Introduction
The p53 tumor suppressor plays critical roles in
preventing malignant transformation. Under stressful conditions
such as genotoxic stress, p53 is activated by multiple
post-translational modifications including phosphorylation and
acetylation (1,2). Activated p53 stimulates transcription
of a variety of downstream genes leading to cell growth arrest, DNA
repair, or apoptosis (1,2).
Triple negative breast cancer [TNBC; negative for
estrogen receptor (ER), progesterone receptor, and HER2/neu]
accounts for 15–20% of all breast cancers and is associated with
the most aggressive clinical outcomes among different subtypes of
breast cancer (3). Thus, there is
an urgent need to find effective targeted therapeutic agents for
treating advanced TNBC. Although TNBC only represents a fraction of
all breast cancers, the majority of the p53 mutations in breast
cancer occur in TNBC (3). These
mutated p53 proteins affect different cellular functions, resulting
in loss of cell cycle control, impairment of pro-apoptotic
signaling pathways and defective cellular responses to DNA damage
(4,5).
As p53 mutations are important in tumor
aggressiveness and poor prognosis, restoring wild-type function of
mutant p53 has been an intensive area of research (4,5).
However, numerous of these attempts remain either in the early
stage of development or have failed in clinical trials (2). It is thus essential to develop novel
compounds that are capable of restoring function of p53 in cancers,
including TNBC and other subtypes of breast cancer.
It is known that histone deacetylase 6 (HDAC6)
inhibits p53 activity and p53-mediated cellular events through
deacetylation of p53 (6). In
addition, in breast cancers, HDAC6 overexpression is also
correlated with increased mobility, migration and invasiveness of
cancer cells (7). Therefore, it
was hypothesized that inhibition of HDAC6 by its new inhibitors
such as ACY-1215 (8) may restore
p53 function in cancer cells.
In addition to mutated p53, the PI3K/Akt pathway is
also dysregulated in the majority of TNBC, and it is known that
these dysregulations cause over-activation of Akt, which leads to
the development of cancer (9). Our
previous studies have shown that a protein deficient in kinase
ataxia telangiectasia's disease, ataxia-telangiectasia, mutated
(ATM), is a stimulator of Akt (10,11).
It was also revealed that a specific inhibitor of the ATM protein
kinase, known as KU-55933, is capable of inhibiting Akt activity in
TNBC and other types of breast cancer cells (9). In addition, our results indicated
that KU-55933 suppressed proliferation of these breast cancer cells
by inducing apoptosis (9).
In the present brief study, the effect of ACY-1215
on p53-mediated cellular events in both MCF-7, a non-TNBC cell line
(Luminal A and ER+) with wild-type p53, and MDA-MB-231,
a TNBC cell line with mutated p53 was first examined. Subsequently,
a novel approach was examined to treat MDA-MB-231 and MCF-7 cells
by combining ACY-1215 with KU-55933 or KU-60019, an analog of
KU-55933. The results were also confirmed in vivo in a mouse
model of TNBC. Our results validated ACY-1215, either as
monotherapy or in combination with KU-55933 or KU-60019, as a
promising new therapeutic agent for TNBC.
Materials and methods
Chemicals and reagents
Antibodies against poly ADP-ribose polymerase
(PARP), p21, acetylated p53 at Lys-382 were obtained from Cell
Signaling Technology, Inc. The anti-β-actin antibody was purchased
from Sigma-Aldrich; Merck KGaA. KU-55933 and KU-60019 were
purchased from Merck KGaA. ACY-1215 was purchased from APeXBIO
Technology LLC.
Cell culture and protein
assessment
MDA-MB-231 (HTB-26) and MCF-7 (HTB-22) cells were
purchased from American Type Culture Collection (ATCC) and were
cultured at 37°C in either RPMI-1640 medium (product no. 10-040-CV)
or DMEM (product no. 10-013-CV; both from Corning, Inc.) medium
supplemented with antibiotics (penicillin and streptomycin; 1:100
dilution; Gibco; Thermo Fisher Scientific, Inc.) and 10% fetal
bovine serum (FBS; cat. no. 26140-079; Gibco; Thermo Fisher
Scientific, Inc.), respectively. Cells were lysed with TGN lysis
buffer containing protease inhibitor cocktails (Roche Diagnostics).
The protein concentration was determined by the BCA method.
Immunoblotting detection of
proteins
Equal amounts of protein (50–80 µg) were subjected
to SDS-PAGE (4–12%) and then transferred to a PVDF membrane. After
blocking (5% milk in TBST for 2 h at RT), PVDF membranes were
incubated with the following primary antibodies: Acetyl-p53
(Lys382) (1:1,000; product no. 2525), p21 Waf1/Cip1 (1:1,000;
product no. 2947), PARP (46D11) (1:1,000; product no. 9532),
acetyl-α-tubulin (K40) (D2063) (1:1,000; product no. 5335; all from
Cell Signaling Technology, Inc), β-actin (1:2,000; product no.
A2228), acetyl-p53 (Lys320) (1:200; product no. 06-1283; both from
Sigma-Aldrich; Merck KGaA), HDAC6 (D-11) (1:100; cat. no. sc-28386)
and p53 (DO-1) (1:500; cat. no. sc-126; both from Santa Cruz
Biotechnology, Inc.) at 4°C overnight, and HRP-conjugated secondary
antibody (1:3,000; product code ab97051 or product code ab97023;
Abcam) for 45 min at room temperature. The images were developed
using SuperSignal™ West Pico PLUS chemiluminescent substrate (cat.
no. 34580; Thermo Fisher Scientific, Inc.) following the
manufacturer's protocol and visualized using Amersham Imager 600.
Densitometric analysis was performed with the Amersham Imager 600
Analysis Software, V 1.0.0, from GE/General Electric.
Flow cytometric assay
Cells were cultured to sub-confluence and then
treated (37°C) with 10 µM ACY-1215 for 24 h. Cells were then
harvested and fixed overnight at −20°C with cold 70% ethanol. Cells
(1×106/ml) were then treated with RNase and stained with
propidium iodide at room temperature in the dark for 30 min (Sigma
Aldrich; Merck KGaA). Cell cycle distribution at the G1 and S
phases was analyzed by flow cytometry (FACSCalibur; and CellQuest
software version 6.0f9; both from BD Biosciences), and cell cycle
arrest was presented as G1/S ratios as previously described
(9).
MTT cell viability assay
Cells (1×106/ml) were seeded in 48- or
96-well plates and cultured (37°C) to sub-confluence. Following
serum starvation for 24 h, the cells were then treated with
ACY-1215, KU-55933 and/or KU-60019 (10 µM each) for 72 h at 37°C.
The ratio of viable cells in each well was determined using a
CellTiter Nonradioactive Cell Proliferation Assay kit (cat. no.
G400; Promega Corporation) according to the manufacturer's
protocol. The Solubilization Solution/Stop Mix (included in the
kit) was then added to the culture wells to solubilize the formazan
product, and the absorbance at 570 nm was recorded using a plate
reader.
IncuCyte cell proliferation assay
Cells were seeded at 40,000/well in 48-well plates
in cell culture medium containing 2% FBS. Following the addition of
chemical compounds (ACY-1215, KU-55933 and/or KU-60019; 10 µM
each), the plates were placed in the IncuCyte S3 Live-Cell Analysis
System (Essen BioScience) where real-time images were captured
every 6 h for 72–96 h at a magnification of ×10 for the entire
experiment. The confluence of each group of cells was evaluated and
plotted with the IncuCyte S3 2019A software (Essen BioScience).
Cell migration assay
MDA-MB-231 cells (1×106/ml) were seeded
in a 6-well plate and were allowed to proliferate. Following the
formation of a confluent monolayer (near 100%), the cells were
scratched with a 200-µl sterile tip. Cells (which were
serum-starved) were then washed with PBS and treated with different
chemical compounds (10 µM ACY-1215 ± KU-55933 or KU-60019; KU55
alone; KU60 alone; as well as a control/vehicle (DMSO): Con; ACY,
KU55, KU60, ACY + KU55, ACY + KU60). Images of the extent of cell
migration (0 and 24 h) were captured with an inverted light
microscope.
Cell invasion assay
Cells (1×106/ml) suspended in serum-free
medium were seeded into the upper well of the invasion chamber
(pore size 8 µm; cat. no. 08-774-122; Corning, Inc.). Medium
containing 10% FBS was added to the bottom well of the chamber to
serve as a chemoattractant. Chemical reagents (Con, ACY-1215, KU55
+ ACY-1215, KU60 + ACY-1215) were added to both the upper and
bottom layer of the chamber. Following incubation at 37°C for 24 h,
the cells on the upper surface of the filters were wiped away with
a cotton swab. The filters were fixed with 2% paraformaldehyde
followed by staining with 0.1% crystal violet for 10 min each at
room temperature. The number of invasive cells were quantified
under a light microscope.
Animal study
A total of 30 female athymic
nude-Foxn1nu mice (10–12 weeks old; Envigo) were
injected at the left and/or right flank with 1×106
MDA-MB-231 cells in PBS-Matrigel (BD Biosciences). The housing
conditions were as follows: 68–74°F; 30–70% humidity; and 12-h
light/dark cycle, with full access to food and water. Experiments
were performed in the date range of 3/12/2020 to 7/10/2020. After
the tumors became visible (~40–50 mm3), the mice with
roughly equal tumor burden were divided into four groups. One group
was intraperitoneally injected with vehicle and the other groups
were injected with 30 mg/kg ACY-1215 (8), 1 mg/kg KU-55933 (12), or the combination. Tumor volumes
were calculated according to the following formula: Volume =
(width)2 × length/2. Width and length of the tumors were
measured using an electronic digital caliper with a resolution of
0.01 mm. Tumor volumes and body weight were recorded weekly and
tumor weight and sizes were measured with the diameter of the
largest tumor at 12 mm. Following four weeks of treatment, the mice
were euthanized via a primary method of carbon dioxide euthanasia
(flow rate of carbon dioxide used was ~30% chamber volume
displaced/min), followed by a secondary method of cervical
dislocation. Tumor samples were then removed. All the procedures
were approved (IACUC Protocol 1905-37107A) by the University of
Minnesota IACUC committee (Minneapolis, MN, USA).
Statistical analysis
Comparisons between two groups were evaluated by a
Student's paired t-test and one-way ANOVA followed by Tukey's post
hoc test was used to compare means of multiple groups with GraphPad
Prism v. 9 (GraphPad Software, Inc.). Data are presented as the
mean ± standard error and P<0.05 was considered to indicate a
statistically significant difference.
Results
ACY-1215 causes increased p53
acetylation and induces p21 in breast cancer cells
As aforementioned, HDAC6 inhibits p53 activity and
p53-mediated cellular events through deacetylation of p53 at Lys382
(6), and ACY-1215 is a newly
developed specific HDAC6-inhibitor (8). Therefore, the effect of ACY-1215 was
first examined on p53 acetylation and induction of p21, a
downstream target of p53, in MCF-7 and MDA-MB-231 cells. First,
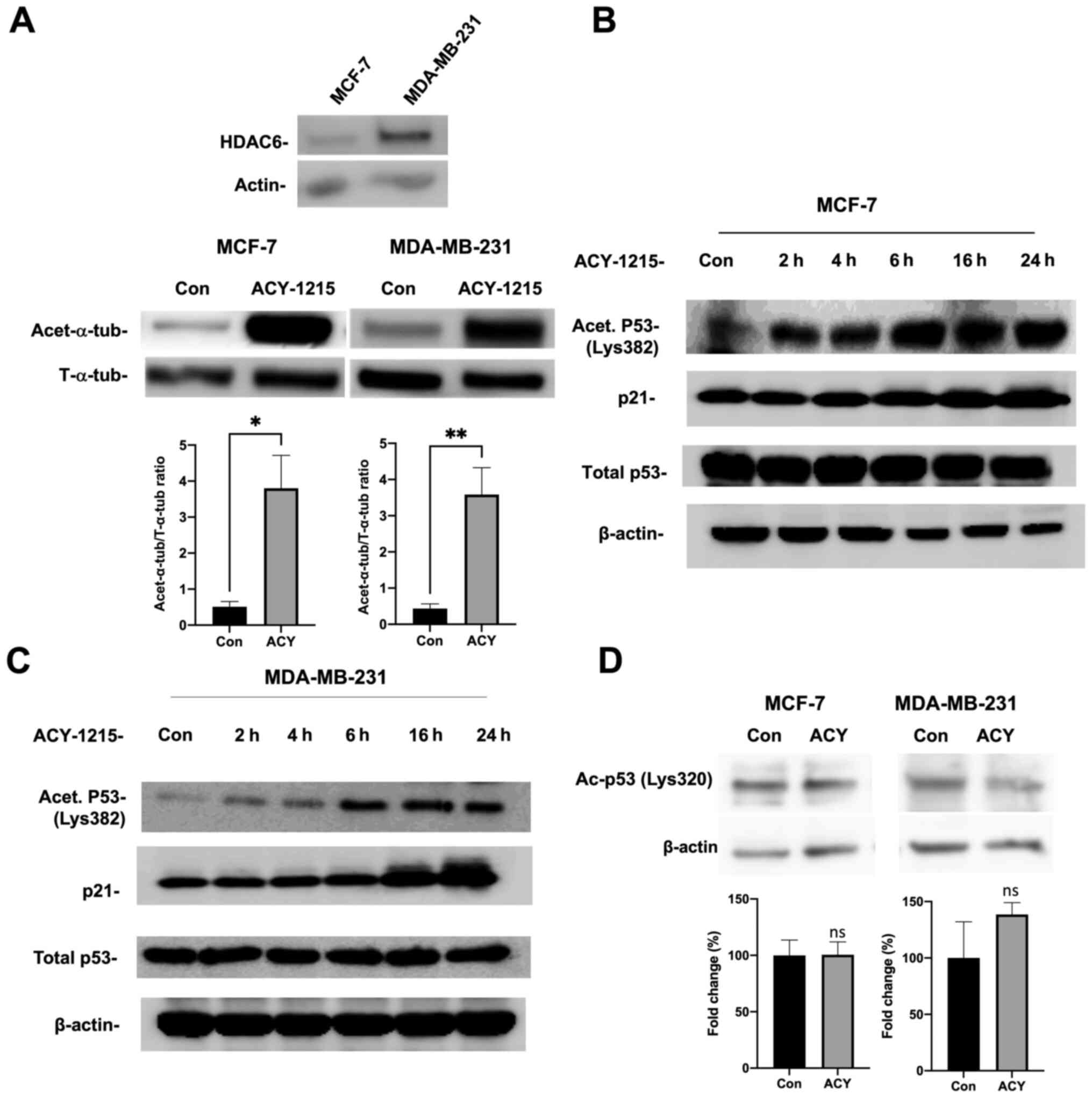
HDAC6 expression in these cell lines was confirmed and ACY-1215
efficacy in the inhibition of HDAC6 was assessed by western
blotting. The well-established substrate, α-tubulin, was identified
as an indirect read out of ACY-1215 inhibiting HDAC6 activity, and
therefore increased acetylated-α-tubulin levels (Fig. 1A). Then, our results showed that
ACY-1215 caused increased acetylation of p53 in MCF-7 cells
(Fig. 1B). Additionally, this
increased acetylation was accompanied by increased expression of
p21, suggesting that ACY-1215 may lead to enhanced transcriptional
activity of p53. Next, MDA-MB-231 cells were treated with ACY-1215
and it was determined that this inhibitor also caused increased
acetylation of p53 (Fig. 1C). To
our surprise, our results showed that this increased acetylation
was also accompanied by increased expression of p21, suggesting
that the inhibition of HDAC6 may lead to enhanced transcriptional
activity of p53 in MDA-MB-231 cells as well. Furthermore, the
effects of ACY-1215 appear to mainly target p53 Lys382, as no
significant differences were observed at Lys320 (Fig. 1D).
ACY-1215 induces apoptosis and G1 cell
cycle arrest in breast cancer cells
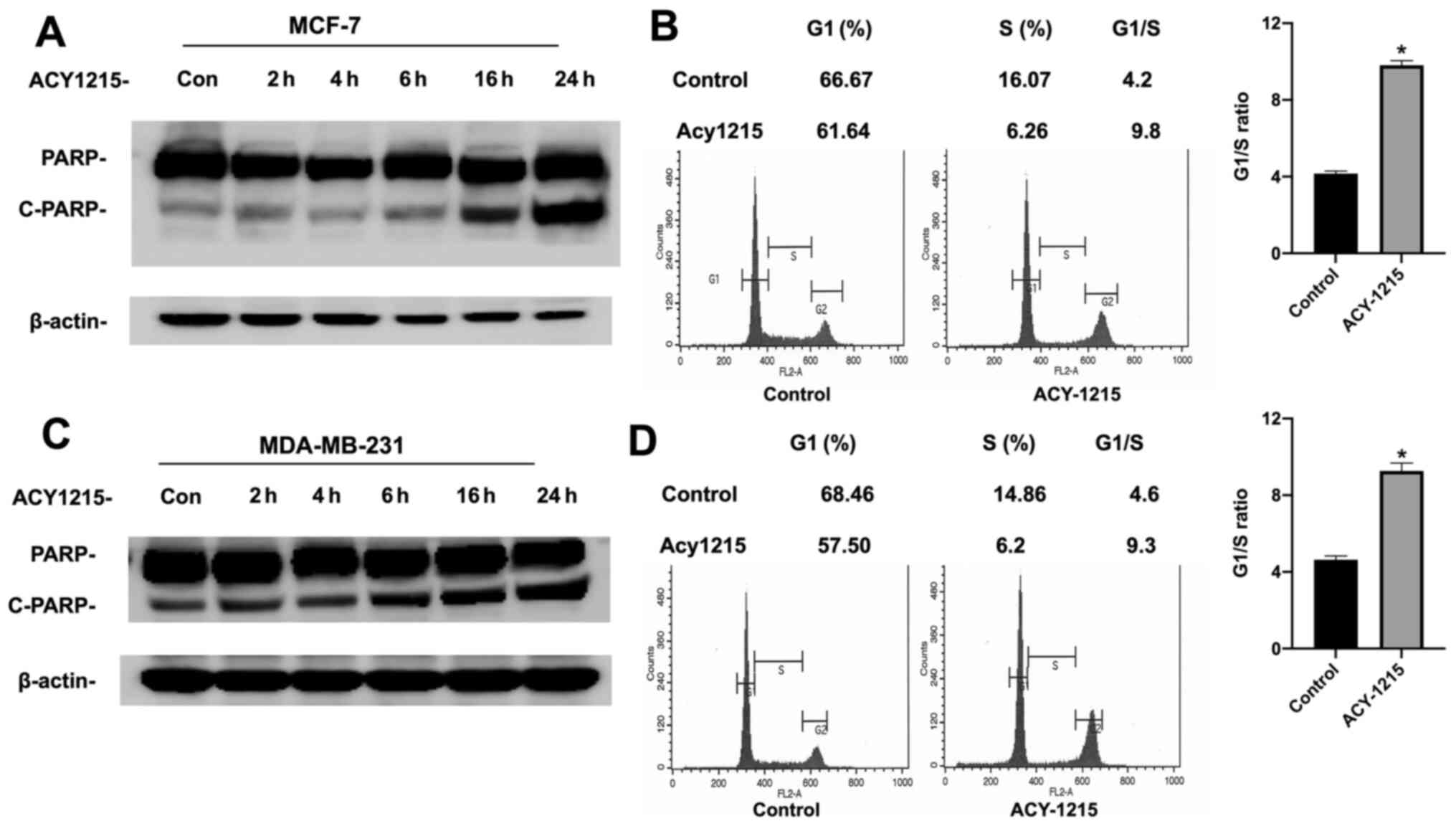
Since p53 controls cellular function by both
inducing cell cycle arrest at the G1 phase and apoptosis in cancer
cell lines, the effect of ACY-1215 on these p53-mediated cellular
events was next examined in both MCF-7 and MDA-MB-231 cells. It was
determined that ACY-1215 treatment resulted in both apoptosis,
revealed by increased cleavage of PARP (Fig. 2A), a substrate of caspase-3, and G1
cell cycle arrest in MCF-7 cells (Fig.
2B). More importantly, it was also observed that ACY-1215
treatment led to increased apoptosis (Fig. 2C) and G1 cell cycle arrest
(Fig. 2D) in MDA-MB-231 cells.
Combination of ACY-1215 and KU-55933
or KU-60019 induces markedly stronger apoptosis and is more
effective in inhibiting MDA-MB-231 cell proliferation
Since the majority of the TNBC cells contain both
p53 mutations and over-activated Akt, and the specific ATM
inhibitors KU-55933 and KU-60019 inhibit Akt activity (13), a novel approach was next examined
to target TNBC cells by combining ACY-1215 with KU-55933 or
KU-60019. It was revealed that while ACY-1215 or KU-55933/KU-60019
as single agent could cause increased cellular apoptosis,
combination of ACY-1215 with KU-55933 or KU-60019 led to markedly
stronger apoptosis in MDA-MB-231 cells (Fig. 3A), suggesting an additive or
synergistic effect of ACY-1215 and KU-55933 or KU-60019 on inducing
apoptosis in MDA-MB-231 cells.
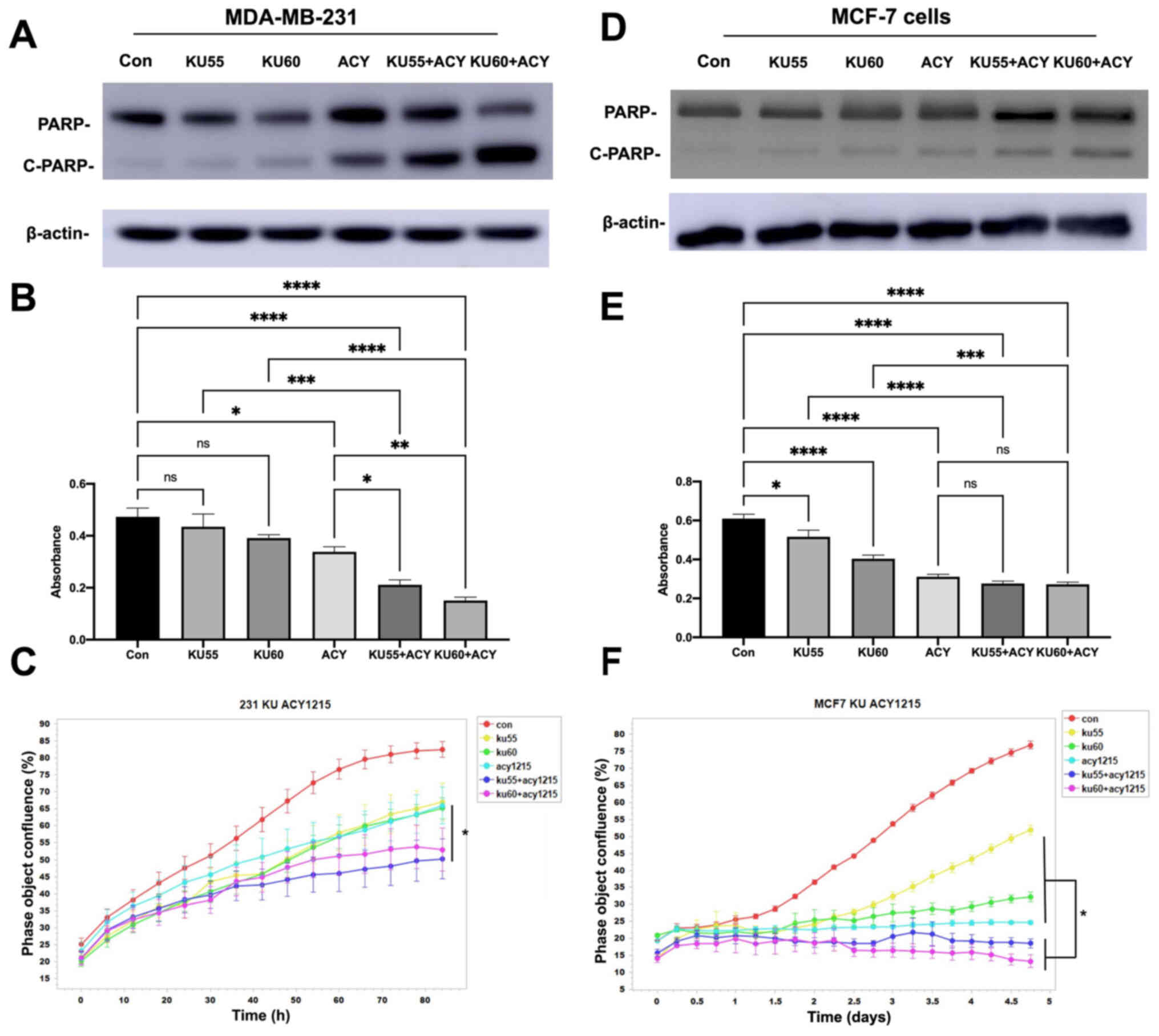 | Figure 3.Combination of ACY-1215 with KU-55933
or KU-60019 has a stronger inhibitory effect on survival and
proliferation than treatment with individual compounds alone. (A)
MDA-MB-231 cells were serum-starved overnight and then treated with
ACY-1215, KU-55933, and/or KU-60019 (10 µM each) for 24 h. Both
floating and attached cells were collected following treatment.
Cells were lysed by TGN lysis buffer, and cell lysates were
subjected to SDS-PAGE. PARP, cleaved PARP, and β-actin were
detected. The results are representative of three individual
experiments. (B) MDA-MB-231 cells were plated on a 48-well plate
and cultured to sub-confluence. Following serum starvation for 24
h, MDA-MB-231 cells were then treated with ACY-1215, KU-55933
and/or KU-60019 (10 µM each) for 72 h and MTT assays were
performed. Results are presented as the absorbance ± SEM (C)
MDA-MB-231 cells were plated on a 48-well plate and cultured in the
presence of 2% FBS and in the absence or presence of ACY-1215,
KU-55933 and/or KU-60019 (10 µM each) for 120 h, and the cell
proliferation was determined by IncuCyte real-time imaging. Data
are expressed as the change in percent confluence from 3–5 repeats.
Treatments are labeled as control (red), KU-55933 (yellow),
KU-60019 (green), ACY-1215 (light blue), KU-55933 + ACY-1215
(blue), and KU-60019 + ACY-1215 (purple). (D) MCF-7 cells were
serum-starved overnight and then treated with ACY-1215, KU-55933,
and/or KU-60019 (10 µM each) for 24 h. Both floating and attached
cells were collected following treatment. Cells were lysed by TGN
lysis buffer, and cell lysates were subjected to SDS-PAGE. PARP,
cleaved PARP, and β-actin were detected. The results are
representative of three individual experiments. (E) MCF-7 cells
were plated on a 48-well plate and cultured to sub-confluence.
Following serum starvation for 24 h, MCF-7 cells were then treated
with ACY-1215, KU-55933 and/or KU-60019 (10 µM each) for 72 h, and
an MTT assay was then performed. Results are presented as the
absorbance ± SEM. (F) MCF-7 cells were plated on a 48-well plate
and cultured in the presence of 2% FBS and in the absence or
presence of ACY-1215, KU-55933 and/or KU-60019 (10 µM each) for 120
h, and the cell proliferation was evaluated by IncuCyte real-time
imaging. Data are expressed as the change in percent confluence
from 3–5 repeats. Treatments are labeled as control (red), KU-55933
(yellow), KU-60019 (green), ACY-1215 (light blue), KU-55933 +
ACY-1215 (blue), and KU-60019+ACY-1215 (purple). *P<0.05,
**P<0.01, ***P<0.001 and ****P<0.0001. |
Next, the combination effect of ACY-1215 with
KU-55933 or KU-60019 on MDA-MB-231 cells proliferation was
determined. First, the effect of ACY-1215 ± KU-55933/KU-60019 on
MDA-MB-231 cells proliferation was examined using MTT assays and a
stronger effect was identified (Fig.
3B). To further examine the effect of these inhibitors, their
effects were tested using the IncuCyte S3 Live-Cell Analysis
System. Following treatment of MDA-MB-231 cells with ACY-1215 and
KU-55933 or KU-60019, the inhibitory effects of either ACY-1215 or
KU-55933 or KU-60019 on proliferation of MDA-MB-231 cells were
again observed, yet the combination of these drugs revealed a
significantly stronger suppressive effect on the proliferation of
MDA-MB-231 cells (Fig. 3C).
Combination of ACY-1215 with KU-55933
or KU-60019 induces markedly stronger apoptosis and inhibits MCF-7
cell proliferation
Next, it was examined whether combination of
ACY-1215 with KU-55933 or KU-60019 could also induce stronger
apoptosis in MCF-7 cells as MCF-7 also display high Akt activity
(9). Similarly, while our results
showed that ACY-1215 or KU-55933/KU-60019 alone could lead to
increased apoptosis or cleavage of PARP, the combination of
ACY-1215 with either KU-55933 or KU-60019 had a markedly stronger
effect on inducing apoptosis in MCF-7 cells (Fig. 3D).
Next, the effect of combination of ACY-1215 with
KU-55933 or KU-60019 on proliferation of MCF-7 cells was also
evaluated using both MTT assay and the IncuCyte S3 Live-Cell
Analysis System. Results from both assays demonstrated that whereas
individual compound including ACY-1215 or KU-55933/KU-60019 could
inhibit MCF-7 cell proliferation, combination of ACY-1215 with
KU-55933 or KU-60019 also had a more potent effect on inhibiting
cell proliferation of MCF-7 cells (Fig. 3E and F).
Noteworthy, the proliferation assays indicated that
MCF-7 cells were slightly more sensitive to the double treatment
compared with MDA-MB-231 cells. Conversely, PARP cleavage was more
prominent in MDA-MB-231 cells than MCF-7 cells. Perhaps, the
different levels of HDAC6 expression (Fig. 1A) may explain the distinct response
of cell proliferation between the two cell lines rendering MCF-7
more sensitive to ACY-1215 than MDA-MB-231. To keep the results
consistent, the cleaved-PARP was evaluated as the indicator of
apoptosis in both MDA-MB-231 and MCF-7 cells. However, MCF-7 is
known to have caspase-3 deficiency and relies on caspase-7 alone
for PARP degradation (14,15). This is likely the reason why MCF-7
exhibited less prominent PARP degradation than MDA-MB-231 for the
double treatment.
Combination of ACY-1215 and KU-55933
or KU-60019 causes markedly stronger inhibition on migration and
invasion of MDA-MB-231 cells
It is known that MDA-MB-231 cells have strong
migration capability in vitro (16). Thus, the effect of combining
ACY-1215 with KU-55933 or KU-60019 on migration and invasion of
MDA-MB-231 cells was examined. Similar to what it was observed in
cell apoptosis and proliferation assays, our results revealed a
markedly stronger inhibitory effect on migration (Fig. 4A) and invasion (Fig. 4B and C) of MDA-MB-231 cells when
both ACY-1215 and KU-55933 or KU-60019 were used to treat the
cells.
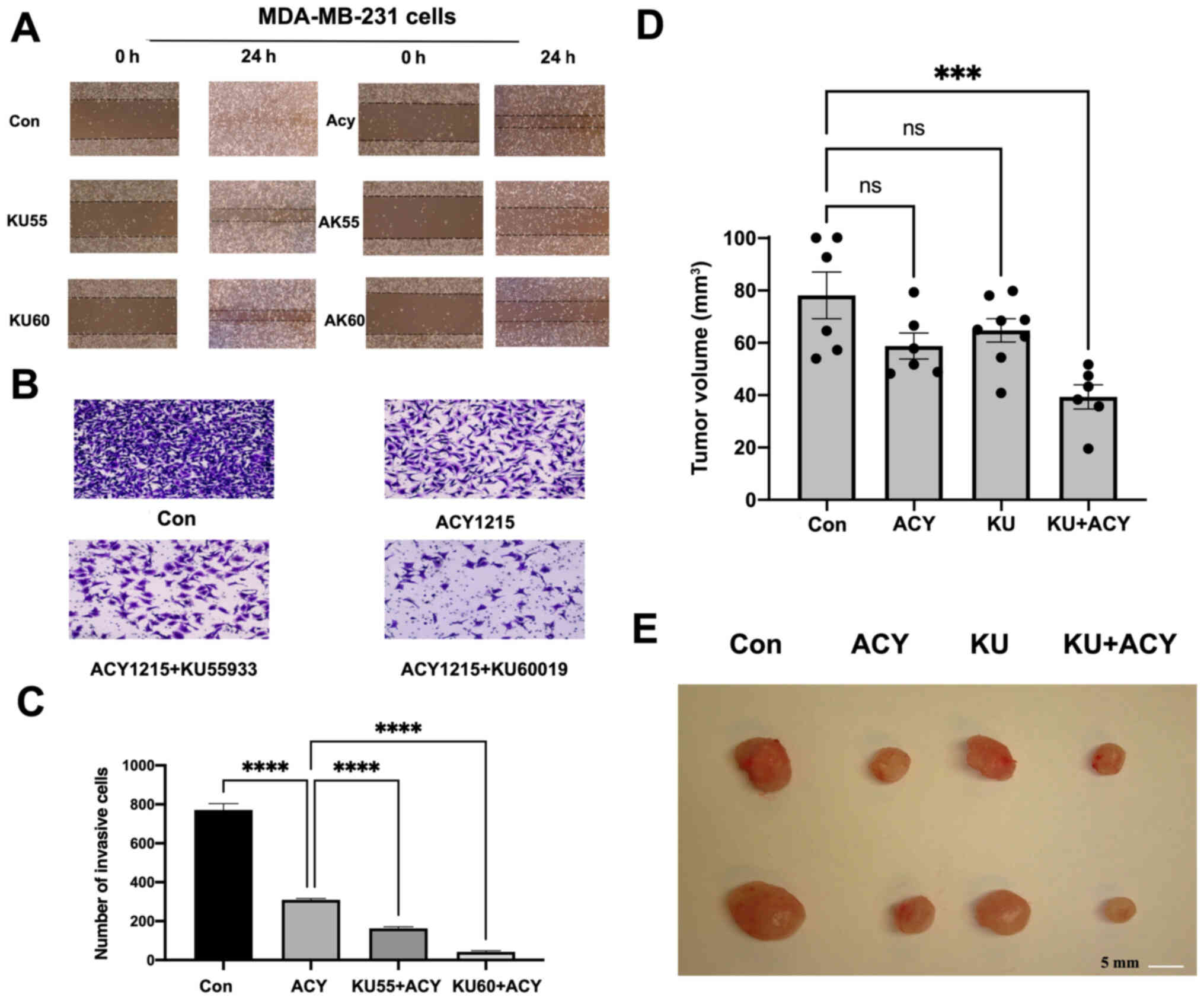 | Figure 4.Combination of ACY-1215 with KU-55933
or KU-60019 has a stronger inhibitory effect on migration and
invasion, and markedly reduces tumor growth in vivo compared
with treatment with individual compounds alone. (A) MDA-MB-231
cells were seeded in a 6-well plate. Following forming a confluent
monolayer, the cells were scratched with a 200-µl sterile tip.
Cells were then treated with ACY-1215 ± KU-55933 or KU-60019 (10 µM
each) as indicated for 24 h. Images of the cell migration were
captured with an inverted microscope (magnification, ×20). The
results are representative of three experiments. (B and C)
MDA-MB-231 cells suspended in serum-free medium were seeded into
the upper well of the invasion chamber. Medium containing 10% FBS
was added to the bottom well of the chamber to serve as a
chemoattractant. ACY-1215 ± KU-55933 or KU-60019 (10 µM each) were
added to both wells of the chamber. Following 24 h of incubation,
the cells on the upper surface of the filters were wiped away. (B)
The filters were stained with crystal violet and images were
captured (magnification, ×100). (C) Number of invasive cells on the
filter were quantified under a microscope, and the number of cells
± SEM from 3 replicates are presented. (D) Female
nude-Foxn1nu mice were injected with
1×106 MDA-MB-231 cells into the left and/or right flanks
as described in the Materials and methods. After the tumors became
visible, the mice were divided into four groups and
intraperitoneally injected with either vehicle or 30 mg/kg body
weight of ACY-1215 per day, 1 mg/kg body weight of KU-55933 per
day, or the same dosages of ACY-1215 plus KU-55933 per day for 5
days per week. The tumor volumes were measured on a weekly basis.
The bar graph indicates the averages of tumor volumes ± SEM within
each group (n=6-8). ***P<0.001. (E) Following 4 weeks of
treatment, the mice were euthanized. The tumors were removed, and
images were captured. Representative images of dissected tumors are
shown. ****P<0.0001. |
Combination of ACY-1215 and KU-55933
causes markedly stronger inhibition on tumor growth than ACY-1215
or KU-55933 alone in mouse tumor xenografts derived from MDA-MB-231
cells
To confirm the in vitro results, the effect
of combining ACY-1215 and KU-55933 versus ACY-1215 or KU-55933
alone on tumor growth in female nude mice inoculated with
MDA-MB-231 cells was further determined. A total of 4 groups of
mice with equal tumor burden were treated with the vehicle,
ACY-1215, KU-55933, or ACY-1215 plus KU-55933 for four weeks.
Treatment with ACY-1215 and/or KU-55933 had no adverse effects on
mice, including food intake and body weight. However, mice treated
with ACY-1215 and KU-55933 had significantly decreased tumor size
compared with mice treated with only ACY-1215 or KU-55933 (Fig. 4D and E) (17).
Discussion
The crucial role of p53 in preventing tumorigenesis
through induction of cell apoptosis, cell cycle arrest(18,19)
and maintenance of genomic stability (20–22)
render reactivation of mutated p53 one of the most promising
therapeutic strategies in cancer therapy. Unfortunately, targeting
mutated p53 protein has revealed to be a challenging task, as thus
far, there are still no reactivators of p53 mutants available for
clinical use (2). However, these
failed attempts made it urgent to develop new compounds that could
successfully restore function of p53 in TNBC and other subtypes of
aggressive cancer.
In the present study, ACY1215 was identified as a
novel activator of mutant p53. It was found that ACY1215 causes
increased acetylation of p53 not only in MCF-7 (non-TNBC with
wild-type p53) but also in MDA-MB-231 cells (TNBC with mutated
p53). More importantly, our results revealed that this increased
acetylation was accompanied by increased expression of p21,
suggesting that ACY1215 may lead to enhanced transcriptional
activity of p53. Consistent with these findings, it was
demonstrated that ACY1215 treatment resulted in G1 cell cycle
arrest and apoptosis in both cell lines.
While ACY-1215 may simply stimulate wild-type p53
transcriptional activity in MCF-7 cells, it may have a distinct and
more profound effect on mutant p53 in MDA-MB-231 cells. While
mutant p53 lost its transcriptional activity as a monomer,
post-translational modifications at its C-terminal, including
acetylation at Lys382, may restore mutant p53 to its normal
conformation as a tetramer so it could bind to its downstream
transcriptional targets, leading to cell cycle arrest and cell
apoptosis (1,2). While our results suggested that the
increased acetylation of p53 at Lys382 caused by ACY1215 is
important for mutant p53 to at least partially recover its
wild-type function in MDA-MB-231 cells, the underlying mechanism as
to how function of mutant p53 is restored by ACY-1215 requires
further investigation.
The reactivation of mutant p53 also poses a great
opportunity in combination therapy with other chemotherapeutic
compounds to target aggressive subtypes of cancer that are
resistant to conventional cancer therapy. As Akt is overactivated
in the majority of TNBC and ATM is a stimulator of Akt in cancer
cells (9), combination of ACY-1215
with KU-55933/KU-60019 was examined on the proliferation of
MDA-MB-231 as well as MCF-7 cells and it was observed that the
combination caused a likely additive or synergistic effect on
inhibition of proliferation of these cells. Our results further
indicated that the suppressive effects on proliferation of these
breast cancer cells are likely caused by enhanced cellular
apoptosis.
Cells undergoing malignant transformation often
exhibit a shift in cellular metabolism from oxidative
phosphorylation to aerobic glycolysis (Warburg effect) to survive
the tumor microenvironment (23).
While p53 and Akt may induce cellular apoptosis through distinct
pathways, i.e. p53/PUMA/Noxa or Akt/Bad/Bax, it is also likely they
may regulate cell apoptosis through a common pathway. Indeed, it
was recently observed that KU-55933 strongly inhibits glucose
uptake and glycolysis in aggressive breast and prostate cancer
cells, including TNBC cells (12).
Our previous results further demonstrated that the ability of
KU-55933 to inhibit glucose uptake and glycolysis is closely
correlated with its induction of apoptosis (10–12).
Interestingly, as a tumor suppressor, p53 is also important in
inhibiting glucose uptake and glycolysis by regulating the
activities of multiple key enzymes involved in the glucose uptake
and glycolysis processes (24,25).
It will be interesting to further explore the mechanisms that are
responsible for the effects of ACY1215 and KU-55933/KU-60019 on
induction of cancer cell apoptosis.
In the present study, it was also demonstrated, for
the first time to the best of our knowledge, that combination of
ACY1215 and KU-55933 may have an additive or synergistic effect on
growth of TNBC-derived breast tumors with no significant side
effects. As TNBC represents one of the most malignant subtypes of
cancer that are resistant to standard of care cancer chemotherapy,
combination of ACY1215 and KU-55933 or their analogs may represent
novel chemotherapeutic agents that are highly effective in
treating/preventing aggressive cancer and have the potential to
replace traditional chemotherapeutic approaches/agents.
In summary, our study has provided novel insights
into the molecular mechanisms underlying the function of ACY-1215
in inhibiting cancer cell proliferation and tumor growth and helped
to identify ACY-1215 as a novel activator of mutant p53 in cancer
cells. Our results have further demonstrated that ACY-1215 may
inhibit proliferation of cancer cells through induction of cancer
cell cycle arrest and apoptosis. Combination of ACY-1215 and
specific ATM inhibitors KU-55933/KU-60019 have also demonstrated
their efficacy in inhibiting cancer cell proliferation and motility
both in vitro and in vivo in cell and tumor models of
TNBC. Thus, these results may pave the way for future clinical
trials testing these compounds in people with high-risk of
developing cancer or patients with various types of aggressive
cancers for which current chemo- or immunotherapy has limited
efficacy.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Cancer Institute
of the National Institutes of Health (grant no. R01CA157012 to
MPC), a Grant-In-Aid Award (to DQY) from the University of
Minnesota, the Paint-The-Town-Pink Research Grant (to SAG) and the
Hormel Foundation.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article, and please contact the
corresponding authors for any materials described in this
study.
Authors' contributions
WC, RS, SR and MJ performed the research. WC, RS and
DQY analyzed the data. SAG, DQY, ABDA, MPC, and YL, supervised the
study. RS and DQY wrote the manuscript. YL, MPC, ABDA and SAG
critically read and reviewed the manuscript. SAG, DQY and MPC
provided funding. DQY and RS confirmed the authenticity of all the
raw data. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
All the procedures followed in mice experiments were
approved by the University of Minnesota IACUC (Minneapolis, MN,
USA).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Halaby MJ and Yang DQ: p53 translational
control: A new facet of p53 regulation and its implication for
tumorigenesis and cancer therapeutics. Gene. 395:1–7. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ji B, Harris BR, Liu Y, Deng Y, Gradilone
SA, Cleary MP, Liu J and Yang DQ: Targeting IRES-Mediated p53
Synthesis for Cancer Diagnosis and Therapeutics. Int J Mol Sci.
18:932017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Network TCGA; Cancer Genome Atlas Network,
: Comprehensive molecular portraits of human breast tumours.
Nature. 490:61–70. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brown CJ, Cheok CF, Verma CS and Lane DP:
Reactivation of p53: From peptides to small molecules. Trends
Pharmacol Sci. 32:53–62. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Muller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ryu HW, Shin DH, Lee DH, Choi J, Han G,
Lee KY and Kwon SH: HDAC6 deacetylates p53 at lysines 381/382 and
differentially coordinates p53-induced apoptosis. Cancer Lett.
391:162–171. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Seidel C, Schnekenburger M, Dicato M and
Diederich M: Histone deacetylase 6 in health and disease.
Epigenomics. 7:103–118. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lorenzo Pisarello M, Masyuk TV, Gradilone
SA, Masyuk AI, Ding JF, Lee PY and LaRusso NF: Combination of a
Histone Deacetylase 6 Inhibitor and a Somatostatin Receptor Agonist
Synergistically Reduces Hepatorenal Cystogenesis in an Animal Model
of Polycystic Liver Disease. Am J Pathol. 188:981–994. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li Y and Yang DQ: The ATM inhibitor
KU-55933 suppresses cell proliferation and induces apoptosis by
blocking Akt in cancer cells with overactivated Akt. Mol Cancer
Ther. 9:113–125. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Halaby MJ, Hibma JC, He J and Yang DQ: ATM
protein kinase mediates full activation of Akt and regulates
glucose transporter 4 translocation by insulin in muscle cells.
Cell Signal. 20:1555–1563. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Halaby MJ, Kastein BK and Yang DQ:
Chloroquine stimulates glucose uptake and glycogen synthase in
muscle cells through activation of Akt. Biochem Biophys Res Commun.
435:708–713. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Harris BRE, Zhang Y, Tao J, Shen R, Zhao
X, Cleary MP, Wang T and Yang DQ: ATM inhibitor KU-55933 induces
apoptosis and inhibits motility by blocking GLUT1-mediated glucose
uptake in aggressive cancer cells with sustained activation of Akt.
FASEB J. 35:e212642021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Golding SE, Rosenberg E, Valerie N,
Hussaini I, Frigerio M, Cockcroft XF, Chong WY, Hummersone M,
Rigoreau L, Menear KA, et al: Improved ATM kinase inhibitor
KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and
ERK prosurvival signaling, and inhibits migration and invasion. Mol
Cancer Ther. 8:2894–2902. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jänicke RU, Sprengart ML, Wati MR and
Porter AG: Caspase-3 is required for DNA fragmentation and
morphological changes associated with apoptosis. J Biol Chem.
273:9357–9360. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang XH, Sladek TL, Liu X, Butler BR,
Froelich CJ and Thor AD: Reconstitution of caspase 3 sensitizes
MCF-7 breast cancer cells to doxorubicin- and etoposide-induced
apoptosis. Cancer Res. 61:348–354. 2001.PubMed/NCBI
|
|
16
|
Zhang B, Shetti D, Fan C and Wei K:
miR-29b-3p promotes progression of MDA-MB-231 triple-negative
breast cancer cells through downregulating TRAF3. Biol Res.
52:382019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Iorns E, Drews-Elger K, Ward TM, Dean S,
Clarke J, Berry D, El Ashry D and Lippman M: A new mouse model for
the study of human breast cancer metastasis. PLoS One.
7:e479952012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Halaby MJ, Harris BR, Miskimins WK, Cleary
MP and Yang DQ: Deregulation of Internal Ribosome Entry
Site-Mediated p53 Translation in Cancer Cells with Defective p53
Response to DNA Damage. Mol Cell Biol. 35:4006–4017. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harris BRE, Wang D, Zhang Y, Ferrari M,
Okon A, Cleary MP, Wagner CR and Yang DQ: Induction of the p53
Tumor Suppressor in Cancer Cells through Inhibition of
Cap-Dependent Translation. Mol Cell Biol. 38:382018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
D'Assoro AB, Barrett SL, Folk C, Negron
VC, Boeneman K, Busby R, Whitehead C, Stivala F, Lingle WL and
Salisbury JL: Amplified centrosomes in breast cancer: A potential
indicator of tumor aggressiveness. Breast Cancer Res Treat.
75:25–34. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
D'Assoro AB, Busby R, Suino K, Delva E,
Almodovar-Mercado GJ, Johnson H, Folk C, Farrugia DJ, Vasile V,
Stivala F, et al: Genotoxic stress leads to centrosome
amplification in breast cancer cell lines that have an inactive
G1/S cell cycle checkpoint. Oncogene. 23:4068–4075. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
D'Assoro AB, Busby R, Acu ID, Quatraro C,
Reinholz MM, Farrugia DJ, Schroeder MA, Allen C, Stivala F, Galanis
E, et al: Impaired p53 function leads to centrosome amplification,
acquired ERalpha phenotypic heterogeneity and distant metastases in
breast cancer MCF-7 ×enografts. Oncogene. 27:3901–3911. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
DeBerardinis RJ, Lum JJ, Hatzivassiliou G
and Thompson CB: The biology of cancer: Metabolic reprogramming
fuels cell growth and proliferation. Cell Metab. 7:11–20. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen JQ and Russo J: Dysregulation of
glucose transport, glycolysis, TCA cycle and glutaminolysis by
oncogenes and tumor suppressors in cancer cells. Biochim Biophys
Acta. 1826:370–384. 2012.PubMed/NCBI
|
|
25
|
Barron CC, Bilan PJ, Tsakiridis T and
Tsiani E: Facilitative glucose transporters: Implications for
cancer detection, prognosis and treatment. Metabolism. 65:124–139.
2016. View Article : Google Scholar : PubMed/NCBI
|