additive, antagonistic or synergistic
pharmacological effects, altering efficacy or producing adverse
effects. PK-based DDIs are caused by changes in absorption,
distribution, metabolism and excretion, leading to altered
bioavailability of a drug and possible unfavorable outcomes. (e.g.,
increased toxicity and reduced treatment efficacy) (10). Metabolism-related DDIs are the most
common PK-based DDIs. Due to the substantial potential for
interaction between PARP inhibitors and other medications that
modulate the activity of metabolic pathways, unwanted clinical
consequences may occur from small changes in drug PK in patients
with cancer (7,8). As such, this may result in an
increased risk of non-compliance, dose reduction or therapy
discontinuation, leading to suboptimal therapy.
The main objective of the present review is to
characterize and summarize the PK parameters and metabolism-related
PK-based DDIs for each PARP inhibitor. In addition, practical
recommendations for managing DDIs during treatment with PARP
inhibitors are provided.
Mechanisms of action of PARP inhibitors
PARPs are a group of enzymes that play a key role in
the DNA repair pathway. Among them, PARP-1, 2 and 3 are the most
extensively studied (2). PARP-1,
accounting for up to 90% of all PARP activity, promotes
single-strand DNA break (SSB) repair via the base excision repair
pathway (2,3). In addition, PARP-1 plays a central
role in microhomology-mediated end joining repair, an error-prone
pathway involved in double-strand DNA break (DSB) repair (4). Beyond DNA repair, PARPs are also
involved in mitosis, transcriptional regulation, cell death,
intracellular metabolism and telomere length (2).
Non-homologous end joining (NHEJ) and homologous
recombination (HR) are two important pathways in the repair of
DSBs. HR [for which breast-related cancer antigen 1/2 (BRCA1/2) are
the first proteins to have been studied] is a high-fidelity repair
pathway, while NHEJ is an error prone pathway that could lead to an
accumulation of genetic aberrations, chromosomal instability, cell
cycle arrest and apoptosis (1,2). If
the HR pathway is altered, NHEJ is left as the only pathway able to
repair the DNA. PARP inhibitors could bind to the nicotinamide
adenine dinucleotide (NAD+) binding pocket of PARP-1,
producing conformational changes that stabilize the binding of
PARP-1 and DNA (5). This process
results in PARP-1 dysfunction, leading to the accumulation of
unrepaired SSBs and inhibiting the progression of replication forks
(RFs) (5). Ultimately, stalled RFs
degrade into highly cytotoxic DSBs. HR proficient cells are able to
repair the DSB and restart replication, while HR deficient cells
(i.e., those with BRCA mutation) are unable to repair the
accumulating DSBs, which may induce cell death. The mechanisms of
action of PARP inhibitors are illustrated in Fig. 1.
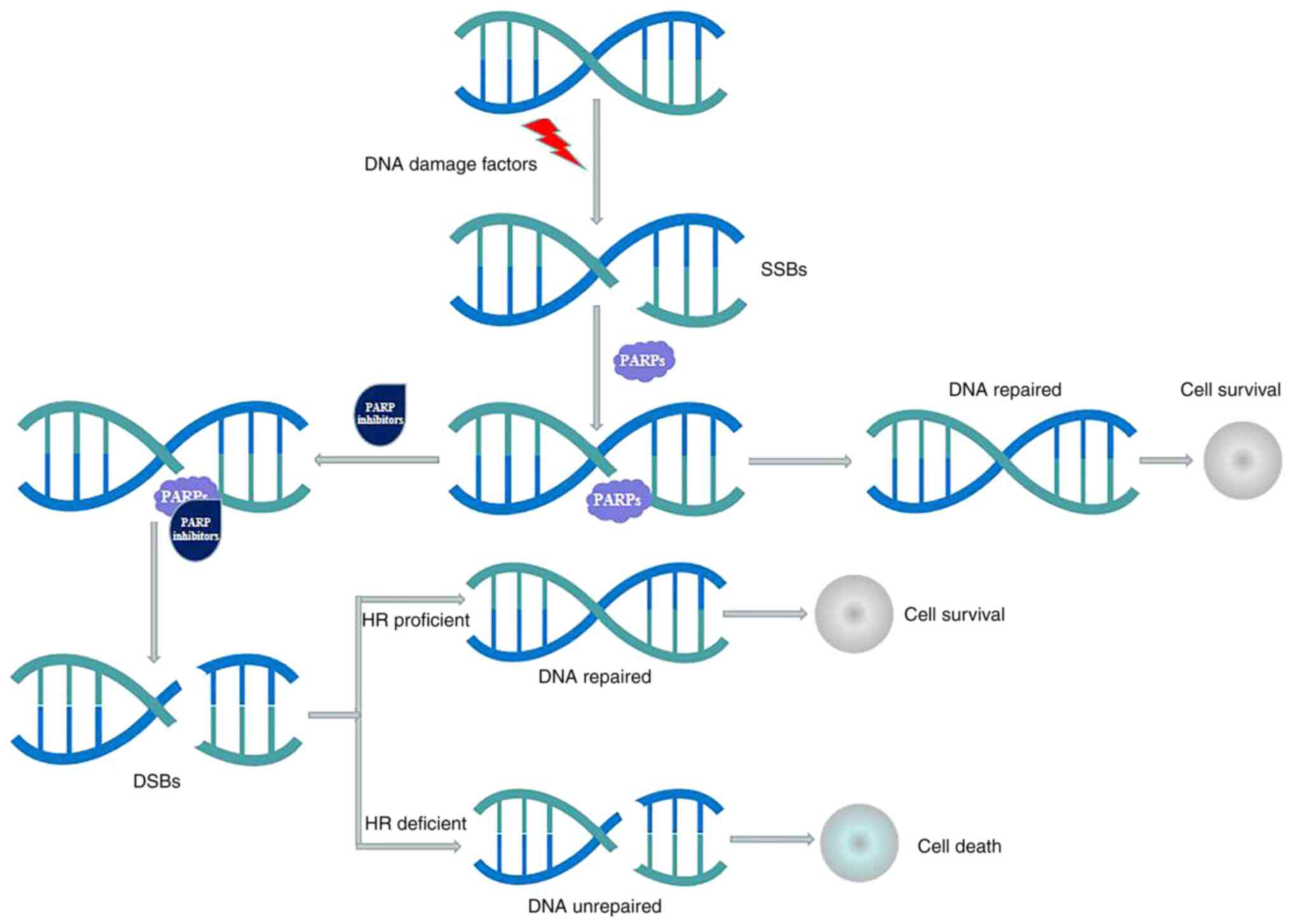 | Figure 1.Mechanisms of action of PARP
inhibitors. SSBs occur frequently in proliferating cells, and SSBs
are repaired mostly by the PARP-dependent base excision repair
pathway. PARP inhibitors may bind to the nicotinamide adenine
dinucleotide binding pocket of PARP-1, producing conformational
changes that stabilize the binding of PARP-1 and DNA. This process
results in PARP-1 dysfunction, leading to the accumulation of
unrepaired SSBs; ultimately, the unrepaired SSBs could be converted
to DSBs. HR proficient cells are able to repair the DSBs and
restart replication, while HR deficient cells are unable to repair
the accumulating DSBs, which may induce cell death. PARP, poly
(ADP-ribose) polymerase; SSB, single-strand DNA break; DSB,
double-strand DNA break; HR, homologous recombination. |
PK parameters of PARP inhibitors
The PARP inhibitors olaparib, niraparib, rucaparib,
talazoparib and veliparib are administered orally. The oral
absorption is rapid, with peak plasma concentration
(Cmax) achieved 0.5 to 3 h after dosing in healthy
subjects and in patients with solid tumors (11–19). The oral
bioavailability is quite different between the five PARP
inhibitors. For instance, in niraparib it is ~73%, whereas in
rucaparib it is 36% (13–15). Numerous factors may contribute to low
oral bioavailability, such as the inability of a drug to cross cell
membranes, poor water solubility and metabolic instability.
Among the five PARP inhibitors, niraparib has the
highest volume of distribution (1,220 liters) (13,14),
potentially indicating a higher tendency to concentrate in tumors
and other tissues rather than in plasma. In terms of the plasma
protein binding rate, >80% of olaparib and niraparib, ~70% of
rucaparib and talazoparib, and only 51% of veliparib is bound to
plasma proteins (11–20).
The five PARP inhibitors undergo slightly different
metabolic pathways: Olaparib, rucaparib and veliparib are primarily
metabolized by the cytochrome P450 (CYP) enzymatic pathway
(11,15,19);
talazoparib undergoes minimal hepatic metabolism, with identified
metabolic pathways, including mono-oxidation, dehydrogenation,
cysteine conjugation and glucuronide conjugation (17,18);
and niraparib is metabolized primarily by carboxylesterases (CEs)
amide hydrolysis to form a major inactive metabolite, and
subsequently undergoes glucuronidation (13,14).
Excretion of the five drugs also varies. Talazoparib
and niraparib both have a long elimination half-life
(T1/2) of 90 and 36 h, respectively (13,17),
olaparib and rucaparib have a moderate T1/2 of 11.9 and
19 h, respectively (11,15), whereas veliparib has a short
T1/2 (5.2 h) (19,20).
This may explain why talazoparib and niraparib are recommended for
administration once daily, while olaparib, rucaparib and veliparib
are medications administered twice daily. Finally, talazoparib and
veliparib are excreted primarily in the urine (17,19–21),
whereas rucaparib is excreted primarily in the feces (15,16).
For olaparib and niraparib, the average percent recovery of the
administered dose is no different in the urine and feces (11,13).
PK parameters for PARP inhibitors are demonstrated in Table I, and metabolic pathways related to
PARP inhibitors are illustrated in Fig. 2.
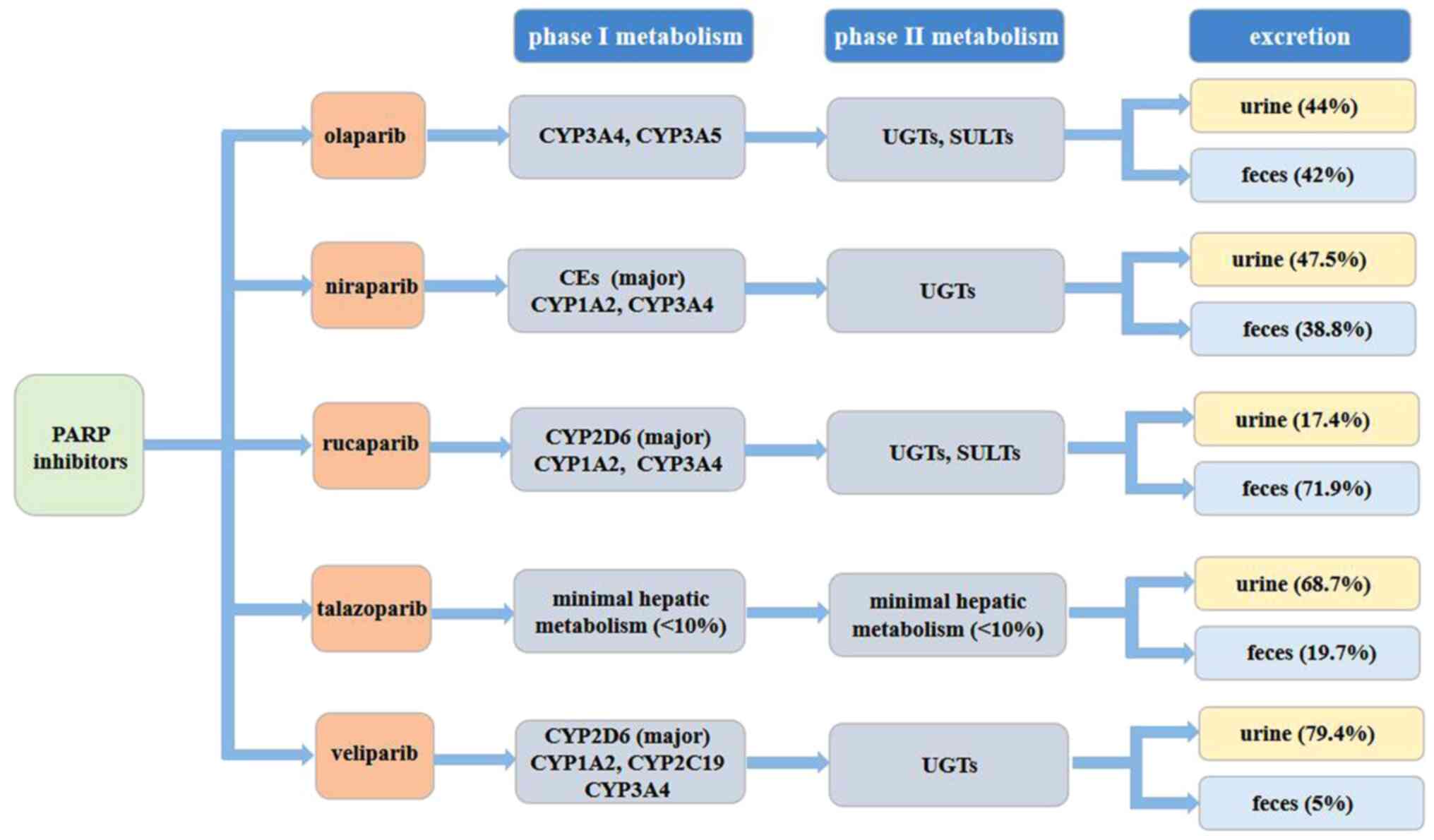 | Figure 2.Metabolic pathways related to PARP
inhibitors. Olaparib, rucaparib and veliparib are primarily
metabolized by the CYP enzymatic pathway, and subsequently, the
metabolites that are produced go under glucuronide or sulfate
conjugation. Talazoparib undergoes minimal hepatic metabolism, and
niraparib is metabolized primarily by carboxylesterases to form a
major inactive metabolite, and subsequently undergoes
glucuronidation. Talazoparib and veliparib are excreted primarily
in the urine, whereas rucaparib is excreted primarily in the feces.
For olaparib and niraparib, the average percent recovery of the
administered dose presents no difference in urine and feces. PARP,
poly (ADP-ribose) polymerase; UGTs, uridine diphosphate
glucuronosyl transferases; SULT, sulfotransferases; CYP, cytochrome
P450. |
 | Table I.PK parameters for poly (ADP-ribose)
polymerase inhibitors. |
Table I.
PK parameters for poly (ADP-ribose)
polymerase inhibitors.
|
|
|
|
|
|
|
|
|
| Excretion |
|---|
|
|
|
|
|
|
|
|
|
|
|
|---|
| PK | Recommended
dose | Tmax,
h | Bioavailability,
% | Volume of
distribution, liters | Plasma protein
binding, % | Metabolism
enzymes | T1/2,
h | Clearance, l/h | Urine, % | Feces, % |
|---|
| Olaparib
(tablet) | 300 mg twice
daily | 1.5 | NA | 158±136 | 82 | CYP3A4, CYP3A5,
UGTs, SULTs, | 14.9±8.2 | 7.4±3.9 | 44 | 42 |
| Olaparib
(capsules) | 400 mg twice
daily | 1-3 | NA | 167±196 | 82 | CYP3A4, CYP3A5,
UGTs, SULTs | 11.9±4.8 | 8.6±7.1 | 44 | 42 |
| Niraparib | 300 mg once
daily | 3 | 73 | 1220±1114 | 83 | CEs (major),
CYP1A2, CYP3A4, UGTs | 36 | 16.2 | 47.5 | 38.8 |
| Rucaparib | 600 mg twice
daily | 1.9 | 36 | 113-262 | 70 | CYP2D6 (major),
CYP1A2, CYP3A4, UGTs, SULTs | 17-19 | 15.3-79.2 | 17.4 | 71.9 |
| Talazoparib | 1 mg once
daily | 1-2 | 40 | 420 | 74 | Minimal hepatic
metabolism (<10%) | 90±58 | 6.45 | 68.7 | 19.7 |
| Veliparib | 400 mg twice
daily | 0.5-1.5 | 73 | 173 | 51 | CYP2D6 (major),
CYP1A2, CYP2C19, CYP3A4, UGTs | 5.2 | 20.9 | 79.4 | 5 |
Metabolism-related PK-based DDIs
Metabolism-related DDIs are the most common type of
PK-based DDIs. Drug metabolizing enzymes are expressed throughout
the body, including in the liver, intestines, kidneys, brain,
heart, lungs and skin. In the small intestine, there are multiple
CYP enzymes (22). An immunoblot
study of microsomes indicated that CYP3A and CYP2C9 represent the
major constituents of the intestinal CYP enzymes, accounting for 80
and 14% of total intestinal CYP enzymes, respectively (23). CYP3A4 was the main CYP3A enzyme,
while CYP3A5 was only detected in certain individuals (24). The remaining detected CYP enzymes,
in decreasing order of abundance, were CYP2C19, CYP2J2 and CYP2D6.
Evidence indicated that a wide variety of orally administered drugs
are metabolized by intestinal CYP enzymes, and that intestinal CYP
enzyme-mediated metabolism could actually eliminate a large
proportion of certain orally administered drugs before they enter
the systemic circulation (24–26). Therefore, orally administered
drugs that are subject to high intestinal metabolism not only
suffer from low oral bioavailability, but they are also more likely
to be susceptible to DDIs (27).
While certain oral drugs are metabolized by both the
intestines and liver, the main site for drug metabolism is the
liver, where both phase I and II metabolic enzymes are expressed in
hepatocytes and the biliary epithelium. Phase I metabolic enzymes
are primarily CYP enzymes, whereas phase II metabolic enzymes
mainly include uridine diphosphate glucuronosyl transferases (UGTs)
and sulfotransferases (SULTs). Unlike in the intestines, the major
metabolic enzyme subfamilies are more evenly spread out across the
liver (28). For phase I
metabolism, CYP3A, CYP1A2, CYP2D6, CYP2C, CYP2B6, CYP2E1 and CYP4F
are all major players (29). For
phase II metabolism, UGT1A, UGT2B and SULT1A1 are the major
metabolic enzymes (30).
Inhibition or induction of any or all of these hepatic enzymes by
co-administered medications or food may lead to increased toxicity
or reduced treatment efficacy (31).
Intestinal drug-metabolizing enzymes affect drug
absorption, while hepatic drug-metabolizing enzymes affect drug
elimination (9,27). Drugs, food and herbal supplements
that compete for metabolism by the same metabolic enzyme, or that
inhibit or induce metabolic enzymes, may mediate DDIs, leading to
an increase or decrease in the serum area under the curve (AUC) of
the enzyme substrate (32).
Increased or decreased exposure by alteration of metabolic enzyme
activity may cause clinically relevant toxic effects or
ineffectiveness of treatment with PARP inhibitors. In addition, as
certain PARP inhibitors could inhibit or induce metabolic enzymes,
they could also influence the exposure of other metabolic
substrates (11,15). The DDIs between PARP inhibitors and
enzyme inhibitors and inducers are listed in Table II. The DDIs between PARP
inhibitors and other enzyme substrates are listed in Table III.
| Table II.DDIs between poly (ADP-ribose)
polymerase inhibitors and enzyme inhibitors and inducers. |
Table II.
DDIs between poly (ADP-ribose)
polymerase inhibitors and enzyme inhibitors and inducers.
| Drugs | Inhibitors | Inducers | AUCR |
CmaxR |
Recommendations | (Refs.) |
|---|
| Olaparib | Itraconazole |
| 2.70 | 1.42 | Avoid
co-administration with strong and moderate CYP3A inhibitors and
consider | (7,11,12,33) |
|
| Fluconazole |
| 2.21 | 1.14 | alternative
medicines with less CYP3A inhibition. If co-administration is
unavoidable, reduce the olaparib dose to 150 mg (capsule) or 100 mg
(tablet) received twice daily for a strong CYP3A inhibitor, and 200
mg (capsule) or 150 mg (tablet) received twice daily for a moderate
CYP3A inhibitor. |
|
|
|
| Rifampin | 0.13 | 0.29 | Co-administration
with strong and moderate CYP3A inducers should be avoided. |
|
|
|
| Efavirenz | 0.40 | 0.69 | If a moderate CYP3A
inducer cannot be avoided, pay attention to the potential decreased
efficacy. |
|
| Niraparib | NA | NA | NA | NA | Co-administration
of CYP enzyme inhibitors or inducers would not cause clinically
significant DDIs. | (13,14) |
| Rucaparib | CYP1A2
inhibitor |
| 1.16 | 1.16 | Concomitant
administration of CYP inhibitors or inducers with rucaparib | (15,16,37) |
|
| CYP2D6
inhibitor |
| 1.00 | 1.01 | is not
restricted. |
|
| Talazoparib | NA | NA | NA | NA | Co-administration
of enzyme inhibitors or inducers would not cause clinically
significant DDIs. | (17,18) |
| Veliparib | NA | NA | NA | NA | Co-administration
of CYP2D6 inhibitors or inducers would not cause clinically
significant DDIs. | (19,21,41) |
| Table III.DDIs between poly (ADP-ribose)
polymerase inhibitors and other enzyme substrates. |
Table III.
DDIs between poly (ADP-ribose)
polymerase inhibitors and other enzyme substrates.
| Drugs | Metabolic
substrates | AUCR |
CmaxR |
Recommendations | (Refs.) |
|---|
| Olaparib | Midazolam | 1.61 | 1.18 | Caution should be
used when olaparib is combined with sensitive CYP3A substrates or
agents with a narrow therapeutic index | (33,34) |
|
| Raltegravir | 1.07 | 1.04 | Restricting the
concomitant use of olaparib with UGT1A1 substrates is not
recommended. |
|
| Niraparib | NA | NA | NA | Caution should be
used when niraparib is combined with CYP1A2 substrates with a
narrow therapeutic index. | (13,14) |
| Rucaparib | Caffeine | 2.55 | 1.00 | Dose adjustments
should be considered for CYP1A2, CYP3A, CYP2C9 and CYP2C19
substrates, | (8,15,16) |
|
| S-warfarin | 1.49 | 1.05 | particularly for
those with a narrow therapeutic index. |
|
|
| Omeprazole | 1.55 | 1.09 |
|
|
|
| Midazolam | 1.38 | 1.13 |
|
|
| Talazoparib | NA | NA | NA | Clinically
significant DDIs appear unlikely to occur between talazoparib and
other CYP or UGT substrates. | (17,18) |
| Veliparib | NA | NA | NA | Veliparib is not
likely to cause any clinically relevant metabolism-related
DDIs. | (19,21) |
Olaparib
Olaparib is primarily metabolized by CYP3A (11,12).
It was previously shown that following administration of a single
radiolabeled dose, unmetabolized olaparib was the major circulating
component (70%) in plasma (11),
and accounted for 15 and 6% of radioactivity in urine and feces,
respectively (11,12). Most of its metabolism is
attributable to oxidation reactions, and subsequently, a number of
metabolites that are produced go under glucuronide or sulfate
conjugation (11,12).
The co-administration of olaparib with itraconazole
was noted to increase the AUC and Cmax of olaparib by
170 and 42%, respectively (7).
Similarly, fluconazole, a moderate CYP3A inhibitor, was predicted
to increase the AUC and Cmax of olaparib by 121 and 14%,
respectively (11). As such, the
concurrent use of strong and moderate CYP3A inhibitors should be
avoided. If a CYP3A inhibitor must be co-administered, the olaparib
dose should be reduced to 150 mg (capsule) or 100 mg (tablet)
administered twice daily for a strong CYP3A inhibitor, or to 200 mg
(capsule) or 150 mg (tablet) received twice daily for a moderate
CYP3A inhibitor (7,11,12).
In addition, grapefruit, grapefruit juice and seville orange juice
should be avoided during olaparib treatment, since they are CYP3A
inhibitors (11,12).
When co-administered with rifampicin, the AUC and
Cmax of olaparib were noted to decrease by 87 and 71%,
respectively (7). Efavirenz, a
moderate CYP3A inducer, was predicted to decrease the AUC and
Cmax of olaparib by ~60 and 31%, respectively (11). Thus, the concurrent use of strong
or moderate CYP3A inducers should also be avoided. If use of a
moderate CYP3A inducer cannot be avoided, there exists a potential
for decreased efficacy of olaparib (7,11,12).
In an in vitro study, olaparib acted as both
an inhibitor and inducer of CYP3A, an inhibitor of UGT1A1 and an
inducer of CYP2B6 (33).
Physiologically based PK (PBPK) modeling predicted that olaparib
could increase the AUC of midazolam (a CYP3A substrate) by 61% and
the Cmax by 18%, and increase the AUC of raltegravir (a
UGT1A1 substrate) by 7% and the Cmax by 4% (34). As a result, caution should be taken
when sensitive CYP3A substrates or agents with a narrow therapeutic
index are combined with olaparib, but restricting the simultaneous
use of olaparib and UGT1A1 substrate is not recommended (11,12,34).
Niraparib
Niraparib is primarily metabolized by CEs to form a
major inactive metabolite (M1) that is subsequently metabolized by
UGTs into minor inactive metabolites (M10) (13,14,35).
The minor pathway of the oxidative metabolism of niraparib is
primarily metabolized by CYP1A2 and CYP3A4, with minor
contributions from CYP2D6 (13,14).
In a PK study, M1 and M10, the subsequently formed M1 glucuronides,
were the major circulating components (36). The influence of CEs or UGT
polymorphisms on niraparib PK was not evaluated, and
co-administration of CYP enzyme inhibitors or inducers is not
expected to cause clinically significant DDIs (13,14).
Neither niraparib nor M1 inhibits CYP or UGT
isoforms, although niraparib is a weak CYP1A2 inducer at high
concentrations (13,14,35).
Therefore, the clinical relevance of a DDI could not be completely
ruled out, and caution should be used when niraparib is combined
with CYP1A2-sensitive substrates, particularly those having a
narrow therapeutic range (14).
Rucaparib
In vitro, rucaparib is primarily metabolized
by CYP2D6 and to a lesser extent by CYP1A2 and CYP3A4, although
with a low metabolic turnover rate; subsequently, the metabolites
undergo sulfation and glucuronidation (15,16).
It was reported that following administration of a single
radiolabeled dose of rucaparib, unmetabolized rucaparib was the
major component and accounted for 64% of the radioactivity in
plasma (37). The major metabolic
pathways for rucaparib are oxidation, N-demethylation,
N-methylation and glucuronidation (15).
In a population PK study, the steady-state
concentrations of rucaparib did not differ significantly across
CYP2D6 or CYP1A2 genotype subgroups (15,16,38).
Concurrent use of a strong CYP1A2 or CYP2D6 inhibitor did not show
significant impact on rucaparib PK. As such, concurrent
administration of CYP inhibitors or inducers with rucaparib is not
restricted (15,16). In vitro, rucaparib has been
revealed to be a moderate inhibitor of CYP1A2, and a weak inhibitor
of CYP2C9, CYP2C19, CYP3A, CYP2C8, CYP2D6 and UGT1A1 (8,15).
Rucaparib has been shown to induce CYP1A2 and downregulate CYP2B6
and CYP3A4 at clinically relevant concentrations (15,16,39).
In a DDI study in patients with cancer, the effects
of a steady dose of rucaparib at 600 mg twice daily on caffeine (a
CYP1A2 substrate), S-warfarin (a CYP2C9 substrate), omeprazole (a
CYP2C19 substrate) and midazolam (a CYP3A substrate) were evaluated
(8). Rucaparib exhibited no effect
on the Cmax of caffeine, although it moderately
increased the AUC by 1.55% (8).
Rucaparib increased the AUC of S-warfarin by 0.49% and the
Cmax by 0.05%, increased the AUC of omeprazole by 0.55%
and the Cmax by 0.09%, and increased the AUC of
midazolam by 0.38% and the Cmax by 0.13% (8). According to the study,
co-administration of rucaparib could increase the systemic exposure
of CYP1A2, CYP3A, CYP2C9 or CYP2C19 substrates, which may increase
the risk of toxicities of these drugs (8). Hence, patients should be
appropriately monitored, and dose adjustments should be considered
for CYP1A2, CYP3A, CYP2C9 and CYP2C19 substrates, particularly for
those with a narrow therapeutic index, if clinically indicated
(8,15,16).
DDI studies to evaluate the effect of rucaparib on
the PK of UGT1A1 substrates have not been established, but a
statement is included in the summary of the product characteristics
(SmPC) to indicate that special caution should be paid when
rucaparib is combined with UGT1A1 substrates (i.e. irinotecan) in
patients with cancer and UGT1A1*28 (15,16).
Talazoparib
Talazoparib undergoes minimal hepatic metabolism
(<10%) (17,18). The identified metabolic pathways
instead include mono-oxidation, dehydrogenation, cysteine
conjugation of mono-desfluoro-talazoparib and glucuronide
conjugation (17,18,40).
Following oral administration of a single radiolabeled dose, no
major circulating metabolites were identified in plasma, and
talazoparib was the only circulating drug-derived entity identified
(17). Therefore, inhibition or
induction of metabolism is unlikely to affect the talazoparib
exposure (17,18).
In vitro, talazoparib has not been revealed
to be an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19,
CYP2D6 or CYP3A4/5, or an inducer of CYP1A2, CYP2B6 or CYP3A4 at
clinically relevant concentrations (17,18).
Furthermore, talazoparib is not an inhibitor of UGT isoforms
(UGT1A1, UGT1A4, UGT1A6, UGT1A9, UGT2B7 and UGT2B15) (17). As such, clinically significant DDIs
are unlikely to occur when talazoparib is combined with other CYP
or UGT substrates (17,18).
Veliparib
Based on a PK study conducted in patients with
cancer, veliparib is metabolized by multiple CYP enzymes, including
CYP1A2, CYP2D6, CYP2C19 and CYP3A4, with CYP2D6 playing a key role
in the formation of M8, the primary active metabolite in humans
(19). It was reported that 79.4%
of the veliparib dose was excreted in the urine as the
unmetabolized drug, indicating that metabolism contributes to at
most 30% of total clearance (19,21).
Veliparib is metabolized by multiple pathways, including oxidation
catalyzed by CYP enzymes and UGT-mediated N-carbamoyl
glucuronidation (21,41). The contribution of CYP enzymes to
total veliparib clearance remains unclear, but may not be
significant. Based on these findings, CYP enzyme polymorphisms or
co-administration of veliparib with CYP enzymes inhibitors or
inducers likely would not cause any clinically relevant
metabolism-related DDIs (19,21,41).
Veliparib has not been demonstrated to inhibit
activities of CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1,
CYP2B6, CYP2C8 and CYP3A4, or to induce the activities of CYP1A2,
CYP2B6, CYP2C9 and CYP3A4 at clinically relevant concentrations
(21). Therefore, veliparib is not
likely to cause any clinically relevant CYP enzyme-related DDIs
(21).
Conclusions
PK-based DDIs occur when one agent influences the
absorption, distribution, metabolism or excretion of another agent.
Altered metabolism is among the most complex of these processes
(42). Of the five aforementioned
PARP inhibitors, olaparib is primarily metabolized by CYP3A
(11), rucaparib has a low
metabolic turnover rate and is metabolized primarily by CYP2D6, and
to a lesser extent by CYP1A2 and CYP3A4 (15), and talazoparib undergoes minimal
hepatic metabolism (17).
Veliparib is metabolized by multiple metabolic enzymes, with CYP2D6
as the major enzyme and nearly 13% of veliparib undergoing hepatic
metabolism by the activity of CYP2D6 (19). Niraparib is primarily metabolized
by CEs, with subsequent metabolism by UGT into inactive metabolites
(13). As the metabolism of these
five PARP inhibitors involves CYP enzymes to varying degrees, each
has a unique set of DDIs. For example, for olaparib, the concurrent
use with strong or moderate CYP3A inhibitors and inducers should be
avoided, or if unavoidable, the dose of olaparib must be adjusted
(7,11,12,33).
Conversely, the co-administration of CYP inhibitors or inducers
with niraparib, rucaparib, talazoparib or veliparib would likely
not cause clinically significant DDIs (13–19,27,37,41). In
addition, PARP inhibitors themselves may cause the inhibition or
induction of CYP enzymes. With the use of olaparib, niraparib or
rucaparib, caution should be exercised when used with sensitive CYP
substrates, particularly those with a narrow therapeutic margin
(11–16,33,34). As talazoparib and veliparib are neither inhibitors
nor inducers of CYP enzymes at clinically relevant concentrations,
clinically significant DDIs appear unlikely to occur in combination
with other CYP substrates (17–19,21).
CYP enzymes are primarily localized in the liver and
small intestines, and as such they could make a major contribution
to the first-pass elimination of substrate drugs after oral
administration (43). There are
both similarities and differences between the hepatic and
intestinal CYP enzymes (44). For
example, while the drug rifampin could induce both hepatic and
intestinal CYP3A, grapefruit juice appears to be selective for
intestinal CYP3A (45). For
certain orally administered drugs, intestinal metabolism could
eliminate a large proportion of the drugs before they are able to
enter the systemic circulation. Orally administered drugs that are
intestinal CYP substrates not only suffer from low oral
bioavailability, but they are also more likely to be susceptible to
DDIs with other CYP substrates, inhibitors or inducers. However,
the hepatic CYP metabolism, intestinal CYP metabolism and
transporters are both involved in the first-pass elimination; thus,
distinguishing the intestinal CYP metabolism related DDIs from the
others could be difficult, and clinical studies regarding DDIs
mediated by intestinal CYP enzymes are at present lacking.
In the liver, drugs are metabolized by phase I and
II drug metabolizing enzymes. Given the predominant role of CYP
enzymes in the metabolism of drugs, the majority of studies
investigating drugs as either culprits or casualties of DDIs
arising from enzyme inhibition or induction have focused on CYP
inhibitors, inducers or substrates (33,38,43,44).
However, for certain drugs, phase II metabolism through UGTs or
SULTs is dominant in their metabolism, and may also be implicated
in DDIs, in particular glucuronidation (46). UGT enzymes catalyze the conjugation
of various endogenous (e.g., bilirubin) and exogenous (e.g., drugs)
compounds, thereby inhibition or induction of UGT enzymes may
significantly alter the elimination of UGT substrates and lead to
clinically significant DDIs (47,48).
While UGT enzymes are involved in the phase II metabolism of the
five aforementioned PARP inhibitors (11–19), the effect of UGT
inhibitors and inducers on the PK of PARP inhibitors has not been
established.
For screening of new drugs for the inhibition of UGT
enzymes, the Food and Drug Administration and European Medicines
Agency DDI guidelines recommend study of the inhibition of UGT
enzymes known to be involved in DDIs, including UGT1A1 and UGT2B7,
if one of the major elimination pathways of the investigational
drug is direct glucuronidation (49,50).
Previous studies have demonstrated that human liver microsomes and
recombinant proteins as the enzyme sources, together with in
vitro-in vivo extrapolation approaches, could predict
the likelihood of interactions arising from UGT enzyme inhibition
in vivo (51–53). Based on in vitro data, olaparib is
an inhibitor of UGT1A1 and rucaparib is a weak inhibitor of UGT1A1,
whereas neither niraparib nor talazoparib are inhibitors of UGT
isoforms (11–18). Clinical studies regarding the effects of PARP
inhibitors on the PK of UGT substrates have not yet been
established, but PBPK modeling predicts that olaparib may increase
the AUC of raltegravir (a UGT1A1 substrate) by 7% and the
Cmax by 4%, which is not considered to be clinically
meaningful (11,12,33).
In addition, a statement is included in the SmPC to reflect that
special caution should be paid when rucaparib is co-administered
with UGT1A1 substrates (15,16).
As niraparib and talazoparib are not inhibitors of UGT isoforms,
clinically significant DDIs appear unlikely to occur when niraparib
and talazoparib are combined with other UGT substrates.
The clinical significance of DDIs depends on several
factors including the PK/PD relationship, the genetic
polymorphisms, the therapeutic index of the victim drug, the
potency and concentration of the inhibitor or inducer, the
bioavailability of the victim drug, whether the victim drug is a
prodrug or an active drug, and the effects of disease on PK and PD
parameters (10). An interaction
should be considered clinically significant if it leads to
unfavorable outcomes such as reduced treatment efficacy or
increased adverse drug reactions (ADRs). However, few DDI studies
are conducted in patient populations to evaluate therapeutic
outcomes, nor are they long enough to completely assess the
development of ADRs.
PK-based DDI studies often use a no effect boundary
of 80–125% to determine whether an interaction is clinically
significant. With this approach, if the AUC is contained completely
between 80 and 125%, the interaction is considered not clinically
significant. However, this default no effect boundary may
occasionally be inappropriate, particularly for medications with a
narrower therapeutic index (10).
For example, for certain medications, a 20% increase in AUC may
lead to severe side effects. Thus, the no effect boundary should be
individualized for a given drug whenever possible with the
exposure-response data.
In order to detect patients at risk from harmful
DDIs, any potential DDIs must be identified. Several methods are
available for reducing the risk of clinically significant
interactions, such as PBPK models and population PK studies
(33,54,55).
Furthermore, to make DDI information more accessible, several DDI
screening software programs and databases have been developed and
are being implemented as clinical decision support tools (56,57).
However, understanding DDIs remains an ongoing challenge and
significant gaps in our knowledge remain. In addition, numerous
studies have concentrated on representative DDIs between two
medicines, but it is quite common for patients to be receiving more
than two medicines at one time. As such, the DDIs could be very
complex and exceedingly difficult to predict. Thus, therapeutic
drug monitoring (TDM) may be a favorable option in managing DDIs
(58,59). For numerous drugs there is a clear
relationship between plasma concentrations, ADRs and treatment
efficacy, and dose adjustments could be made if plasma
concentrations are outside of the therapeutic range (60). Furthermore, TDM has the advantage
of monitoring drug treatment continuously over long periods of
time, which may bring about improved treatment outcomes (61). Further research is required to
confirm the clinical relevance of TDM as a tool in DDI
management.
Ultimately, in order to achieve the improved
management of DDIs, clinicians and clinical pharmacists should be
consulted to perform a complete assessment of the DDI risk for a
given patient, to give recommendations to reduce these risks and to
arrange subsequent patient monitoring measures.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
DZ and JW designed the study. DZ and XL performed
the literature search. DZ drafted and revised the manuscript. All
the authors have read and approved the final manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Liang X, Wu P, Yang Q, Xie Y, He C, Yin L,
Yin Z, Yue G, Zou Y, Li L, et al: An update of new small-molecule
anticancer drugs approved from 2015 to 2020. Eur J Med Chem.
220:1134732021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tew WP, Lacchetti C, Ellis A, Maxian K,
Banerjee S, Bookman M, Jones MB, Lee JM, Lheureux S, Liu JF, et al:
PARP inhibitors in the management of ovarian cancer: ASCO
guideline. J Clin Oncol. 38:3468–3493. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mirza MR, Coleman RL, González-Martín A,
Moore KN, Colombo N, Ray-Coquard I and Pignata S: The forefront of
ovarian cancer therapy: Update on PARP inhibitors. Ann Oncol.
31:1148–1159. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Valabrega G, Scotto G, Tuninetti V, Pani A
and Scaglione F: Differences in PARP inhibitors for the treatment
of ovarian cancer: Mechanisms of action, pharmacology, safety, and
efficacy. Int J Mol Sci. 22:42032021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miller RE, Leary A, Scott CL, Serra V,
Lord CJ, Bowtell D, Chang DK, Garsed DW, Jonkers J, Ledermann JA,
et al: ESMO recommendations on predictive biomarker testing for
homologous recombination deficiency and PARP inhibitor benefit in
ovarian cancer. Ann Oncol. 31:1606–1622. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rolfo C, Swaisland H, Leunen K, Rutten A,
Soetekouw P, Slater S, Verheul HM, Fielding A, So K, Bannister W
and Dean E: Effect of food on the pharmacokinetics of olaparib
after oral dosing of the capsule formulation in patients with
advanced solid tumors. Adv Ther. 32:510–522. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dirix L, Swaisland H, Verheul HM, Rottey
S, Leunen K, Jerusalem G, Rolfo C, Nielsen D, Molife LR, Kristeleit
R, et al: Effect of itraconazole and rifampin on the
pharmacokinetics of olaparib in patients with advanced solid
tumors: Results of Two Phase I open-label studies. Clin Ther.
38:2286–2299. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xiao JJ, Nowak D, Ramlau R,
Tomaszewska-Kiecana M, Wysocki PJ, Isaacson J, Beltman J, Nash E,
Kaczanowski R, Arold G and Watkins S: Evaluation of drug-drug
interactions of rucaparib and CYP1A2, CYP2C9, CYP2C19, CYP3A, and
P-gp substrates in patients with an advanced solid tumor. Clin
Transl Sci. 12:58–65. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
van Leeuwen RW, van Gelder T, Mathijssen
RH and Jansman FG: Drug-drug interactions with tyrosine-kinase
inhibitors: A clinical perspective. Lancet Oncol. 15:e315–e326.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tannenbaum C and Sheehan NL: Understanding
and preventing drug-drug and drug-gene interactions. Expert Rev
Clin Pharmacol. 7:533–544. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
US Food and Drug Administration: Label.
https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/206162s011lbl.pdfJune
25–2021
|
|
12
|
European Medicines Agency: Product
information. https://www.ema.europa.eu/en/documents/product-information/lynparza-epar-product-information_en.pdfJune
25–2021
|
|
13
|
US Food and Drug Administration Label.
https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/208447s022s024lbl.pdfJune
25–2021
|
|
14
|
European Medicines Agency: Product
information. https://www.ema.europa.eu/en/documents/product-information/zejula-epar-product-information_en.pdfJune
25–2021
|
|
15
|
US Food and Drug Administration Label.
https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/209115s008lbl.pdfJune
25–2021
|
|
16
|
European Medicines Agency: Product
information. https://www.ema.europa.eu/en/documents/product-information/rubraca-epar-product-information_en.pdfJune
25–2021
|
|
17
|
US Food and Drug Administration Label.
https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/211651s006lbl.pdfJune
25–2021
|
|
18
|
European Medicines Agency: Product
information. https://www.ema.europa.eu/en/documents/product-information/talzenna-epar-product-information_en.pdfJune
25–2021
|
|
19
|
LoRusso PM, Li J, Burger A, Heilbrun LK,
Sausville EA, Boerner SA, Smith D, Pilat MJ, Zhang J, Tolaney SM,
et al: Phase I safety, pharmacokinetic, and pharmacodynamic study
of the poly(ADP-ribose) polymerase (PARP) inhibitor veliparib
(ABT-888) in combination with Irinotecan in patients with advanced
solid tumors. Clin Cancer Res. 22:3227–3237. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mittica G, Ghisoni E, Giannone G, Genta S,
Aglietta M, Sapino A and Valabrega G: PARP inhibitors in ovarian
cancer. Recent Pat Anticancer Drug Discov. 13:392–410. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li X, Delzer J, Voorman R, de Morais SM
and Lao Y: Disposition and drug-drug interaction potential of
veliparib (ABT-888), a novel and potent inhibitor of
poly(ADP-ribose) polymerase. Drug Metab Dispos. 39:1161–1169. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Teo YL, Ho HK and Chan A:
Metabolism-related pharmacokinetic drug-drug interactions with
tyrosine kinase inhibitors: Current understanding, challenges and
recommendations. Br J Clin Pharmacol. 79:241–253. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Paine MF, Hart HL, Ludington SS, Haining
RL, Rettie AE and Zeldin DC: The human intestinal cytochrome P450
‘pie’. Drug Metab Dispos. 34:880–886. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xie F, Ding X and Zhang QY: An update on
the role of intestinal cytochrome P450 enzymes in drug disposition.
Acta Pharm Sin B. 6:374–383. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Klomp F, Wenzel C, Drozdzik M and Oswald
S: Drug-drug interactions involving intestinal and Hepatic CYP1A
Enzymes. Pharmaceutics. 12:12012020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
van Herwaarden AE, van Waterschoot RA and
Schinkel AH: How important is intestinal cytochrome P450 3A
metabolism? Trends Pharmacol Sci. 30:223–227. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Scripture CD and Figg WD: Drug
interactions in cancer therapy. Nat Rev Cancer. 6:546–558. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Manikandan P and Nagini S: Cytochrome P450
structure, function and clinical significance: A review. Curr Drug
Targets. 19:38–54. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Almazroo OA, Miah MK and Venkataramanan R:
Drug metabolism in the liver. Clin Liver Dis. 21:1–20. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
An S, Jeon M, Kennedy EL and Kyoung M:
Phase-separated condensates of metabolic complexes in living cells:
Purinosome and glucosome. Methods Enzymol. 628:1–17. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Roberts AG and Gibbs ME: Mechanisms and
the clinical relevance of complex drug-drug interactions. Clin
Pharmacol. 10:123–134. 2018.PubMed/NCBI
|
|
32
|
Hussaarts KGAM, Veerman GDM, Jansman FGA,
van Gelder T, Mathijssen RHJ and van Leeuwen RWF: Clinically
relevant drug interactions with multikinase inhibitors: A review.
Ther Adv Med Oncol. 11:17588359188183472019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
McCormick A, Swaisland H, Reddy VP,
Learoyd M and Scarfe G: In vitro evaluation of the inhibition and
induction potential of olaparib, a potent poly(ADP-ribose)
polymerase inhibitor, on cytochrome P450. Xenobiotica. 48:555–564.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pilla Reddy V, Bui K, Scarfe G, Zhou D and
Learoyd M: Physiologically based Pharmacokinetic modeling for
olaparib dosing recommendations: Bridging formulations, drug
interactions, and patient populations. Clin Pharmacol Ther.
105:229–241. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Scott LJ: Niraparib: First global
approval. Drugs. 77:1029–1034. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
van Andel L, Zhang Z, Lu S, Kansra V,
Agarwal S, Hughes L, Tibben MM, Gebretensae A, Lucas L, Hillebrand
MJX, et al: Human mass balance study and metabolite profiling of
14C-niraparib, a novel poly(ADP-Ribose) polymerase
(PARP)-1 and PARP-2 inhibitor, in patients with advanced cancer.
Invest New Drugs. 35:751–765. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liao M, Watkins S, Nash E, Isaacson J,
Etter J, Beltman J, Fan R, Shen L, Mutlib A, Kemeny V, et al:
Evaluation of absorption, distribution, metabolism, and excretion
of [(14)C]-rucaparib, a poly(ADP-ribose) polymerase inhibitor, in
patients with advanced solid tumors. Invest New Drugs. 38:765–775.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liao M, Jaw-Tsai S, Beltman J, Simmons AD,
Harding TC and Xiao JJ: Evaluation of in vitro absorption,
distribution, metabolism, and excretion and assessment of drug-drug
interaction of rucaparib, an orally potent poly(ADP-ribose)
polymerase inhibitor. Xenobiotica. 50:1032–1042. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Syed YY: Rucaparib: First global approval.
Drugs. 77:585–592. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hoy SM: Talazoparib: First global
approval. Drugs. 78:1939–1946. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Niu J, Scheuerell C, Mehrotra S, Karan S,
Puhalla S, Kiesel BF, Ji J, Chu E, Gopalakrishnan M, Ivaturi V, et
al: Parent-metabolite pharmacokinetic modeling and pharmacodynamics
of veliparib (ABT-888), a PARP inhibitor, in patients with BRCA
1/2-mutated cancer or PARP-sensitive tumor types. J Clin Pharmacol.
57:977–987. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yu J, Petrie ID, Levy RH and
Ragueneau-Majlessi I: Mechanisms and clinical significance of
pharmacokinetic-based drug-drug interactions with drugs approved by
the U.S. Food and drug administration in 2017. Drug Metab Dispos.
47:135–144. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Preskorn SH: Drug-drug interactions (DDIs)
in psychiatric practice, part 9: Interactions mediated by
drug-metabolizing cytochrome P450 enzymes. J Psychiatr Pract.
26:126–134. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mouly S, Lloret-Linares C, Sellier PO,
Sene D and Bergmann JF: Is the clinical relevance of drug-food and
drug-herb interactions limited to grapefruit juice and Saint-John's
Wort? Pharmacol Res. 118:82–92. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Thelen K and Dressman JB: Cytochrome
P450-mediated metabolism in the human gut wall. J Pharm Pharmacol.
61:541–558. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rowland A, Miners JO and Mackenzie PI: The
UDP-glucuronosyltransferases: Their role in drug metabolism and
detoxification. Int J Biochem Cell Biol. 45:1121–1132. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Miners JO, Chau N, Rowland A, Burns K,
McKinnon RA, Mackenzie PI, Tucker GT, Knights KM and Kichenadasse
G: Inhibition of human UDP-glucuronosyltransferase enzymes by
lapatinib, pazopanib, regorafenib and sorafenib: Implications for
hyperbilirubinemia. Biochem Pharmacol. 129:85–95. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Miners JO, Rowland A, Novak JJ, Lapham K
and Goosen TC: Evidence-based strategies for the characterisation
of human drug and chemical glucuronidation in vitro and
UDP-glucuronosyltransferase reaction phenotyping. Pharmacol Ther.
218:1076892021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
US Food and Drug Administration: Guidance
for industry. drug interaction studies-study design, data analysis,
implications for dosing, and labelling recommendations. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformatiom/Guidances/default/htmJune
5–2021.
|
|
50
|
European Medicines Agency: Guideline on
the investigation of drug interactions. ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdfJune
5–2021
|
|
51
|
Cheng X, Lv X, Qu H, Li D, Hu M, Guo W, Ge
G and Dong R: Comparison of the inhibition potentials of icotinib
and erlotinib against human UDP-glucuronosyltransferase 1A1. Acta
Pharm Sin B. 7:657–664. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang Z, Wang X, Wang Z, Jia Y, Feng Y,
Jiang L, Xia Y, Cao J and Liu Y: In vitro inhibition of human
UDP-glucuronosyltransferase (UGT) 1A1 by osimertinib, and
prediction of in vivo drug-drug interactions. Toxicol Lett.
348:10–17. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Korprasertthaworn P, Chau N, Nair PC,
Rowland A and Miners JO: Inhibition of human
UDP-glucuronosyltransferase (UGT) enzymes by kinase inhibitors:
Effects of dabrafenib, ibrutinib, nintedanib, trametinib and BIBF
1202. Biochem Pharmacol. 169:1136162019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Min JS and Bae SK: Prediction of drug-drug
interaction potential using physiologically based pharmacokinetic
modeling. Arch Pharm Res. 40:1356–1379. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Falcão A, Fuseau E, Nunes T, Almeida L and
Soares-da-Silva P: Pharmacokinetics, drug interactions and
exposure-response relationship of eslicarbazepine acetate in adult
patients with partial-onset seizures: Population pharmacokinetic
and pharmacokinetic/pharmacodynamic analyses. CNS Drugs. 26:79–91.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zakrzewski-Jakubiak H, Doan J, Lamoureux
P, Singh D, Turgeon J and Tannenbaum C: Detection and prevention of
drug-drug interactions in the hospitalized elderly: Utility of new
cytochrome p450-based software. Am J Geriatr Pharmacother.
9:461–470. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Roblek T, Vaupotic T, Mrhar A and Lainscak
M: Drug-drug interaction software in clinical practice: A
systematic review. Eur J Clin Pharmacol. 71:131–142. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Solassol I, Pinguet F and Quantin X: FDA-
and EMA-approved tyrosine kinase inhibitors in advanced
EGFR-Mutated Non-Small cell lung cancer: Safety, tolerability,
plasma concentration monitoring, and management. Biomolecules.
9:6682019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Janssen JM, Dorlo TPC, Steeghs N, Beijnen
JH, Hanff LM, van Eijkelenburg NKA, van der Lugt J, Zwaan CM and
Huitema ADR: Pharmacokinetic targets for therapeutic drug
monitoring of small molecule kinase inhibitors in pediatric
oncology. Clin Pharmacol Ther. 108:494–505. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Di Francia R, De Monaco A, Saggese M,
Iaccarino G, Crisci S, Frigeri F, De Filippi R, Berretta M and
Pinto A: Pharmacological profile and pharmacogenomics of
anti-cancer drugs used for targeted therapy. Curr Cancer Drug
Targets. 18:499–511. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Cardoso E, Csajka C, Schneider MP and
Widmer N: Effect of adherence on pharmacokinetic/pharmacodynamic
relationships of oral targeted anticancer drugs. Clin
Pharmacokinet. 57:1–6. 2018. View Article : Google Scholar : PubMed/NCBI
|














