Introduction
The phosphatidylinositol 3-kinase/mammalian target
of rapamycin (PI3K/mTOR) signaling pathway plays an important role
in cellular proliferation, growth, and survival by integrating
signals from growth factors, cytokines, and other environmental
sources (1,2). PI3K/mTOR inhibitors are therapeutic
agents developed for various types of human tumors (3). These inhibitors include PI3K
inhibitors, Akt inhibitors, mTOR complex 1 (mTORC1) inhibitors, and
dual PI3K/mTOR inhibitors that inhibit PI3K catalytic isoforms,
mTORC1, and mTOR complex 2 (mTORC2). Dual PI3K/mTOR inhibitors are
more potent because they suppress the feedback re-activation that
limits the efficacy of mTORC1 inhibitors (4,5).
Studies of dual PI3K/mTOR inhibitors indicate that
several molecular mechanisms affect antitumor activity. In the
PI3K/mTOR signaling pathway, mutations in the PIK3CA gene,
which encodes the PI3Kα isoform, and the loss of phosphatase and
tensin homolog (PTEN) increase sensitivity to dual PI3K/mTOR
inhibitors (6). In contrast, various
biological processes, including compensatory signaling pathways
(7,8), the epithelial-mesenchymal transition
(9), and drug efflux with ATP
binding cassette transporters (10)
mediate cellular resistance to dual PI3K/mTOR inhibitors. Despite
these discoveries, strategies to overcome resistance mechanisms and
develop biomarkers associated with clinical outcomes are not
established. Therefore, the identification of the molecular
determinants that affect the antitumor activity of these inhibitors
is necessary to maximize the clinical outcomes for dual PI3K/mTOR
inhibitor treatments (3).
The PI3K/mTOR signaling pathway is activated in
specific canine tumors, including hemangiosarcoma (11,12),
mammary carcinoma (12,13), glioma (12), lymphoma (12), mast cell tumor (12,14),
osteosarcoma (15,16), and melanoma (17–19).
Dual PI3K/mTOR inhibitors decreased cell viability and induced
apoptosis, primarily in melanoma and hemangiosarcoma in
vitro (19,20). However, the molecular mechanisms
involved in the cellular resistance are not clear in any canine
tumor.
The aim of the present study was to investigate the
in vitro antitumor activity of gedatolisib, a dual PI3K/mTOR
inhibitor, against various canine tumors, and to explore the
molecular determinants involved in the cellular sensitivity to
gedatolisib. The antitumor activity of gedatolisib on cell
viability, protein phosphorylation, and cell cycle distribution was
assessed using 12 canine tumor cell lines from six types of tumor.
In addition, the involvement of serum-and-glucocorticoid-regulated
kinase 1 (SGK1), PIK3CA, and ATP-binding cassette, subfamily
B, member 1 (ABCB1) was investigated in gedatolisib resistance.
Materials and methods
Cell lines and culture
Two canine osteosarcoma cell lines (POS and HMPOS)
(21,22), four canine urinary bladder
transitional cell carcinoma (TCC; canine equivalent of
muscle-invading bladder cancer in humans) cell lines [MCTCC, LCTCC,
MegTCC, and MomoTCC, which was established at our laboratory
(Laboratory of Veterinary Surgery, Department of Clinical Sciences,
Graduate School of Veterinary Medicine, Sapporo, Japan) and
erroneously named as MonoTCC in a previous study] (23,24), two
canine malignant melanoma cell lines [CMeC, which was provided by
the University of Tokyo (Tokyo, Japan) and MCM-N1, which was
purchased by DS Pharma Biochemical Co., Ltd.] (25,26), two
canine histiocytic sarcoma cell lines (DH82, which was purchased by
DS Pharma Biochemical Co., Ltd. and CHS-4, which was provided by
the University of Tokyo) (27,28), a
canine mast cell tumor cell line (CoMS, which was established at
our laboratory) (29), a canine lung
adenocarcinoma cell line [CLAC, which was provided by Azabu
University, (Sagamihara, Japan)] (30), and a human embryonic kidney cell line
(293T, which was provided by Laboratory of Comparative Pathology,
Department of Clinical Sciences, Faculty of Veterinary Medicine,
Hokkaido University) were used in the present study. Each cell line
was maintained in Dulbecco's modified Eagle's medium (DMEM; Thermo
Fisher Scientific, Inc.) or RPMI-1640 medium (Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS;
Nichirei Biosciences, Inc.), 100 IU/ml penicillin G (Wako Pure
Chemical Industries, Ltd.), and 100 µg/ml streptomycin (Wako Pure
Chemical Industries, Ltd.) at 37°C and 5% CO2.
Inhibitors
Two mTORC1 inhibitors, rapamycin and everolimus,
were purchased from Adipogen Life Sciences and AdooQ Bioscience,
respectively. Two dual PI3K/mTOR inhibitors (gedatolisib and
PF-04691502), one Akt inhibitor (MK-2206), and one PI3K inhibitor
(BKM120) were purchased from AdooQ Bioscience. ABCB1 inhibitors,
cyclosporin A and tariquidar, were purchased from Wako Pure
Chemical Industries, Ltd. and MedChemExpress, respectively. A
receptor tyrosine kinase inhibitor, toceranib, was purchased from
Toronto Research Chemicals. All inhibitors were dissolved in
dimethyl sulfoxide (DMSO) and stored in aliquots at −30°C.
Cell viability assay
Cells were seeded at densities of
0.3–2×103 cells/well in 96-well plates (the density of
the cells was based on the growth rate of each cell line), allowed
to attach overnight, and treated with 0.1% DMSO (control) or
various concentrations (0.1 nM-10 µM) of each inhibitor for 96 h.
The cell viability was determined using a WST-1 assay kit (Takara
Bio, Inc.). The final concentration of premix WST-1 was 9% (10 µl
premix WST-1/100 µl culture medium), and the incubation period was
4 h at 37°C with 5% CO2. The absorbance was measured at
450 and 620 nm using a Multiskan FC microplate spectrophotometer
(Thermo Fisher Scientific, Inc.).
Immunoblotting
Cell lysis, sodium dodecyl sulfate-polyacrylamide
gel electrophoresis, and immunoblotting were performed as
previously described with minor modifications (31,32). The
protein concentrations of each lysate were quantified using a
Protein Quantification kit (Takara Bio, Inc.). The primary
antibodies and secondary antibody used in the present study are
listed in Table SI. The proteins
were visualized using the Western BLoT Ultra-Sensitive HRP
Substrate (Takara Bio, Inc.) and detected using an ImageQuant
LAS-4000 mini system (GE Healthcare; Cytiva).
To prepare the cell lysate, 6×104 cells
were seeded in 6 cm dishes and cultured for 48 h at 37°C and 5%
CO2. For evaluation of the dose-dependent suppression of
gedatolisib, the cells were treated with 0.1% DMSO or various
concentrations (0.1 nM-10 µM) of gedatolisib for 24 h. For
evaluation of the time-dependent suppression of gedatolisib, the
cells were treated with 0.1% DMSO or gedatolisib (100 nM or 10 µM)
for 0, 4, 8, 24 and 48 h. For immunoblotting analysis of p-NDRG1
(Thr346), the cells were treated with 0.1% DMSO or 100 nM
gedatolisib for 4 h.
Cell cycle analysis
Cells were seeded at 1–4×105 cells/10-cm
dish and incubated for 72 h at 37°C and 5% CO2. The
cells were then treated with DMSO or gedatolisib (0.1, 1, or 10 µM)
for 24 h, collected using 0.25% trypsin-ethylenediaminetetraacetic
acid (EDTA) in phosphate-buffered saline (PBS), and fixed with 70%
ethanol overnight at −30°C. The fixed cells were treated with RNase
A (10 µg/ml in PBS; EMD Millipore) for 30 min at 37°C and stained
with 50 µg/ml propidium iodide (PI) in PBS for 20 min at 25°C. The
DNA content of the cells was measured using a FACSVerse system (BD
Biosciences) equipped with a 488-nm argon laser and 527/32 and
700/54 nm bandpass filters. The percentage of cells at each stage
of the cell cycle was calculated by manually gating the histograms.
All data were analyzed using the FACSuite software 1.0 (BD
Biosciences).
Immunocytochemistry
Immunocytochemistry was performed as previously
described with minor modifications (33). Cells (0.2×105) were
cultured in an 8-well culture slide (Iwaki®) with 400 µl
of the medium containing 10% FBS. After treatment with 100 nM
gedatolisib for 24 h, the cells were fixed with pre-warmed PBS
containing 4% paraformaldehyde and 4% sucrose at 37°C for 10 min.
The cells were permeabilized with 0.2% Triton X-100 (ICN
Biomedicals) in PBS for 10 min and blocked with 5% FBS in PBS at
25°C for 30 min. The antibodies and probes used this assay are
presented in Table SI.
Establishment of SGK1-overexpressing
cell lines
The canine SGK1 sequence was identified using
cDNA from the CMeC cell line and transfected into the MegTCC cell
line using a lentiviral system. The primers used for cell line
establishment and the specific conditions of the polymerase chain
reaction (PCR) are summarized in Tables
SII and SIII, respectively.
Canine SGK1 was amplified using KOD -Plus- Ver. 2 (Toyobo
Life Science) and the fragment was ligated to the pTA2 vector
(Toyobo Life Science). This recombinant plasmid was introduced into
DH5α competent cells (Takara Bio, Inc.), purified using a
NucleoSpin Plasmid EasyPure kit (Takara Bio, Inc.), and sequenced
by the Kazusa DNA Research Institute (Chiba, Japan).
The recombinant plasmid was digested with
XhoI and BamHI (Takara Bio, Inc.), and the digested
fragment was cloned into CSII-CMV-MCS-IRES2-Bsd (developed by Dr H.
Miyoshi, Keio University, Tokyo, Japan). The plasmid was provided
by the Riken BioResource Research Center through the National
Bio-Resource Project of the Ministry of Education, Culture, Sports,
Science and Technology, Tokyo, Japan. The plasmid (4.6 µg) and
packing plasmids (2.7 µg each) were co-transfected into 293T cells
using Lipofectamine 3000 (Thermo Fisher Scientific, Inc.) for 15
min at 25°C. Then, the cells were further incubated for 4 h at 37°C
with 5% CO2 and medium was replaced with fresh
medium.
After co-transfection of the plasmids, the culture
supernatant was collected after 24 and 48 h, and the lentiviral
vector particles were concentrated using a Lenti-X™ Concentrator
(Takara Bio, Inc.). MegTCC cells were infected with the lentiviral
vector using polybrene (Nacalai Tesque, Inc.) and cultured in DMEM
supplemented with 10% FBS for 48 h at 37°C with 5% CO2.
The cells were exposed with 10 µg/ml blasticidin (Wako Pure
Chemical Industries, Ltd.) for 48 h at 37°C with 5% CO2,
and growing cells were collected and maintained in DMEM
supplemented with 10% FBS with 10 µg/ml blasticidin for further
passage. After at least 5 times passages, the cells were used for
subsequent experimentations.
As SGK1 function is post-transcriptionally regulated
(34), the mutated SGK1
(SGK1 Δ60 S422D), which encodes the constitutively active
form of SGK1 (35,36), was constructed. Inverse PCR was
performed using a KOD -Plus- Mutagenesis kit (Toyobo Life Science),
and this construct was introduced into competent cells. The same
procedure as for SGK1 was performed for transfection, virus
production, and infection of the mutated SGK1.
Gene mutation analysis in PIK3CA
The partial sequence of PIK3CA was amplified
using the cDNA from all previously described canine cell lines. The
primers and the specific conditions of the PCR reactions are
summarized in Tables SII and
SIII, respectively. Ex Taq DNA
polymerase (Takara Bio, Inc.) was used for the PCR reaction. The
PCR products were purified using a PCR clean-up gel extraction kit
(Takara Bio, Inc.). Direct sequencing using the purified PCR
products was performed at the Kazusa DNA Research Institute. The
predicted amino acid sequences of PIK3CA were aligned using
the Clustal W program (37).
Rhodamine-123 uptake/efflux assay
Cells were seeded at 1–3×105 cells/10-cm
dish and incubated for 96 h at 37°C and 5% CO2. The
cells were treated with rhodamine-123 (1.3 µM; Wako Pure Chemical
Industries, Ltd.) for 90 min at 37°C. The cells evaluated for
rhodamine-123 efflux were also incubated without rhodamine-123 for
90 min. These cells were collected using 0.25% trypsin-EDTA in PBS
and stained with 50 µg/ml PI in PBS for 5 min at 4°C. Rhodamine-123
and PI fluorescence intensity were measured with FACSVerse. Debris
and dead cells were eliminated by forward vs. side scatter gating
based on light and the PI intensity, respectively.
Gene accession numbers
The sequence data were submitted to the DNA Data
Bank of Japan database (http://getentry.ddbj.nig.ac.jp) under the accession
nos. LC424654-LC424655 and LC424656-LC424679 for SGK1 and
PIK3CA, respectively.
Statistical analysis
All statistical analyses were performed using
GraphPad Prism software 7.04 (GraphPad Software, Inc.). For the
cellular viability assay, IC50 values were calculated by
fitting dose-response curves to a three-parameter variable slope
sigmoidal dose-response model. For cell cycle analysis, only the
G0/G1 phase was compared between groups and
treatments. Significant differences between groups and treatments
were analyzed using the Mann-Whitney U test and multiple
t-tests (statistically significant differences in multiple
comparisons were corrected using the Bonferroni-Dunn method),
respectively. The sample number in each group was described in
figure legends. P-values of <0.05 were considered to indicate a
statistically significant difference.
Results
Gedatolisib decreases cell viability
in various types of canine tumor cell lines
Cell viability against the dual PI3K/mTOR inhibitor
gedatolisib compared with other PI3K/mTOR signaling pathway
inhibitors including rapamycin, everolimus, PF-04691502, MK-2206,
and BKM120 in 12 canine tumor cell lines from 6 types of tumor were
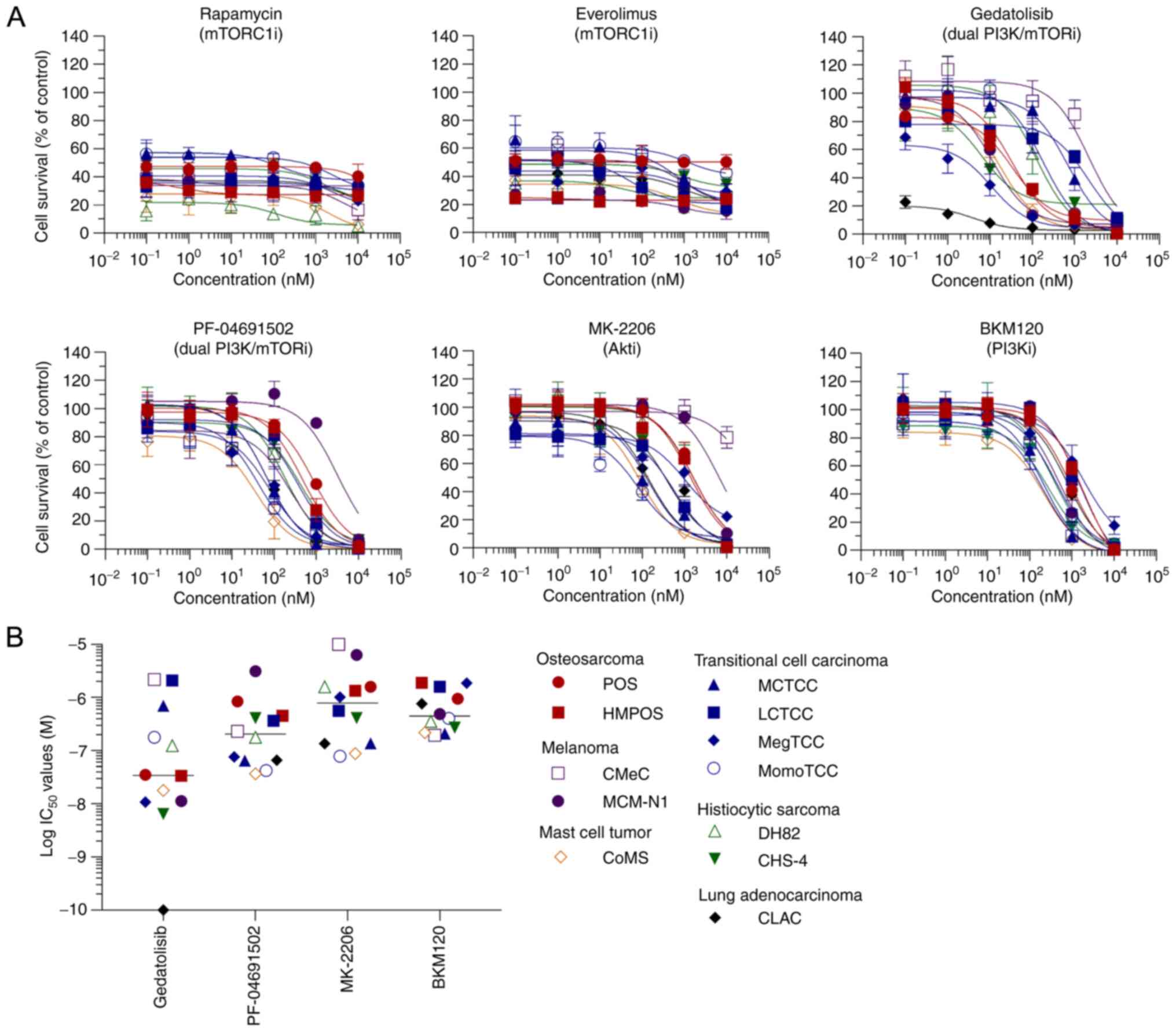
evaluated (Fig. 1). All inhibitors,
except for the mTORC1 inhibitors, decreased cell viability in a
dose-dependent manner (Fig. 2A).
Gedatolisib exhibited a marked decrease in cell viability with
lower median IC50 values than the other inhibitors and
IC50 values of <1 µM in 10 of the 12 cell lines
(Fig. 2B).
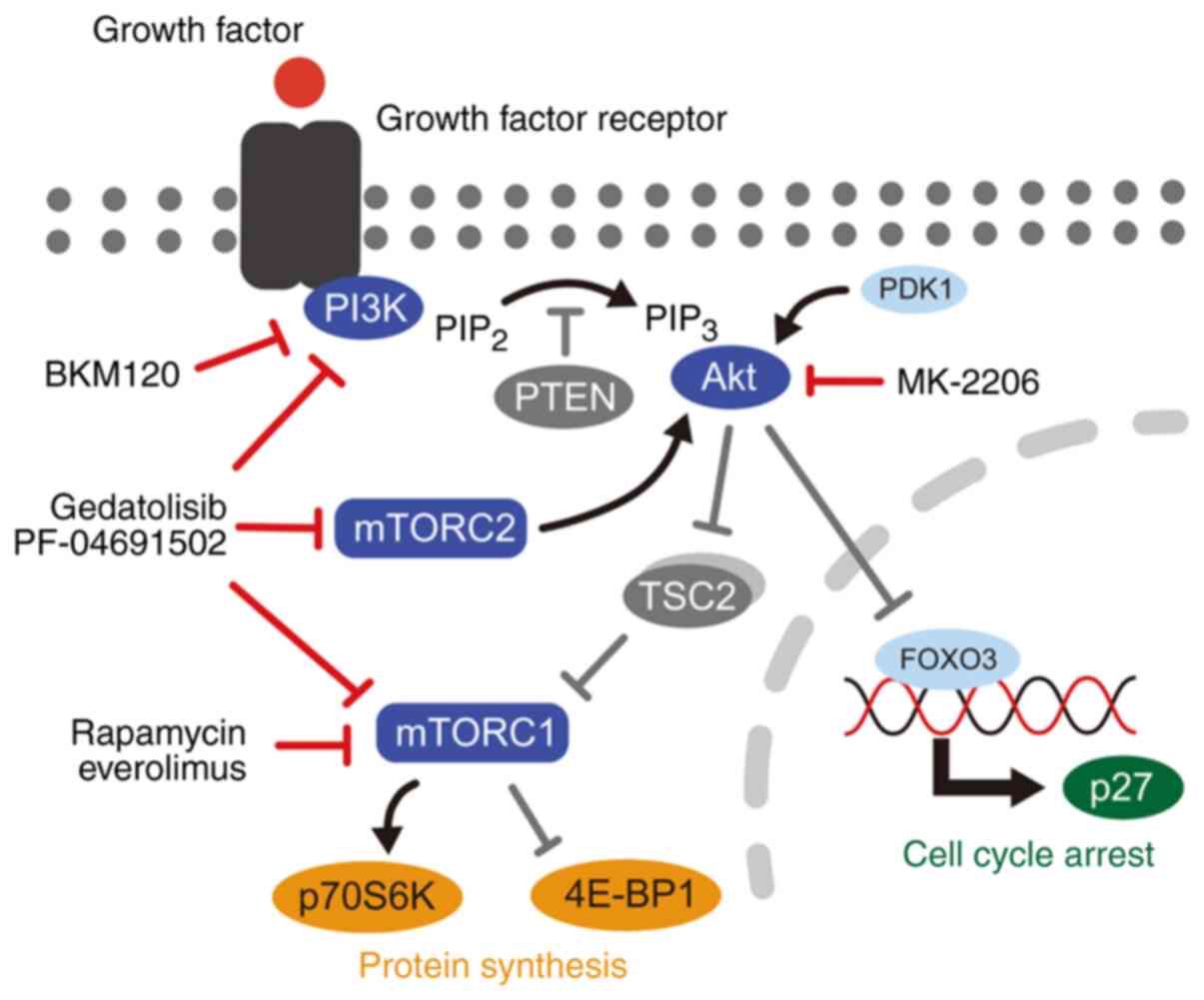 | Figure 1.Schematic diagram of the PI3K/mTOR
signaling pathway. PI3K/mTOR inhibitors used in the present study,
included rapamycin, everolimus, gedatolisib, PF-04691502, MK-2206,
and BKM120. PI3K, phosphatidylinositol 3-kinase; PIP2,
phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol
3,4,5-triphosphate; PTEN, phosphatase and tensin homolog; TSC2,
tuberous sclerosis complex 2; mTORC, mammalian target of rapamycin
complex; p70S6K, p70S6 kinase; 4E-BP1, 4E-binding protein 1; FOXO3,
forkhead box O3. |
PI3K/mTOR signaling pathway is
activated in canine tumor cell lines
To reveal the mode of action of gedatolisib and
explore the molecular determinants affecting cellular sensitivity
to this inhibitor, the basal expression and phosphorylation of
PI3K/mTOR signaling pathway molecules were examined, by verifying
the presence or absence of protein expression and phosphorylation
rather than semi-quantitative comparison. The phosphorylation of
p70S6 kinase (p70S6K) was clearly observed in all cell lines except
in MegTCC, MomoTCC, MCM-N1 and CoMS cells. The phosphorylation of
4E-binding protein 1 (4E-BP1) was clearly observed in TCCs, MCM-N1,
and CLAC cells. It was observed that mTORC1 and its substrates
(p70S6K and/or 4E-BP1) were phosphorylated in all cell lines, which
suggested that mTORC1 is active in these cell lines (Fig. 3A). The expression of PTEN and
tuberous sclerosis complex 2 (TSC2) was examined because the
deficiency of these suppressive regulators of the PI3K/mTOR
signaling pathway has been revealed to increase cell sensitivity to
PI3K/mTOR inhibitors (6,38,39). The
PTEN expression was not observed in five cell lines (POS, HMPOS,
MCM-N1, DH82, and CHS-4). However, there were no significant
differences in the IC50 values between PTEN-positive and
PTEN-negative/low cell lines (Fig.
3B). TSC2 expression was observed in all cell lines, which
indicated that TSC2 expression did not sensitize the low
IC50 cell lines against gedatolisib.
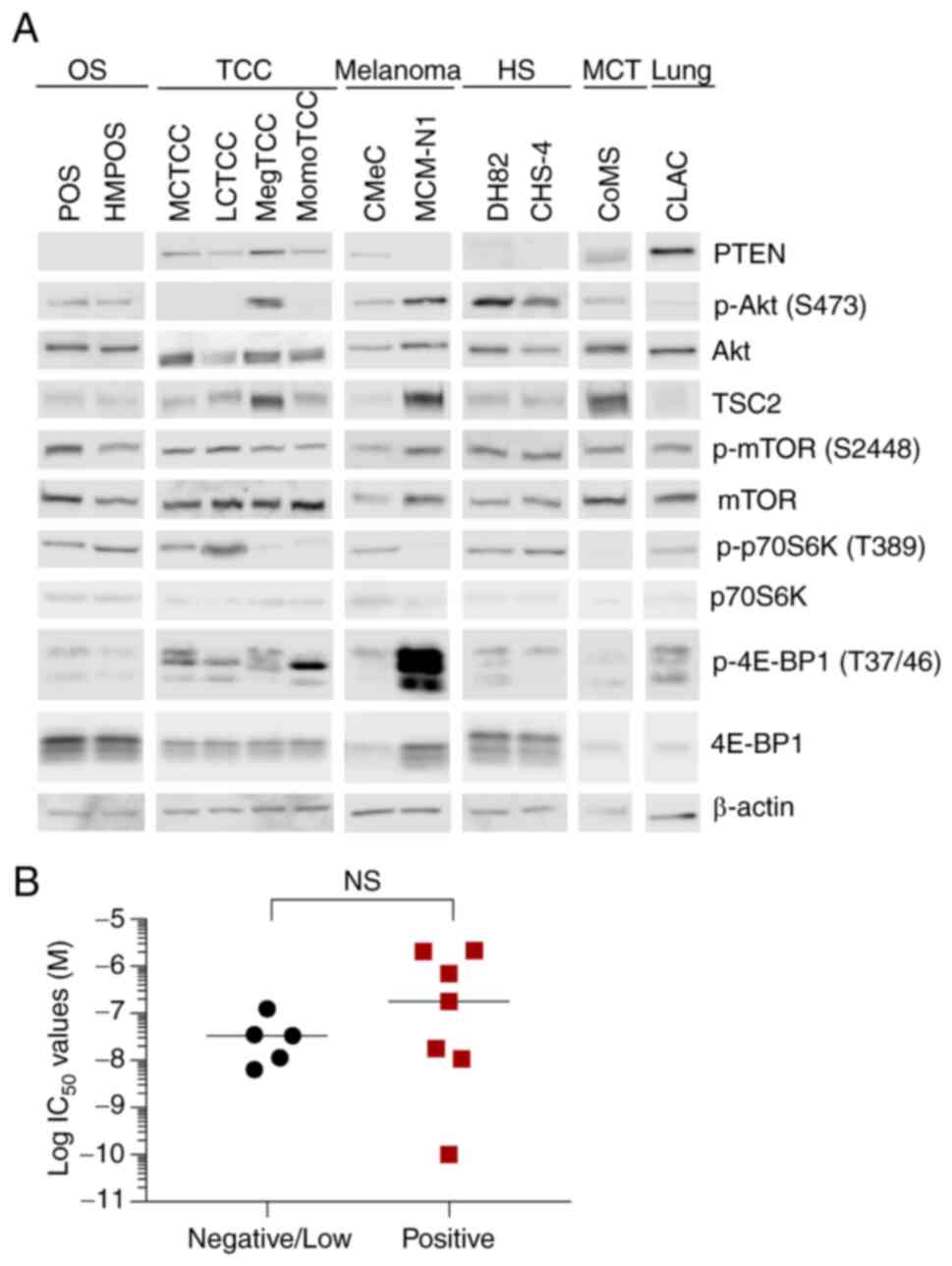 | Figure 3.Basal expression/phosphorylation
levels of PI3K/mTOR signaling pathway molecules in canine tumor
cell lines. The subtle bands of PTEN in DH82 and CHS-4 cell lines
were interpreted as artifacts due to their molecular weight. The
reason of these artifacts is unknown. (A) Immunoblotting analysis
of PTEN, p-Akt (Ser473), Akt, TSC2, p-mTOR (Ser2448), mTOR,
p-p70S6K (Thr389), p-4E-BP1 (Thr37/46), and β-actin in canine tumor
cell lines. The samples were run based on tumor type and grouping
of images was from different gels. The samples of CLAC cells were
run with other samples. (B) IC50 values of gedatolisib
in PTEN-positive (n=7) and PTEN-negative/low (n=5) canine tumor
cell lines. No significant differences were observed between the
groups in the IC50 values of gedatolisib (P=0.43,
Mann-Whitney U test). PI3K, phosphatidylinositol 3-kinase; mTOR,
mammalian target of rapamycin; PTEN, phosphatase and tensin
homolog; p-, phosphorylated; TSC2, tuberous sclerosis complex 2;
p70S6K, p70S6 kinase; 4E-BP1, 4E-binding protein 1; OS,
osteosarcoma; TCC, transitional cell carcinoma; HS, histiocytic
sarcoma; MCT, mast cell tumor; NS, not significant. |
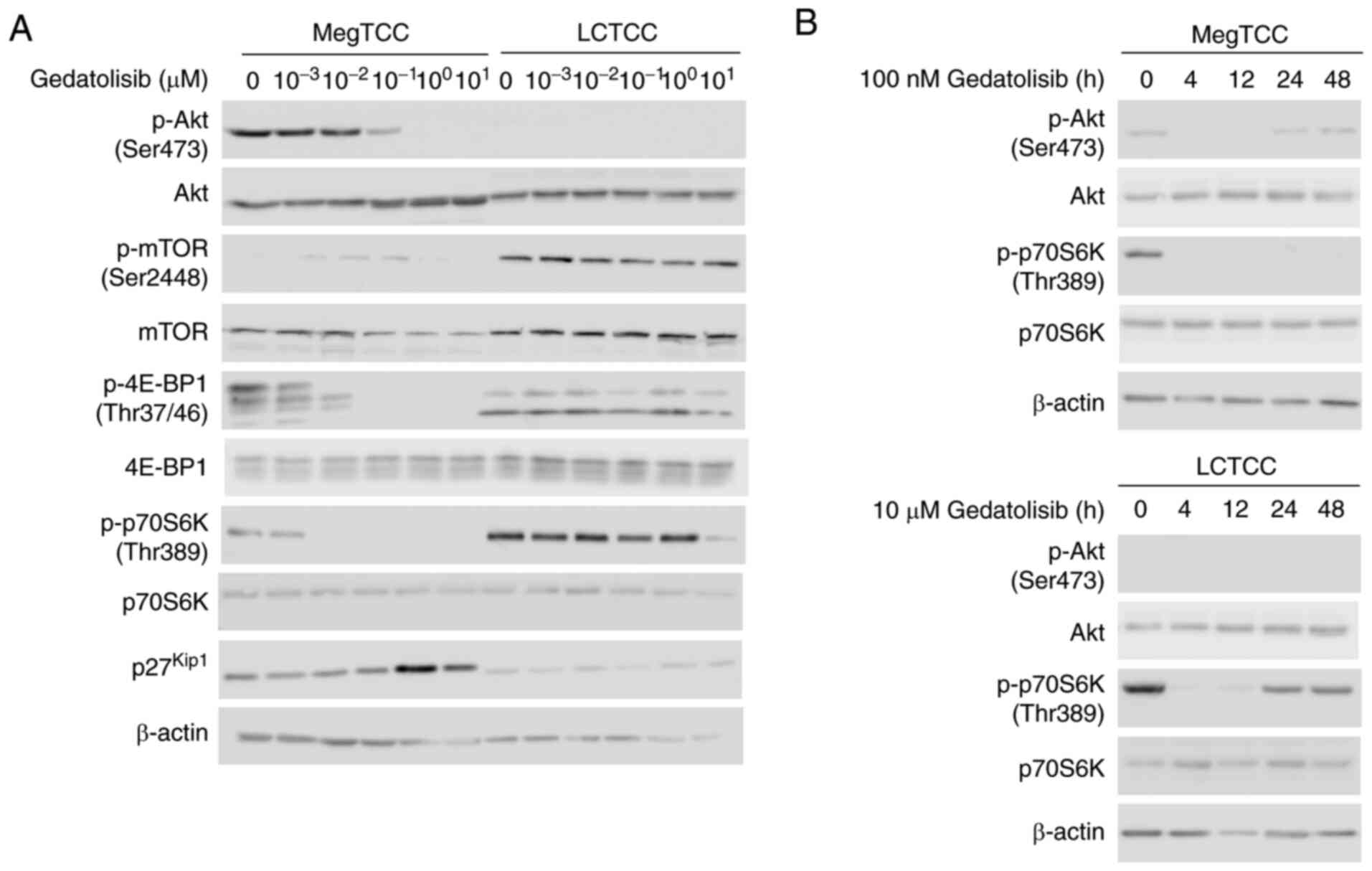
Gedatolisib inhibits the
phosphorylation of PI3K/mTOR signaling pathway molecules in MegTCC
and LCTCC cells
The inhibitory effect of gedatolisib on the
phosphorylation of PI3K/mTOR signaling pathway molecules in
gedatolisib-sensitive MegTCC cells and gedatolisib-resistant LCTCC
cells was evaluated. In MegTCC cells, gedatolisib inhibited the
phosphorylation of Akt, 4E-BP1, and p70S6K but induced the
expression of p27kip1 in a dose-dependent manner. These
changes were not detected in LCTCC cells (Fig. 4A). The time-course inhibition of
p70S6K phosphorylation was observed and it was demonstrated that
gedatolisib inhibited p70S6K phosphorylation in LCTCC cells;
however, this inhibition was transient and a higher concentration
of gedatolisib was required than that in MegTCC cells (Fig. 4B).
Gedatolisib induces
G0/G1 cell cycle arrest in the canine tumor
cell lines
The effects of gedatolisib on cell cycle
distribution were assessed in the canine tumor cell lines.
Gedatolisib significantly increased the percentage of cells in the
G0/G1 phase in MegTCC and LCTCC cells, but
LCTCC cells were more resistant to G0/G1 cell
cycle arrest (Fig. 5A). In the
100-nM gedatolisib treatment, a significant increase in the
percentage of cells in the G0/G1 phase was
observed in five cell lines (CLAC, CHS-4, MegTCC, MCM-N1, and
HMPOS), in which the IC50 values were <100 nM
(Fig. 5B). To explore the molecular
determinants involved in the cellular sensitivity to gedatolisib,
the canine tumor cell lines were divided into two groups, namely
the low-IC50 cell lines (IC50 values <100
nM) and high-IC50 cell lines (IC50 values
>100 nM). The percentage increase of cells in the
G0/G1 phase in the low-IC50 cell
lines was significantly higher than in the high-IC50
cell lines (Fig. 5C). The
localization of p27kip1 was examined to demonstrate the
mechanism of the G0/G1 cell cycle arrest. In
MegTCC cells, p27kip1 expression was primarily observed
in the nucleus (Fig. 5D).
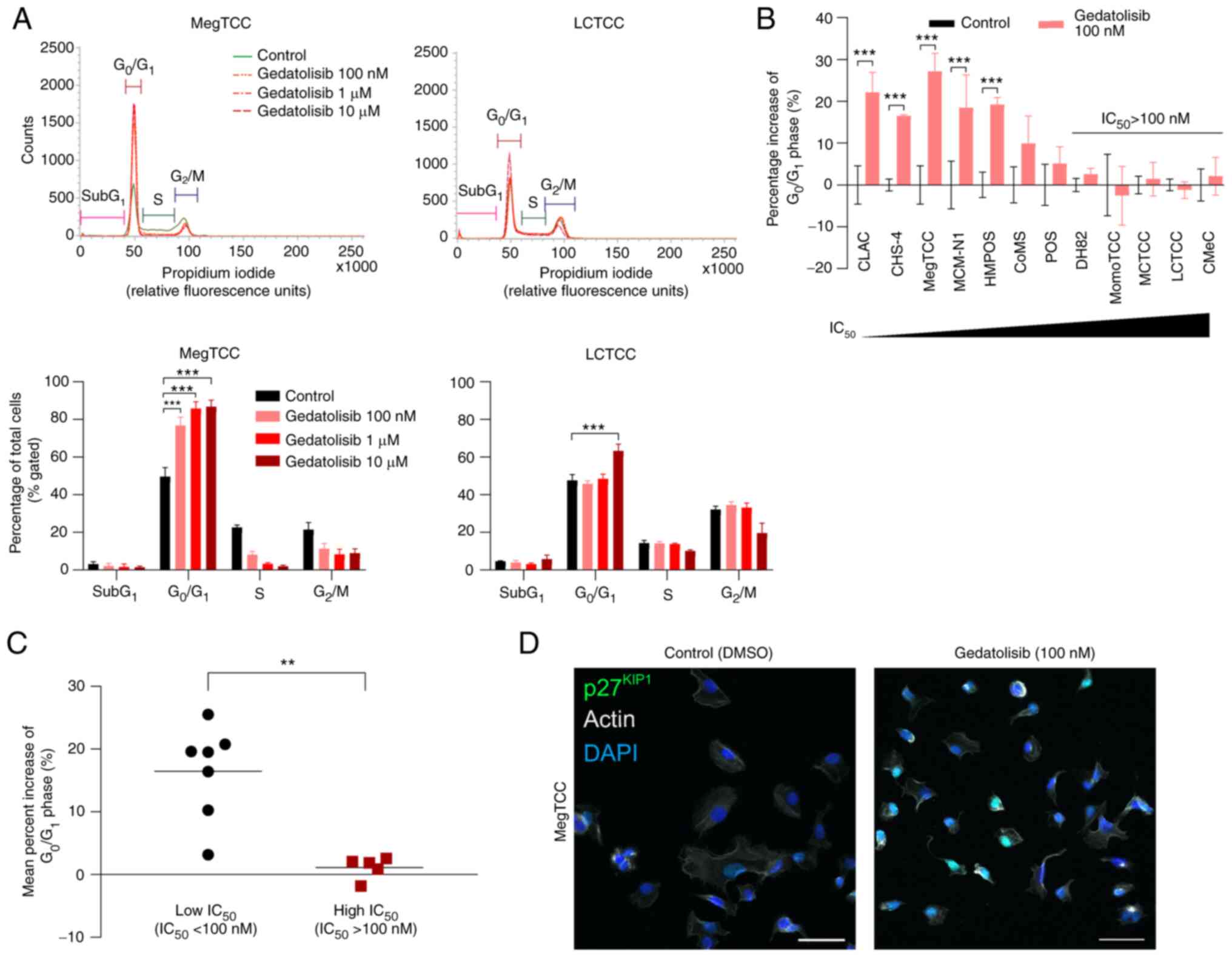 | Figure 5.Gedatolisib induces
G0/G1 cell cycle arrest in canine tumor cell
lines. (A) The cell cycle distribution (top panels) was evaluated
using flow cytometry after 24 h of treatment with 100 nM, 1, 10 µM
gedatolisib, or DMSO (control) in MegTCC and LCTCC cells, with the
percentage of each cell cycle phase (bottom panels) analyzed. Data
are presented as the mean ± SD from three independent experiments.
(B) Percentage increase of cells in the G0/G1
phase after 24 h of treatment with 100 nM gedatolisib in canine
tumor cell lines. Data are presented as the mean ± SD from three
independent experiments. (C) The mean percent increase of cells in
the G0/G1 phase in the low (IC50
values <100 nM, n=7) and high (IC50 values >100
nM, n=5) IC50 cell lines. Each point represents the
individual mean from three independent experiments, with bars
representing the median. (D) p27kip1 localization in
MegTCC cells treated with 100 nM gedatolisib for 24 h. These cells
were stained with the anti-p27kip1 antibody (green) and
counterstained with DAPI (blue) and actin (gray). Scale bars, 50
µm. **P<0.01 and ***P<0.001 (for A and B, multiple t-tests;
for C, Mann-Whitney U test). DMSO, dimethyl sulfoxide. |
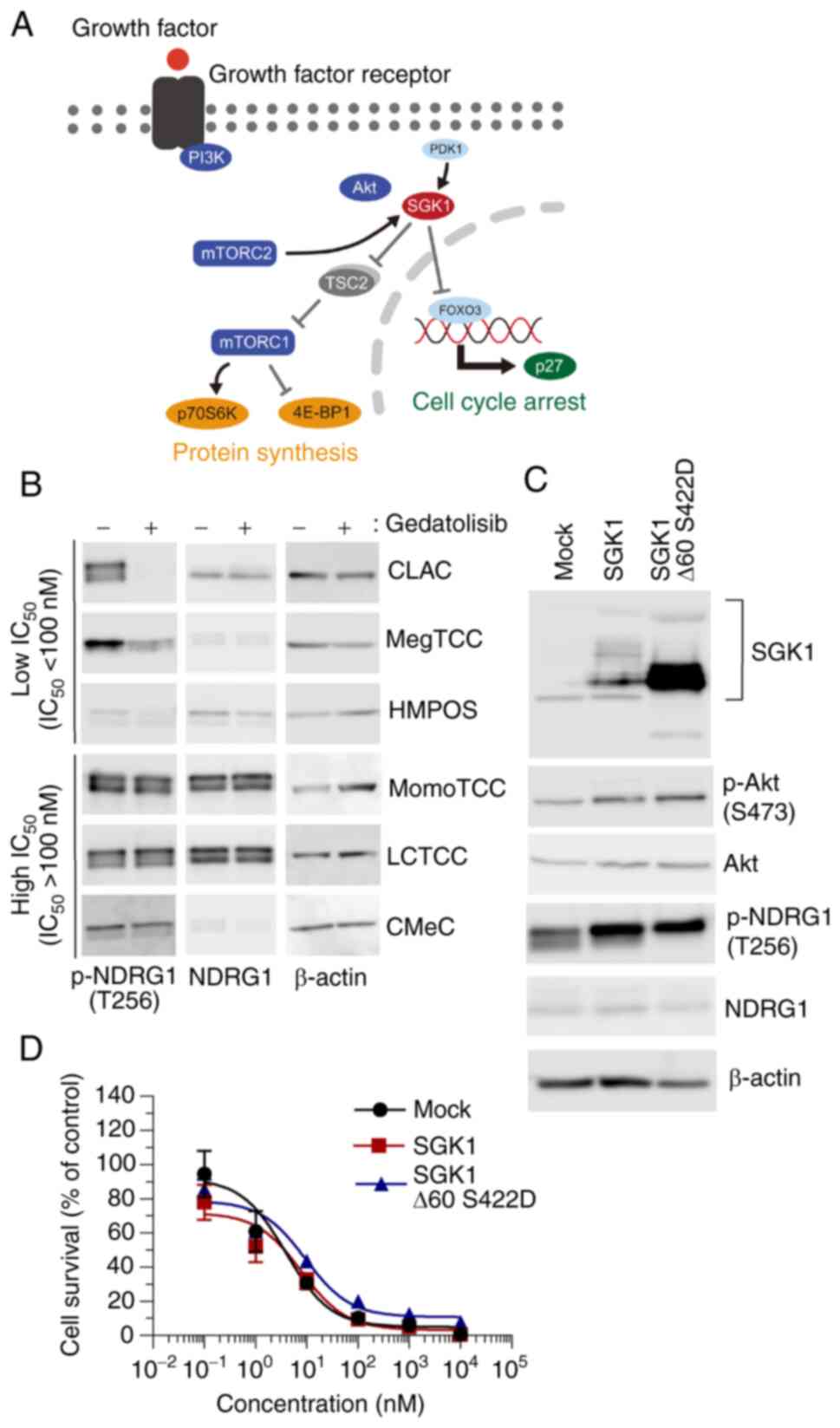
Overexpression of SGK1 does not confer
resistance to gedatolisib in MegTCC cells
As LCTCC cells resisted the inhibition of mTORC1 and
G0/G1 cell cycle arrest, it was hypothesized
that the molecular determinants were not downstream molecules from
Akt, but compensatory signaling pathways, upstream molecules from
Akt, or drug efflux pumps. The compensatory signaling pathways were
investigated and the involvement of SGK1, which has a similar
function to that of Akt (Fig. 6A)
(34) was evaluated because
overexpressed SGK1 has been revealed to confer resistance to PI3Kα
and Akt inhibitors (40,41).
In CLAC and MegTCC cells, which exhibited low
IC50 to gedatolisib, the phosphorylation level of N-myc
downstream-regulated 1 (NDRG1), a substrate of both SGK1 and Akt
(40), was low with gedatolisib
treatment (Fig. 6B), which suggested
that the activities of Akt and SGK1 were low. To the best of our
knowledge, this is the first study that identified the canine
SGK1 sequence (Fig. S1) and
established MegTCC cells expressing SGK1 to reveal the contribution
of SGK1. The expression of SGK1 was observed in MegTCC cells
transfected with wild-type and mutated SGK1, which was
constitutively the active form of SGK1 (Fig. 6C) (35,36).
Neither the overexpression of SGK1 nor that of its mutated form was
observed to confer cellular resistance to gedatolisib (Fig. 6D).
Mutations E545K and H1047R in PIK3CA
are not observed in canine tumor cell lines
To investigate the contribution of PI3K, the
presence of gene mutations E545K and H1047R in PIK3CA, which
increase the sensitivity of tumor cells to dual PI3K/mTOR
inhibitors, were examined (6).
Consequently, gene mutations E545K and H1047R in PIK3CA were
not observed in any of the canine tumor cell lines, which indicated
that these mutations did not sensitize the low-IC50 cell
lines against gedatolisib (Fig.
S2).
ABCB1 inhibition enhances the
antitumor activity of gedatolisib in LCTCC cells
The functional integrity of ABCB1 was evaluated
using rhodamine-123, a well-established ABCB1 substrate (42). It was demonstrated that the percent
decrease of the mean fluorescence intensity in the
high-IC50 cell lines was significantly higher than in
the low-IC50 cell lines (Fig.
7A and B).
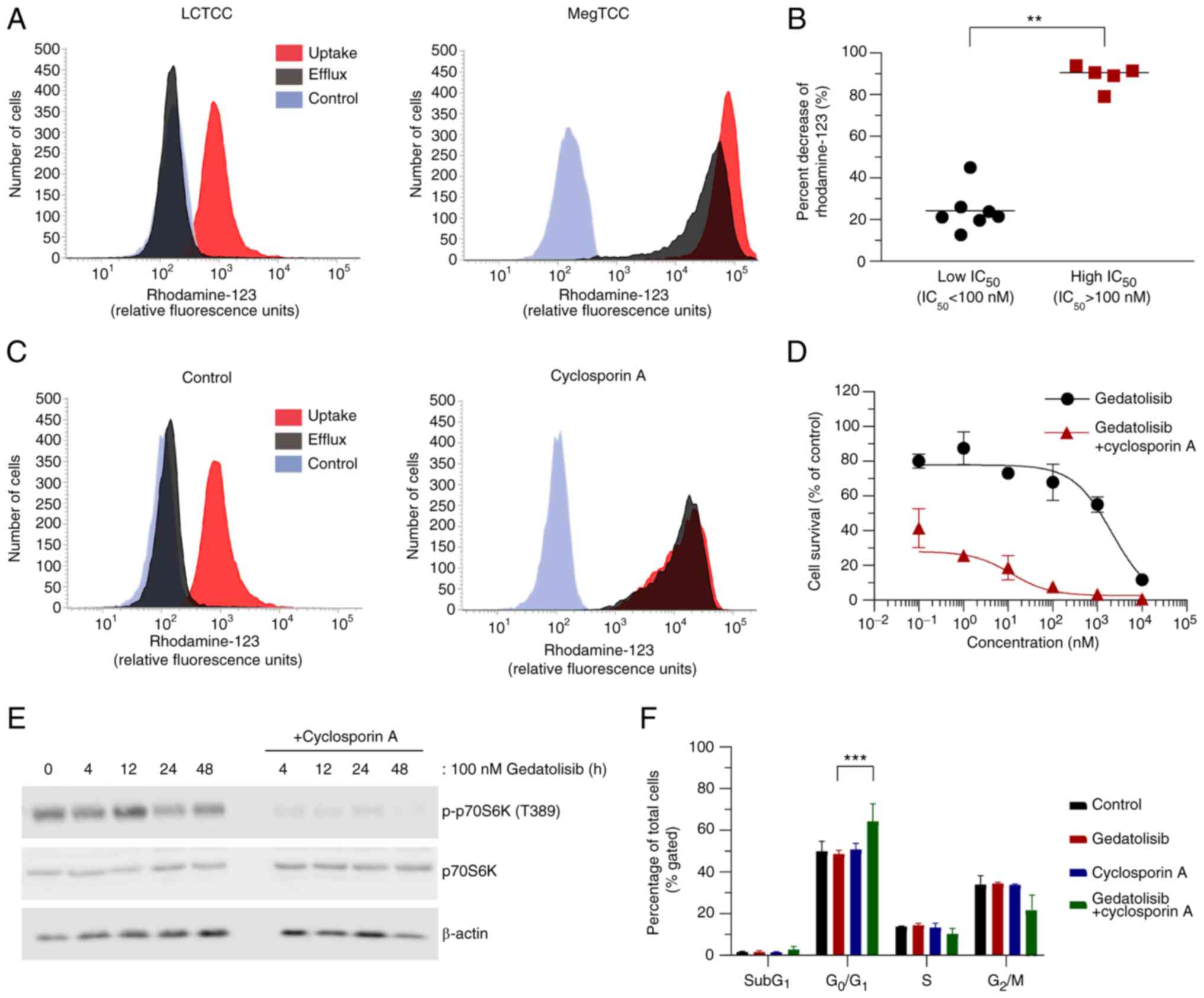 | Figure 7.ABCB1 inhibition enhances the
antitumor activity of gedatolisib in LCTCC cells. (A)
Representative histograms of rhodamine-123 uptake (red) and efflux
(black) in the low (MegTCC) and high (LCTCC) IC50 cell
lines. The gray histograms indicate cell auto-fluorescence. (B)
Percent decrease in rhodamine-123 intensity in canine tumor cell
lines. Each plot represents the mean percent decrease of
rhodamine-123 intensity from three independent experiments, with
bars representing the median (five high IC50 and seven low
IC50 cell lines). (C) Histograms of rhodamine-123
uptake/efflux in the absence (control) or presence of 5 µM
cyclosporin A treatment in LCTCC cells. (D) Dose-response curves of
LCTCC cells treated with increasing concentrations of gedatolisib
in the absence or presence of 5 µM cyclosporin A. The results are
presented as the mean ± SD of five replicates. (E) Immunoblotting
analysis of LCTCC cells treated with gedatolisib (100 nM), with or
without cyclosporin A (5 µM). (F) Cell cycle analysis of LCTCC
cells treated with gedatolisib (100 nM), cyclosporin A (5 µM), or
both agents for 24 h. Data are presented as the mean ± SD from
three independent experiments. **P<0.01 and ***P<0.001 (for
B, Mann-Whitney U test; for F, multiple t-tests). ABCB1,
ATP-binding cassette, subfamily B, member 1; p-, phosphorylated;
p70S6K, p70S6 kinase. |
To reveal the importance of ABCB1 in gedatolisib
resistance, the ABCB1 function was inhibited using cyclosporin A
(43). The dose-response curve of
cyclosporin A in LCTCC is presented in Fig. S3. Treatment with cyclosporin A
increased the mean fluorescence intensity of rhodamine-123, which
demonstrated inhibition of ABCB1 function in LCTCC cells (Fig. 7C). Furthermore, ABCB1 inhibition
enhanced the decrease in cell viability (Fig. 7D), PI3K/mTOR signaling pathway
inhibition (Fig. 7E), and induction
of G0/G1 cell cycle arrest (Fig. 7F) that resulted from gedatolisib
treatment.
To demonstrate the effect of ABCB1 inhibition,
tariquidar, an ABCB1 inhibitor (44), and toceranib, a receptor tyrosine
kinase inhibitor with a similar structure to sunitinib (45), were evaluated. Toceranib was selected
because it is frequently used to treat various canine tumors
(46), and its analog sunitinib has
a weaker ABCB1 inhibitory effect than did cyclosporin A (47). Consequently, tariquidar enhanced the
decrease of cell viability against gedatolisib with a potent ABCB1
inhibitory effect. However, toceranib did not produce this
enhancement and caused less ABCB1 inhibition than tariquidar
(Fig. S3). These results indicated
that ABCB1 inhibition enhanced the antitumor activity of
gedatolisib in LCTCC cells.
Discussion
In the present study, the PI3K/mTOR signaling
pathway inhibitors decreased cell viability in the canine tumor
cell lines derived from six types of tumors. These results were
consistent with the results of previous studies that revealed that
PI3K/mTOR signaling pathway inhibitors decreased cell viability in
various types of tumor cells (10,12).
However, the question remains whether PI3K/mTOR signaling pathway
inhibitors selectively exhibit antitumor activity to tumor cells.
The PI3K/mTOR signaling pathway is involved in numerous cellular
functions in non-tumor and tumor cells (1). As the PI3K/mTOR signaling pathway is
aberrantly activated in tumor cells, PI3K/mTOR signaling pathway
inhibitors have been revealed to selectively decrease cell
viability in tumor cells (3). In
fact, BEZ235, which is a dual PI3K/mTOR inhibitor, exhibited
anti-leukemic activities in acute myeloid leukemia cells without
affecting normal hematopoiesis ex vivo (48). Although the effect of PI3K/mTOR
signaling pathway inhibitors in non-tumor cells remains unclear in
this study, the present results suggest that PI3K/mTOR signaling
pathway inhibitors decrease cell viability against various types of
canine tumor cells.
Gedatolisib decreased cell viability in most of the
canine tumor cell lines. The IC50 values in 10 of the 12
cell lines were <1 µM, which are similar to those in human
tumors (non-small cell lung cancer and breast cancer) currently in
clinical trials (3,10). The two cell lines with high
IC50 values were from TCC and melanoma and they
exhibited high ABCB1 activity. In a previous study, gedatolisib
decreased cell viability of 50 diverse human tumor cell lines.
There were resistant cell lines from multiple tumor types and ABCB1
inhibition enhanced the anti-proliferative activity of gedatolisib
(10). The present results were
consistent with these results, which suggests that canine tumors
have a similar sensitivity and resistance mechanism as human
tumors.
Overexpression of SGK1 did not confer resistance to
gedatolisib and the gene mutations in PIK3CA were not
observed in all canine tumor cell lines. Previous studies revealed
that overexpressed SGK1 conferred resistance to PI3Kα and Akt
inhibitors (40,41). However, the involvement of SGK1 in
the cellular resistance to dual PI3K/mTOR inhibitors remained
unclear and the results in this study could not show the
involvement of SGK1 in the cellular resistance to gedatolisib. As
for PIK3CA, several studies have reported the involvement in
the cellular sensitivity to the dual PI3K/mTOR inhibitor, BEZ235,
in human tumor cell lines (6). Since
the gene mutation in PIK3CA was not observed in all the cell
lines used in the present study, the involvement of PIK3CA
remains unclear in canine tumor cell lines. To reveal its
involvement, further research with gene modification is
required.
In the present study, high ABCB1 activity in the
resistant cell lines was observed. ABCB1 inhibition in those cells
resulted in the suppression of the PI3K/mTOR signaling pathway and
induction of a G0/G1-cell cycle arrest. To
the best of our knowledge, this is the first study to explore the
molecular mechanisms involved in the cellular resistance in canine
tumors. These results were consisted with ones in human tumor cell
lines (10), suggesting that ABCB1
was involved in the resistance of gedatolisib, decreasing the
suppression of the PI3K/mTOR signaling pathway in both human and
canine tumor cell lines. In human tumors, however, compensatory
signaling pathways are reported as resistance mechanisms; the
signaling pathways cross-talk with and compensate each other
(7,8,49). In
the present study, the contribution of other signaling pathways in
gedatolisib resistance was not clear. As ABCB1 expression is
regulated by several signaling pathways and transcriptional factors
(50), it may be possible that other
signaling pathways are involved in the resistance mechanisms to
gedatolisib via ABCB1 expression.
ABCB1 inhibition enhanced cell viability reduction
due to gedatolisib in canine tumor cell lines. The same effect
occurs in human tumor cell lines (10), which suggests the following two
possibilities. Firstly, ABCB1 expression could be a predictive
marker for the efficacy of gedatolisib. In a clinical study,
stathmin, a regulatory protein for microtubule dynamics, was used
as a biomarker for PI3K/mTOR signaling activation. However,
stathmin levels did not correlate with the efficacy of gedatolisib
(51). Evaluation of the ABCB1
expression level in tumor tissue would be beneficial to identify
the predictive marker. Secondly, ABCB1 could be a therapeutic
target to overcome gedatolisib resistance. In the present study,
ABCB1 was inhibited using three compounds and it was discovered
that the third generation ABCB1 inhibitor, tariquidar, exhibited
the highest inhibition of ABCB1 (Fig.
S3). Although ABCB1 inhibition by cyclosporin A and
multi-kinase inhibitors has been proposed (52), the third generation ABCB1 inhibitors
may be selected for further studies due to their inhibitory
activity.
At least three limitations are acknowledged in the
present study. Firstly, the in vivo activity of gedatolisib
against canine tumors remains unclear. It is necessary to evaluate
the in vivo activity and tolerability of gedatolisib using
an animal model. Secondly, although 12 cell lines from six types of
tumor were used, the number of tumor types and cell lines was
limited. The antitumor activity in other types of tumors is worth
evaluating. Thirdly, potential mechanisms involved in cellular
sensitivity to gedatolisib were explored. However, the contribution
of other possible mechanisms (e.g., other signaling pathways, pHe,
nutrient depletion, and hypoxia) was not elucidated. Further
investigation for the resistance mechanisms is warranted.
In conclusion, the dual PI3K/mTOR inhibitor
gedatolisib potently inhibited the activation of the PI3K/mTOR
signaling pathway, decreased cell viability, and induced a
G0/G1 cell cycle arrest in the canine tumor
cell lines. These effects were enhanced by ABCB1 inhibition.
Collectively, these novel results support the potential usage of
gedatolisib for canine tumors and suggest that ABCB1 plays an
important role in the cellular resistance to gedatolisib.
Supplementary Material
Supporting Data
Acknowledgements
We would like to thank Dr Y. Hoshino (Division of
Small Animal Surgery, Department of Veterinary Medicine, Faculty of
Agriculture, Iwate University), Dr H. Sahara (Laboratory of
Biology, Azabu University School of Veterinary Medicine,
Sagamihara, Japan), and Dr T. Kimura (Laboratory of Comparative
Pathology, Department of Clinical Sciences, Faculty of Veterinary
Medicine, Hokkaido University) for providing TCC cell lines (MCTCC,
LCTCC, MegTCC, and MomoTCC), a lung adenocarcinoma cell line
(CLAC), and 293T cells, respectively. We are grateful to Dr H.
Miyoshi (Keio University) for providing the lentiviral vector.
Funding
This research was supported by Grant-In-Aid for Graduate
Students 2017, Program for Leading Graduate Schools, Hokkaido
University and Grant-in-Aid for Scientific Research (grant no.
17K15375) from Japan Society for the Promotion of Science.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
KH, SK and YM conceived and designed the
experiments. YM and TS performed the experiments. YM analyzed the
data. KH, SK, and MO supervised the experiments. YM wrote the draft
manuscript. KH and MO reviewed and edited the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
PI3K
|
phosphatidylinositol 3-kinase
|
|
mTOR
|
mammalian target of rapamycin
|
|
SGK1
|
serum-and-glucocorticoid- induced
kinase 1
|
|
ABCB1
|
ATP-binding cassette, subfamily B,
member 1
|
|
mTORC1
|
mTOR complex 1
|
|
mTORC2
|
mTOR complex 2
|
|
PTEN
|
phosphatase and tensin homolog
|
|
TCC
|
transitional cell carcinoma
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
FBS
|
fetal bovine serum
|
|
DMSO
|
dimethyl sulfoxide
|
|
EDTA
|
ethylenediaminetetraacetic acid
|
|
PBS
|
phosphate-buffered saline
|
|
PI
|
propidium iodide
|
|
PCR
|
polymerase chain reaction
|
|
TSC2
|
tuberous sclerosis complex 2
|
|
NDRG1
|
N-myc downstream-regulated 1
|
References
|
1
|
Thorpe LM, Yuzugullu H and Zhao JJ: PI3K
in cancer: Divergent roles of isoforms, modes of activation and
therapeutic targeting. Nat Rev Cancer. 15:7–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fruman DA and Rommel C: PI3K and cancer:
Lessons, challenges and opportunities. Nat Rev Drug Discov.
13:140–156. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Janku F, Yap TA and Meric-Bernstam F:
Targeting the PI3K pathway in cancer: Are we making headway? Nat
Rev Clin Oncol. 15:273–291. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wander SA, Hennessy BT and Slingerland JM:
Next-generation mTOR inhibitors in clinical oncology: How pathway
complexity informs therapeutic strategy. J Clin Invest.
121:1231–1241. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Courtney KD, Corcoran RB and Engelman JA:
The PI3K pathway as drug target in human cancer. J Clin Oncol.
28:1075–1083. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Weigelt B and Downward J: Genomic
determinants of P13K pathway inhibitor response in cancer. Front
Oncol. 2:1092012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Serra V, Scaltriti M, Prudkin L, Eichhorn
PJA, Ibrahim YH, Chandarlapaty S, Markman B, Rodriguez O, Guzman M,
Rodriguez S, et al: PI3K inhibition results in enhanced HER
signaling and acquired ERK dependency in HER2-overexpressing breast
cancer. Oncogene. 30:2547–2557. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Britschgi A, Andraos R, Brinkhaus H,
Klebba I, Romanet V, Muller U, Murakami M, Radimerski T and
Bentires-Alj M: JAK2/STAT5 inhibition circumvents resistance to
PI3K/mTOR blockade: A rationale for cotargeting these pathways in
metastatic breast cancer. Cancer Cell. 22:796–811. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Heavey S, Dowling P, Moore G, Barr MP,
Kelly N, Maher SG, Cuffe S, Finn SP, O'Byrne KJ and Gately K:
Development and characterisation of a panel of
phosphatidylinositide 3-kinase-mammalian target of rapamycin
inhibitor resistant lung cancer cell lines. Sci Rep. 8:16522018.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mallon R, Feldberg LR, Lucas J, Chaudhary
I, Dehnhardt C, Santos ED, Chen Z, dos Santos O, Ayral-Kaloustian
S, Venkatesan A and Hollander I: Antitumor efficacy of PKI-587, a
highly potent dual PI3K/mTOR kinase inhibitor. Clin Cancer Res.
17:3193–3203. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Murai A, Abou Asa S, Kodama A, Hirata A,
Yanai T and Sakai H: Constitutive phosphorylation of the
mTORC2/Akt/4E-BP1 pathway in newly derived canine hemangiosarcoma
cell lines. BMC Vet Res. 8:1282012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen YT, Tan KA, Pang LY and Argyle DJ:
The class I PI3K/Akt pathway is critical for cancer cell survival
in dogs and offers an opportunity for therapeutic intervention. BMC
Vet Res. 8:732012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Delgado L, Gärtner E and Pereira PD:
Activation of mammalian target of rapamycin in canine mammary
carcinomas: An immunohistochemical study. J Comp Pathol.
152:138–144. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rodriguez S, Fadlalla K, Graham T, Tameru
B, Fermin CD and Samuel T: Immunohistochemical evaluation of AKT
protein activation in canine mast cell tumours. J Comp Pathol.
147:171–176. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gordon IK, Ye F and Kent MS: Evaluation of
the mammalian target of rapamycin pathway and the effect of
rapamycin on target expression and cellular proliferation in
osteosarcoma cells from dogs. Am J Vet Res. 69:1079–1084. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Paoloni MC, Mazcko C, Fox E, Fan T, Lana
S, Kisseberth W, Vail DM, Nuckolls K, Osborne T, Yalkowsy S, et al:
Rapamycin pharmacokinetic and pharmacodynamic relationships in
osteosarcoma: A comparative oncology study in dogs. PLoS One.
5:e110132010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kent MS, Collins CJ and Ye F: Activation
of the AKT and mammalian target of rapamycin pathways and the
inhibitory effects of rapamycin on those pathways in canine
malignant melanoma cell lines. Am J Vet Res. 70:263–269. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fowles JS, Denton CL and Gustafson DL:
Comparative analysis of MAPK and PI3K/AKT pathway activation and
inhibition in human and canine melanoma. Vet Comp Oncol.
13:288–304. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wei BR, Michael HT, Halsey CH, Peer CJ,
Adhikari A, Dwyer JE, Hoover SB, El Meskini R, Kozlov S, Weaver
Ohler Z, et al: Synergistic targeted inhibition of MEK and dual
PI3K/mTOR diminishes viability and inhibits tumor growth of canine
melanoma underscoring its utility as a preclinical model for human
mucosal melanoma. Pigment Cell Melanoma Res. 29:643–655. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pyuen AA, Meuten T, Rose BJ and Thamm DH:
In vitro effects of PI3K/mTOR inhibition in canine hemangiosarcoma.
PLoS One. 13:e02006342018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kadosawa T, Nozaki K, Sasaki N and
Takeuchi A: Establishment and characterization of a new cell line
from a canine osteosarcoma. J Vet Med Sci. 56:1167–1169. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Barroga EF, Kadosawa T, Okumura M and
Fujinaga T: Establishment and characterization of the growth and
pulmonary metastasis of a highly lung metastasizing cell line from
canine osteosarcoma in nude mice. J Vet Med Sci. 61:361–367. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yamazaki H, Iwano T, Otsuka S, Kagawa Y,
Hoshino Y, Hosoya K, Okumura M and Takagi S: SiRNA knockdown of the
DEK nuclear protein mRNA enhances apoptosis and chemosensitivity of
canine transitional cell carcinoma cells. Vet J. 204:60–65. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Takagi S, Kitamura T, Hosaka Y, Ohsaki T,
Bosnakovski D, Kadosawa T, Okumura M and Fujinaga T: Molecular
cloning of canine membrane-anchored inhibitor of matrix
metalloproteinase, RECK. J Vet Med Sci. 67:385–391. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Inoue K, Ohashi E, Kadosawa T, Hong SH,
Matsunaga S, Mochizuki M, Nishimura R and Sasaki N: Establishment
and characterization of four canine melanoma cell lines. J Vet Med
Sci. 66:1437–1440. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ohashi E, Inoue K, Kagechika H, Hong SH,
Nakagawa T, Takahashi T, Mochizuki M, Nishimura R and Sasaki N:
Effect of natural and synthetic retinoids on the proliferation and
differentiation of three canine melanoma cell lines. J Vet Med Sci.
64:169–172. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wellman ML, Krakowka S, Jacobs RM and
Kociba GJ: A macrophage-monocyte cell line from a dog with
malignant histiocytosis. In Vitro Cell Dev Biol. 24:223–229. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Azakami D, Bonkobara M, Washizu T, Iida A,
Kondo M, Kato R, Niikura Y, Iwaki S, Tamahara S, Matsuki N and Ono
K: Establishment and biological characterization of canine
histiocytic sarcoma cell lines. J Vet Med Sci. 68:1343–1346. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ishiguro T, Kadosawa T, Mori K, Takagi S,
Okumura M and Fujinaga T: Establishment and characterization of a
new canine mast cell tumor cell line. J Vet Med Sci. 63:1031–1034.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nemoto Y, Maruo T, Sato T, Deguchi T, Ito
T, Sugiyama H, Ishikawa T, Madarame H, Watanabe T, Shida T and
Sahara H: Identification of cancer stem cells derived from a canine
lung adenocarcinoma cell line. Vet Pathol. 48:1029–1034. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Murase Y, Konnai S, Yamada S, Githaka N,
Isezaki M, Ito T, Takano A, Ando S, Kawabata H, Murata S and Ohashi
K: An investigation of binding ability of Ixodes persulcatus
Schulze Salp15 with Lyme disease spirochetes. Insect Biochem Mol
Biol. 60:59–67. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yamada S, Konnai S, Imamura S, Ito T,
Onuma M and Ohashi K: Cloning and characterization of Rhipicephalus
appendiculatus voraxin alpha and its effect as anti-tick vaccine.
Vaccine. 27:5989–5997. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yamasaki T, Suzuki A, Shimizu T, Watarai
M, Hasebe R and Horiuchi M: Characterization of intracellular
localization of PrP(Sc) in prion-infected cells using a mAb that
recognizes the region consisting of aa 119–127 of mouse PrP. J Gen
Virol. 93:668–680. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Talarico C, Dattilo V, D'Antona L, Menniti
M, Bianco C, Ortuso F, Alcaro S, Schenone S, Perrotti N and Amato
R: SGK1, the new player in the game of resistance: Chemo-radio
molecular target and strategy for inhibition. Cell Physiol Biochem.
39:1863–1876. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Park J, Leong MLL, Buse P, Maiyar AC,
Firestone GL and Hemmings BA: Serum and glucocorticoid-inducible
kinase (SGK) is a target of the PI 3-kinase-stimulated signaling
pathway. EMBO J. 18:3024–3033. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
García-Martínez JM and Alessi DR: mTOR
complex 2 (mTORC2) controls hydrophobic motif phosphorylation and
activation of serum- and glucocorticoid-induced protein kinase 1
(SGK1). Biochem J. 416:375–385. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Thompson JD, Higgins DG and Gibson TJ:
CLUSTAL-W: Improving the sensitivity of progressive multiple
sequence alignment through sequence weighting, position-specific
gap penalties and weight matrix choice. Nucleic Acids Res.
22:4673–4680. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Santiskulvong C, Konecny GE, Fekete M,
Chen KYM, Karam A, Mulholland D, Eng C, Wu H, Song M and Dorigo O:
Dual targeting of phosphoinositide 3-kinase and mammalian target of
rapamycin using NVP-BEZ235 as a novel therapeutic approach in human
ovarian carcinoma. Clin Cancer Res. 17:2373–2384. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huynh H, Hao HX, Chan SL, Chen D, Ong R,
Soo KC, Pochanard P, Yang D, Ruddy D, Liu M, et al: Loss of
Tuberous Sclerosis Complex 2 (TSC2) is frequent in hepatocellular
carcinoma and predicts response to mTORC1 inhibitor everolimus. Mol
Cancer Ther. 14:1224–1235. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Castel P, Ellis H, Bago R, Toska E, Razavi
P, Carmona FJ, Kannan S, Verma CS, Dickler M, Chandarlapaty S, et
al: PDK1-SGK1 signaling sustains AKT-independent mTORC1 activation
and confers resistance to PI3Kα inhibition. Cancer Cell.
30:229–242. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sommer EM, Dry H, Cross D, Guichard S,
Davies BR and Alessi DR: Elevated SGK1 predicts resistance of
breast cancer cells to Akt inhibitors. Biochem J. 452:499–508.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee JS, Paull K, Alvarez M, Hose C, Monks
A, Grever M, Fojo AT and Bates SE: Rhodamine efflux patterns
predict P-glycoprotein substrates in the National-Cancer-Institute
drug screen. Mol. Pharmacol. 46:627–638. 1994.PubMed/NCBI
|
|
43
|
Kathawala RJ, Gupta P, Ashby CR and Chen
ZS: The modulation of ABC transporter-mediated multidrug resistance
in cancer: A review of the past decade. Drug Resist Update.
18:1–17. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fox E and Bates SE: Tariquidar (XR9576): A
P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer
Ther. 7:447–459. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
London CA, Hannah AL, Zadovoskaya R, Chien
MB, Kollias-Baker C, Rosenberg M, Downing S, Post G, Boucher J,
Shenoy N, et al: Phase I dose-escalating study of SU11654, a small
molecule receptor tyrosine kinase inhibitor, in dogs with
spontaneous malignancies. Clin Cancer Res. 9:2755–2768.
2003.PubMed/NCBI
|
|
46
|
London CA: Small molecule inhibitors in
veterinary oncology practice. Vet Clin North Am Small Anim Pract.
44:893–908. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shukla S, Robey RW, Bates SE and Ambudkar
SV: Sunitinib (Sutent, SU11248), a small-molecule receptor tyrosine
kinase inhibitor, blocks function of the ATP-binding cassette (ABC)
transporters P-Glycoprotein (ABCB1) and ABCG2. Drug Metab Dispos.
37:359–365. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chapuis N, Tamburini J, Green AS, Vignon
C, Bardet V, Neyret A, Pannetier M, Willems L, Park S, Macone A, et
al: Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as
a new therapeutic strategy for acute myeloid leukemia. Clin Cancer
Res. 16:5424–5435. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mendoza MC, Er EE and Blenis J: The
Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends
Biochem Sci. 36:320–328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lopes-Rodrigues V, Seca H, Sousa D, Sousa
E, Lima RT and Vasconcelos MH: The network of P-glycoprotein and
microRNAs interactions. Int J Cancer. 135:253–263. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
del Campo JM, Birrer M, Davis C, Fujiwara
K, Gollerkeri A, Gore M, Houk B, Lau S, Poveda A, González-Martín
A, et al: A randomized phase II non-comparative study of
PF-04691502 and gedatolisib (PF-05212384) in patients with
recurrent endometrial cancer. Gynecol Oncol. 142:62–69. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chen ZL, Shi TL, Zhang L, Zhu PL, Deng MY,
Huang C, Hu TT, Jiang L and Li J: Mammalian drug efflux
transporters of the ATP binding cassette (ABC) family in multidrug
resistance: A review of the past decade. Cancer Lett. 370:153–164.
2016. View Article : Google Scholar : PubMed/NCBI
|