Introduction
Primary effusion lymphoma (PEL), also referred to as
body cavity-based lymphoma, is classified as a non-Hodgkin's B cell
lymphoma that develops in immunocompromised individuals, such as in
patients with acquired immunodeficiency syndrome (AIDS) or those
who have undergone organ transplantation and are prescribed
immunosuppressants (1,2). PEL cells are infected with Kaposi's
sarcoma-associated herpesvirus [KSHV, also known as human
herpesvirus-8 (HHV-8)] and often with Epstein-Barr virus (EBV).
KSHV is the causative agent of Kaposi's sarcoma, PEL and
multicentric Castleman disease (3). During a latent infection, the KSHV
genome circularizes to form a double-stranded episome in the
nucleus of PEL cells. KSHV establishes a latent infection in PEL
cells and expresses several viral molecules, including viral
FLIP/open reading frame (ORF)71, viral cyclin/ORF72,
latency-associated nuclear antigen (LANA)/ORF73, kaposin/K12, viral
interleukin-6/K2 and various microRNAs. These molecules, including
several lytic phase-related molecules, dysregulate nuclear
factor-κB (NF-κB), mitogen-activated protein kinases (MAPKs),
Wnt/β-catenin, AKT, p53, Jak/STAT and interferon signaling to
maintain the malignant phenotype and to ensure PEL cell survival,
cell cycle progression, apoptosis inhibition and immune escape
(4,5).
The authors previously reported that KSHV
LANA-induced β-catenin stabilization, which is mediated by the
interaction of LANA with glycogen synthase kinase (GSK)-3β, leads
to the activation of Wnt/β-catenin signaling in infected cells
(6–9). In the absence of Wnt signaling,
cytoplasmic β-catenin forms a complex with Axin, adenomatous
polyposis coli (APC) and GSK-3β in the cytoplasm. The Axin-APC
complex functions as a platform for the interaction of GSK-3β and
β-catenin, and subsequently, GSK-3β phosphorylates β-catenin.
Phosphorylated β-catenin is conjugated with polyubiquitin and is
then degraded by the 26S proteasome. Although GSK-3β is primarily
localized in the cytoplasm, a small proportion of GSK-3β is known
to re-localize into the nucleus during the S phase. It was also
found that LANA increased the number of cells in the S phase, and
interacted with nuclear GSK-3β, resulting in the depletion of
cytoplasmic GSK-3β. Therefore, β-catenin was stabilized in the
cytoplasm of PEL and Kaposi's sarcoma cells, and the
transcriptional activation of Wnt signaling target genes were
stimulated in KSHV-infected cells.
The fullerene C60 (also known as
buckminsterfullerene C60) is a unique spherical carbon
molecule (10), which has been
utilized as the material for bioactive substances, semiconductors
and drug delivery. Since fullerene C60 is poorly soluble
in H2O, numerous water-soluble fullerene derivatives
have been synthesized by the conjugation of various hydrophilic
groups to a fullerene C60 core. The addition of these
hydrophilic groups (e.g., pyrrolidinium or pyridinium moiety)
confers novel biological activities to fullerene and may facilitate
its use in numerous biomedical applications including in drug
delivery (11,12), gene delivery (13), DNA photocleaving (14), the extinction of reactive oxygen
species (ROS) (15), the
generation of ROS (16) and
anti-bacterial activity (17). In
addition, fullerene derivatives exhibit antiviral activities by
inhibiting human immunodeficiency virus (HIV)-1 protease (18), HIV-1 reverse transcriptase (RT)
(19), hepatitis C virus (HCV) RNA
polymerase (NS5B) (19) and
influenza virus endonuclease (20).
Water-soluble cationic and anionic fullerene
derivatives may be utilized as novel compounds for the treatment of
PEL. The authors previously developed a pyrrolidinium-type
fullerene derivative (1,1,1′,1′-tetramethyl
[60]fullerenodipyrrolidinium diiodide) and found that this
derivative induced the generation of ROS and inhibited viral
enzymes (16,19,20)
(referred to as derivative #1 in Fig.
1). It was also found that this pyrrolidinium-type fullerene
exerts antitumor effects against PEL (21). In addition, in PEL cells, this
pyrrolidinium-type fullerene (derivative #1) reduced the
phosphorylation of AKT at Ser473 and inhibited the phosphorylation
of procaspase-9 at Ser196. Ser473-phosphorylated AKT (i.e.,
activated AKT) is known to phosphorylate procaspase-9 at Ser196,
which inactivates procaspase-9 (22). Thus, derivative #1 induces the
apoptosis of PEL cells via caspase-9 activation and AKT
inactivation. Since derivative #1 has a cation-functionalized
moiety (pyrrolidinium) and has several beneficial biological
properties, other cationic fullerene derivatives were synthesized
(Fig. 1) (16,17,19,21,23–25).
In the present study, the inhibitory activities of these
derivatives on PEL cell growth were analyzed and the underlying
molecular mechanism(s) leading to PEL cell growth inhibition were
evaluated.
Materials and methods
Agents, cell lines and cell
culture
Fullerene derivatives #1, #2 (16,19,21),
#3 (23), #5 (24,25),
#7 (24), #8 (25) and #9 (25) were synthesized and purified
according to previously reported methods. Derivative #4 was
synthesized via 1,3-dipolar cycloaddition reactions of azomethine
ylide generated from 4-dimetylaminobenzaldehyde and 4-pycolylamine,
followed by methylation with methyl iodide. Derivative #6 was
prepared by butylation of the precursor used for the synthesis of
derivative #5, with butyl iodide instead of methyl iodide. All the
fullerene derivatives were dissolved in DMSO at a concentration of
10 mM and were used as stock solutions. KSHV-positive PEL cell
lines (BCBL1, BC2, BC3, HBL6 and JSC1) and KSHV-negative lymphoma
cell lines (Ramos, Raji and DG75) were kindly provided by Dr S.D.
Hayward (Johns Hopkins University School of Medicine, Baltimore,
MD, USA) (6) and were cultured in
RPMI-1640 medium with 10% fetal bovine serum (FBS). The Ramos,
Raji, and DG75 are classified into Burkitt's lymphoma, and Raji
cells are EBV-positive. The HeLa cell line (RCB0007) was provided
by the RIKEN BioResource Research Center. Peripheral blood
mononuclear cells (PBMCs; cat. no. 1007, Astarte Biologics, LLC;
http://cellero.com/products/pbmc/)
were cultured in RPMI-1640 medium with 20% FBS.
Cell proliferation assay
PEL cells, KSHV-negative B-lymphoma cells and PBMCs
(1×104 cells/well) were seeded in a 96-well plate and
cultured in medium with or without each compound at 37°C for 24 h.
The viable cell number was determined using Cell Count Reagent SF
(Nacalai Tesque, Inc.) as previously described (26). The SF reagent (10 µl) was mixed
with 90 µl cell-medium suspension in a well, and the mixture was
incubated at 37°C for 2 h. The optical density of each sample was
measured at 450 nm on a microplate spectrophotometer (Tecan M200;
Tecan Group, Ltd.) and is expressed as a percentage (the absorbance
of untreated cells was defined as 100%). The 50% cytotoxic
concentration (CC50) is defined as the concentration of
compound that reduces cell viability by 50%. The CC50
value of each fullerene derivative was calculated using non-linear
regression using GraphPad Prism 7 (GraphPad Software, Inc.).
Western blot analysis and
antibodies
Western blot analysis was performed as previously
described (26). For sample
preparation, the cells (3×106) were solubilized in 300
µl SDS sample buffer (containing 1% 2-mercaptoethanol, 0.2 mM NaF,
1 mM β-glycerophosphate, 1 µg/ml aprotinin and 0.25 mM PMSF),
boiled for 5 min, and sonicated for 15 sec with a probe type
sonicator for chromosomal DNA disruption. The protein concentration
of the sample was determined using a Protein Assay BCA kit (Nacalai
Tesque, Inc.) or by the measurement of the absorbance at 280 nm on
a microplate spectrophotometer (Tecan M200). The resulting lysate
(15 or 20 µg) was applied to SDS-PAGE on 8, 10 or 12%
polyacrylamide gel followed by western blotting. Proteins were
transferred onto a ClearTrans nitrocellulose membrane (FUJIFILM
Wako) and the membrane was incubated with blocking buffer, 3%
non-fat dry milk and 0.1% Tween-20 in PBS, for 1 h at room
temperature. The membrane was incubated with primary antibodies
(1,000-fold dilution) diluted with Can Get Signal Immunoreaction
Enhancer Solution (Toyobo Life Science) at 4°C overnight (or at
room temperature for 2 h) and subsequently with a secondary
antibody, HRP-conjugated anti-mouse (NA931V) or anti-rabbit IgG
antibody (NA934V) (Cytiva) (2,000- or 4,000-fold dilution) at room
temperature for 1 h. The membrane was then mixed with ECL Western
Blotting Detection Reagents (Cytiva) and visualized with X-ray
film. In the densitometric analysis of western blotting, the band
intensities of the detected proteins were measured using ImageJ
software version 1.52a (National Instituters of Health). The band
intensities of the detected protein (or the phosphorylated protein)
were normalized to those of GAPDH (or the unphosphorylated
protein). The normalized values are presented at the bottom of the
blots.
The following antibodies were used for western
blotting, immunofluorescence assay (IFA) and co-immunoprecipitation
(co-IP): Anti-Thr202/Tyr204-phospho-ERK1/2 (cat. no. 4370),
caspase-3 (cat. no. 9662), Ser9-phospho-GSK-3β (cat. no. 5558),
Ser33/37/Thr41-phospho-β-catenin (cat. no. 9561),
Ser473-phospho-AKT (cat. no. 4060), Tyr705-phospho-STAT3 (cat. no.
9145), Akt (cat. no. 9272) (all from Cell Signaling Technology,
Inc.); anti-Thr180/Tyr182-phospho-p38 MAPK (cat. no. 612281), p38
MAPK (cat. no. 612168), panERK (cat. no. 610123), GSK-3β (cat. no.
610202), β-catenin (cat. no. 610154), IκBα (cat. no. 610691), NF-κB
(cat. no. 610868) (all from BD Biosciences); anti-Axin (cat. no.
sc-293190), β-actin (cat. no. sc-69879), cyclin D1 (cat. no.
sc-718) (all from Santa Cruz Biotechnology, Inc.); anti-Flag
(DDDDK)-tag (cat. no. 185-3L), HA-tag (cat. no. M180-3) and
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (cat. no. M171-3)
(all from MBL International Co.). FK2 antibodies that were
previously established (27) were
used to detect polyubiquitin conjugates. The anti-LANA rabbit
polyclonal antibody, which recognizes the N-terminal region (amino
acids 1-275) of LANA, was also established in the authors'
laboratory. As the present study had a research resource
limitation, several western blotting (as indicated in the specific
figure legends) experiments were performed twice using independent
samples.
Caspase assay
The cells (4×105 cells/ml) were incubated
with 20 µM of derivative #5 for 8 h, and caspase-3/7 activities
were measured using the Caspase-Glo Assay with a
luciferin-conjugated polypeptide substrate (Promega Corporation).
In accordance with a previous study by the authors, the activation
of caspases in PEL cells was detected at 6-12 h following the
addition of anticancer drugs (26), and the drug-treatment time was set
at 8 h. The luminescence was measured on a Tecan M200 luminescence
microplate reader (Tecan Group, Ltd.). The caspase activities of
the untreated cells were defined as 1.0.
Lactate dehydrogenase (LDH) assay
The cells (1×104 cells/well) were seeded
in a 96-well plate and cultured in medium with 20 or 50 µM
derivative #5 for 24 h. The dead cell-derived LDH in the culture
supernatants was measured using the Cytotoxicity LDH Assay kit
(Nacalai Tesque, Inc.). The optical density of each sample was
measured at 490 nm using a microplate spectrophotometer (Tecan
M200; Tecan Group, Ltd.) and is expressed as a percentage. The
absorbance of cells treated with a detergent solution (positive
control) that is contained in the LDH Assay kit was defined as
100%.
Flow cytometry
To analyze the cell cycle, the DNA amount was
measured using flow cytometry. Cells were fixed in ethanol for 1 h
at −20°C and the fixed cells were then incubated with 20 µg/ml
propidium iodide (Nacalai Tesque, Inc.) and 1,000 µg/ml RNaseA
(Takara Bio Inc.) in PBS for 10 min at 37°C. The cells washed with
PBS were subjected to analysis using a LSRFortessa Flow Cytometer
(BD Biosciences). Dead cells were excluded by gating on forward
scatter and side scatter profiles.
Luciferase reporter assay
Wnt/β-catenin-dependent transcriptional activity was
evaluated with the luciferase reporter assay using GL3-OT as
previously described (6,7). The BC3 PEL cells (4×105)
were transfected with 0.4 µg GL3-OT (TCF4 reporter plasmid) and 0.1
µg of pSV-β-Gal plasmid (Promega Corporation) using ScreenFect A
plus (FUJIFILM Wako Pure Chemical Corporation) according to the
manufacturer's instructions. GL3-OT was kindly provided by Dr K.
Kinzler and Dr B. Vogelstein (Johns Hopkins University School of
Medicine, Baltimore, MD, USA). The transfected cells were incubated
for 24 h at 37°C in medium containing 20 µM of derivative #5. The
cells were lysed by resuspension with 0.1 ml lysis buffer A [50 mM
Tris-HCl (pH 7.8), 0.05% Nonidet P-40 (NP-40) and 1 mM DTT] and
twice freeze-thaw treatment. Cell lysates were subjected to the
Firefly luciferase and β-gal assays, as previously described
(6,26). The luciferase activity was measured
using a GloMax 20/20 luminometer (Promega Corporation). The Firefly
luciferase activity was normalized to β-gal activity. The value of
luciferase activity/β-gal activity of DMSO-treated cells was
defined as 1.0.
Cycloheximide (CHX) chase
analysis
The cells (5×105 cells/well) were seeded
in a 6-well plate and cultured in medium with 20 µM derivative #5
or DMSO in the presence of 50 µg/ml CHX (Nacalai Tesque, Inc.) for
0, 2, 4 and 6 h. Harvested cells were solubilized in 100 µl SDS
sample buffer and subjected to western blot analysis using
anti-β-catenin antibody.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted and purified from
1×106 cells using RNAiso Plus (Takara Bio Inc.).
First-strand cDNA was synthesized for 15 min at 37°C from 80 ng
total RNA using the ReverTra Ace qPCR RT kit (cat. no. FSQ-101;
Toyobo Life Science). Real-time PCR was performed with the
THUNDERBIRD SYBR qPCR Mix (cat. no. QPS-201; Toyobo Life Science)
using the primer sets listed in Table
I. The program of PCR was as follows: 95°C 3 min
(pre-denaturation); followed by 40 cycles of 95°C for 10 sec
(denaturation), 55°C for 30 sec (annealing and extension). The
results were normalized to GAPDH using the 2−∆∆Cq method
(28).
 | Table I.Primers used for reverse
transcription-quantitative PCR. |
Table I.
Primers used for reverse
transcription-quantitative PCR.
| Gene | Forward
sequence | Reverse
sequence |
|---|
| β-catenin |
5′-TTGATGGGCTGCCAGATCTG-3′ |
5′-CTTTCTGAGATACCAGCCCAC-3′ |
| vIL-6 |
5′-GGTCGGTTCACTGCTGGTATC-3′ |
5′-ATGCCGGTACGGTAACAGAG-3′ |
| GAPDH |
5′-TGACCACAGTCCATGCCATC-3′ |
5′-GGGGAGATTCAGTGTGGTGG-3′ |
| LANA |
5′-TCCCGCAACACCTTTA-3′ |
5′-CGGAGACACAGGATGG-3′ |
| k-bZip |
5′-AAGTCTCTTGGACAAG-3′ | 5′-TGAGCATGGCAGATGT
−3′ |
| RTA |
5′-ATAATCCGAATGCACACATCTTCCACCAC-3′ |
5′-TCGTCGGCCTCTCGGACGAAACTGA-3′ |
Co-IP assay
In the co-IP experiments with overexpressed LANA and
GSK-3β, HeLa cells (1×106 cells) were transfected with
2.5 µg Flag-LANA (dR) (6) and 2.5
µg of HA-GSK-3β (6) plasmids for
15 h at 35°C using the calcium-phosphate method described in the
study by Chen and Okayama (29).
The transfection medium was changed to fresh normal medium, and the
cells were cultured for 24 h at 37°C. Subsequently, the cells were
treated with 20 µM of derivative #5 for 10 h. Cell extracts were
prepared with 0.8 ml of lysis buffer B [50 mM Tris-HCl (pH 7.8), 50
mM NaCl, 1% glycerol, 1 mM DTT and 0.05% NP-40]. In order to
immunoprecipitate HA-GSK-3β, the extracts (0.8 ml) were incubated
for 1 h at 4°C with 20 µl (bead volume) protein G Sepharose beads
(Merck KGaA) immobilized with 1 µg anti-HA antibody. The beads were
washed three times with lysis buffer B and resuspended in 40 µl SDS
sample buffer with 6% 2-ME. The suspension was boiled for 5 min and
centrifuged at 11,000 × g (12,000 rpm) for 0.5 min to precipitate
the beads. The supernatant was collected by the small-bore gel
loading tip (cat. no. 010-Q; Thermo Fisher Scientific, Inc.). In
order to detect the interaction of Flag-LANA and HA-GSK-3β, 20 µl
supernatant was subjected to SDS-PAGE on 8% polyacrylamide gel
followed by western blot analysis with anti-Flag antibody, as
described above.
In the co-IP assay for endogenous β-catenin, BC3 PEL
cells (1×107 cells) were cultured in medium with 20 µM
of derivative #5 and 10 µM of MG132 for 2 or 4 h. The harvested
cells were lysed with 0.8 ml of RIPA buffer [50 mM Tris-HCl (pH
7.8), 0.1% SDS, 0.5% sodium deoxycholate, 1% NP-40, 150 mM NaCl, 1
mM NEM and 1 mM DTT]. In order to immunoprecipitate endogenous
β-catenin, the cell lysates were incubated for 1 h at 4°C with 20
µl protein G Sepharose beads (Merck KGaA) immobilized with 1 µg
anti-β-catenin antibody. The procedures for preparation of the
immunoprecipitates and SDS-PAGE were the same as those described
above. In order to detect the polyubiquitin-conjugated β-catenin,
the immunoprecipitates were probed by western blot analysis with
FK2 antibody, as previously described (27).
Measurement of proteasome
activity
The proteasome activity was measured as previously
described (26). Briefly, cells
(1×106) were lysed in 0.2 ml buffer containing 50 mM
Tris-HCl (pH 7.6), 1 mM MgCl2, 0.1 mM EDTA, 1% glycerol,
1 mM DTT, 0.2 mM ATP and 0.2% NP-40, and cells were homogenized
with 27 G needles. The lysate protein concentrations were measured
using the BCA method using a Protein Assay Bicinchoninate kit
(Nacalai Tesque, Inc.). The proteasome activity in the cell lysates
were assessed with the fluorogenic peptide,
Suc-Leu-Leu-Val-Tyr-4-methylcoumaryl7-amide (MCA) (Peptide
Institute, Inc.). The AMC fluorescence intensity (excitation, 380
nm; emission, 460 nm) was determined using a Tecan M200 microplate
spectrofluorometer (Tecan Group, Ltd.).
Animal experiments
Animal experiments were approved (approval no.
18-039) by the Animal Experimentation Committee of Kyoto
Pharmaceutical University (Kyoto, Japan) and conducted in
accordance with the Guidelines for the Care and Use of Laboratory
Animals of the Science Council of Japan. To establish a
PEL-xenograft mouse model, BCBL1 cells as a transplant tumor cell
and SCID mice as recipient mice were used (26,30,31).
C.B-17 IcrHsd-Prkcd SCID male mice aged 5 weeks were purchased from
SHIMIZU Laboratory Supplies Co., Ltd. The mice were kept under
standard laboratory conditions (temperature 22±2°C; relative
humidity 50±10%; 12-h light/dark cycle) with access to food and
water ad libitum. BCBL1 cells were injected
intraperitoneally into the SCID mice twice (body weight, 20-24 g).
A BCBL1 cell suspension with 1 ml PBS (4.5×107
cells/mouse) was injected at 10 days, and a BCBL1 cell suspension
(3.8×107 cells/mouse) was additionally injected at 1 day
prior to the commencement of fullerene derivative administration.
Derivative #5 (or DMSO) dissolved in corn oil was administered into
the intraperitoneal region at a dose of 20 mg/kg body weight every
2 days for the first 1 week and subsequently every 3 days for
following 2 weeks. The DMSO-administered normal mice (n=3),
derivative #5-administered normal mice (n=3), DMSO-administered
PEL-xenografted mice (n=3), and derivative #5-administered
PEL-xenografted mice (n=3) were observed, and their body weight was
measured each day for 3 weeks. All mice were euthanized on day 21,
and their ascites and organs were collected. The ascites collected
from each mouse was centrifuged at 300 × g (1,400 rpm) for 5 min,
and tumor cells were precipitated to determine the weight volume of
the tumor cells. All mice that reached the study endpoint were
euthanized by cervical dislocation under 2-3% isoflurane
anesthesia. The humane endpoints were determined to be when the
xenograft tumor diameter was >20 mm, the xenograft tumor reached
>20% of the animal body weight, body weight loss >20%
occurred due to tumor growth, and signs of immobility, the
inability to eat, ulceration, infection, or necrosis were observed.
Death was verified by observation of pupil dilation as well as
ceasing of breath and heartbeat.
IFA and phase-contrast image
In the IFA to detect the localization of LANA and
GSK-3β, HeLa cells (2×105 cells) grown on a glass slide
were transfected with 3 µg Flag-LANA (dR) and 2 µg HA-GSK-3β
plasmids for 15 h at 35°C according to the method described in the
study by Chen and Okayama (29).
The transfection medium was changed, and cells were cultured in
normal medium for 24 h at 37°C. Subsequently, the cells were
treated with 20 µM of derivative #5 for 10 h and were fixed with 4%
paraformaldehyde for 15 min at 4°C and permeabilized with 0.1%
Triton X-100 in PBS for 10 min at room temperature. In the animal
experiment, ascites and BCBL1 cells were fixed on a glass slide
with 4% paraformaldehyde at room temperature for 1 h and
permeabilized with 0.1% Triton X-100 in PBS. The cells were then
incubated in PBS-T containing 10% FBS and the primary antibody
(anti-HA, anti-Flag or anti-LANA antibody) (500-fold dilution) at
room temperature for 1 h. The cells were further treated with the
secondary antibody, Alexa Fluor 488-labeled donkey anti-mouse (cat.
no. A32766), Alexa Fluor 594-labeled donkey anti-rabbit (cat. no.
A32754), or Alexa Fluor 488-labeled donkey anti-rabbit IgG antibody
(cat. no. A32790) (Thermo Fisher Scientific, Inc.) (3,000-fold
dilution) at room temperature for 1 h. The stained samples were
embedded in Fluoro-KEEPER Antifade Reagent, Non-Hardening Type with
DAPI (12745–74) (Nacalai Tesque, Inc.) and were observed under a
confocal LSM 800 microscope (Carl Zeiss AG) using LSM software ZEN
3.3 lite/blue edition (Carl Zeiss AG).
Phase-contrast images were obtained using an
inverted Olympus IX-71 microscope (Olympus Corporation). Cells
seeded in a 6-cm dish were cultured in media, and the
phase-contrast images of living cells were obtained under an
inverted microscope with the 10X objective using analysis software
DP2-BSW (Olympus Corporation).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 7 (GraphPad Software, Inc.). Data are presented as the mean ±
SD, and the standard deviation was determined by analyzing the data
from at least three experiments. Statistical differences between
groups were determined using one-way analysis of variance (ANOVA)
followed by Dunnett's test for multiple comparisons or the
two-tailed Student's t-test. A value of P<0.05 was considered to
indicate a statistically significant difference.
Results
Effects of eight fullerene derivatives
on PEL cell viability
First of all, the effects of two pyrrolidinium-type
(derivatives #1 and #2), six pyridinium-type (derivatives #3 and #5
to #9), and one anilinium-type (derivative #4) fullerene
derivatives on PEL cell viability were analyzed (Fig. 2A-I). KSHV-infected PEL cell lines
(HBL6, BCBL1 and BC3) and KSHV-uninfected B-lymphoma cell lines
(DG75 and Ramos) were cultured in medium with or without the
fullerene derivative. After 24 h, the number of viable cells was
quantified. The tested derivatives presented various viability
profiles against each B-lymphoma cell line. Derivative #1 increased
the viability of the DG75 and Ramos cells at the concentrations of
6.25 and 12.5 µM compared with the controls, and derivatives #3 and
#4 increased the viability of the BC3 PEL cells at the
concentrations of 6.25 µM. Although the viability of the
KSHV-uninfected DG75 and Ramos cells decreased by 0-15% at the high
concentration (25 µM) of derivatives #8 and #9, the uninfected
cells were less sensitive to 25 µM of these derivatives compared
with the PEL cells. On the other hand, derivatives #3 and #4
decreased the viability of all cell lines at 25 µM. The PEL cell
lines exhibited sensitivity to derivatives #5 and #6, whereas the
KSHV-negative cells were less sensitive to these derivatives
(Fig. 2A-I). In particular,
derivative #5 (3-[5′-
(ethoxycarbonyl)-1′,5′-dihydro-2′H-[5,6]fullereno-C60-Ih-[1,9-c]pyrrol-2′-yl]-1-methylpyridinium
iodide) markedly inhibited the proliferation of PEL cell lines
compared with KSHV-uninfected B-cell lines. To assess the accurate
CC50 of derivative #5 and the specificity for PEL, the
cytotoxic effects of derivative #5 on various B-cell lines were
analyzed (Table II). The results
revealed that derivative #5 reduced the number of viable PEL cells
(BCBL1, BC3, BC2 and JSC1) compared with the uninfected cells
(DG75, Ramos and Raji) (Fig. 2J).
Indeed, derivative #5 was the most potent inhibitor of PEL cell
viability. The cytotoxic effects of derivatives #1 to #9 on
B-lymphoma cells are summarized in Table II. Derivative #5 was active
against PEL cell lines (CC50, 6-17 µM), while the
KSHV-uninfected B cell lines were insensitive to derivative #5
(CC50, >35 µM). Moreover, the cytotoxic effects of
derivative #5 on human PBMCs were evaluated. Although 5 µM
derivative #5 decreased the viability of the PBMCs by 6.4%, marked
toxicity was not detected in <25 µM derivative #5-treated PBMCs
(Fig. 2K). Since derivative #5 was
the most potent inhibitor of PEL cell viability, the underlying
molecular mechanism(s) contributing to this inhibition were
analyzed.
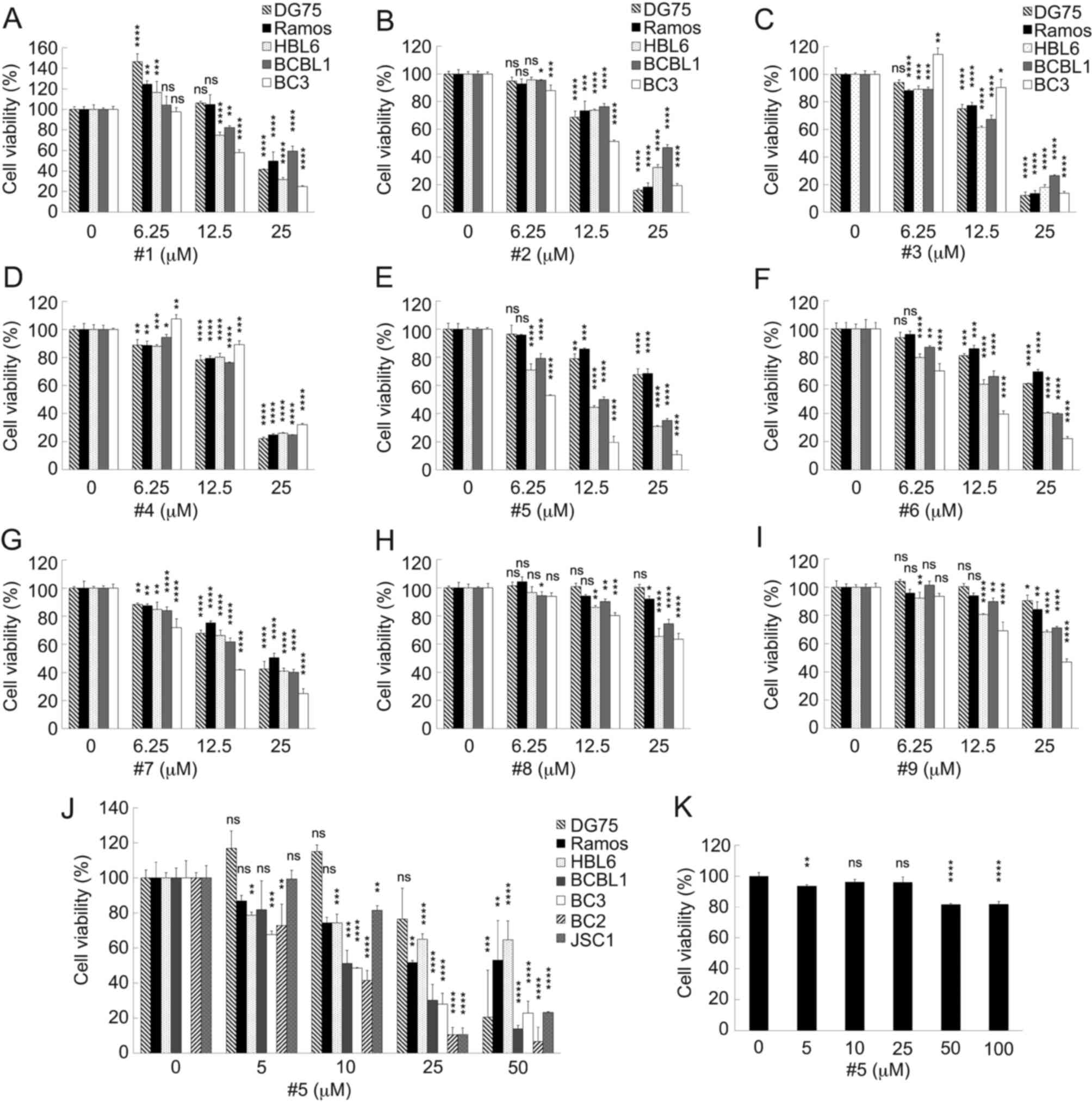 | Figure 2.Effects of the fullerene derivatives
on the viability of KSHV-positive PEL cells and KSHV-negative B
cell lines. (A-I) Effects of the fullerene derivatives on the
viability of KSHV-positive PEL cells and KSHV-negative B cell
lines. KSHV-positive PEL cells (BCBL1, BC3 and HBL6) and
KSHV-negative cells (Ramos and DG75) were incubated with the
indicated concentrations of the fullerene derivatives for 24 h and
the number of viable cells was assessed. The cell viability of the
respective untreated cells was defined as 100%. (J) The effects of
derivative #5 on the viability of KSHV-positive PEL cells (BCBL1,
BC2, BC3 and JSC1) and KSHV-negative B cells (Ramos, DG75 and
Raji). Cells were incubated with derivative #5 for 24 h. The
viability of the respective untreated cells was defined as 100%.
(K) Effects of derivative #5 on human PBMCs. The human PBMCs were
treated with various concentrations of derivative #5 for 24 h and
the number of viable cells was measured. The cell viability of the
untreated cells was defined as 100%. (A-K) Data are based on three
independent experiments. The results are presented as the mean ±
SD. KSHV, Kaposi's sarcoma-associated herpesvirus; PEL, primary
effusion lymphoma; PBMCs, peripheral blood mononuclear cells.
*P<0.05, **P<0.005, ***P<0.0005 and ****P<0.00005,
statistically significant difference compared with the control
group (or DMSO-treated group). ns, not significant. |
| Table II.Cytotoxic effects of fullerene
derivative #5 on B-lymphoma cell lines. |
Table II.
Cytotoxic effects of fullerene
derivative #5 on B-lymphoma cell lines.
|
|
CC50a (µM)b |
|---|
|
|
|
|---|
|
| KSHV (−) cell
lines | KSHV (+) cell
lines |
|---|
|
|
|
|
|---|
| Derivative | DG75 | Ramos | Raji | HBL6 | BCBL1 | BC3 | BC2 | JSC1 |
|---|
| #1 | 23.4 | 24.9 | DN | 19.7 | >25 | 15.5 | DN | DN |
| #2 | 16.9 | 17.8 | DN | 19.7 | 23.2 | 13.0 | DN | DN |
| #3 | 17.2 | 17.7 | DN | 15.5 | 17.2 | 18.9 | DN | DN |
| #4 | 18.3 | 18.8 | DN | 19.0 | 18.1 | 20.6 | DN | DN |
| #5c | >50 | 36.9 | >50 | 10.5 | 10.9 | 9.6 | 5.9 | 16.7 |
| #6 | >25 | >25 | DN | 16.6 | 17.8 | 9.6 | DN | DN |
| #7 | 18.8 | >25 | DN | 18.1 | 15.8 | 9.7 | DN | DN |
| #8 | >25 | >25 | DN | >25 | >25 | >25 | DN | DN |
| #9 | >25 | >25 | DN | >25 | >25 | 19.3 | DN | DN |
Derivative #5 induces the suppression
of PEL cell growth and induces the formation of cell clumps
The present study then examined whether the
cytotoxic effects of derivative #5 on PEL cells were due to
apoptosis. KSHV-infected PEL cell lines (BC2, JSC1, BC3 and BCBL1)
and KSHV-uninfected B-lymphoma cell lines (DG75 and Raji) were
treated with 20 µM of derivative #5 for 8-24 h and cell extracts
from harvested cells were examined by western blot analysis. An
increase in cleaved (i.e., activated) caspase-3 was not detected in
the PEL cell lines (BC2, JSC1, BC3 and BCBL1) treated with 20 µM of
derivative #5 (Fig. 3A). Moreover,
the activation of caspase-3/7 activities were monitored in the
Raji, BC2, BC3 and BCBL1 cells treated with derivative #5 using a
colorimetric assay (Fig. 3B).
However, no increase in caspase-3/7 peptidase activities was
detected in the cells treated with derivative #5. In order to
further explore the mechanisms through which derivative #5
decreases PEL cell viability, the number of dead cells were
measured following treatment with derivative #5 using an LDH assay
and flow cytometry (Fig. 3D and
E). LDH assay revealed that treatment with 50 µM derivative #5
increased the number of dead BCBL1 and BC3 cells by ~7-15%,
although significant differences were not detected with 20 µM of
the derivative #5 (Fig. 3C). To
analyze the cell cycle and the sub-G1 phase including dead cells,
the DNA amount of PEL cells (BC3 and BCBL1) treated with derivative
#5 was measured using flow cytometry (Fig. 3D and E). As was expected,
derivative #5 increased the number of cells at the sub-G1 phase in
a concentration-dependent manner. These data indicated that
derivative #5 induced caspase-independent cell death and decreased
PEL cell viability.
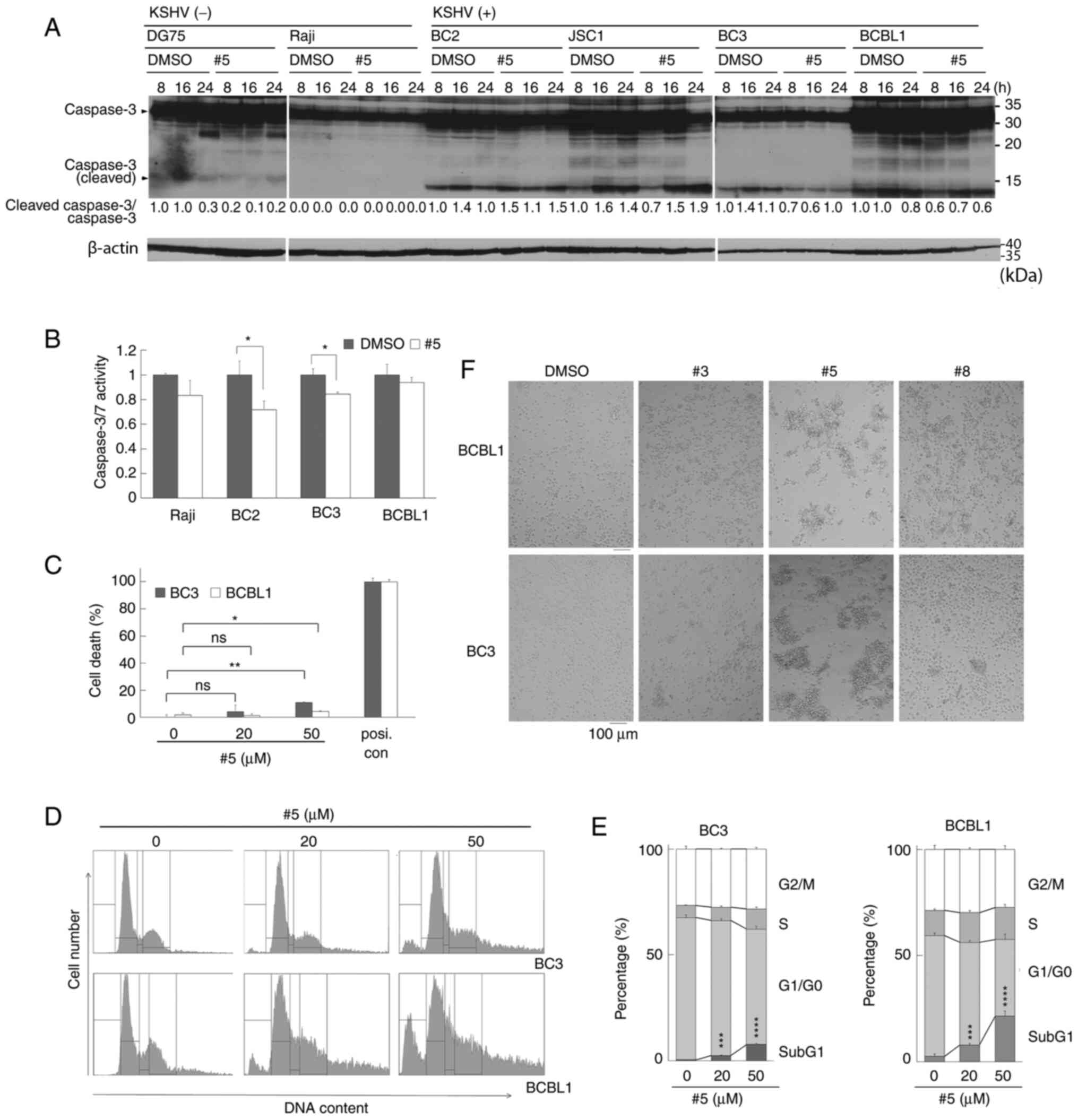 | Figure 3.Treatment with derivative #5 induces
PEL cell clumping. (A) Western blot analysis using anti-caspase-3
antibody. PEL cells (BC2, JSC1, BC3 and BCBL1) and uninfected DG75
and Raji cells were incubated with 20 µM of derivative #5 or DMSO
(vehicle) for 8-24 h. The western blotting experiments the results
of which are depicted were performed twice using independent
samples. (B) The proteolytic activities of caspase-3/7 in PEL cells
(BC2, BC3 and BCBL1) and KSHV-uninfected Raji cells treated with
derivative #5. Cells were incubated with 20 µM of derivative #5 for
8 h, and the activities of caspase-3/7 in cell lysates were
measured using a luciferin-conjugated polypeptide substrate.
Caspase-3/7 activity in untreated cells was defined as 1.0 relative
light unit. (C) Quantification of dead cells using LDH assay. Cells
were incubated with 20 µM of derivative #5 for 24 h, and the number
of dead cells was measured using LDH assay. The absorbance of cells
treated with a detergent (positive control) was defined as 100%. (B
and C) Data are representative of at least three independent
experiments. (D) Derivative #5 increased the sub-G1 fraction in PEL
cells in a concentration-dependent manner. To analyze the cell
cycle and the sub-G1 including dead cells, the DNA amount of
derivative #5-treated PEL cells were measured using flow cytometry.
(E) The distribution of cell cycle phases was calculated based on
the flow cytometer data. (F) The increase in cell clumping induced
by treatment with derivatives #3, #5 and #8. Cells were cultured
with 20 µM of derivative #5 for 24 h. Phase-contrast images were
obtained using an inverted microscope. *P<0.05, **P<0.005,
***P<0.0005 and ****P<0.00005, statistically significant
difference compared with the control group (or DMSO-treated group).
ns, not significant; PEL, primary effusion lymphoma; LDH, lactate
dehydrogenase. |
Subsequently, the effects of derivatives #5, #3 and
#8 on the morphology of BCBL1 and BC3 PEL cells were examined
(Fig. 3F). The morphology of the
BC2 PEL cells and KSHV-uninfected Ramos cells is illustrated in
Fig. S1. Derivatives #5, #3 and
#8 led to the formation of cell clumps in all tested cells; of
note, derivative #5 prominently induced cell-to-cell clumping
(Figs. 3F and S1). Among the tested derivatives,
derivative #5 induced the largest cell clumps, suggesting the
association between cell clumping formation and cell death by
derivative #5.
Derivative #5 suppresses Wnt/β-catenin
signaling in PEL cells through the destabilization of
β-catenin
NF-κB (26,32,33),
AKT (21,34), p38 MAPK (34,35),
ERK (35,36) and Wnt/β-catenin (4–9)
signaling pathways are activated in a number of PEL cell lines,
which are necessary for PEL cell survival and proliferation.
Therefore, the present study examined whether derivative #5
affected these pathways. The KSHV-infected PEL cell lines (BC2,
JSC1, BC3 and BCBL1) were treated with derivative #5 for 8-24 h,
and cell lysates from harvested cells were subjected to western
blot analysis. No changes were detected in the phosphorylation
status of AKT, ERK1/2, p38 MAPK and GSK-3β (Fig. 4A). Moreover, the levels of
T705-phospho-STAT3, NF-κB and IκBα were not changed in derivative
#5-treated PEL cells (Fig. S2A).
However, the expression level of β-catenin was decreased in the
derivative #5-treated PEL cell lines. The HBL6 cells exhibited a
low expression of β-catenin compared with the other PEL cell lines
(Fig. S2B). This may be due to
differences in the genetic background or differentiation status of
the tested cells. In order to confirm the suppression of
Wnt/β-catenin signaling in derivative #5-treated PEL cells, a
reporter assay was performed using the TCF4-β-catenin reporter
(GL3-OT) plasmid. As was expected, the Wnt/β-catenin
transcriptional activity of the BC3 cells was significantly
decreased to 60% upon treatment with derivative #5 (Fig. 4B). Since the cyclin D1 gene (CCND1)
is one of the target genes of Wnt/β-catenin signaling, the
expression of cyclin D1 was monitored in the derivative #5-treated
PEL cells. As was expected, treatment with derivative #5 decreased
cyclin D1 expression in the PEL cells (Fig. 4A).
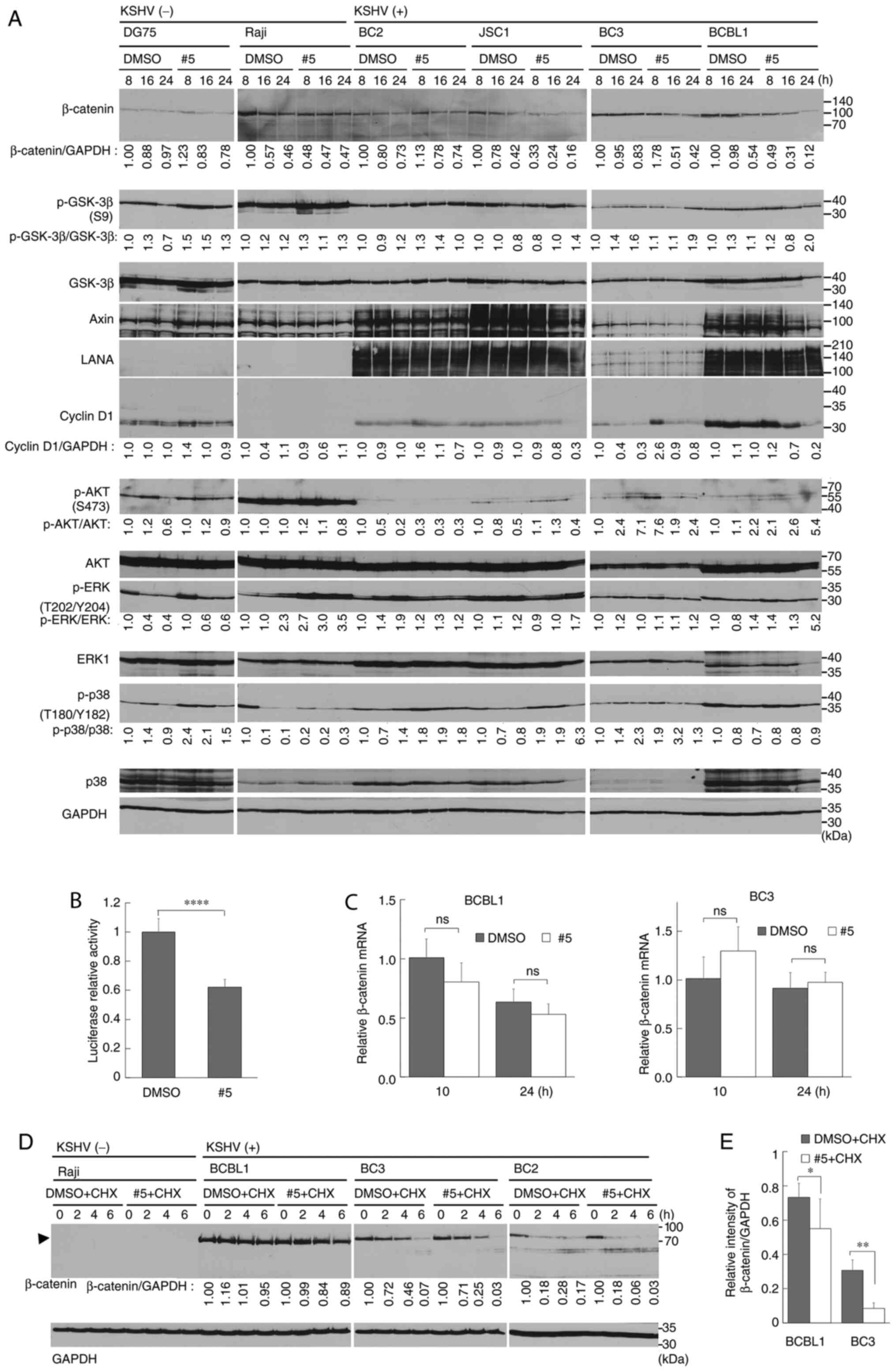 | Figure 4.Derivative #5 downregulates Wnt
signaling in PEL cells via β-catenin destabilization. (A)
Downregulation of β-catenin by treatment of PEL cells with
derivative #5. PEL (BC2, JSC1, BC3 and BCBL1) and KSHV-uninfected
(Raji and DG75) cells were treated with 20 µM of derivative #5 for
8-24 h, and cell lysates were examined using western blot analysis.
Derivative #5-treated and DMSO-treated cells are denoted as #5 and
DMSO, respectively. The values of β-catenin/GAPDH are presented at
the bottom of the blots. The value of each DMSO-treated cell line
(treated for 8 h) was defined as 1.0. The western blotting
experiments the results of which are depicted were performed twice
using independent samples. (B) Derivative #5-mediated inhibition of
Wnt signaling-induced transcriptional activation. BC3 cells were
transfected with the TCF4-β-catenin reporter plasmid (GL3-OT), and
transfected cells were cultured in media containing 20 µM of
derivative #5 or DMSO for 24 h. The luciferase activity of
DMSO-treated transfected BC3 cells was defined as 1.0. (C) Effect
of derivative #5 on β-catenin mRNA expression. Cells were treated
with 20 µM of derivative #5, and total RNA was subjected to reverse
transcription-quantitative PCR. The values obtained from
DMSO-treated cells (treated for 10 h) were defined as 1.0. (B and
C) Results are presented as the mean ± SD (n=3). (D) Effect of
derivative #5 on β-catenin stabilization. Cells were cultured for
0-6 h in media with 20 µM derivative #5 in the presence of 50 µg/ml
CHX. The values of β-catenin/GAPDH in DMSO-treated cells at 0 h was
defined as 1.0. (E) Quantitative analysis of β-catenin expression
in derivative #5-treated PEL cells. Cells were incubated with 20 µM
derivative #5 in the presence of 50 µg/ml CHX for 0 or 6 h. Cell
lysates prepared by three independent experiments were examined
using western blot analysis. The blotting images are shown in
Fig. S3. The value of derivative
#5 (or DMSO)-treated cells for 0 h was defined as 1.0. *P<0.05,
**P<0.005 and ****P<0.00005, statistically significant
difference compared with the control group (or DMSO-treated group).
ns, not significant; PEL, primary effusion lymphoma; GSK-3β,
glycogen synthase kinase 3β; LANA, latency-associated nuclear
antigen; CHX, cycloheximide. |
In order to determine whether the derivative
#5-mediated downregulation of β-catenin is due to its
transcriptional inactivation or its protein destabilization, the
mRNA expression levels of β-catenin and β-catenin protein stability
were examined in derivative #5-treated PEL cells. The mRNA
expression of β-catenin in the BC3 or BCBL1 cells treated with
derivative #5 was monitored using RT-qPCR (Fig. 4C). Derivative #5 treatment slightly
reduced the mRNA expression of β-catenin in the BCBL1 cells.
Subsequently, a CHX chase analysis was conducted in order to
determine whether derivative #5 induced the destabilization of
β-catenin. CHX was used to inhibit de novo protein synthesis. In
this experiment, the cells were cultured in medium with or without
derivative #5 in the presence of 50 µg/ml CHX. The cells were
harvested at 2-6 h. The results indicated that derivative #5
induced the destabilization of β-catenin protein in PEL cells as
compared with the vehicle control (Fig. 4D). To obtain further evidence, the
quantitative analysis of β-catenin expression in derivative
#5-treated PEL cells was performed using three independent
experiments. The BCBL1 and BC3 cells were incubated with 20 µM
derivative #5 (or DMSO) with 50 µg/ml CHX for 0 or 6 h. The
destabilization of β-catenin was then examined using western blot
analysis (Fig. S3). The data
indicated that derivative #5 induced β-catenin destabilization in
PEL cells (Fig. 4E).
Derivative #5 enhances the
phosphorylation of β-catenin at Ser33/Ser37/Thr41
The mechanism of β-catenin degradation consists of
three sequential steps (37,38):
i) The phosphorylation of β-catenin at Ser33/Ser37/Thr41 by GSK-3β;
ii) the polyubiquitination of Ser33/Ser37/Thr41-phosphorylated
β-catenin by E3 ubiquitin ligase; and iii) the degradation of
polyubiquitinated β-catenin by the 26S proteasome. The present
study therefore attempted to determine which step was affected by
derivative #5, which leads to β-catenin destabilization (Fig. 5A-E). It was found that the levels
of GSK-3β and Ser9-phosphorylated GSK-3β (i.e., the inactive form
of GSK-3β) were not altered in the B-lymphoma and PEL cells
following treatment with derivative #5 (Fig. 4A). Thus, the present study also
evaluated whether derivative #5 affected 26S proteasome activity.
No differences in the protease activity of the 26S proteasome were
found between the derivative #5-treated and untreated cells
(Fig. 5A). On the other hand, it
was previously found that KSHV LANA interacted with GSK-3β in the
nucleus, leading to β-catenin stabilization and subsequent
activation of Wnt/β-catenin signaling in KSHV-infected cells
including PEL cells (6–9). GSK-3β is localized primarily in the
cytoplasm, and GSK-3β phosphorylates the cytoplasmic β-catenin for
the destabilization of β-catenin. GSK-3β is known to enter the
nucleus during the S phase, and LANA increases the number of cells
in the S phase and binds to nuclear GSK-3β (6–9).
Therefore, the present study examined the effects of derivative #5
on GSK-3β-mediated β-catenin destabilization, as well as
LANA-mediated β-catenin stabilization. As was expected,
co-transfection of plasmids encoding GSK-3β and β-catenin resulted
in the loss of detectable β-catenin in the HeLa cells (Fig. 5B). Furthermore, in the presence of
LANA, the ability of GSK-3β to promote β-catenin destabilization
was markedly decreased, and β-catenin was easily detectable. These
results are in agreement with those of previous studies (6,7).
This LANA-mediated β-catenin stabilization was partially overcome
by treatment with derivative #5. Moreover, in the absence of LANA
and GSK-3β, β-catenin was also destabilized by derivative #5.
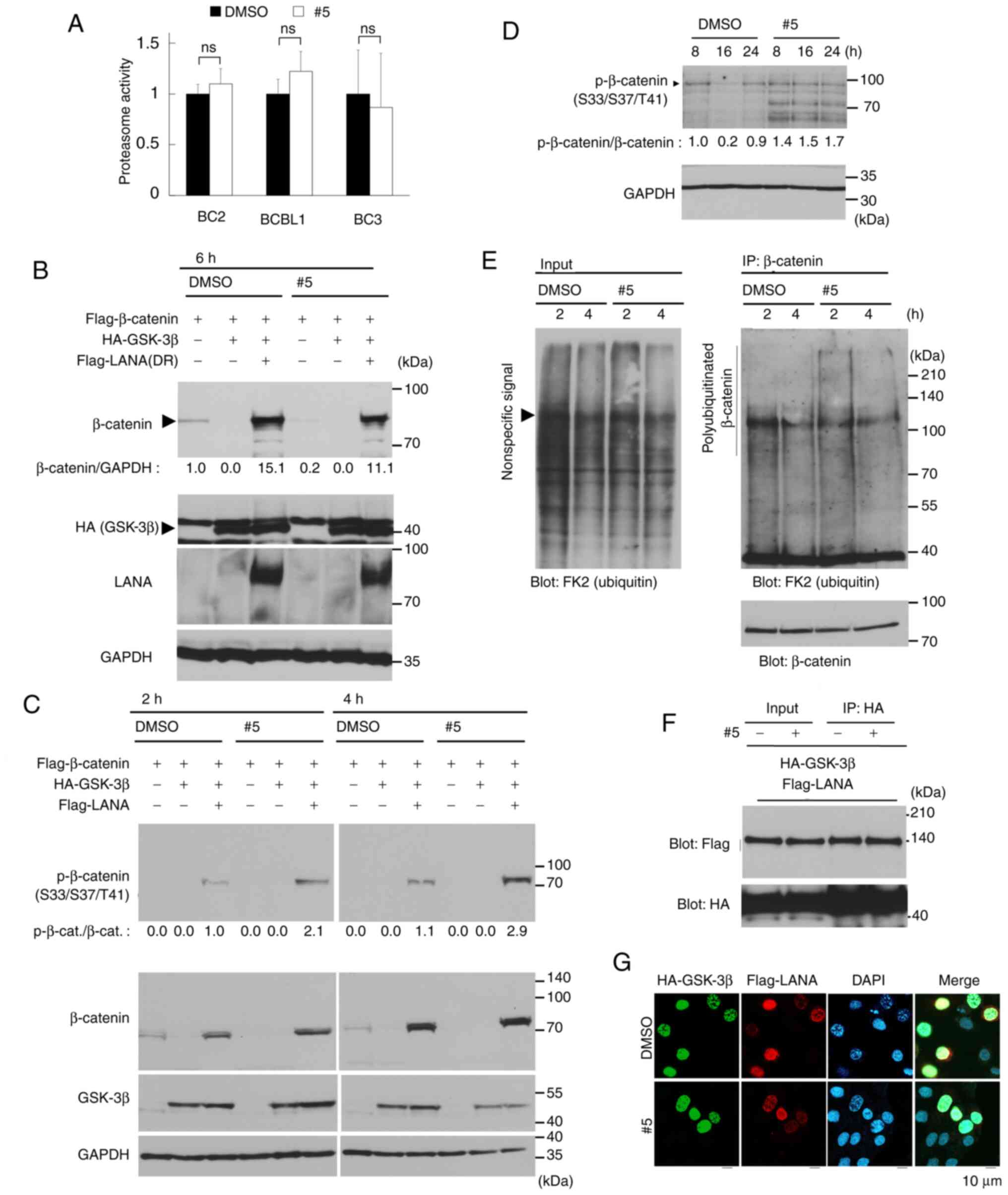 | Figure 5.Derivative #5 induces β-catenin
destabilization by increasing the phosphorylation of β-catenin at
Ser33/Ser37/Thr41. (A) Effects of derivative #5 on the proteasomal
chymotrypsin-like activity in PEL cells. Cells were cultured with
20 µM derivative #5 or DMSO for 24 h. The proteasome activities of
the cell lysates were evaluated using a fluorometric assay with a
synthetic peptide. The proteasome activity of DMSO-treated cells
was defined as 1.0. The results are presented as the mean ± SD
(n=3). (B) Derivative #5 destabilized β-catenin and overcame
LANA-mediated β-catenin stabilization. HeLa cells were
co-transfected with 1 µg Flag-β-catenin, 0.2 µg HA-GSK-3β, 0.3 µg
Flag-LANA (dR) and empty pCIneo (for adjusting to 1.5 µg as the
total amount) plasmids. The cells were then treated with 20 µM
derivative #5 and 100 µg/ml cycloheximide for 6 h. (C) Derivative
#5 induced the phosphorylation of β-catenin at Ser33/Ser37/Thr41.
HeLa cells were co-transfected with 1 µg Flag-β-catenin, 0.2 µg
HA-GSK-3β, 0.3 µg Flag-LANA (dR) and empty plasmids. The cells were
then treated with 20 µM derivative #5 and 100 µg/ml cycloheximide.
(D) Derivative #5 induced the phosphorylation of β-catenin at
Ser33/Ser37/Thr41 in BC3 PEL cells. The cell lysates, which were
same as the lysates used in Fig.
4A, were used. The lysates were prepared from cells treated
with 20 µM derivative #5 and subjected to western blot analysis.
The data of band intensities of β-catenin in Fig. 4A were used to calculate the
p-β-catenin/β-catenin ratio. (E) Derivative #5 induced the
polyubiquitination of endogenous β-catenin. BC3 cells were treated
with 20 µM derivative #5 and 10 µM MG132 for 2 or 4 h. The cell
extracts were incubated with anti-β-catenin antibody-immobilized
beads, and immunoprecipitated (IP) β-catenin was subjected to
western blot analysis with anti-polyubiquitin antibody (FK2) to
detect the polyubiquitinated β-catenin. The western blotting
experiments the results of which are depicted were performed twice
using independent samples. (F) Effect of derivative #5 on the
interaction between KSHV LANA and GSK-3β. HeLa cells were
co-transfected with Flag-LANA (dR) and HA-GSK-3β and then treated
with 20 µM derivative #5 for 10 h. HA-GSK-3β was immunoprecipitated
from cell extracts with anti-HA antibody-immobilized beads (IP:
HA), and the immunoprecipitate was probed by anti-Flag antibody.
(G) Effect of derivative #5 on nuclear colocalization of GSK-3β and
LANA. HeLa cells were co-transfected with Flag-LANA (dR) and
HA-GSK-3β plasmids and treated with 20 µM derivative #5 for 10 h.
The localization of Flag-LANA (red) and HA-GSK-3β (green) were
analyzed by IFA. ns, not significant; PEL, primary effusion
lymphoma; GSK-3β, glycogen synthase kinase 3β; LANA,
latency-associated nuclear antigen. |
Subsequently, the effects of derivative #5 on the
phosphorylation and polyubiquitination of β-catenin were analyzed.
Of note, the data indicated that derivative #5 induced the
Ser33/Ser37/Thr41 phosphorylation of exogenous and endogenous
β-catenin (Fig. 5C and D). The
destabilization of β-catenin was detected by 6 h of treatment with
derivative #5 (Fig. 5B); however,
the destabilization of β-catenin was not detected by 2 and 4 h of
treatment with derivative #5 (Fig.
5C). These results may be due to different durations of
treatment with derivative #5. As indicated in Fig. 4A, treatment with derivative #5 for
2-4 h was not sufficient to induce β-catenin destabilization. In
addition to phosphorylation, derivative #5 increased the endogenous
polyubiquitination of β-catenin in BC3 cells (Fig. 5E). Since LANA interacts with GSK-3β
in the nucleus, leading to β-catenin stabilization in PEL cells,
the present study determined whether derivative #5 affected the
interaction and the nuclear co-localization of LANA and GSK-3β. The
Co-IP data confirmed the interaction of GSK-3β and LANA in
co-transfected cells (Fig. 5F).
However, derivative #5-treatment did not affect the interaction of
GSK-3β and LANA. In addition to the interaction, the effect of
derivative #5 on the nuclear co-localization of GSK-3β and LANA was
evaluated by IFA (Fig. 5G). The
data revealed the nuclear co-localization of GSK-3β (green) and
LANA (red) in co-transfected cells; however, derivative #5 did not
affect the nuclear co-localization of GSK-3β and LANA. The data
demonstrated that GSK-3β markedly induced β-catenin
destabilization, and KSHV LANA stabilized β-catenin even in the
presence of GSK-3β. Moreover, it was found that derivative #5
induced the phosphorylation and polyubiquitination of β-catenin,
which overcame LANA-mediated β-catenin stabilization. However,
derivative #5 did not affect the interaction and the nuclear
co-localization of GSK-3β and LANA.
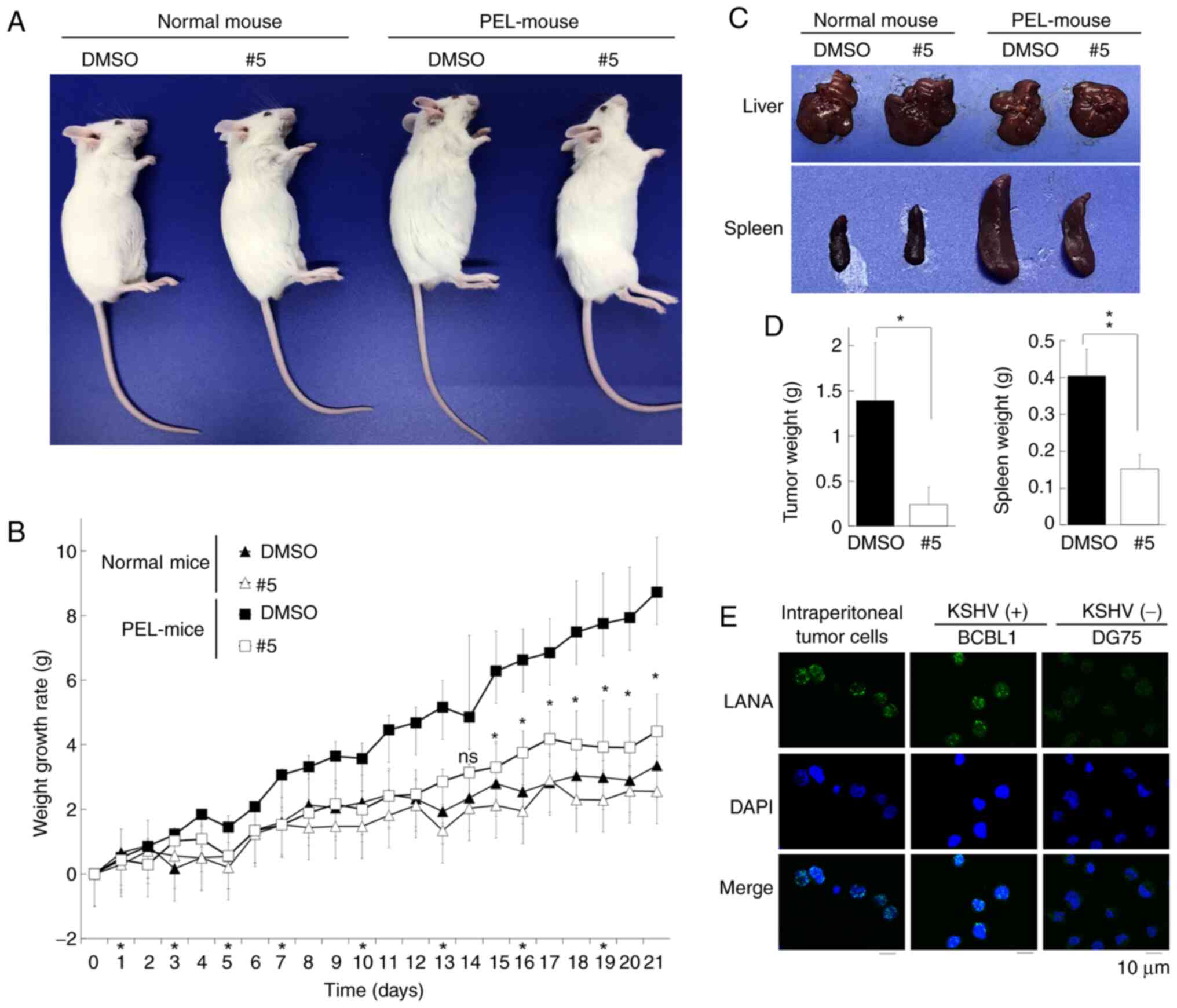
Derivative #5 abrogates PEL tumor cell
development in SCID mice
Since derivative #5 was cytotoxic against PEL cell
lines (Fig. 1), the present study
then investigated whether derivative #5 exerted cytotoxic effects
against xenograft PEL cells in SCID mice. BCBL1 PEL cells were
injected intraperitoneally into SCID mice twice (10 days and 1 day
prior to the commencement of derivative #5 administration), and
PEL-xenografted mice (PEL-mice) were established. The corn oil
emulsions of derivative #5 or the vehicle control (DMSO) were
injected intraperitoneally into PEL-mice or normal mice once per 2
(or 3) days for 3 weeks. The PEL-xenografted mice with and without
derivative #5 administration differed significantly in their gross
appearance (Fig. 6A and B): The
abdomen of the DMSO-treated PEL-mice exhibited abdominal expansion,
whereas the derivative #5-treated mice had an apparently normal
body shape. The increase in the body weight of the derivative
#5-treated PEL-mice was less marked than that of the DMSO-treated
PEL-mice. Moreover, the body weight curves of the derivative #5-
and DMSO-treated non-xenografted mice (normal mice) exhibited a
similar trend. These data indicated that derivative #5 had a low
toxicity in both the mouse model and the in vitro model
using PBMCs (Fig. 2K). Murine
autopsies demonstrated that the spleens of DMSO-treated PEL-mice
exhibited distention compared to spleens from the derivative
#5-treated PEL-mice (Fig. 6C). It
has been previously reported that PEL-xenografted SCID mice exhibit
spleen distention (26), which is
in agreement with the present data. The weight of the spleen in the
derivative #5-treated group was ~0.15 g, which was lower (~0.4 g)
than that of the DMSO-treated group (Fig. 6D). By contrast, the livers of the
derivative #5- and DMSO-treated mice appeared normal and were
similar in morphology. In addition, the weight of the tumor cells
in the ascites of the derivative #5-treated group was significantly
lower than that of the DMSO-treated group (Fig. 6D). IFA confirmed that the tumor
cells in the ascites of DMSO-treated PEL-mice were derived from
administered BCBL1 cells as these tumor cells expressed LANA, a
marker of KSHV latent infection (Fig.
6E). These results indicated that xenograft BCBL1-derived tumor
cells developed in the ascites of DMSO-treated control mice, and
that derivative #5 prevented BCBL1-derived tumor cell development
in the ascites.
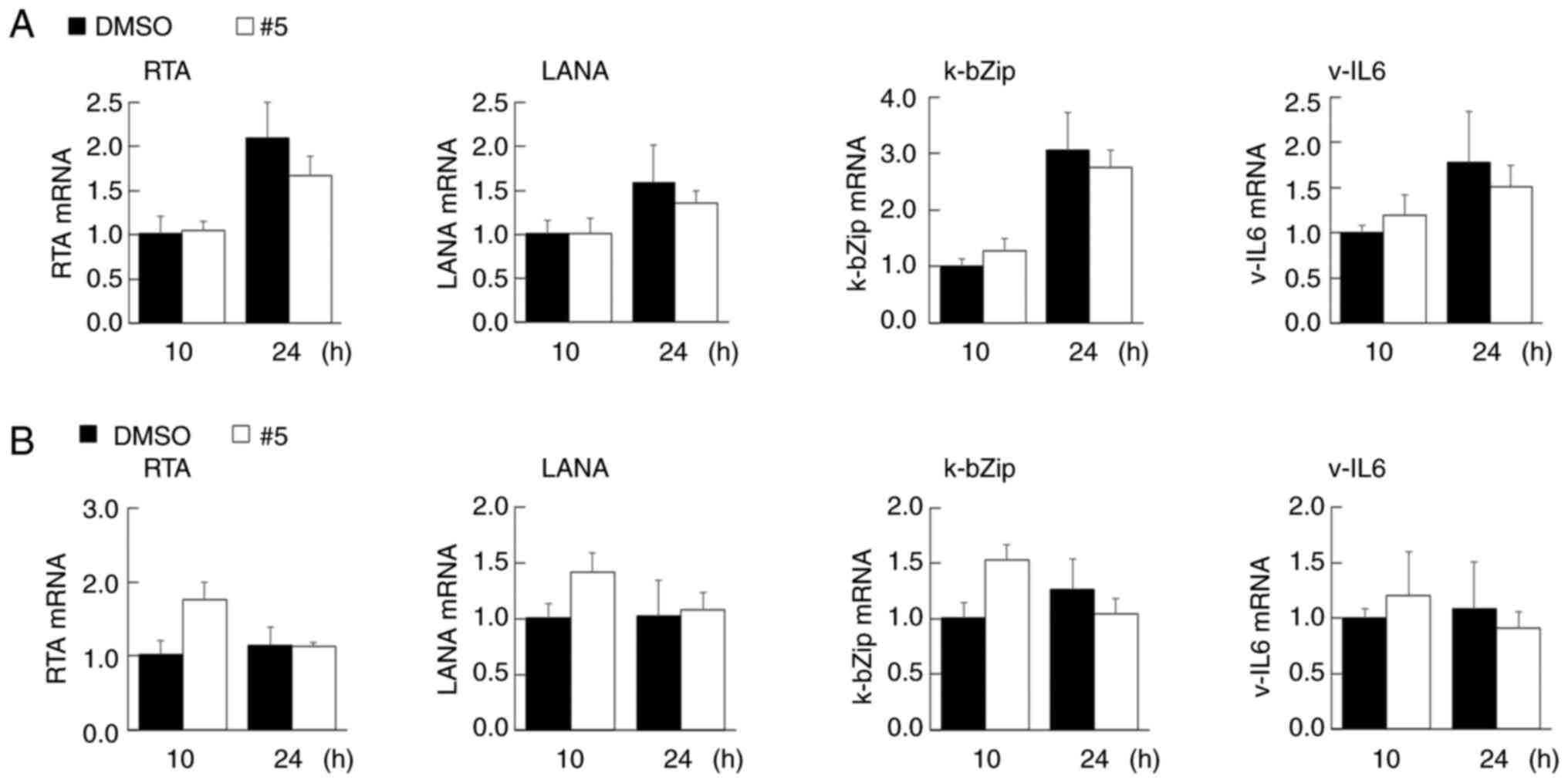
Derivative #5 does not affect the KSHV
latent infection of PEL cells and does not induce lytic
replication
In KSHV lytic replication (4,5),
virions are produced in PEL cells and are subsequently released,
resulting in cell death. During reactivation, lytic genes are
expressed in an orderly manner and are sequentially expressed. The
lytic genes are divided into three transcriptional stages:
Immediate early, early and late. KSHV lytic replication is
triggered by the expression of the replication transcription
activator (RTA)/ORF50, which is an immediate early gene product and
viral transcription factor that transcriptionally activates the
early genes, such as k-bZip (5).
In the present study, since the viability of the PEL cells were
decreased to ~50% following treatment with derivative #5 (Fig. 2E), it was then determined whether
derivative #5 induces lytic replication in KSHV latently infected
BC3 (Fig. 7A) and BCBL1 (Fig. 7B) cells. It was found that
treatment with 20 µM of derivative #5 did not induce the mRNA
expression of the immediate early gene, RTA/ORF50, nor that of the
early gene, k-bZip. Furthermore, in the BC3 and BCBL1 cells,
derivative #5 did not influence the transcription of the latent
genes, LANA and v-IL6. These results indicated that derivative #5
suppressed proliferation of PEL cells without the production of
nascent virus.
Discussion
The authors previously reported that a
pyrrolidinium-type fullerene (derivative #1) induced the generation
of ROS (16) and exhibited
anti-viral activities by inhibiting viral enzymes such as HIV RT
(19), HCV NS5B (19) and influenza virus endonuclease
(20). It was also found that
derivative #1 suppressed PEL cell proliferation by interfering with
the phosphorylation of AKT and procaspase-9 (21). Therefore, the present study focused
on the cation-functionalized moiety of derivative #1 and evaluated
other cationic fullerene derivatives containing pyrrolidinium,
pyridinium or anilinium (derivatives #2-#9) (16,17,19,21,23–25).
All the derivatives suppressed the growth of not only PEL cell
lines, but also that of KSHV-uninfected B cell lines. In addition,
derivatives #5, #6, #8 and #9 tended to decrease the number of
viable KSHV-infected PEL cells compared with the KSHV-uninfected
cells (Fig. 2). In addition, among
the tested derivatives, derivatives #5 and #6 exerted potent growth
inhibitory effects against PEL cells. Considering the consensus
structure of derivative #5 and #6, it was hypothesized that the
pyrrolidine ring of fullerene needs to possess one
3-N-methylpyridinium (or 3-N-ethylpyridinium) moiety and one
COOC2H5 moiety at the appropriate position in
order to exert selective and potent antitumor activity against PEL
cells.
To reveal the mechanisms through which derivative #5
decreases PEL cell viability, the cleavage and activation of
caspases was evaluated. However, both were not detected in PEL
cells treated with derivative #5 (Fig.
3A and B). The cell cycle analysis and LDH assay disclosed that
derivative #5 increased the number of dead cells and the cells with
a sub-G1 DNA content (Fig. 3C-E).
These findings suggested that the derivative #5 induced
caspase-independent cell death and decreased PEL cell viability. On
the other hand, derivatives #5, #3 and #8 formed cell clumps of
KSHV-infected PEL and uninfected B cells. Of note, derivative #5
prominently induced cell-to-cell clumping (Figs. 3F and S1). It was hypothesized that there may
be an association between cell clumping formation and cell death
induced by derivative #5. However, the mechanisms of
fullerene-dependent clumping remain unclear and warrant further
investigation in future studies.
In the present study, it was discovered that
derivative #5 exhibited preferential cytotoxic activity against
KSHV latently infected PEL cells compared with KSHV-uninfected B
cells. KSHV constitutively activates Wnt/β-catenin signaling in PEL
cells (4–9), which may be related to the higher
sensitivity of PEL cells to derivative #5. The KSHV-uninfected
cells were less sensitive to derivative #5, which may be explained
by their Wnt/β-catenin signaling-independent cell proliferation.
The results indicated that derivative #5 suppressed Wnt/β-catenin
signaling in PEL cells thorough the destabilization of β-catenin.
To the best of our knowledge, this is the first study to describe
the dysregulation of Wnt/β-catenin signaling by a water-soluble
fullerene derivative.
Wnt signaling is involved in several critical
developmental processes and in tumorigenesis. The Wnt/β-catenin
pathway regulates the availability of nuclear β-catenin (37) (Fig.
8). In the absence of Wnt signaling, β-catenin is held in a
complex with Axin, APC, and GSK-3β. The Axin-APC complex functions
as a platform for the association of GSK-3β and β-catenin.
Phosphorylated β-catenin is conjugated to polyubiquitin and then
degraded by the 26S proteasome (37,38).
The Wnt signaling cascade is triggered when the Wnt ligand binds to
the Frizzled receptor, which leads to downregulation of the
Axin-APC-GSK-3β complex through Dvl, LRP and FRAT. Subsequently
β-catenin is stabilized and it translocates to the nucleus and
forms a complex with the transcription factor TCF4. The complex of
β-catenin and TCF4 transcriptionally activates specific target
genes (CCND1, c-MYC, etc.) (39,40).
Wnt/β-catenin signaling is upregulated in various tumors, including
colorectal cancer, which has mutations in APC and β-catenin
(41,42). These mutations ultimately result in
the stabilization of β-catenin. The authors previously found that
KSHV LANA stabilizes β-catenin. In KSHV-infected cells, β-catenin
is stabilized by the following mechanisms: i) GSK-3β enters the
nucleus during S-phase; ii) LANA binds to nuclear GSK-3β; iii) LANA
inhibits nuclear export of GSK-3β and GSK-3β accumulates in the
nucleus; and iv) the reduced cytoplasmic abundance of GSK-3β allows
for the stabilization and nuclear entry of β-catenin (6–9). The
present study examined the mechanisms underlying derivative
#5-mediated β-catenin destabilization. Derivative #5 did not affect
the mRNA expression of β-catenin (Fig.
4C). Moreover, derivative #5 had no effect on the level of
GSK-3β, on phosphorylation of GSK-3β (Fig. 4A), on 26S proteasome activity
(Fig. 5A) and on the interaction
of GSK-3β and LANA (Fig. 5F). The
data also confirmed that GSK-3β induced β-catenin destabilization,
and LANA stabilized β-catenin even in the presence of GSK-3β.
Notably, derivative #5 induced the phosphorylation of β-catenin at
Ser33/Ser37/Thr41 (Fig. 5C and D),
which resulted in the polyubiquitination of β-catenin and
subsequent proteasomal degradation. The data revealed that
derivative #5 overcame LANA-mediated β-catenin stabilization via
the enhancement of β-catenin phosphorylation, which leads to
β-catenin polyubiquitination and degradation.
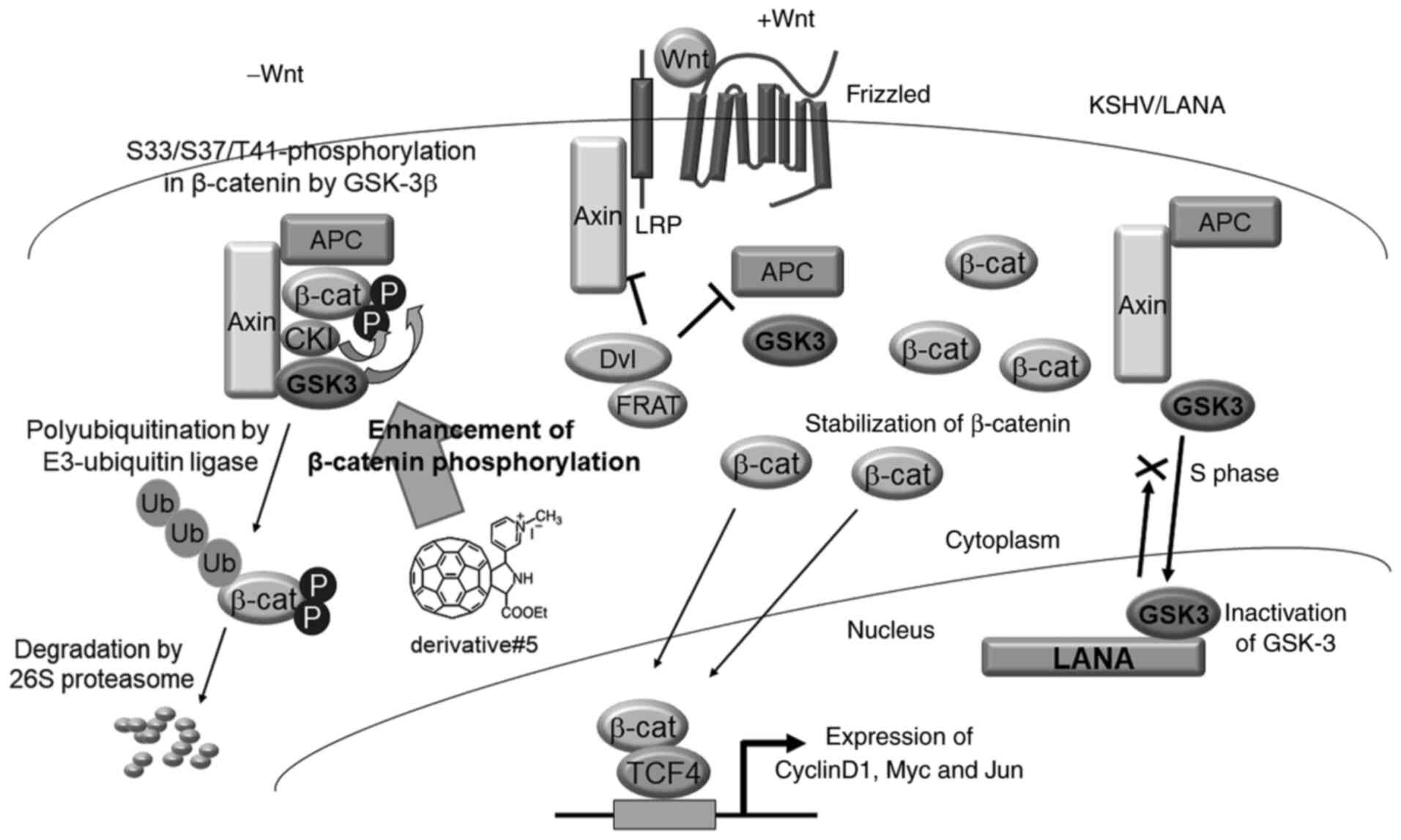 | Figure 8.Model of derivative #5-mediated
downregulation of Wnt/β-catenin signaling. Under normal conditions
or in the absence of Wnt ligand, Wnt/β-catenin signaling is
downregulated, as β-catenin is degraded in the following sequential
fashion: i) The phosphorylation of β-catenin at Ser33/Ser37/Thr41
by GSK-3β induces the formation of a complex containing β-catenin,
Axin and APC; ii) the polyubiquitination of the phosphorylated
β-catenin by E3 ubiquitin ligase; and iii) the degradation of
polyubiquitinated β-catenin by the 26S proteasome. In the presence
of Wnt ligand, β-catenin is stabilized by Dvl- and LRP-mediated
disassociation of a complex containing GSK-3β, Axin and APC. In
KSHV-infected cells, KSHV LANA interacts with GSK-3β in the
nucleus, leading to β-catenin stabilization and subsequent
activation of Wnt/β-catenin signaling. Derivative #5 induces the
phosphorylation-dependent destabilization of β-catenin, leading to
downregulation of Wnt/β-catenin signaling. APC, adenomatous
polyposis coli; CK1, casein kinase 1; β-cat, β-catenin; circled P,
phospho; Dvl, Dishevelled; FRAT, frequently rearranged in advanced
T-cell lymphomas; GSK3, glycogen synthase kinase-3β; LANA,
latency-associated nuclear antigen; circled Ub, ubiquitin; LRP,
low-density lipoprotein receptor-related protein and TCF4, T-cell
factor and lymphoid enhancer factor. |
The phosphorylation of β-catenin is essential for
polyubiquitination mediated by the E3 ubiquitin ligase,
SCFβTrCP. SCFβTrCP recognizes and binds the
consensus sequence (D-pS-G-XX-pS) on substrates (43). GSK-3β phosphorylates Ser and Thr
residues in the phosphorylation consensus sequence (S/T-XXX-pS/pT).
Before GSK-3β-mediated phosphorylation, the downstream Ser or Thr
in the sequence (S/T-XXX-S/T) has to first be phosphorylated by a
priming kinase. The priming kinase for GSK-3β is casein kinase 1
(CK1), which phosphorylates Ser45 in β-catenin. Following the
CK1-mediated phosphorylation of β-catenin at Ser45, GSK-3β
phosphorylates the Ser33, Ser37 and Thr41 residues in β-catenin
(44,45). Since the present study demonstrated
that derivative #5 increased both the phosphorylation of β-catenin
at Ser33/Ser37/Thr41 (Fig. 5C and
D) and the polyubiquitination of β-catenin (Fig. 5E), it was thus hypothesized that
derivative #5 would enhance GSK-3β-mediated β-catenin
phosphorylation. In addition, it was hypothesized that derivative
#5 may affect the priming phosphorylation (i.e., Ser45 of
β-catenin) by CK1.
In order to improve the aqueous solubility of
fullerene, various hydrophilic groups were added to the fullerene
core. It has been reported that water-soluble fullerene derivatives
exhibit unique and characteristic pharmacological effects, as
mentioned in the Introduction section. In the present study,
fullerene derivatives containing a pyrrolidinium or pyridinium
moiety as the hydrophilic group were generated, and nine cationic
fullerene derivatives were evaluated. It was previously found that
derivatives #1 and #5 displayed HIV RT inhibitory activity
(24,25). Moreover, derivative #7 inhibited
both HIV RT and HIV protease (24). Derivatives #1 and #5 also exerted
influenza virus endonuclease (20)
and HCV NS5B inhibitory activities (19), respectively. Derivatives #1, #3 and
#5 suppressed cancer cell proliferation (21,23).
Furthermore, it was disclosed that derivatives #1 and #5
dysregulated the AKT (21) and
Wnt/β-catenin (the present study) signaling pathways, respectively.
Based on the aforementioned biological properties of the
pyrrolidinium/pyridinium-type fullerene derivatives, these
derivatives may be utilized as novel compounds for the treatment of
various types of cancer.
The present study aimed to find the effective
anticancer compounds for PEL and to disclose underlying the
molecular mechanisms leading to their anticancer effects. To
achieve this, nine fullerene derivatives were synthesized. The
strength of the study lies in the novelty of compounds. The authors
designed and synthesized these fullerene derivatives with a yield
of 50-100 mg per compound. However, the present study had a
research resource limitation; the authors could not unrestrictedly
use derivative #5. Thus, several western blotting experiments for
quantitative analysis (Figs. 3A,
4A, 5B-E and S2) could not be repeated more than three
times. However, these data were confirmed twice using western blot
analysis using independent samples. Further studies to identify the
simple and high-yielding synthetic approach for fullerene
derivatives are currently underway.
In conclusion, the present study demonstrated that
the pyridinium-type fullerene derivative #5 (3- [5′-
(ethoxycarbonyl) - 1′, 5′- dihydro- 2′H- [5, 6] fullereno-
C60- Ih- [1, 9- c] pyrrol- 2′- yl] - 1-
methylpyridinium iodide) suppressed Wnt/β-catenin signaling in PEL
cells via β-catenin downregulation, which resulted in a decrease in
PEL cell viability. These biological functions of derivative #5
serve as an anti-proliferative effector in PEL cells. Therefore,
KSHV-infected PEL cells may be more sensitive to the
anti-proliferative effect of derivative #5 than KSHV-uninfected
cells, indicating that derivative #5 may serve as an effective
treatment for PEL and KSHV-associated cancers.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported in part by the JSPS Grant-in-Aid
for Scientific Research (18K06642).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
AK, MM, TW, YS, TO, TM and MF designed the
experiments. AK performed the experiments and collected the data.
AK and MM analyzed and interpreted the data. TY, SN, TA, TO and TM
designed and synthesized the fullerene derivatives. AK drafted the
manuscript, and TO and MF reviewed and edited the manuscript. TO
and MF confirm the authenticity of all the raw data. All the
authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
All experimental methods were performed in
accordance with relevant guidelines and regulations. The animal
protocol was approved by the committee on the Ethics of Animal
Research of Kyoto Pharmaceutical University (Approval no. 18-039),
and all experiments were performed in accordance with the National
Institutes of Health Guide for Care and Use of Laboratory
Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AIDS
|
acquired immunodeficiency
syndrome
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
HCV
|
hepatitis C virus
|
|
HIV
|
human immunodeficiency virus
|
|
HHV-8
|
human herpes virus-8
|
|
IκB
|
inhibitor of NF-κB
|
|
KSHV
|
Kaposi's sarcoma-associated
herpesvirus
|
|
LANA
|
latency-associated nuclear
antigen
|
|
p38 MAPK
|
p38 mitogen-activated protein
kinase
|
|
MEK
|
mitogen-activated protein kinase
kinase
|
|
PEL
|
primary effusion lymphoma
|
|
RT
|
reverse transcriptase
|
References
|
1
|
Russo JJ, Bohenzky RA, Chien MC, Chen J,
Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y and
Moore PS: Nucleotide sequence of the Kaposi sarcoma-associated
herpesvirus (HHV8). Proc Natl Acad Sci USA. 93:14862–14867. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nador RG, Cesarman E, Chadburn A, Dawson
DB, Ansari MQ, Sald J and Knowles DM: Primary effusion lymphoma: A
distinct clinicopathologic entity associated with the Kaposi's
sarcoma-associated herpes virus. Blood. 88:645–656. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chang Y, Cesarman E, Pessin MS, Lee F,
Culpepper J, Knowles DM and Moore PS: Identification of
herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma.
Science. 266:1865–1869. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Watanabe T, Sugimoto A, Hosokawa K and
Fujimuro M: Signal transduction pathways associated with
KSHV-related tumors. Human Herpesviruses. Kawaguchi Y, Mori Y and
Kimura H: Springer; Berlin/Heidelberg: pp. 321–355. 2018,
View Article : Google Scholar
|
|
5
|
Damania B and Cesarman E: Kaposi's
sarcoma-associated herpesvirus. Fields Virology. 6th edition. Knipe
DM and Howley PM: Lippincott Williams & Wilkins; pp. 2080–2128.
2013
|
|
6
|
Fujimuro M, Wu FY, ApRhys C, Kajumbula H,
Young DB, Hayward GS and Hayward SD: A novel viral mechanism for
dysregulation of beta-catenin in Kaposis sarcoma-associated
herpesvirus latency. Nat Med. 9:300–306. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fujimuro M and Hayward SD: The
latency-associated nuclear antigen of Kaposi's sarcoma-associated
herpesvirus manipulates the activity of glycogen synthase
kinase-3beta. J Virol. 77:8019–8030. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fujimuro M, Liu J, Zhu J, Yokosawa H and
Hayward SD: Regulation of the interaction between glycogen synthase
kinase 3 and the Kaposi's sarcoma-associated herpesvirus
latency-associated nuclear antigen. J Virol. 79:10429–10441. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hayward SD, Liu J and Fujimuro M: Notch
and Wnt signaling: Mimicry and manipulation by gamma herpesviruses.
Sci STKE. 2006:re42006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kroto HW, Heath JR, O'Brien SC, Curl RF
and Smalley RE: C60: Buckminsterfullerene. Nature.
318:162–163. 1985. View Article : Google Scholar
|
|
11
|
Zakharian TY, Seryshev A, Sitharaman B,
Gilbert BE, Knight V and Wilson LJ: A fullerene-paclitaxel
chemotherapeutic: Synthesis, characterization, and study of
biological activity in tissue culture. J Am Chem Soc.
127:12508–12509. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chaudhuri P, Paraskar A, Soni S, Mashelkar
RA and Sengupta S: Fullerenol-cytotoxic conjugates for cancer
chemotherapy. ACS Nano. 3:2505–2514. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Isobe H, Nakanishi W, Tomita N, Jinno S,
Okayama H and Nakamura E: Nonviral gene delivery by tetraamino
fullerene. Mol Pharm. 3:124–134. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tokuyama H, Yamago S, Nakamura E, Shiraki
T and Sugiura Y: Photoinduced biochemical activity of fullerene
carboxylic acid. J Am Chem Soc. 115:7918–7919. 1993. View Article : Google Scholar
|
|
15
|
Okuda K, Mashino T and Hirobe M:
Superoxide radical quenching and cytochrome c peroxidase-like
activity of C60-dimalonic acid,
C64(COOH)4. Bioorg Med Chem Lett. 6:539–542.
1996. View Article : Google Scholar
|
|
16
|
Nishizawa C, Hashimoto N, Yokoo S,
Funakoshi-Tago M, Kasahara T, Takahashi K, Nakamura S and Mashino
T: Pyrrolidinium-type fullerene derivative-induced apoptosis by the
generation of reactive oxygen species in HL-60 cells. Free Radical
Res. 43:1240–1247. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mashino T, Nishikawa D, Takahashi K, Usui
N, Yamori T, Seki M, Endo T and Mochizuki M: Antibacterial and
antiproliferative activity of cationic fullerene derivatives.
Bioorg Med Chem Lett. 13:4395–4397. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Friedman SH, DeCamp DL, Sijbesma RP,
Srdanov G, Wudl F and Kenyon GL: Inhibition of the HIV-1 protease
by fullerene derivatives: Model building studies and experimental
verification. J Am Chem Soc. 115:6506–6509. 1993. View Article : Google Scholar
|
|
19
|
Mashino T, Shimotohno K, Ikegami N,
Nishikawa D, Okuda K, Takahashi K, Nakamura S and Mochizuki M:
Human immunodeficiency virus-reverse transcriptase inhibition and
hepatitis C virus RNA-dependent RNA polymerase inhibition
activities of fullerene derivative. Bioorg Med Chem Lett.
15:1107–1109. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shoji M, Takahashi E, Hatakeyama D, Iwai
Y, Morita Y, Shirayama R, Echigo N, Kido H, Nakamura S, Mashino T,
et al: Anti-influenza activity of C60 fullerene
derivatives. PLoS One. 8:e663372013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Watanabe T, Nakamura S, Ono T, Ui S, Yagi
S, Kagawa H, Watanabe H, Ohe T, Mashino T and Fujimuro M:
Pyrrolidinium fullerene induces apoptosis by activation of
procaspase-9 via suppression of Akt in primary effusion lymphoma.
Biochem Biophys Res Commun. 451:93–100. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cardone MH, Roy N, Stennicke HR, Salvasen
GS, Franke TF, Stanbridge E, Frisch S and Reed JC: Regulation of
cell death protease caspase-9 by phosphorylation. Science.
282:1318–1321. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yasuno T, Ohe T, Ikeda H, Takahashi K,
Nakamura S and Mashino T: Synthesis and antitumor activity of novel
pyridinium fullerene derivatives. Int J Nanomedicine. 14:6325–6337.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yasuno T, Ohe T, Kataoka H, Hashimoto K,
Ishikawa Y, Furukawa K, Tateishi Y, Kobayashi T, Takahashi K,
Nakamura S and Mashino T: Fullerene derivatives as dual inhibitors
of HIV-1 reverse transcriptase and protease. Bioorg Med Chem Lett.
31:1276752021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yasuno T, Ohe T, Takahashi K, Nakamura S
and Mashino T: The human immunodeficiency virus-reverse
transcriptase inhibition activity of novel pyridine/pyridinium-type
fullerene derivatives. Bioorg Med Chem Lett. 25:3226–3229. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shigemi Z, Furukawa Y, Hosokawa K, Minami
S, Matsuhiro J, Nakata S, Watanabe T, Kagawa H, Nakagawa K, Takeda
H and Fujimuro M: Diallyl trisulfide induces apoptosis by
suppressing NF-κB signaling through destabilization of TRAF6 in
primary effusion lymphoma. Int J Oncol. 48:293–304. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fujimuro M, Sawada H and Yokosawa H:
Production and characterization of monoclonal antibodies specific
to multi-ubiquitin chains of polyubiquitinated proteins. FEBS Lett.
349:172–180. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen C and Okayama H: High-efficiency
transformation of mammalian cells by plasmid DNA. Mol Cell Biol.
7:2745–2752. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Takahashi-Makise N, Suzu S, Hiyoshi M,
Ohsugi T, Katano H, Umezawa K and Okada S: Biscoclaurine alkaloid
cepharanthine inhibits the growth of primary effusion lymphoma in
vitro and in vivo and induces apoptosis via suppression of the
NF-kappaB pathway. Int J Cancer. 125:1464–1472. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim YJ, Kim Y, Kumar A, Kim CW, Toth Z,
Cho NH and Lee HR: Kaposi's sarcoma-associated herpesvirus
latency-associated nuclear antigen dysregulates expression of MCL-1
by targeting FBW7. PLoS Pathog. 17:e10091792021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Saji C, Higashi C, Niinaka Y, Yamada K,
Noguchi K and Fujimuro M: Proteasome inhibitors induce apoptosis
and reduce viral replication in primary effusion lymphoma cells.
Biochem Biophys Res Commun. 415:573–578. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Higashi C, Saji C, Yamada K, Kagawa H,
Ohga R, Taira T and Fujimuro M: The effects of heat shock protein
90 inhibitors on apoptosis and viral replication in primary
effusion lymphoma cells. Biol Pharm Bull. 35:725–730. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ishiura Y, Ishimaru H, Watanabe T and
Fujimuro M: Sulforaphane exhibits cytotoxic effects against primary
effusion lymphoma cells by suppressing p38MAPK and AKT
phosphorylation. Bio Pharm Biol. 42:2109–2112. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Moriguchi M, Watanabe T, Kadota A and
Fujimuro M: Capsaicin induces apoptosis in KSHV-pisitive primary
effusion lymphoma by suppressing ERK and p38 MAPK signaling and
IL-6 expression. Front Oncol. 9:832019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wakao K, Watanabe T, Takadama T, Ui S,
Shigemi Z, Kagawa H, Higashi C, Ohga R, Taira T and Fujimuro M:
Sangivamycin induces apoptosis by suppressing Erk signaling in
primary effusion lymphoma cells. Biochem Biophys Res Commun.
444:135–140. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Willert K and Jones KA: Wnt signaling: Is
the party in the nucleus? Genes Dev. 20:1394–1404. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kimelman D and Xu W: beta-catenin
destruction complex: Insights and questions from a structural
perspective. Oncogene. 25:7482–7491. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Albrecht LV, Tejeda-Muñoz N and De
Robertis EM: Cell biology of canonical Wnt signaling. Annu Rev Cell
Dev Biol. 37:369–389. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hoppler S and Kavanagh CL: Wnt signalling:
variety at the core. J Cell Sci. 120:385–393. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Segditsas S and Tomlinson I: Colorectal
cancer and genetic alterations in the Wnt pathway. Oncogene.
25:7531–7537. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Clements WM, Wang J, Sarnaik A, Kim OJ,
MacDonald J, Fenoglio-Preiser C, Groden J and Lowy AM: beta-Catenin
mutation is a frequent cause of Wnt pathway activation in gastric
cancer. Cancer Res. 62:3503–3506. 2002.PubMed/NCBI
|
|
43
|
Nakayama KI and Nakayama K: Ubiquitin
ligases: Cell-cycle control and cancer. Nat Rev Cancer. 6:369–381.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu C, Li Y, Semenov M, Han C, Baeg GH,
Tan Y, Zhang Z, Lin X and He X: Control of beta-catenin
phosphorylation/degradation by a dual-kinase mechanism. Cell.
108:837–847. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Amit S, Hatzubai A, Birman Y, Andersen JS,
Ben-Shushan E, Mann M, Ben-Neriah Y and Alkalay I: Axin-mediated
CKI phosphorylation of β-catenin at Ser 45: A molecular switch for
the Wnt pathway. Genes Dev. 16:1066–1076. 2002. View Article : Google Scholar : PubMed/NCBIPubMed/NCBI
|