Introduction
Inhibins, which members of the transforming growth
factor-β superfamily, are heterodimers consisting of a common
α-subunit and one of two homologous β-subunits (βA or βB) (1,2).
Inhibin suppresses the production and secretion of
follicle-stimulating hormone in classical endocrine function and
acts in an autocrine and paracrine manner in reproductive tissues
(3,4). Studies in transgenic mice have
identified a tumor-suppressor role of inhibin-α (5,6).
Inhibin-α gene expression is inhibited in malignant sites of
high-grade prostate cancer tissue, but inhibin-α is not expressed
in poorly differentiated tumor cells. Accordingly, unlike ovarian
granulocyte tumor cells, inhibin-α gene is expressed at low levels
in poorly differentiated prostate cancer (7). DNA methylation may be responsible for
this transcriptional silencing (8).
Methylation is catalyzed by enzymes and is involved
in heavy metal modification, gene expression regulation, protein
function regulation and RNA processing. Aberrant methylation of CpG
islands that are not normally methylated occurs relatively
frequently in immortalized and transformed cells, and this
phenomenon is associated with inappropriate silencing of human
tumor suppressor genes (9,10). This modification is particularly
associated with CpG islands in the promoter region and has
important regulatory effects on gene expression (11,12).
CpG island methylation is an epigenetic change involved in
tumorigenesis via transcriptional inactivation (13). Epigenetic changes, including DNA
methylation and histone modifications, represent important
mechanisms for cancer suppressor gene silencing and affect
chromatin structure and genomic stability (14). The DNA methylation characteristics
of skin metastatic tumor tissue can provide predictive evidence of
therapeutic response to immune checkpoint suppression in patients
with stage IV metastatic melanoma (15). Latent methylation component-based
detection of DNA methylation has been indicated to be a tool for
developing classifiers and evaluating treatment response in
patients with cancer undergoing targeted immunotherapy (16). DNA methylation at CpG sites in
cancer is an epigenetic change. Hypermethylation limits immune
checkpoint blockade therapy by inhibiting the endogenous interferon
response in cancer cell recognition (17). Conversely, hypomethylation
indicates the expression of programmed death ligand 1 and
inhibitory cytokines accompanied by epithelial-mesenchymal changes,
which may contribute to immunosuppression (18). Melanoma pathogenesis is facilitated
through numerous genomic alterations in various components of the
mitogen-activated protein kinase and phosphoinositol-3-kinase
(PI3K) pathways (19), and these
pathways have been examined for potential use as biomarkers in
advanced melanoma treatment and prognosis strategies (20).
Transcriptional silencing of the inhibin-α gene has
been confirmed in human prostate cancer, gastric cancer and
leukemia (21–23). The primary causes of the silencing
were methylation, mutation and heterozygous loss of chromosomal
abnormalities in the inhibin-α promoter. The human inhibin α-gene
is located at the q33-q36 region of chromosome 2, and a deletion of
2q has been observed in numerous human tumors, including prostate
cancer (24–26). Inhibin serves an important role in
hormonal regulation; however, there has been no investigation of
its methylation, mutations and chromosomal abnormalities in human
melanoma cells.
Therefore, the methylation status, mutations and
loss of heterozygosity (LOH) of the inhibin-α gene promoter, as
well as the regulation of inhibin-α gene expression by an inhibitor
of DNA methylation [5-aza-2′-deoxycytidine (5-Aza-dC)] in human
melanoma cells were investigated. In addition, the association
between a mutation in the inhibin-α 5′-untranslated region (5′-UTR)
and the expression of phosphatidylinositol
3,4,5-trisphosphate-dependent Rac exchanger 2 (PREX2) and
phosphatase and tensin homolog (PTEN) as well as AKT/PI3K signaling
was examined.
Materials and methods
Cell culture
G361, SK-MEL-3, SK-MEL-5, SK-MEL-24 and SK-MEL-28
human melanoma cell lines were purchased from the American Tissue
Culture Collection. SNU-668 cells (inhibin-α positive control
cells) were supplied from Korean Cell Line Bank; Korean Cell Line
Research Foundation. G361 and SNU-668 cells were cultured in
RPMI-1640 medium (Corning, Inc.), and SK-MEL-3, SK-MEL-5, SK-MEL-24
and SK-MEL-28 cells were cultured in Dulbecco's modified Eagle
medium (Corning, Inc.) containing 10% fetal bovine serum (Gibco;
Thermo Fisher Scientific, Inc.) and antibiotic-antimycotic solution
(Gibco; Thermo Fisher Scientific, Inc.) at 37°C in a humidified
atmosphere with 5% CO2 and 95% air.
Bisulfite modification
Genomic DNA was extracted using Wizard Genomic DNA
purification kit (cat. no. A1120; Promega Corporation). DNA (2 µg)
methylation was performed using a bisulfite conversion kit (cat.
no. D5001; EZ DNA methylation kit; Zymo Research Corp.) according
to the manufacturer's protocol. DNA was resuspended in 20 µl water
and either used immediately or stored at −20°C.
Detection of methylation
Methylation was assessed via polymerase chain
reaction (PCR) and sequence analysis of the bisulfite-treated DNA.
The bisulfite reaction converted unmethylated cytosine to uracil,
whereas methylated cytosine remained unchanged. The inhibin-α
5′-UTR region was amplified using nested PCR with primers designed
for the bisulfite-converted sequence, as previously described
(26). The primer sequences are
listed in Table I. Primer
sequences methylation-specific PCR (MSP) 1 and 2 were used for the
first round of PCR, whereas primer sequences MSP 3 and 4 were used
for the second round. The first round of PCR was performed in a
25-µl reaction mixture consisting of 2 µl bisulfite-converted DNA,
1X PCR buffer (10 mM Tris-HCl pH 8.3, 50 mM KCl, 1.5 mM
MgCl2), 0.2 mM dNTPs, 10 pmol of each primer (1 and 2)
and 1 unit AmpliTaq Gold DNA polymerase (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The first round of PCR cycle conditions
were as follows: 95°C for 15 min, followed by five cycles at 95°C
for 1 min, 50°C for 2 min and 72°C for 3 min and 30 cycles at 95°C
for 1 min, 55°C for 2 min and 72°C for 2 min, with a final
incubation step at 72°C for 10 min. A 2-µl sample of the first PCR
product was amplified in a 25-µl reaction mixture as
aforementioned, except for primers 3 and 4 being used. The second
round PCR conditions included 35 cycles at 95°C for 1 min, 60°C for
2 min and 72°C for 2 min. The PCR products were electrophoresed on
1.5% agarose containing 0.5 µg/ml ethidium bromide, gel-purified,
ligated into the pCR2.1-TOPO vector, and cloned using the TOPO TA
Cloning Kit according to the manufacturer's protocol (cat. no.
45-0641; Invitrogen; Thermo Fisher Scientific, Inc.). For each PCR,
10 clones were sequenced, and the methylation status at each of the
seven CpGs was determined. Sequencing analysis was performed by
BIONICS Co., Ltd. using a high throughput DNA analyzer (3730XL DNA
Analyzer; Applied Biosystems; Thermo Fisher Scientific, Inc.)
according to the Sanger method.
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Type of analysis of
inhibin-α gene | Primer name | Orientation | Sequence
(5′-3′) | Product size
(bp) | Annealing
temperature (°C) | GenBank accession
number or locus map on the chromosome |
|---|
| Methylation | MSP 1 | Forward |
GATAAGAGTTTAGATTGGTTTTATTGGTT | 681 | 50, 55 | AF272341.1 |
|
| MSP 2 | Reverse |
ACACCATAACTCACCTAACCCTACTAATAA |
|
|
|
|
| MSP 3 | Forward |
GAAGGTGTTGTATGTTTGTATGTGTGAGTT | 275 | 60 |
|
|
| MSP 4 | Reverse |
ACCCCTTCTACCAAAATCTACCCAAAA |
|
|
|
| Mutation | 5′-UTR and exon
1 | Forward |
GACTGGGGAAGACTGGATGA | 240 | 57 | NM_002191.2 |
|
|
| Reverse |
TCACCTTGGCCAGAACAAGT |
|
|
|
|
| Exon 2 | Forward |
AGCAGCCTCCAATAGCTCTG | 396 | 57 | NM_002191.2 |
|
|
| Reverse |
AGCTCCTGGAAGGAGATGTTC |
|
|
|
| LOH | 2q32-q33 | Forward |
TAAAGCCTAGTGGAAGATCATC | 198 | 55 | D2S389 |
|
|
| Reverse |
GCTGAGTTAACAGTTATCAACAATT |
|
|
|
|
| 2q33-q36 | Forward |
AAACTGAGATTTGTCTAAGGGG | 155 | 55 | D2S128 |
|
|
| Reverse |
AGCCAGGAATTTTTGCTATT |
|
|
|
| RT-PCR | Inhibin-α | Forward |
AGGAAGAGGAGGATGTCTCC | 823 | 50 | NM_002191.4 |
|
|
| Reverse |
GAGTAACCTCCATCCGAGGT |
|
|
|
|
| β-actin | Forward |
CTTCTACAATGAGCTGCGTG | 305 | 55 | NM_001101.3 |
|
|
| Reverse |
TCATGAGGTAGTCAGTCAGG |
|
|
|
| qPCR | Inhibin-α | Forward |
CTCGGATGGAGGTTACTCTTTCAA | 88 | 60 | NM_002191.4 |
|
|
| Reverse |
GAAGACCCCCCACCCTTAGA |
|
|
|
|
| β-actin | Forward |
GCGAGAAGATGACCCAGATC | 77 | 60 | NM_001101.3 |
|
|
| Reverse |
GGATAGCACAGCCTGGATAG |
|
|
|
DNA analysis
DNA was isolated from cultured cells using standard
methods. Two regions of the inhibin-α gene were amplified from
genomic DNA using PCR with specific oligonucleotide primers as
previously described (27). The
first region of 240 bp (fragment A), which includes 140 bp of
5′-UTR and 100 bp of exon 1, and the second region of 396 bp
(fragment B), which comprises part of exon 2, was amplified using
the primers listed in Table I.
Genomic DNA (200 ng) was amplified in a 50-µl reaction mixture
containing 1X PCR buffer, 2 mM MgCl2, 2.5% DMSO, 0.2 mM
of each dNTP, 20 pmol of each specific primer and 1.5 unit of
AmpliTaq Gold DNA polymerase. The conditions for the amplification
were as follows: Denaturation at 95°C for 14 min; followed by
denaturation at 95°C for 40 sec, annealing at 57°C for 30 sec and
extension at 72°C for 1 min for 35 cycles; and a final extension at
72°C for 7 min. The polymorphism −16C->T in the 5′-UTR was
screened via restriction enzyme analysis with SpeI (New
England Biolabs, Inc.). Briefly, fragment A was amplified using
PCR, and 5 µl of the purified PCR product were digested overnight
at 37°C with 5 units SpeI, electrophoresed on 8% acrylamide
gels, stained with 0.5 µg/ml ethidium bromide, and visualized using
a Gel Doc 1000 Gel Documentation System (Bio-Rad Laboratories,
Inc.). The presence of a 240-bp fragment indicated a homozygous
wild-type variant, whereas the presence of two 120-bp fragments
corresponded to the homozygous variant T. The substitution of
769G->A in exon 2 was analyzed via digestion of fragment B with
different restriction enzymes. In brief, 5 µl of the purified PCR
product were digested overnight at 37°C with 5 units of
BsrFI (New England Biolabs, Inc.) and analyzed as
aforementioned. The restriction site that renders two fragments of
340 and 56 bp was abolished in the variant allele. In addition, 5
ml of the purified PCR product was digested overnight at 37°C with
5 units of Fnu4HI (New England Biolabs, Inc.),
electrophoresed on 15% acrylamide gels, stained with 0.5 µg/ml
ethidium bromide, and visualized by image analysis. The 396-bp
fragment rendered four fragments of 153, 107, 51 and 25 bp, among
other fragments of lower molecular weight in the wild-type allele,
whereas the allele with a 769G->A substitution rendered four
fragments of 153, 107, 76 and 51 bp, among other fragments of lower
molecular weight.
LOH analysis
LOH was determined using microsatellite markers on
2q32-q33 and 2q33-q36, as previously described (26). The oligonucleotide primer sequences
are listed in Table I. PCR was
performed in a 20-µl reaction mixture consisting of 200 ng DNA, 1X
PCR buffer, 0.2 mM of each dNTP, 10 pmol of each primer and 1 unit
of AmpliTaq Gold DNA polymerase. The conditions for amplification
were as follows: Denaturation at 95°C for 14 min; followed by
denaturation at 95°C for 1 min, annealing at 55°C for 1 min and
extension at 72°C for 1 min for 35 cycles; and final extension at
72°C for 10 min. Subsequently, 10 µl PCR products were mixed with
10 ml of stop solution containing 95% formamide, 10 mM NaOH, 0.25%
bromophenol blue and 0.25% xylene cyanol FF. The mixture was
denatured at 95°C for 5 min, placed on ice for 5 min,
electrophoresed on 12% acrylamide gels containing 10% glycerol with
1X Tris/Borate/EDTA buffer, and stained with 0.5 µg/ml ethidium
bromide. LOH was defined as a reduction in the intensity of the
signal of a single allele by >50% in the tumor cell DNA as
assessed by direct visualization, compared with DNA of peripheral
blood lymphocytes (22,23). Allelic imbalance was evaluated when
a difference in the running pattern of the products was
observed.
5-Aza-dC treatment
Cells were seeded at a density of 5×105
cells/100 mm culture dish. The cells were allowed to attach for 24
h and then exposed to different concentrations (0, 0.1, 0.5, 2, 5,
and 10 µM) of the DNA methylation inhibitor 5-Aza-dC
(Sigma-Aldrich; Merck KGaA) for 5 days at 37°C. The medium and the
drug were replaced every 2 days. At the end of the treatment
period, the medium was removed, and the cell pellets were subjected
to reverse transcription-quantitative PCR (RT-qPCR) and immunoblot
analysis.
Cell proliferation assay
For the cell viability assay, cells were seeded at
5×104 cells/well in 6-well plates and exposed to various
concentrations (0, 0.1, 0.5, 2, 5 and 10 µM) of 5-Aza-dC for 5 days
at 37°C. To evaluate cell doubling time following exposure to 5 µM
5-Aza-dC, the cells were seeded at 2×104 cells/well in
12-well plates containing culture medium. Cell number was
determined daily using the trypan blue exclusion assay (0.4% trypan
blue solution; 10 µl diluted cells combined with 10 µl trypan blue
solution) for 5 consecutive days. Untreated cells were analyzed
under similar conditions as a control. The cell number was counted
under a light microscope (magnification, ×100). The average cell
number from two plates was determined, and the mean cell numbers
were plotted to calculate the cell population doubling times.
RNA isolation and RT-qPCR
Total RNA was purified from cultured cells using the
TRIzol® reagent method, according to the manufacturer's
protocol (Invitrogen; Thermo Fisher Scientific, Inc.). First-strand
cDNA synthesis was performed using 1 µg total RNA and
Oligo(dT)15 primers in a reverse transcription system
kit (cat. no. A3500; Promega Corporation), according to the
manufacturer's protocol. The primer sequences are listed in
Table I.
PCR was performed using 2 µl cDNA in a 50-µl
reaction mixture containing 1X PCR buffer, 200 µM of each dNTP, 20
pmol of each inhibin-α primer and 2 units of AmpliTaq Gold DNA
polymerase. The reactions were carried out in a thermal cycler with
an initial denaturation step at 95°C for 14 min; followed by 35
cycles (25 cycles for β-actin) of denaturation at 95°C for 1 min,
primer annealing at 50°C (inhibin-α) and 55°C (β-actin) for 1 min;
and a final extension step at 72°C for 1 min. The reaction was
terminated at 72°C for 10 min, and samples were stored at 4°C.
Next, 10 µl of PCR products were separated by electrophoresis on a
1.5% agarose gel containing ethidium bromide (0.5 µg/ml) and
visualized using image analysis.
RT-qPCR was performed on a StepOnePlus Real-Time PCR
System with the Power SYBR Green PCR Master Mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The gene-specific
primer sequences used are listed in Table I. PCR was performed using 1 µl cDNA
in a 20-µl reaction mixture containing 10 µl Power SYBR Green PCR
Master Mix, 2 µl primers and 7 µl PCR-grade water. The reaction
conditions included denaturation at 95°C for 10 min, followed by 40
cycles at 95°C for 15 sec and 60°C for 1 min. The crossing points
of the target genes with β-actin were obtained, and the relative
amounts were quantified using the 2−ΔΔCq method
(28).
Fluorescein isothiocyanate (FITC)-flow
cytometric analysis of inhibin-α protein
Cultured cells were detached using 0.05%
trypsin-EDTA solution. After being washed with cold
phosphate-buffered saline (PBS), the cells were incubated with an
anti-inhibin-α mouse monoclonal antibody (1:50; cat. no. sc-365439;
Santa Cruz Biotechnology, Inc.) or normal mouse serum (1:50; cat.
no. sc-45051; Santa Cruz Biotechnology, Inc.) as a negative control
for 30 min at 4°C. After three washes with cold PBS, the cells were
stained with a FITC-labeled mouse antibody (1:50; sc-516140; Santa
Cruz Biotechnology, Inc.) for 30 min at 4°C. Washing was repeated
in the same manner, and cell-surface immunofluorescence was
analyzed using a FACSCalibur instrument with the CellQuest software
6.0 (both from BD Biosciences).
Immunoblot analysis
Cells were washed with cold PBS and then lysed using
cell lysis buffer (Cell Signaling Technology, Inc.) containing 1 mM
PMSF (Sigma-Aldrich; Merck KGaA). Protein concentration was
determined using the BCA protein assay (Pierce; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Briefly, 10 µg of protein was fractionated by 10% SDS-PAGE and then
transferred onto a nitrocellulose membrane (Amersham; Cytiva). The
membranes were blocked with 1% bovine serum albumin (Sigma-Aldrich)
for 1 h at room temperature and then incubated overnight at 4°C
with antibodies against AKT (cat. no. 4685), phosphorylated (p)-AKT
(cat. no. 9271), PTEN (cat. no. 9559) (all from Cell Signaling
Technology, Inc.), p-PI3K (cat. no. ab182651), PREX2 (cat. no.
ab169027) (both from Abcam), PI3K (cat. no. sc-1637; Santa Cruz
Biotechnology, Inc.) and β-actin (cat. no. A1978; Sigma-Aldrich;
Merck KGaA), which were all diluted at 1:1,000 (1:5,000 for
β-actin) with Tris-buffered saline containing 0.05% Tween 20
(TBS-T). After washing with TBS-T for 1 h, the membranes were
incubated for 1 h at room temperature with anti-rabbit (cat. no.
7074) and anti-mouse (cat. no. 7076) horseradish
peroxidase-conjugated secondary antibodies (Cell Signaling
Technology, Inc.) diluted at 1:2,500 (1:10,000 for β-actin only) in
TBS-T. The membranes were subsequently washed with TBS-T for 1 h,
and proteins were detected using Amersham ECL Prime Western
Blotting Detection Reagent (Cytiva). Protein levels were analyzed
using an Amersham Imager 600 (Cytiva). Protein band densities were
measured using the ImageJ analysis software (version 1.44; National
Institutes of Health).
Statistical analysis
Data are presented as the mean ± SEM of three
independent samples. Data were compared using one-way analysis of
variance followed by Tukey's post-hoc test. Statistical analyses
were performed using GraphPad Prism 5 software (GraphPad Software
Inc.). *P<0.05 and **P<0.01 were considered to indicate a
statistically significant difference.
Results
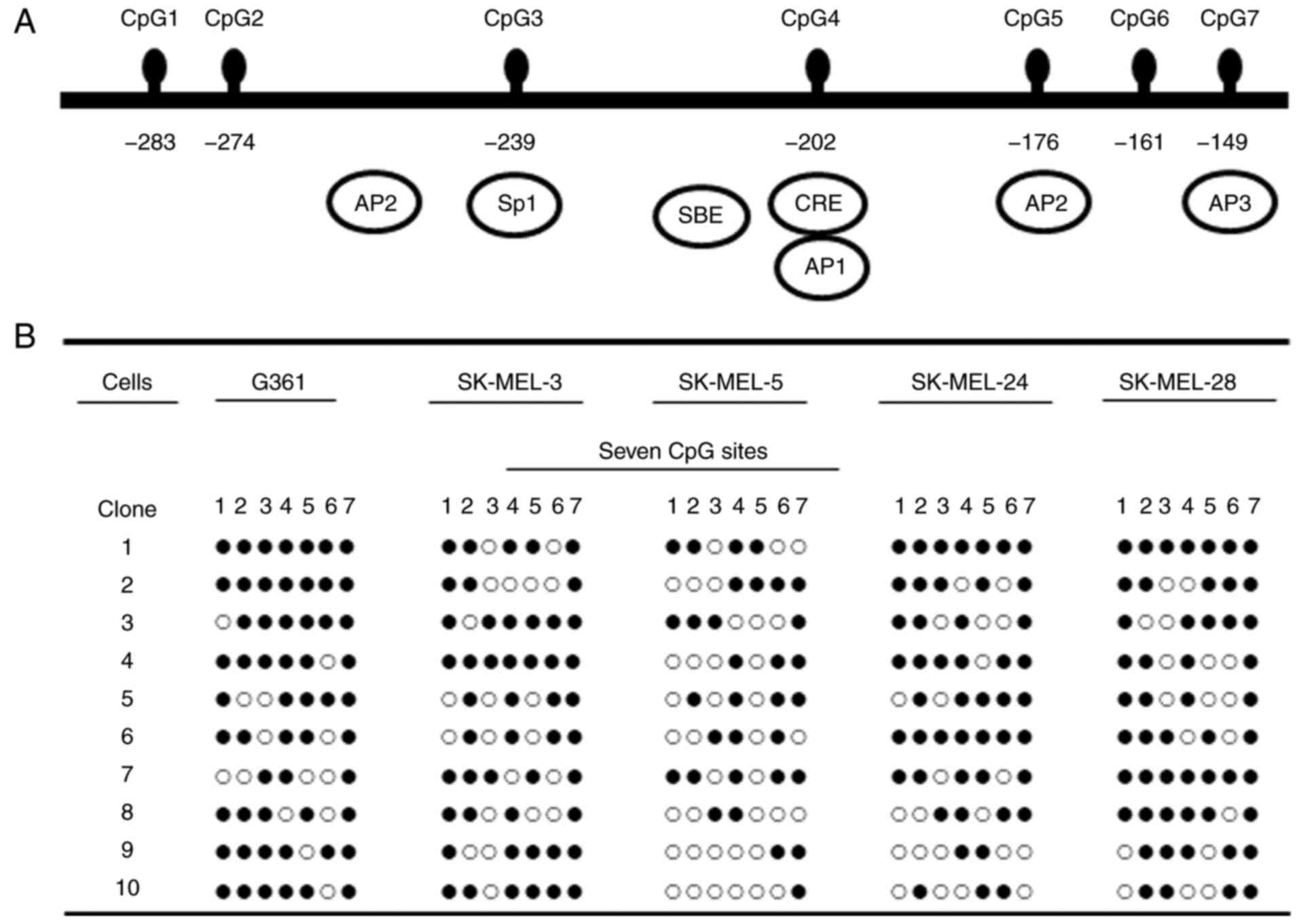
Methylation status of the inhibin-α
gene promoter in human melanoma cells
The methylation state of the inhibin-α gene was
investigated in human melanoma cells. The methylation status of the
CpG sites of the inhibin-α promoter was analyzed via MSP of
bisulfite-treated DNA, and clones were analyzed by sequencing.
Methylation was determined for the seven CpG sites in the 135 bp
regions from −149 to −284 of the ATG site in the human inhibin-α
gene promoter via bisulfite DNA sequencing (Fig. 1A). The inhibin-α promoter was
hypermethylated in G361, SK-MEL-3, SK-MEL-24 and SK-MEL-28 cells
and moderately methylated in SK-MEL-5 cells (Fig. 1B).
Mutations and LOH in the inhibin-α
gene of human melanoma cells
Two polymorphic sites in the inhibin-α gene of human
melanoma cells, namely −16C->T in the 5′-UTR and 769G->A in
exon 2, were assessed for mutations. Genomic DNA was amplified by
PCR. PCR products were digested with restriction enzyme, and
electrophoresed on acrylamide gels, stained with ethidium bromide
and photographed. The PCR product (fragment A) including
nucleotide-16 was digested using SpeI enzyme (Fig. 2A). Polymorphisms were examined
within the 5′-UTR and exon 1 and were used to divide the cell lines
into the following two groups: i) The CC genotype (SK-MEL-3 cells);
and ii) the CT genotype (G361, SK-MEL-5, SK-MEL-24, and SK-MEL-28
cells). Substitution of 769G->A in exon 2 was detected via
digestion by a restriction enzyme. A PCR product comprising
nucleotide 769, fragment B, was digested with BsrFI enzyme,
but was not detected in human melanoma cells (Fig. 2B). Fragment B was digested using
Fnu4HI enzyme, and a single base change at 769G->A of
exon 2 was observed in SK-MEL-3 cells (Fig. 2C). Analysis of the 2q chromosome
arm investigated microsatellite markers on 2q32-33 (D2S389) and
2q33-36 (D2S128). Furthermore, 2q32-33 was revealed to display
allelic imbalance in G361, SK-MEL-3, SK-MEL-24 and SK-MEL-28 cells.
LOH at 2q33-36 was observed in G361 and SK-MEL-3 cells, and allelic
imbalance was identified in SK-MEL-5 cells (Fig. 2D).
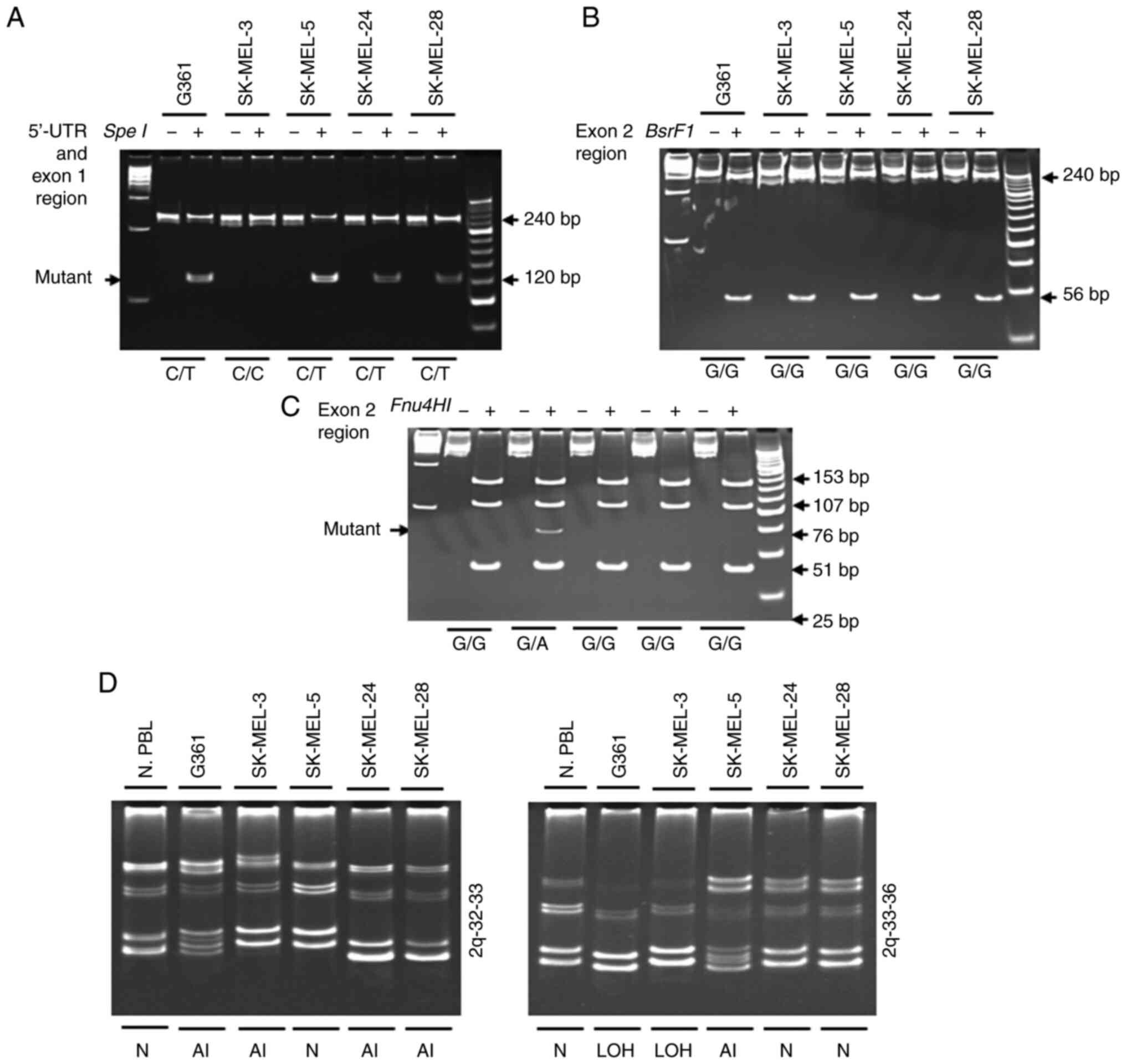 | Figure 2.Analysis of the −16C>T
polymorphism in the 5′-UTR region and the 769G->A substitution
in exon 2 via restriction enzyme digestion. (A) Fragment A was
digested using SpeI. The presence of a 240-bp fragment
indicated a variant homozygous for C, whereas the presence of two
fragments of 120 bp each corresponded to a variant homozygous for
T. (B) Fragment B was digested using BsrFI. Digestion of the
396-bp PCR product yielded two fragments of 340 and 56 bp in the
wild-type allele G, whereas the mutated A allele remained
non-cleaved. (C) Fragment B was digested using FnuHI.
Digestion of the 396-bp PCR product yielded four fragments of 153,
107, 51 and 25 bp (among others of lower molecular weight) in the
wild-type allele, whereas the allele with the 769G->A
substitution yielded four fragments of 153, 107, 76 and 51 bp
(among others of lower molecular weight). PCR products were
incubated with (+) or without (−) restriction enzymes. (D) LOH
analysis of 2q (2q32-33 and 2q33-36) in human melanoma cells.
Genomic DNA was amplified using PCR for the analysis of the 2q
chromosomal region. N, normal; LOH, loss of heterozygosity; AI,
allelic imbalance; N. PBL; normal peripheral blood leucocytes; UTR,
untranslated region. |
Gene mutations in human melanoma
cells
The BRAF gene was mutated in G361, SK-MEL-28
(V599E), SK-MEL-3, SK-MEL-5 and SK-MEL-24 (V600E) cells. The p53
gene was mutated in SK-MEL-3 (exon 8) and SK-MEL-28 (exon 5) cells;
however, the wild-type gene was identified in G361, SK-MEL-5 and
SK-MEL-24 cells. The N-ras gene was not mutated in any melanoma
cells (Table II).
| Table II.Gene mutations in human melanoma
cells. |
Table II.
Gene mutations in human melanoma
cells.
|
| Gene mutations |
|---|
|
|
|
|---|
|
| Inhibin-α | BRAF | PTEN | p53 | N-ras | (Refs.) |
|---|
| Human melanoma
cells | Exon 1 | Exon 2 |
|
|
|
|
|
|---|
| G361 | Mutant | Wild-type | V599E | Wild-type | Wild-type | Wild-type | (53,54) |
| SK-MEL-3 | Wild | Mutant | V600E | Wild-type | R267W | Wild-type | (55) |
| SK-MEL-5 | Mutant | Wild-type | V600E | Not known | Wild-type | Wild-type | (55) |
| SK-MEL-24 | Mutant | Wild-type | V600E | del | Wild-type | Wild-type | (56) |
| SK-MEL-28 | Mutant | Wild-type | V599E | A499G | L145R | Wild-type | (53,54) |
Effects of 5-Aza-dC on the cell growth
suppression and doubling time of human melanoma cells
Human melanoma cells were treated with various
concentrations of 5-Aza-dC for 5 days. Cell viability was
determined using an automatic counter. 5-Aza-dC notably inhibited
the proliferation of melanoma cells in a dose-dependent manner,
compared with that of the control. The doubling time of human
melanoma cells was increased by 1.07-1.75-fold (Table III).
| Table III.Effects of 5-Aza-dC on cell
proliferation in human melanoma cells. |
Table III.
Effects of 5-Aza-dC on cell
proliferation in human melanoma cells.
|
|
| 5-Aza-dC
treatment |
|
|---|
|
|
|
|
|
|---|
|
|
| Viability, % | Doubling time,
h |
|
|---|
|
|
|
|
|
|
|---|
| Human melanoma cell
lines |
IC50 | 0 µM | 0.1 µM | 0.5 µM | 2 µM | 5 µM | 10 µM | 0 µM | 5 µM | Fold growth
suppression |
|---|
| G361 | 0.60 | 100±0.0 | 92.4±3.1 | 75.0±3.0 | 59.1±2.6 | 54.7±2.3 | 47.9±2.9 | 22.4 | 34.4 | 1.54 |
| SK-MEL-3 | 0.78 | 100±0.0 | 94.2±2.0 | 78.7±3.8 | 66.9±2.4 | 58.0±3.0 | 54.0±4.1 | 68.0 | 72.7 | 1.07 |
| SK-MEL-5 | 0.24 | 100±0.0 | 84.0±2.7 | 53.0±2.2 | 42.6±2.3 | 33.4±1.9 | 29.5±1.3 | 37.9 | 41.6 | 1.10 |
| SK-MEL-24 | 0.19 | 100±0.0 | 84.2±1.6 | 48.1±2.8 | 33.9±4.7 | 30.3±3.0 | 28.7±4.1 | 28.8 | 50.5 | 1.75 |
| SK-MEL-28 | 0.17 | 100±0.0 | 83.4±3.8 | 46.0±3.2 | 29.0±2.3 | 23.2±2.1 | 23.1±1.5 | 30.7 | 53.0 | 1.73 |
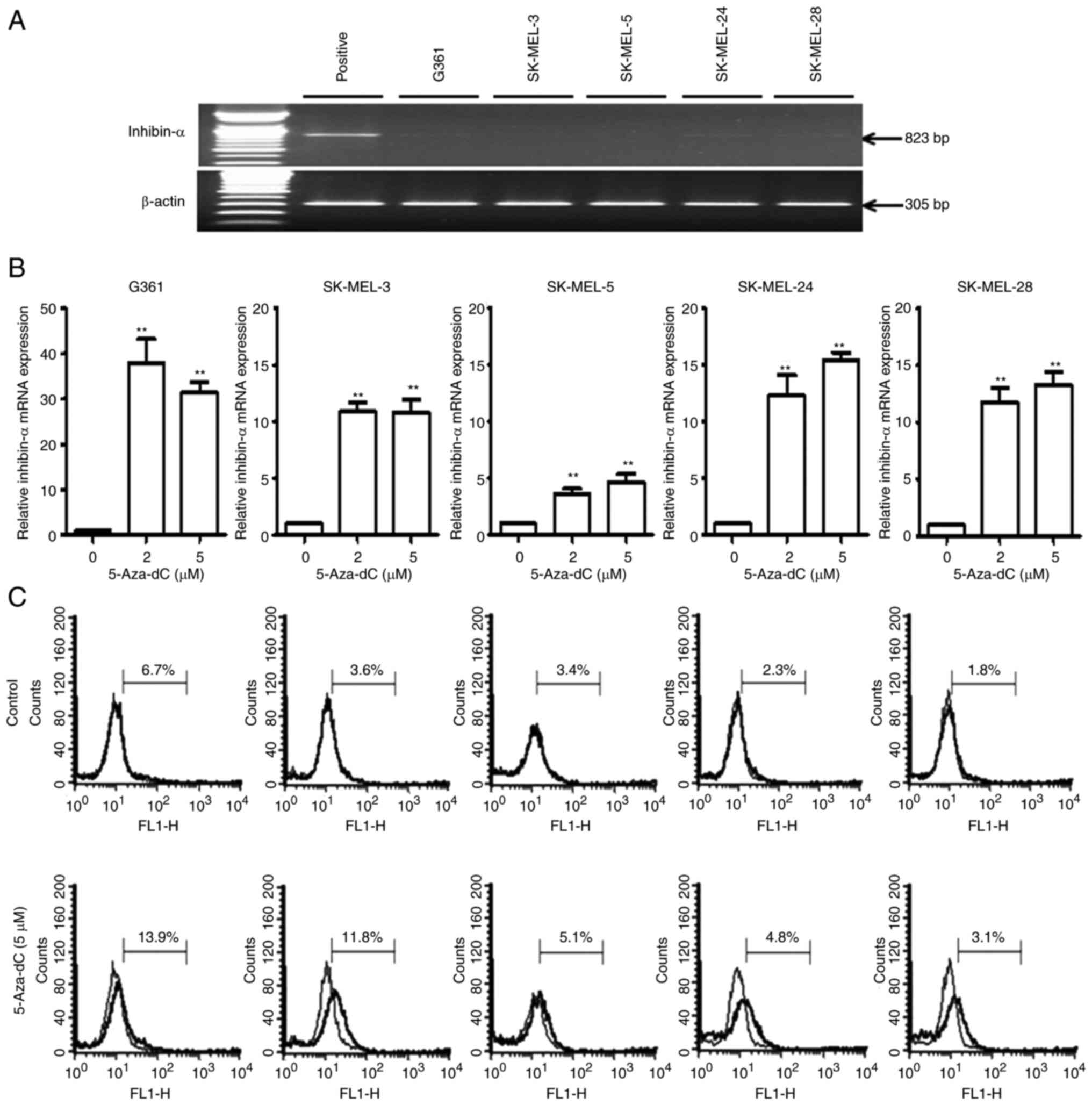
Effect of 5-Aza-dC treatment on
inhibin-α mRNA and protein levels in human melanoma cells
RT-qPCR analysis revealed low or undetectable mRNA
expression levels of inhibin-α in all melanoma cells (Fig. 3A). To evaluate the role of
methylation in the inactivation of the inhibin-α gene promoter in
human melanoma cells, a DNA methyltransferase inhibitor, 5-Aza-dC,
was used. Human melanoma cells were treated with 2 and 5 µM of
5-Aza-dC, and inhibin-α mRNA and protein levels were evaluated
using RT-qPCR and flow cytometric analysis with FITC staining,
respectively. 5-Aza-dC increased the mRNA levels of inhibin-α by
3.6-37.9-fold (Fig. 3B) and its
protein levels by 1.5-3.3-fold (Fig.
3C).
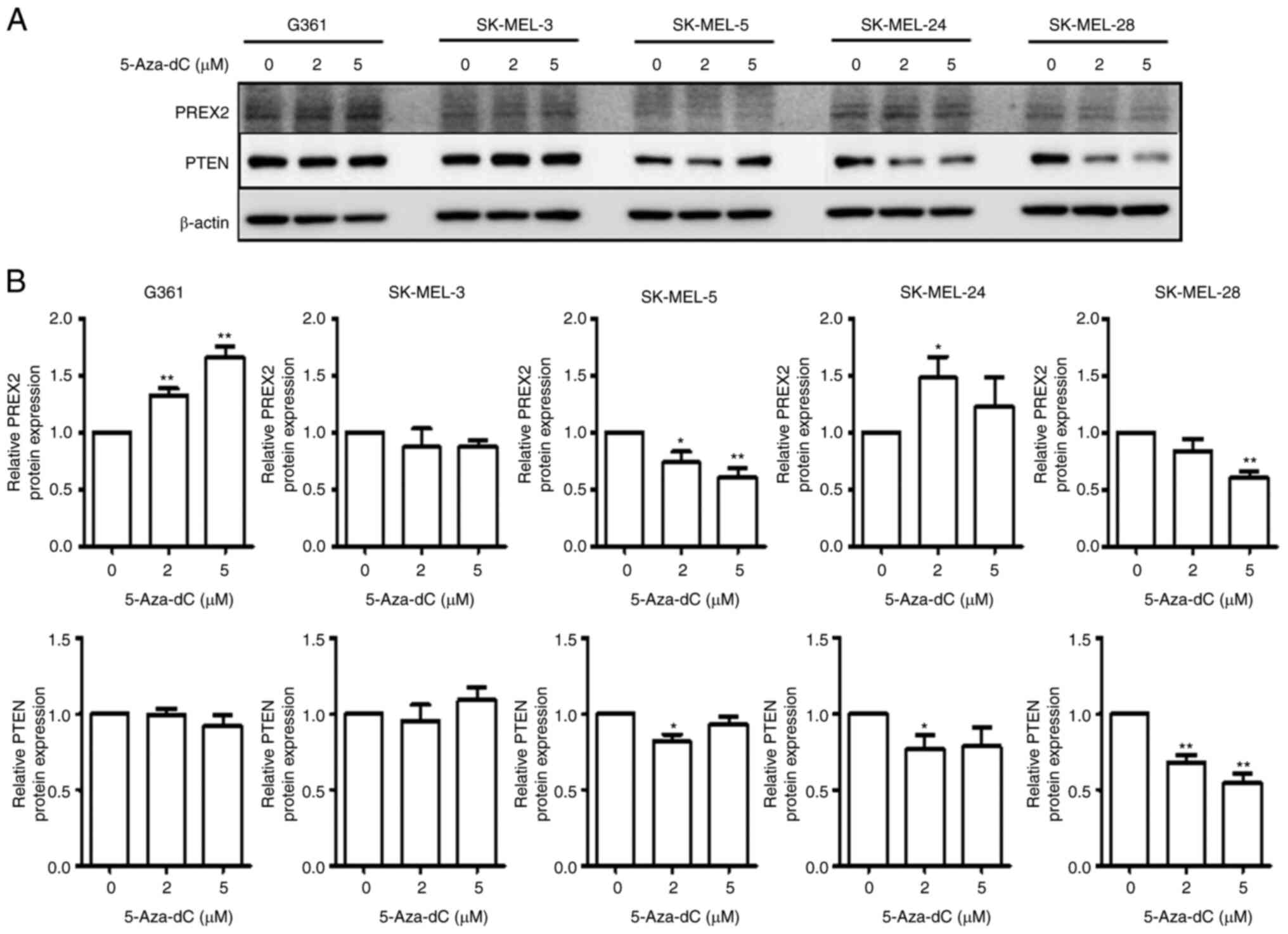
Effect of 5-Aza-dC treatment on PREX2
and PTEN expression in human melanoma cells
After treatment with 5-Aza-dC, protein expression
was determined using immunoblot analysis. PREX2 protein expression
increased in G361 and SK-MEL-24 cells, but decreased in SK-MEL-3,
SK-MEL-5 and SK-MEL-28 cells. PTEN protein expression was mostly
decreased in melanoma cells (Fig. 4A
and B).
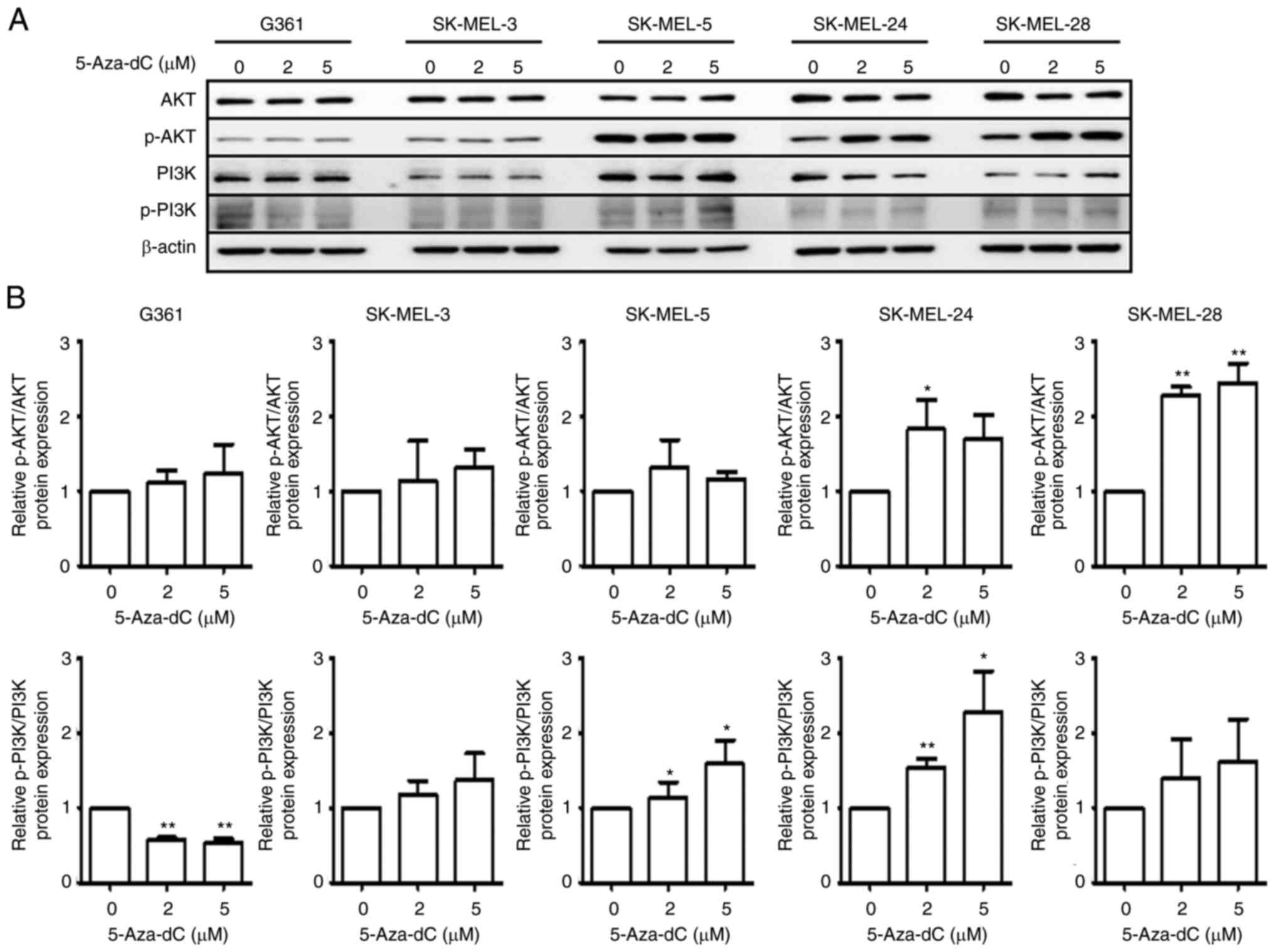
Effect of 5-Aza-dC treatment on AKT
and PI3K phosphorylation levels in human melanoma cells
The protein expression of AKT and PI3K in
5-Aza-dC-treated cells was evaluated via immunoblot analysis. AKT
phosphorylation was increased in all melanoma cells, and PI3K
phosphorylation was enhanced in melanoma cells except for G361
cells (Fig. 5A and B).
Discussion
Epigenetic changes, represented by promoter CpG
island hypermethylation and histone alterations, are important
carcinogenic mechanisms observed in almost all types of cancer
(13,14). A total of ~60–70% of human genes
have CpG islands in their promoters, and certain of these genes are
hypermethylated to block the expression of the corresponding genes
(29). Accordingly, the tumor
suppressor function is lost, thereby promoting the growth of tumor
cells. In the majority of cancers, in addition to local changes,
such as promoter CpG island hypermethylation, genome-wide
demethylation is often simultaneously observed. Such genomic
hypomethylation is closely related to chromosomal instability
(30,31). Melanoma was analyzed in a tumor
model to investigate the association between DNA methylation of the
MMP-9 gene and its overexpression at the transcription and protein
levels, and hypermethylation was observed in the CpG-2 region of
the MMP-9 gene (32). Aberrant DNA
methylation is important for epigenomic regulation in melanoma
formation and progression (33).
Research on DNA methylation has advanced its potential use as a
diagnostic, prognostic and therapeutic biomarker for cancer
(34). In the present study, the
degree of methylation was assessed in seven CpG sites in the
inhibin-α gene promoter of human melanoma cells. A total of seven
CpG sites in the 135 bp region in the inhibin-α gene promoter
revealed significant hypermethylation (G361, SK-MEL-3, SK-MEL-24
and SK-MEL-28 cells) or moderate methylation (SK-MEL-5 cells) in
human melanoma cells. Previously, primary prostate carcinoma has
been revealed to present hypermethylation at the CpG1-4 and CpG7
sites, compared with non-malignant cells. The CpG6 site exhibited
lower methylation levels, whereas the CpG5 site was unmethylated in
non-malignant and malignant samples (26). The findings of the present study
suggested that methylation of the CpG sites occurred in human
melanoma cells and may be related to the cause of transcriptional
silencing.
Mutations are frequently involved in the
transcriptional silencing of cancer suppressor genes (35). A mutation at the −16-bp site of the
5′-UTR was found to be heterozygous in melanoma (G361, SK-MEL-5,
SK-MEL-24 and SK-MEL-28) cells, and a mutation at 769G->A in
exon 2 was identified in SK-MEL-3 cells, suggesting that human
melanoma cells may be mostly affected by the −16>T allele
variant. The present study revealed the occurrence of LOH in the 2q
chromosome arm with one microsatellite marker at 2q32-36 in human
melanoma cells. The 2q32-33 region displayed allelic imbalance in
G361, SK-MEL-3, SK-MEL-24 and SK-MEL-28 cells. LOH at 2q33-36 was
observed in G361 and SK-MEL-3 cells, and allelic imbalance was
revealed in SK-MEL-5 cells; there were no cells with LOH both at
2q32-33 and 2q33-36. Chromosome 2q has been indicated to exhibit
changes in prostate carcinoma and pediatric adrenocortical tumors
(36–38). In addition, a 2q deletion has been
revealed to indicate poor prognosis in the advanced stage of
bladder carcinoma and head-and-neck squamous cell carcinoma
(39,40). Taken together, the results of the
present study indicated that hypermethylation and mutations in the
inhibin-α gene and changes in its LOH may be associated with
transcriptional silencing in human melanoma cells.
In the experiments of the present study, after
treatment with the demethylating agent 5-Aza-dC, it was determined
whether inhibin-α gene expression was reactivated. The mRNA and
protein expression of inhibin-α in melanoma cells was increased
after 5-Aza-dC treatment. Furthermore, an association was observed
between inhibin-α mRNA and protein levels in human melanoma cells.
In human cancer cells, inhibin-α mRNA expression has been
demonstrated to be reactivated following treatment with 5-Aza-dC
(21–23). Collectively, the results of the
present study revealed that inhibin-α gene expression was increased
by 5-Aza-dC in a dose-dependent manner. Interestingly, cells with a
hypermethylated inhibin-α promoter exhibited a high mRNA and
protein expression following 5-Aza-dC treatment, whereas cells that
were moderately methylated exhibited low 5-Aza-dC gene expression.
Among the examined melanoma cells, G361 and SK-MEL-3 cells, which
exhibited LOH, presented higher mRNA and protein expression levels
of inhibin-α than SK-MEL-5, SK-MEL-24 and SK-MEL-28 cells. This
finding suggests that LOH is involved in demethylation.
Treatment with 5-Aza-dC decreased cell proliferation
in a dose-dependent manner and substantially delayed the doubling
time of surviving melanoma cells. These results suggested that
inhibin-α has an important cellular function. Additionally,
5-Aza-dC affects the expression levels of several genes involved in
cell cycle regulation, apoptosis and survival in hepatocellular
carcinoma and leukemia cells (41,42).
PREX2 binds to PTEN through the guanine nucleotide
exchange factor domain, thereby inhibiting PTEN activity. When PTEN
activity is inhibited, PREX2 activates the downstream PI3K
signaling pathway (43,44). PREX2 regulates pancreatic cancer
cell proliferation, invasion and migration, presumably at least
through modulation of the activity of PTEN and the PI3K signaling
pathway (45). PTEN is a tumor
suppressor gene that is frequently mutated or deleted. PTEN has
been identified to inhibit cell proliferation and promote apoptosis
in numerous cancer cell types, including breast cancer cells and
HepG2 cells (46,47). Downregulation, inhibition or
inactivation of PTEN increases mitochondrial ATP production. In
addition, PTEN induces activation of the proteolytic cell apoptosis
cascade by inhibiting the AKT/PI3K signaling pathway (48,49).
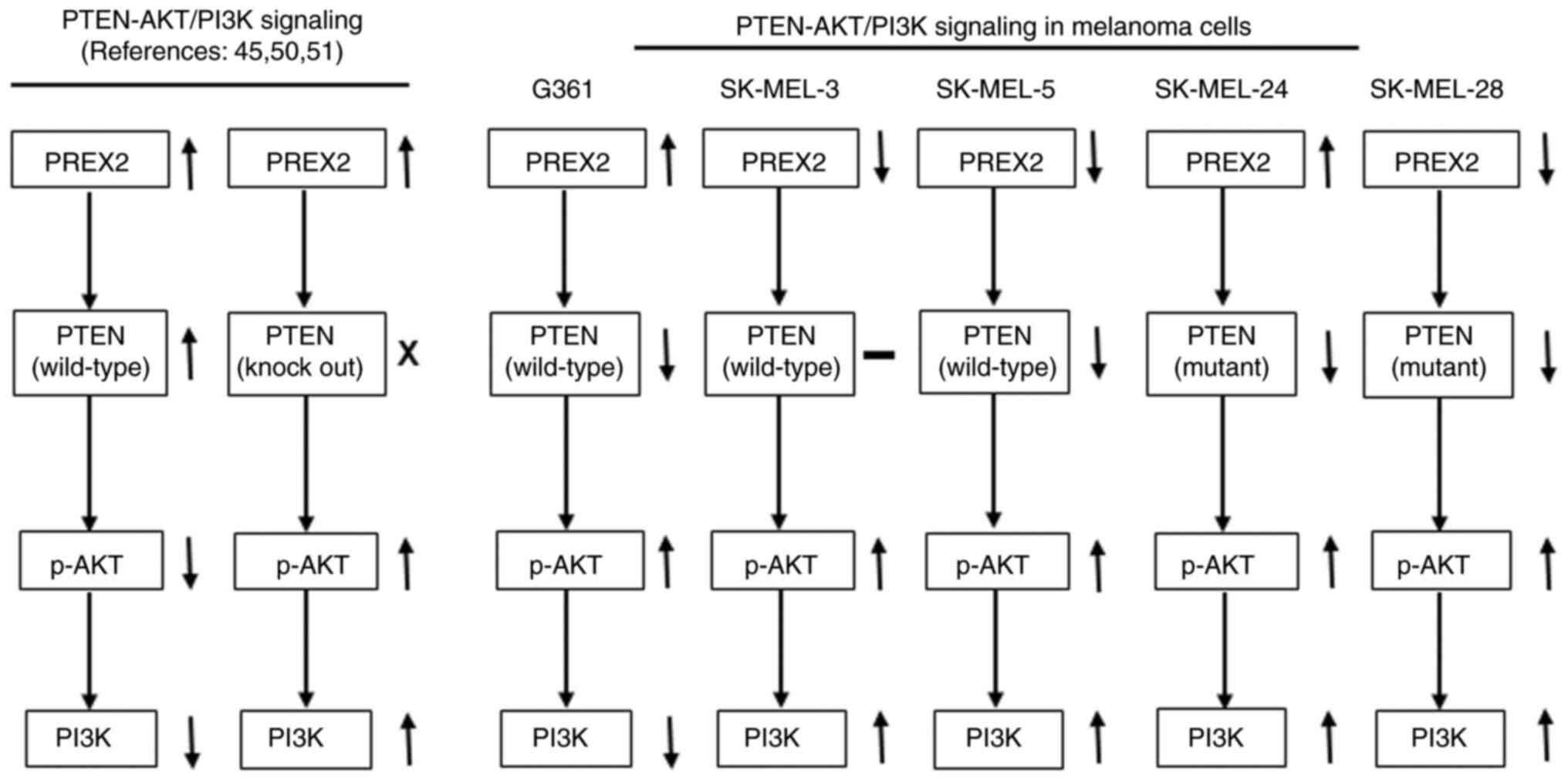
In the present study, PREX2 protein expression
increased in G361 and SK-MEL-24 cells and decreased in SK-MEL-3,
SK-MEL-5 and SK-MEL-28 cells after 5-Aza-dC treatment. PTEN protein
expression mostly decreased in all melanoma cells. However, AKT and
PI3K phosphorylation increased in most melanoma cells. These
findings suggested that the AKT/PI3K signaling pathway was possibly
affected by the PTEN expression pattern. Additionally, PREX2
overexpression has been indicated to decrease PTEN activity and
increase AKT phosphorylation and PI3K signaling pathway activity.
By contrast, knockdown of PREX2 enhanced PTEN activity and
inhibited PI3K signaling pathway activity (45,50).
A previous study indicated that wild-type mouse embryonic
fibroblasts exhibited increased PTEN expression and decreased AKT
phosphorylation. When PTEN was knocked out, PREX2 expression was
slightly increased, PTEN was not expressed, and AKT phosphorylation
was increased. However, when PREX2 was knocked out, PREX2 was not
expressed, and AKT phosphorylation was decreased (51). PTEN is methylated by protein
arginine N-methyltransferase 6 in the cytoplasm, and methylated
PTEN can migrate from the cytoplasm to the nucleus, suggesting that
it is difficult for PTEN to inhibit AKT/PI3K signaling (52). In the present study, the expression
of PTEN and that of genes involved in AKT/PI3K signaling were
compared following treatment with a demethylating agent. Based on
the findings of the present study, it remains difficult to
establish a direct association between inhibin-α methylation and
PTEN expression as well as AKT/PI3K signaling.
In conclusion, the present study revealed the
methylation, mutation and LOH of the inhibin-α gene in human
melanoma cells. Inhibin-α gene expression was reactivated by a
demethylating agent, and cell proliferation was inhibited. However,
PREX2 expression either weakly increased or decreased, whereas
AKT/PI3K signaling increased potentially owing to the decreased
PTEN expression (Fig. 6). In
addition, mutations in the tumor suppressor genes inhibin-α, PTEN
and p53 were not associated with transcriptional silencing, gene
expression and cell growth. Overall, mutations in the inhibin-α and
BRAF genes appear to be frequent in melanoma cells. Therefore,
further studies are required to determine the importance of
biomarkers for the tumor suppressor gene inhibin-α.
Acknowledgements
Not applicable.
Funding
The present study was supported by Basic Science Research
Program through the National Research Foundation of Korea (NRF)
funded by the Ministry of Education (grant no.
NRF-2017R1D1A1B03027993).
Availability of data and materials
The datasets used and/or analyzed in the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HK and YIK performed the experiments. HK, HJA and
YIK analyzed the data and wrote the manuscript. HJA and YIK
contributed to the study concept and design of the project. HJA and
YIK confirm the authenticity of all the raw data. All authors
reviewed the results. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Vale W, Rivier J, Vaughan J, McClintock R,
Corrigan A, Woo W, Karr D and Spiess J: Purification and
characterization of an FSH releasing protein from porcine ovarian
follicular fluid. Nature. 321:776–779. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mathews LS: Activin receptors and cellular
signaling by the receptor serine kinase family. Endocr Rev.
15:310–325. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Namwanje M and Brown CW: Activins and
inhibins: Roles in development, physiology, and disease. Cold
Spring Harb Perspect Biol. 8:a0218812016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mather JP, Moore A and Li RH: Activins,
inhibins, and follistatins: Further thoughts on a growing family of
regulators. Proc Soc Exp Biol Med. 215:209–222. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Matzuk MM, Finegold MJ, Su JG, Hsueh AJ
and Bradley A: Alpha-inhibin is a tumour-suppressor gene with
gonadal specificity in mice. Nature. 360:313–319. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kumar TR, Donehower LA, Bradley A and
Matzuk MM: Transgenic mouse models for tumour-suppressor genes. J
Intern Med. 238:233–238. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mellor SL, Richards MG, Pedersen JS,
Robertson DM and Risbridger GP: Loss of the expression and
localization of inhibin alpha-subunit in high grade prostate
cancer. J Clin Endocrinol Metab. 83:969–975. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Curradi M, Izzo A, Badaracco G and
Landsberger N: Molecular mechanisms of gene silencing mediated by
DNA methylation. Mol Cell Biol. 22:3157–3173. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Esteller M: CpG island hypermethylation
and tumor suppressor genes: A booming present, a brighter future.
Oncogene. 21:5427–5440. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bird AP: CpG-rich islands and the function
of DNA methylation. Nature. 321:209–213. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bird A: The essentials of DNA methylation.
Cell. 70:5–8. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Teodoridis JM, Strathdee G and Brown R:
Epigenetic silencing mediated by CpG island methylation: Potential
as a therapeutic target and as a biomarker. Drug Resist Updat.
7:267–278. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhao Z and Shilatifard A: Epigenetic
modifications of histones in cancer. Genome Biol. 20:2452019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Filipski K, Scherer M, Zeiner KN, Bucher
A, Kleemann J, Jurmeister P, Hartung TI, Meissner M, Plate KH,
Fenton TR, et al: DNA methylation-based prediction of response to
immune checkpoint inhibition in metastatic melanoma. J Immunother
Cancer. 9:e0022262021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lutsik P, Slawski M, Gasparoni G, Vedeneev
N, Hein M and Walter J: MeDeCom: Discovery and quantification of
latent components of heterogeneous methylomes. Genome Biol.
18:552017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Perrier A, Didelot A, Laurent-Puig P,
Blons H and Garinet S: Epigenetic mechanisms of resistance to
immune checkpoint inhibitors. Biomolecules. 10:10612020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Emran AA, Chatterjee A, Rodger EJ, Tiffen
JC, Gallagher SJ, Eccles MR and Hersey P: Targeting DNA Methylation
and EZH2 activity to overcome melanoma resistance to immunotherapy.
Trends Immunol. 40:328–344. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Leonardi GC, Falzone L, Salemi R, Zanghì
A, Spandidos DA, McCubrey JA, Candido S and Libra M: Cutaneous
melanoma: From pathogenesis to therapy (Review). Int J Oncol.
52:1071–1080. 2018.PubMed/NCBI
|
|
20
|
Leonardi GC, Candido S, Falzone L,
Spandidos DA and Libra M: Cutaneous melanoma and the immunotherapy
revolution (Review). Int J Oncol. 57:609–618. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Balanathan P, Ball EM, Wang H, Harris SE,
Shelling AN and Risbridger GP: Epigenetic regulation of inhibin
alpha-subunit gene in prostate cancer cell lines. J Mol Endocrinol.
32:55–67. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim YI, Shim J, Kim BH, Lee SJ, Lee HK,
Cho C and Cho BN: Transcriptional silencing of the inhibin-α gene
in human gastric carcinoma cells. Int J Oncol. 41:690–700. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim YI, Park SW, Kwon HS, Yang HS, Cho SY,
Kim YJ and Lee HJ: Inhibin-α gene mutations and mRNA levels in
human lymphoid and myeloid leukemia cells. Int J Oncol.
50:1403–1412. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Barton DE, Yang-Feng TL, Mason AJ, Seeburg
PH and Francke U: Mapping of genes for inhibin subunits alpha, beta
A, and-beta B on human and mouse chromosomes and studies of jsd
mice. Genomics. 5:91–99. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Watson RH, Roy WJ Jr, Aavis M, Hitchcock A
and Campbell IG: Loss of heterozygosity at the alpha-inhibin locus
on chromosome 2q is not a feature of human granulosa cell tumors.
Gynecol Oncol. 65:387–390. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schmitt JF, Millar DS, Pedersen JS, Clark
SL, Venter DJ, Frydenberg M, Molloy PL and Risbridger GP:
Hypermethylation of the inhibin alpha-subunit gene in prostate
carcinoma. Mol Endocrinol. 16:213–220. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sundblad V, Chiauzzi VA, Andreone L, Campo
S, Charreau EH and Dain L: Controversial role of inhibin
alpha-subunit gene in the aetiology of premature ovarian failure.
Hum Reprod. 21:1154–1160. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gu H, Smith ZD, Bock C, Boyle P, Gnirke A
and Meissner A: Preparation of reduced representation bisulfite
sequencing libraries for genome-scale DNA methylation profiling.
Nat Protoc. 6:468–481. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kulis M and Esteller M: DNA methylation
and cancer. Adv Genet. 70:27–56. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Moarii M, Boeva V, Vert JP and Reyal F:
Changes in correlation between promoter methylation and gene
expression in cancer. BMC Genomics. 16:8732015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Falzone L, Salemi R, Travali S, Scalisi A,
McCubrey JA, Candido S and Libra M: MMP-9 overexpression is
associated with intragenic hypermethylation of MMP9 gene in
melanoma. Aging (Albany NY). 8:933–944. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wouters J, Vizoso M, Martinez-Cardus A,
Carmona FJ, Govaere O, Laguna T, Joseph J, Dynoodt P, Aura C, Foth
M, et al: Comprehensive DNA methylation study identifies novel
progression-related and prognostic markers for cutaneous melanoma.
BMC Med. 15:1012017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Micevic G, Theodosakis N and Bosenberg M:
Aberrant DNA methylation in melanoma: Biomarker and therapeutic
opportunities. Clin Epigenetics. 9:342017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kazanets A, Shorstova T, Hilmi K, Marques
M and Witcher M: Epigenetic silencing of tumor suppressor genes:
Paradigms, puzzles, and potential. Biochim Biophys Acta.
1865:275–288. 2016.PubMed/NCBI
|
|
36
|
Alers JC, Rochat J, Krijtenburg PJ, Hop
WC, Kranse R, Rosenberg C, Tanke HJ, Schröder FH and van Dekken H:
Identification of genetic markers for prostatic cancer progression.
Lab Invest. 80:931–942. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Suarez BK, Lin J, Burmester JK, Broman KW,
Weber JL, Banerjee TK, Goddard KA, Witte JS, Elston RC and Catalona
WJ: A genome screen of multiplex sibships with prostate cancer. Am
J Hum Genet. 66:933–944. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Longui CA, Lemos-Marini SH, Figueiredo B,
Mendonca BB, Castro M, Liberatore R Jr, Watanabe C, Lancellotti CL,
Rocha MN, Melo MB, et al: Inhibin alpha-subunit (INHA) gene and
locus changes in paediatric adrenocortical tumours from TP53 R337H
mutation heterozygote carriers. J Med Genet. 41:354–359. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ransom DT, Barnett TC, Bot J, de Boer B,
Metcalf C, Davidson JA and Turbett GR: Loss of heterozygosity on
chromosome 2q: Possibly a poor prognostic factor in head and neck
cancer. Head Neck. 20:404–410. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhao J, Richter J, Wagner U, Roth B,
Schraml P, Zellweger T, Ackermann D, Schmid U, Moch H, Mihatsch MJ,
et al: Chromosomal imbalances in noninvasive papillary bladder
neoplasms (pTa). Cancer Res. 59:4658–4661. 1999.PubMed/NCBI
|
|
41
|
Valdez BC, Li Y, Murray D, Corn P,
Champlin RE and Andersson BS: 5-Aza-2′-deoxycytidine sensitizes
busulfan-resistant myeloid leukemia cells by regulating expression
of genes involved in cell cycle checkpoint and apoptosis. Leuk Res.
34:364–372. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sanaei M, Kavoosi F and Ghasemi A:
Investigation of the effect of 5-Aza-2′-deoxycytidine on p15INK4,
p16INK4, p18INK4, and p19INK4 genes expression, cell growth
inhibition, and apoptosis induction in hepatocellular carcinoma
PLC/PRF/5 Cell Line. Adv Biomed Res. 9:332020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rosenfeldt H, Vazquez-Prado J and Gutkind
JS: P-REX2, a novel PI-3-kinase sensitive Rac exchange factor. FEBS
Lett. 572:167–171. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pandiella A and Montero JC: Molecular
pathways: P-Rex in cancer. Clin Cancer Res. 19:4564–4569. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yang J, Gong X, Ouyang L, He W, Xiao R and
Tan L: PREX2 promotes the proliferation, invasion and migration of
pancreatic cancer cells by modulating the PI3K signaling pathway.
Oncol Lett. 12:1139–1143. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yang ZF, Yi JL, Li XR, Xie DX, Liao XF and
Ma X: PTEN induces apoptosis and up-regulates p53 expression in
HepG2 cells. Zhonghua Gan Zang Bing Za Zhi. 12:745–748. 2004.(In
Chinese). PubMed/NCBI
|
|
47
|
Wu J, Gao H, Ge W and He J: Over
expression of PTEN induces apoptosis and prevents cell
proliferation in breast cancer cells. Acta Biochim Pol. 67:515–519.
2020.PubMed/NCBI
|
|
48
|
Liu Y, Cao Y, Sun S, Zhu J, Gao S, Pang J,
Zhu D and Sun Z: Transforming growth factor-beta1 upregulation
triggers pulmonary artery smooth muscle cell proliferation and
apoptosis imbalance in rats with hypoxic pulmonary hypertension via
the PTEN/AKT pathways. Int J Biochem Cell Biol. 77:141–154. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu Y, Yan J, Sun C, Li G, Li S, Zhang L,
Di C, Gan L, Wang Y, Zhou R, et al: Ameliorating mitochondrial
dysfunction restores carbon ion-induced cognitive deficits via
co-activation of NRF2 and PINK1 signaling pathway. Redox Biol.
17:143–157. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
He S, Lin J, Yu S and Sun S: Upregulation
of PREX2 promotes the proliferation and migration of hepatocellular
carcinoma cells via PTEN-AKT signaling. Oncol Lett. 11:2223–2228.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Mense SM, Barrows D, Hodakoski C,
Steinbach N, Schoenfeld D, Su W, Hopkins BD, Su T, Fine B,
Hibshoosh H and Parsons R: PTEN inhibits PREX2-catalyzed activation
of RAC1 to restrain tumor cell invasion. Sci Signal. 8:ra322015.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Feng J, Dang Y, Zhang W, Zhao X, Zhang C,
Hou Z, Jin Y, McNutt MA, Marks AR and Yin Y: PTEN arginine
methylation by PRMT6 suppresses PI3K-AKT signaling and modulates
pre-mRNA splicing. Proc Natl Acad Sci USA. 116:6868–6877. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sasaki Y, Niu C, Makino R, Kudo C, Sun C,
Watanabe H, Matsunaga J, Takahashi K, Tagami H, Aiba S and Horii A:
BRAF point mutations in primary melanoma show different prevalences
by subtype. J Invest Dermatol. 123:177–183. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Aronchik I, Kundu A, Quirit JG and
Firestone GL: The antiproliferative response of indole-3-carbinol
in human melanoma cells is triggered by an interaction with NEDD4-1
and disruption of wild-type PTEN degradation. Mol Cancer Res.
12:1621–1634. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Pap M, Bátor J and Szeberényi J:
Sensitivity of human malignant melanoma cell lines to newcastle
disease virus. Anticancer Res. 35:5401–5406. 2015.PubMed/NCBI
|
|
56
|
Yamashita T, Tokino T, Tonoki H, Moriuchi
T, Jin HY, Omori F and Jimbow K: Induction of apoptosis in melanoma
cell lines by p53 and its related proteins. J Invest Dermatol.
117:914–919. 2001. View Article : Google Scholar : PubMed/NCBI
|