Introduction
A variety of chemotherapeutic agents and radiation
therapies are used to treat malignant tumors, including colorectal
cancer. A number of these therapies induce DNA damage in cancer
cells, which results in genomic instability and ultimately leads to
cancer cell death. For example, 5-fluorouracil (5-FU) damages DNA
by inserting 5-fluoro-2′-deoxyuridine 5′-triphosphate into DNA
(1). Platinum drugs such as
cisplatin and oxaliplatin damage DNA by forming cross-linked
structures (2). Furthermore,
radiation therapy induces DNA double-strand breaks, resulting in
marked DNA damage in cancer cells (3). However, the problem with all of these
therapies is that their continuous use renders the cells resistant
to the therapies and decreases their effectiveness (4). The present study focused on the DNA
damage response (DDR) of the cells as a factor causing this
resistance.
In both normal and cancer cells, various DDR
proteins are activated to maintain genomic stability, in turn
activating cell activities, including cell cycle arrest, apoptosis
and premature senescence (5).
Dysfunction of these systems results in DNA damage accumulation,
genomic instability and eventually inability of the cells to
survive. In cancer cells, on the other hand, DNA damage from cancer
therapies is similarly repaired by the DDR, but this results in
re-stabilization and survival of cancer cells and, ultimately, in
treatment failure (6).
DDR and cell cycle arrest due to DNA damage occur at
cell cycle checkpoints. In mammals, there are two main checkpoints,
G1 and G2, which serve an important role in
cell survival. The former depends on the ATM serine/threonine
kinase (ATM)/p53/p21 signaling pathway, while the latter depends on
both the ATM/p53/p21 and ATM/ataxia telangiectasia mutated and
rad3-related (ATR)/checkpoint kinase 1 (Chk1)/cell division cycle
25 (Cdc25) signaling pathways (7–11).
Given that most cancer cells have genetic mutations in p53
(12–14), their survival after DNA damage
depends on the function of the G2 checkpoint mediated by
the ATM/ATR/Chk1/Cdc25 signaling pathway (15–17).
Therefore, agents that can inhibit the G2 checkpoint may
be promising for inducing synthetic lethality in p53-deficient
cancer cells and for chemosensitization of known cancer therapies
or reversal of resistance.
The agent used in the present study, AZD6738, is a
cell cycle checkpoint inhibitor classified as an ATR inhibitor
(18). It is a specific inhibitor
of ATR that works by inhibiting phosphorylation of Chk1 (Ser345)
(19,20). Preclinical studies have reported
its potentiating effect on pancreatic cancer cells when combined
with gemcitabine (21) and on
treatment sensitivity when combined with radiation (22,23).
In addition, phase I and II trials are ongoing in clinical practice
(24–26). However, to the best of our
knowledge, its efficacy in combination with 5-FU, the mainstay
chemotherapeutic agent for colorectal cancer, is still unclear.
5-FU has a damaging effect on DNA (1), and it was hypothesized that its
effect was likely to be enhanced when AZD6738 was used in
combination. The aim of the present study was to confirm the
potentiating effect of AZD6738 with 5-FU. To the best of our
knowledge, the present study was the first to investigate the
effect of AZD6738 in combination with 5-FU. At present, AZD6738 is
undergoing clinical trials; it has been reported to be used in
combination with radiotherapy (27), and it has been reported to be
effective in combination with olaparib in ovarian cancer (28). If AZD6738 is shown to be effective
in combination with 5-FU, it is expected to have early clinical
applications in colorectal cancer, and furthermore, it is expected
to prolong overall survival. In addition, if this research
progresses, it is expected to be applied to colorectal cancer that
has become resistant to 5-FU by DDR.
Materials and methods
Cell culture
The human colorectal cancer cell lines (HT29, SW480,
HCT116 and DLD-1) were obtained from American Type Culture
Collection. HT29 has been authenticated (no. KBN0811) using short
tandem repeat DNA analysis by the Japanese Collection of Research
Bioresources Cell Bank. All cells were cultured in DMEM
(MilliporeSigma) supplemented with 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin/streptomycin. All cells were
cultured at 37°C with 5% CO2.
Reagents
AZD6738 (AstraZeneca) was used at a final
concentration of 0.5 µM in all cell lines, unless otherwise
indicated. This concentration (0.5 µM) of AZD6738 had no effect on
cell proliferation at 37°C for 72 h (Fig. S1A). Unless otherwise indicated,
5-FU (FUJIFILM Wako Pure Chemical Corporation) was used at a final
concentration of 5 µM (for HT29 and HCT116 cells) or 25 µM (for
SW480 and DLD-1 cells), which are the half-maximal inhibitory
concentrations at 37°C for 72 h (Fig.
S1B). The concentration of nocodazole (MilliporeSigma) was 300
nM. Distilled water was used as a control in WST-1 assay (Fig. 3), western blotting (Figs. 2A and 3A) and cell cycle analysis (Fig. 1A).
Water-soluble tetrazolium 1 (WST-1)
cell viability assay
The viability of HT29, SW480, HCT116 and DLD-1 cells
was determined WST-1 assay. The viability of the cells was
determined using the Premix WST-1 Cell Viability Assay System
(Takara Bio, Inc.) according to the manufacturer's protocols. Cell
lines were seeded in 96-well plates (1.0×104/well) in
100 µl medium and allowed to attach overnight. Once the cells had
attached, they were treated with 5-FU and/or AZD6738 at 37°C for 72
h. The concentration of 5-FU was 0–1,000 µM and the concentration
of AZD6738 was 0–10 µM. After 72 h, 10 µl WST-1 reagent was added
to the plates followed by an additional incubation for 1 h. The
absorbance reading in each well was measured using a microplate
reader (SpectraMax ABC; Molecular Devices, LLC) at a wavelength of
450 nm.
Western blotting
Collected HT29 cells were suspended in SDS sample
buffer (87.5 mmol/l Tris-HCl, pH 6.8, 9% glycerol, 2.75% SDS,
0.003% bromophenol blue and 150 mmol/l dithiothreitol;
concentration, 1×106 cells/150 µl), and treated at 98°C
for 5 min. For each lane, 14 µl sample was dispensed, proteins in
the lysates were separated on 4–20% Mini-PROTEAN TGX Precast gels
(Bio-Rad Laboratories, Inc.) and transferred to nitrocellulose
membranes. The membranes were blocked in 5% skim milk (BD
Biosciences) at room temperature for 30 min. The membranes were
incubated overnight with the primary antibodies at 4°C, followed by
1 h of incubation with the secondary antibodies at 4°C. Primary
antibodies against the following proteins were used for western
blotting: Chk1 (dilution, 1:1,000; cat. no. C9358; MilliporeSigma),
phospho-Chk1 Ser345 (dilution, 1:1,000; cat. no. 2348; Cell
Signaling Technology, Inc.), Apoptosis Western Blot Cocktail
(dilution, 1:250; cat. no. ab136812; Abcam) for cleaved caspase-3,
H2A.X variant histone (H2AX; dilution, 1:1,000; cat. no. ab11175;
Abcam), phosphorylated form of H2AX (γH2AX; dilution, 1:2,000; cat.
no. 05–636; Merck KGaA) and β-actin (dilution, 1:1,000; cat. no.
3700; Cell Signaling Technology, Inc.). The secondary antibody for
Chk1, γH2AX and β-actin was HRP-conjugated goat anti-mouse
immunoglobulins (1:2,000; cat. no. P0447; Agilent Technologies,
Inc.). The secondary antibody for phospho-Chk1 and H2AX was
HRP-conjugated goat anti-rabbit immunoglobulins (dilution, 1:1,000;
cat. no. P0448; Agilent Technologies, Inc.). The secondary antibody
for cleaved caspase-3 was HRP-conjugated secondary antibody
cocktail of the Apoptosis Western Blot Cocktail (dilution, 1:100;
cat. no. ab136812; Abcam). The protein-antibody complexes were
visualized with a SuperSignal West Pico Chemiluminescent Substrate,
SuperSignal West Femto Chemiluminescent Substrate (pChk1) or Pierce
ECL Western Blotting Substrate (Chk1, β-actin, γH2AX, H2AX,
Cleaved-Caspase-3) (all from Thermo Fisher Scientific, Inc.). The
immunoreactive protein bands were detected using an ImageQuant
LAS-4000mini (Cytiva). The results were semi-quantified by
densitometry analysis using ImageJ software version 1.53 (National
Institutes of Health).
Cell cycle analysis
HT29 Cells were harvested at 0, 24, 48 and 72 h
after treatment and fixed with 70% ethanol at −20°C overnight. Cell
pellets were washed once with PBS and DNA was stained using the
Cycletest Plus DNA Reagent Kit (cat. no. 340242; BD Biosciences)
following the manufacturer's instructions. The counts of cell cycle
distribution were evaluated by PI staining, and all samples were
analyzed using a FACSCanto II flow cytometer (BD Biosciences) at a
wavelength of 488 nm with the appropriate software (BD FACSDiva
Software ver. 8.0.2; BD Biosciences).
Measurement of M phase cells
HT29 cells were treated with 5-FU and/or AZD6738 for
24 h at 37°C, followed by treatment with nocodazole (300 nM) for
another 24 h at 37°C. Nocodazole was added to prevent cells from
exiting mitosis. Subsequently, the cells were fixed with 70%
ethanol at −20°C overnight. Cell pellets were washed once with PBS
and stained with an antibody against phospho-histone H3 at S10
(dilution, 1:100; cat. no. 06–570; MilliporeSigma) for 3 h,
followed by 30 min of incubation with an Alexa Fluor 488 secondary
antibody (dilution, 1:50; cat. no. ab150077; Abcam). DNA was
counterstained with a BD Cycletest Plus DNA Reagent Kit (cat. no.
340242; BD Biosciences). All samples were analyzed using a
FACSCanto II flow cytometer (BD Biosciences) at a wavelength of 488
nm with appropriate software (BD FACSDiva Software ver. 8.0.2; BD
Biosciences).
Animals
The present study used female mice, referring to
previous studies (21,29). A total of 20 female BALB/c nu-nu
mice were purchased from Japan SLC, Inc. The animals were housed in
standard Plexiglas cages in a room maintained at a constant
temperature (20–26°C) and humidity (40–60%) under a 12 h light/dark
cycle. Mice had access to autoclaved chow and water ad
libitum. The time interval between injection and the end of the
experiment was 6 weeks. All experiments were conducted according to
the Guidelines for Animal Experiments of the Nagoya City University
Graduate School of Medical Sciences and approved by the Animal Care
and Use Committee of the Nagoya City University Graduate School of
Medical Sciences (Nagoya, Japan).
HT29 human colorectal cancer cells (5×106
in 200 µl PBS) were injected subcutaneously into the right flank of
each mouse (8 weeks old; weight range, 17.0-21.1 g/mouse). Once the
tumor volume surpassed ~100 mm3, the mice were randomly
divided into two groups (5-FU, and 5-FU and AZD6738). Based on the
results of the experiments in vitro and with reference to
previous reports (30,31), five mice were used in each group.
In the 5-FU group, 5-FU (FUJIFILM Wako Pure Chemical Corporation)
was dissolved in saline solution (Otsuka Pharmaceutical Factory,
Inc.) at 2.5 mg/ml and administered to the mice at 25 mg/kg/day
intraperitoneally (5 times a week for 3 weeks). The solvent (10%
DMSO, 40% propylene glycol and 50% deionized sterile water) was
administered at 10 ml/kg/day by oral gavage (5 times a week for 3
weeks). In the 5-FU and AZD6738 group, 5-FU was dissolved in saline
solution (Otsuka Pharmaceutical Factory, Inc.) at 2.5 mg/ml and
administered to the mice at 25 mg/kg/day intraperitoneally (5 times
a week for 3 weeks). AZD6738 (AstraZeneca) was dissolved in 10%
DMSO, 40% propylene glycol and 50% deionized sterile water at 2.5
mg/ml and administered to the mice at 25 mg/kg/day by oral gavage
(5 times a week for 3 weeks). The tumor volume (mm3) was
calculated as follows: (longest tumor diameter) × (shortest tumor
diameter)2/2. Finally, the tumors were harvested from
mice and fixed in 10% formaldehyde at 4°C for 24 h. There was no
significant difference in the weight of the mice between the two
groups. To investigate the toxicity of AZD6738 alone, the same
experiment was also performed on the control group and AZD6738
group. There were five mice per group. In the control group,
instead of AZD6738 solution, the solvent was administered at 10
ml/kg/day by oral gavage. There was no significant difference in
the weight between these two groups either. Before harvesting the
tumor, all animals were euthanized by cervical dislocation under
2.0-2.5% isoflurane inhalation anesthesia using isoflurane
inhalation solution. Animal death was confirmed by the loss of
signs, such as heartbeat and response to toe pinch. The graying of
the mucous membranes and rigor mortis of mice were also confirmed.
The following were used as humane endpoints to determine that mice
should be euthanized: Total tumor volume >10% of body weight,
tumor diameter >20 mm, tumor ulceration/necrosis, gait
disturbance, and impaired water and food intake.
Immunohistochemistry
Formalin-fixed (4% paraformaldehyde at 4°C for 6 h),
paraffin-embedded sections (3-µm-thick) were mounted on
3-aminopropyltriethoxylsilane-coated slides. The sections were
deparaffinized with xylene and hydrated with ethanol at 100% twice,
90, 80 and 70% for 5 min each. After washing with running water,
samples were soaked in 10 nM citric acid buffer and boiled using a
microwave (10 min; 600 watts). Subsequently, the slides were soaked
in 100% methanol and 0.3% hydrogen peroxide mixed solution for 30
min to block endogenous peroxidase activity, and blocked with 4%
Block Ace Powder (cat. no. UKB80; DS Pharma Biomedical Co., Ltd.)
for 10 min in a humidity box at room temperature. The sections were
stained with a primary antibody against γH2AX (cat. no. 05–636;
dilution, 1:500; Merck KGaA) overnight at 4°C followed by
anti-mouse EnVision+/HRP-labeled polymer (cat. no. K4001; dilution,
1:1,500; Dako; Agilent Technologies, Inc.) as secondary antibody
for 45 min at room temperature. 3,3-diaminobenzidine substrate
(cat. no. K3467; Dako; Agilent Technologies, Inc.) was used as the
chromogen for detection for 10 min at room temperature. Hematoxylin
was used for counterstaining for 30 sec at room temperature. The
result was presented as the mean percentage of γH2AX-positive cells
± SD per high-power field. Ten fields of view were examined for
each tumor. The slides were imaged with a fluorescence microscope
(BZ-X710; Keyence Corporation) and examined using the BZ-X710
Analyzer (1.4.0.1) software (Keyence Corporation) (32).
Statistical analysis
All statistical analyses were performed using JMP
software version 14.3.0 (SAS Institute, Inc.). All data are
presented as the mean ± standard deviation. Comparisons between two
groups were performed using Student's t-test (unpaired t-test).
Comparisons among more than two groups were performed using one-way
analysis of variance followed by Tukey's test. P<0.05 was
considered to indicate a statistically significant difference.
Results
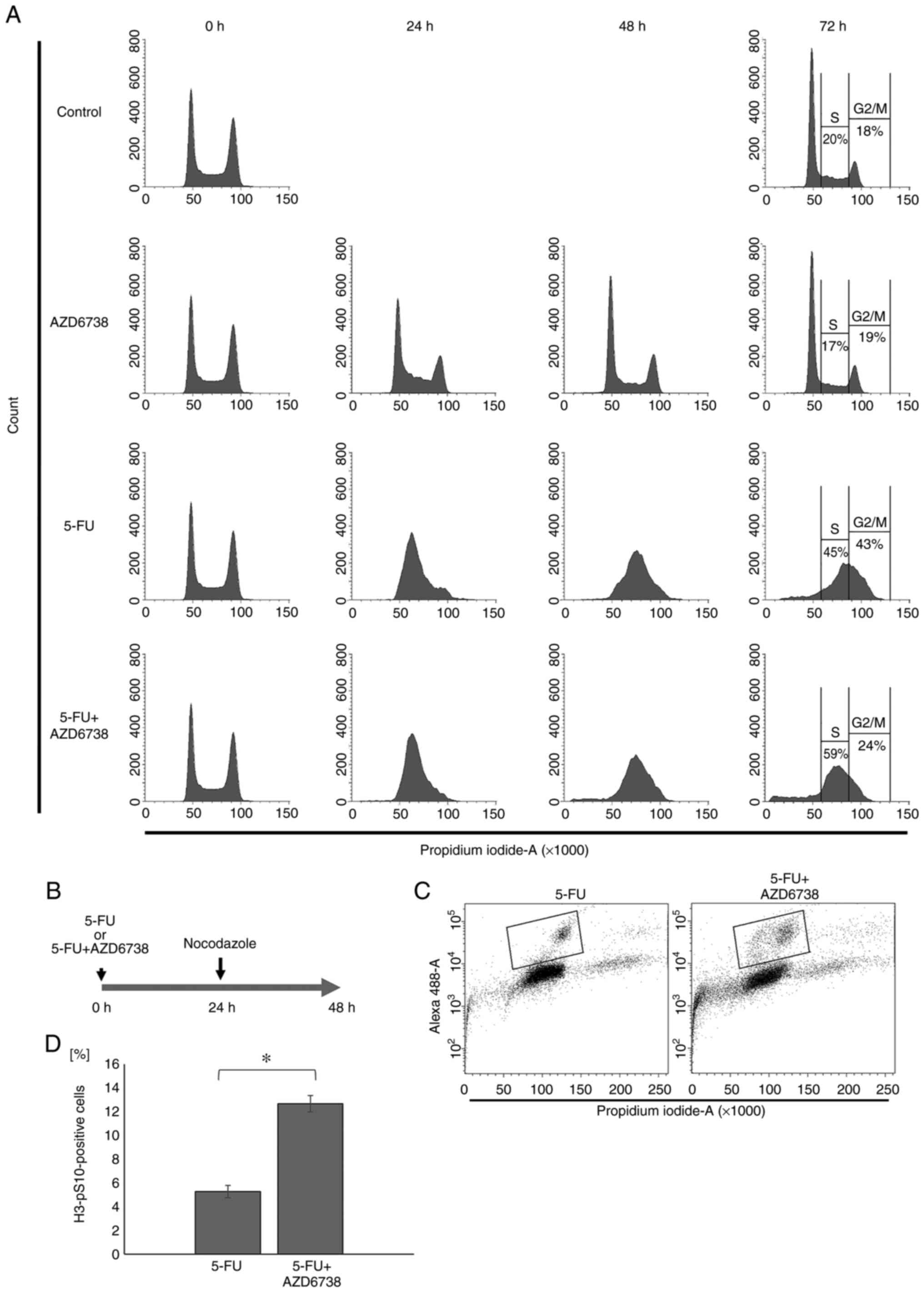
AZD6738 abrogates 5-FU-induced
activation of the G2/M checkpoint
First, to investigate whether the combination of
AZD6738 and 5-FU resulted in cell cycle perturbations in HT29
cells, which lack functional p53 (33), the cell cycle profiles were
evaluated by flow cytometry at 24, 48 and 72 h after treatment with
AZD6738 (0.5 µM), 5-FU (5 µM), their combination or control
(Fig. 1A). At 24 and 48 h, the
cell cycle was similar in the 5-FU group and the 5-FU+AZD6738
combination group, but at 72 h, the percentage of cells in the S
phase was increased in the 5-FU+AZD6738 combination group compared
with the 5-FU alone group. The AZD6738 alone group was comparable
to the control group in the cell cycle.
To confirm the percentage of cells that have entered
the M phase, nocodazole was used as shown in Fig. 1B. Phospho-histone H3 at S10 was
used as a marker for mitotic cells. The percentage of cells
positive for phospho-histone H3 at S10 was measured by flow
cytometry. The mitotic cells were increased significantly in the
presence of AZD6738 (P<0.05; Fig.
1C and D). These results indicated that AZD6738 inhibited
5-FU-induced activation of the G2 checkpoint.
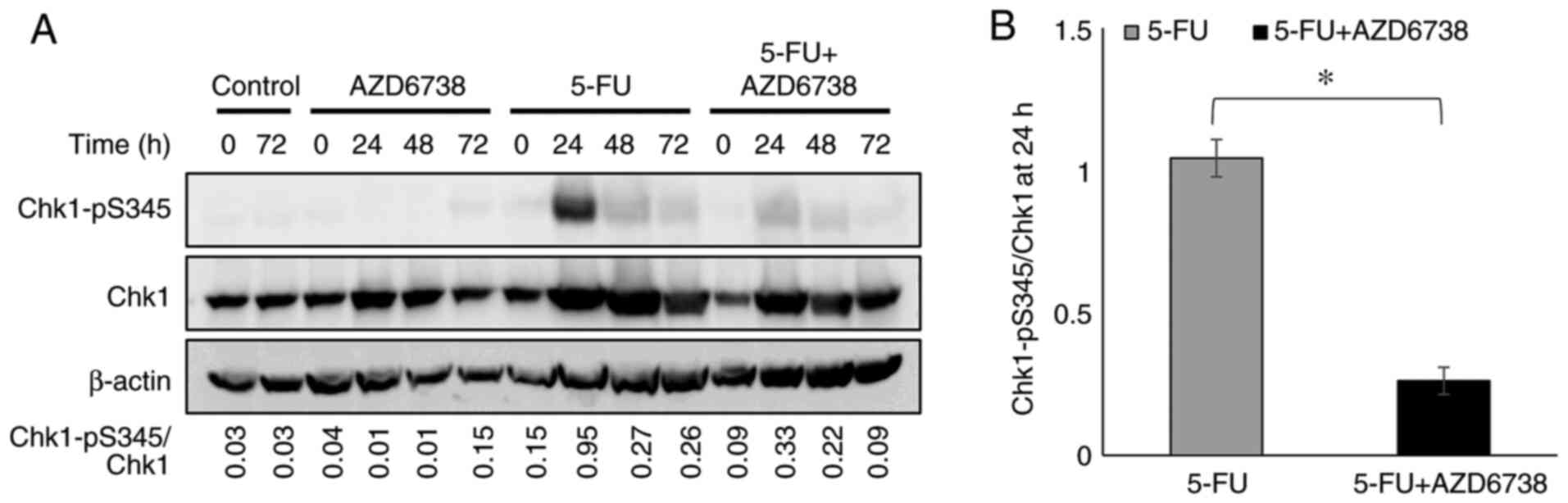
AZD6738 reduces the level of
phosphorylated Chk1
AZD6738 inhibits ATR by inhibiting the
phosphorylation of Chk1 (19,20).
If 5-FU induces DNA damage, Chk1 phosphorylation at the
G2 checkpoint occurs as a consequence (34). The present study examined whether
this effect is inhibited by AZD6738 in HT29 cells. After 5-FU
monotherapy, Chk1 phosphorylation at S345 was strongly detected at
24 h, whereas the 5-FU/AZD6738 combination suppressed the
phosphorylation of Chk1 (Fig.
2).
AZD6738/5-FU combination treatment
suppresses the survival of colorectal cancer cells
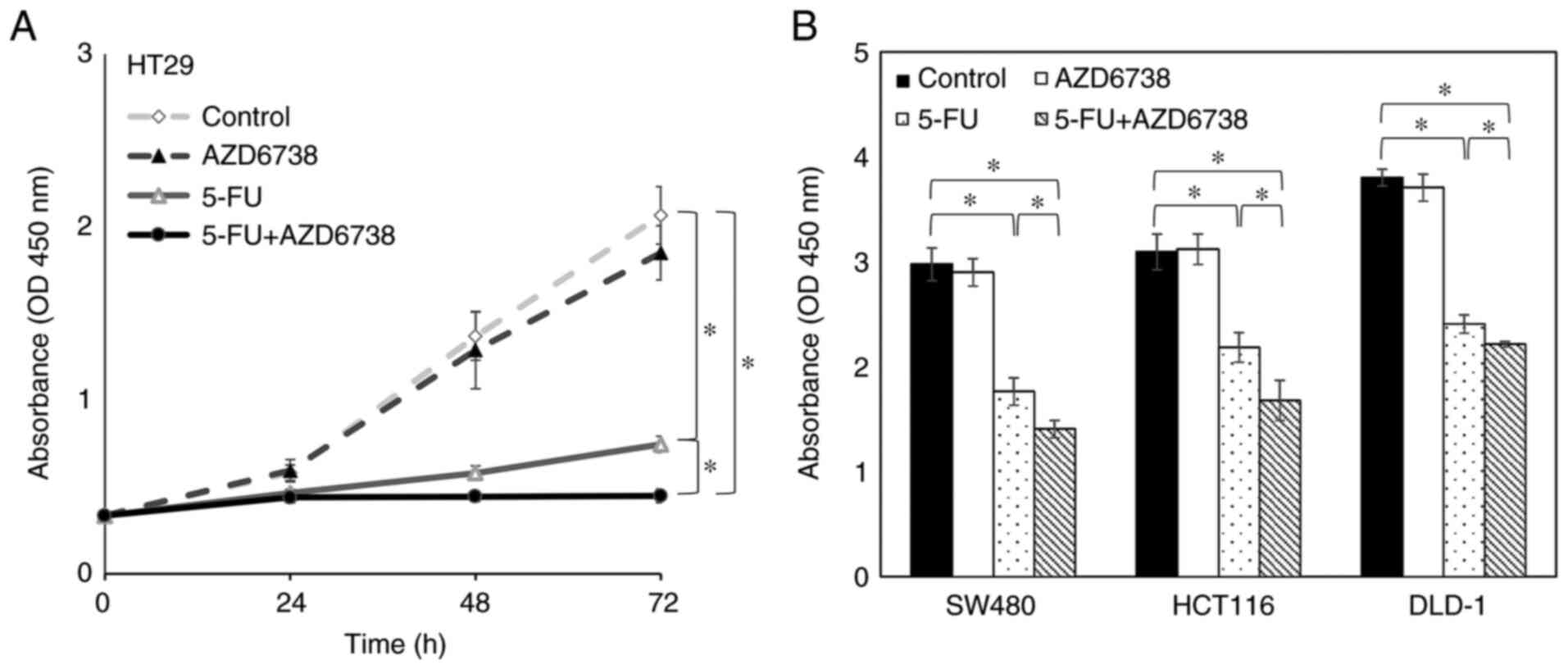
Next, to examine whether inhibition of the
G2 checkpoint suppresses cell survival, a WST-1 assay
was performed using HT29 cells and the synergistic effect of
AZD6738 and 5-FU on cell survival at 24, 48 and 72 h was
investigated. At 24 and 48 h, there was no significant difference
between cells treated with 5-FU alone and those treated with both
AZD6738 and 5-FU. However, at 72 h, cell survival was significantly
decreased in the AZD6738/5-FU combination group compared with the
5-FU alone group (P<0.05; Fig.
3A).
The present study subsequently investigated the
synergistic effect of AZD6738 and 5-FU on the survival of other
p53-deficient colorectal cancer cells. SW480, HCT116 and DLD-1
cells were treated with both 5-FU and AZD6738 for 72 h, which
effectively inhibited cell survival compared with 5-FU alone in all
three cell lines (P<0.05; Fig.
3B).
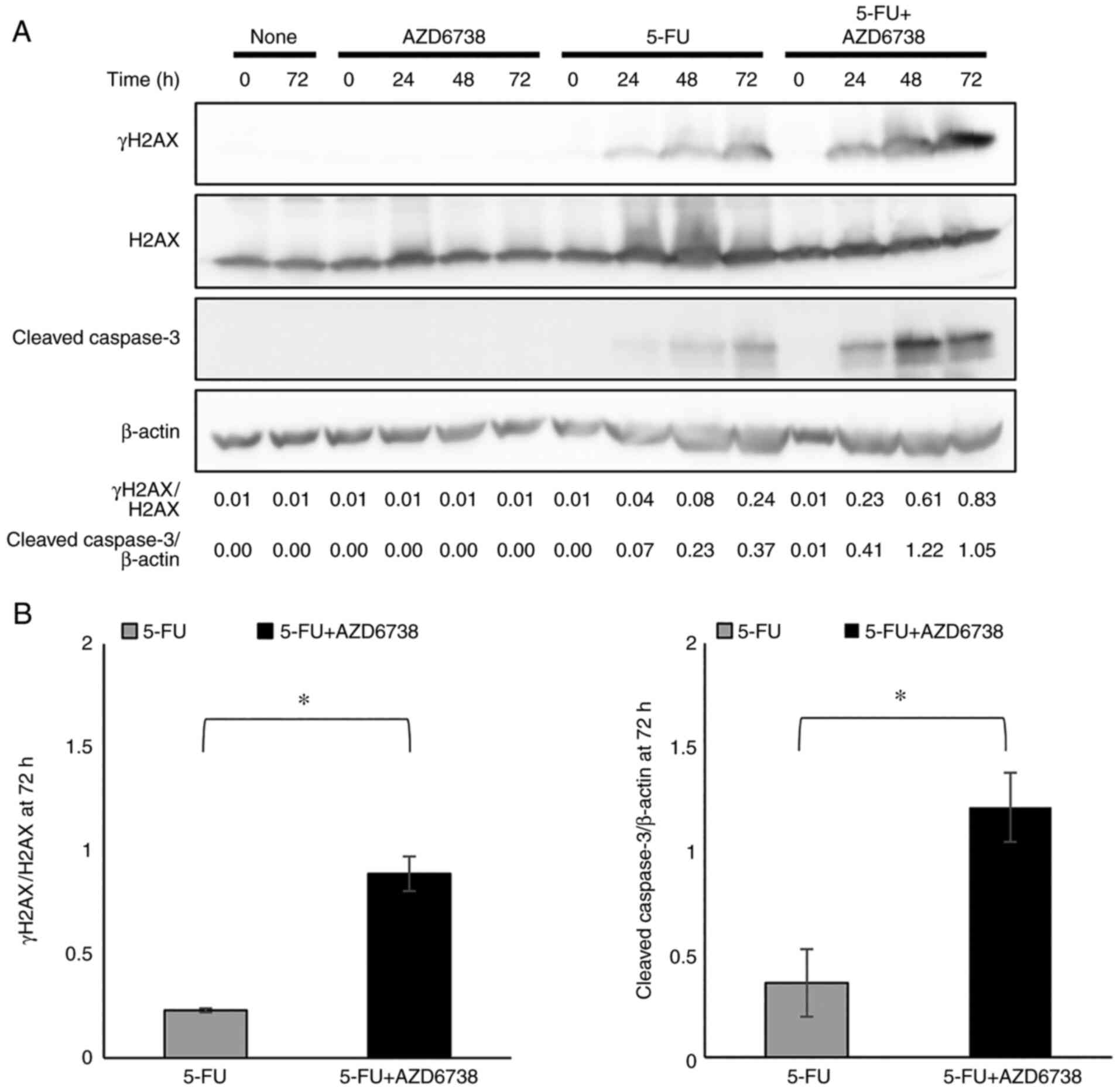
AZD6738 enhances 5-FU-mediated
apoptosis and DNA damage
The present study examined whether the combination
of AZD6738 and 5-FU increases cell death. The levels of cleaved
caspase-3, a marker of apoptosis, and those of γH2AX, a marker of
DNA damage, were increased after the combination treatment compared
with 5-FU monotherapy at 72 h (Fig.
4). These results indicated that the treatment of 5-FU combined
with AZD6738 caused accumulation of DNA damage and increased
apoptosis.
AZD6738 enhances 5-FU-induced
inhibition of tumor growth and DNA damage in mouse xenograft
models
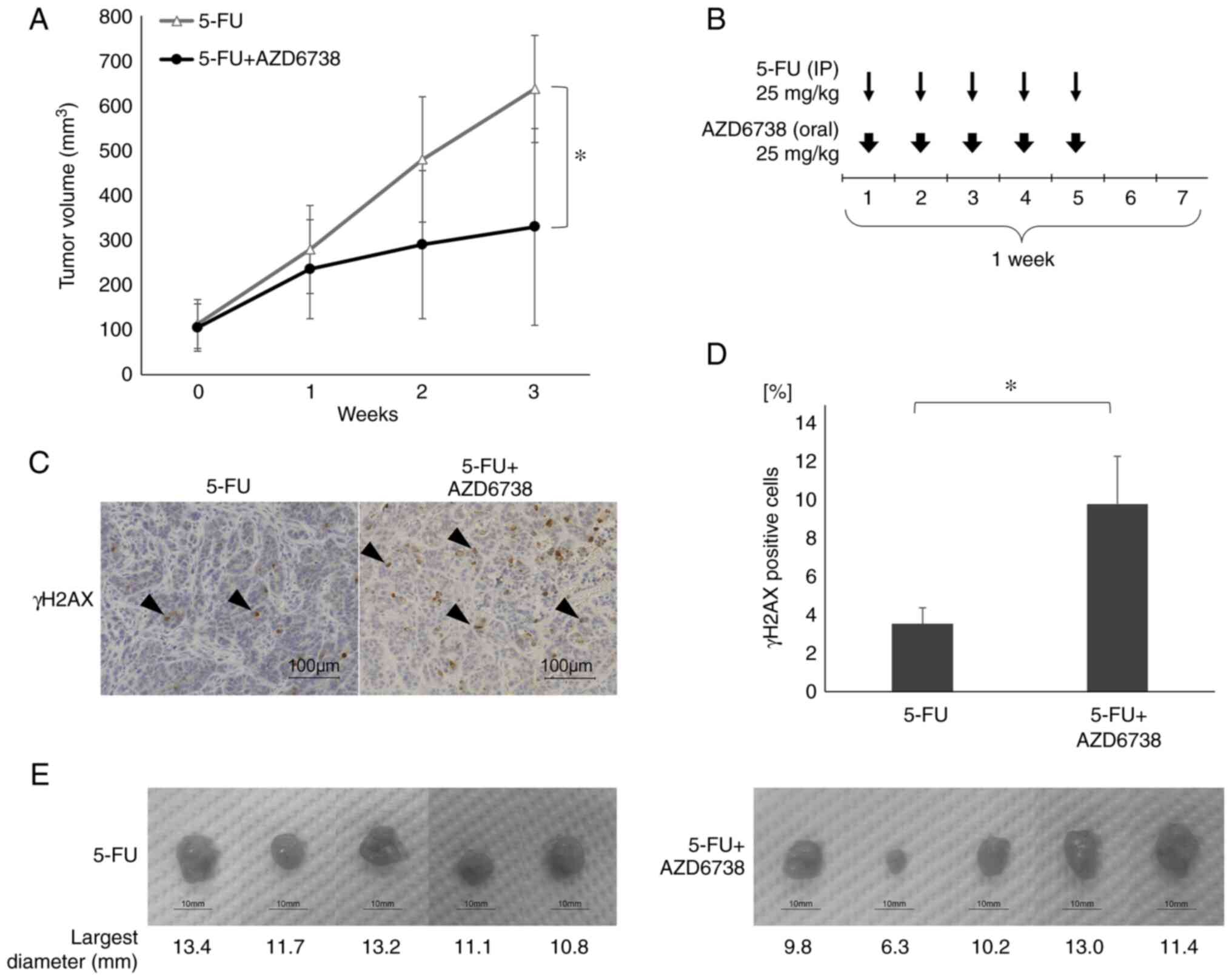
To determine whether AZD6738 enhances the
therapeutic effect of 5-FU in vivo, a HT29 subcutaneous
xenograft model was developed in nude mice. Once the tumors had
developed, each treatment was administered for 3 weeks. As shown in
Fig. 5, the 5-FU/AZD6738
combination treatment resulted in significantly greater inhibition
of tumor growth compared with 5-FU (25 mg/kg 5 days/week)
monotherapy at 3 weeks (P<0.05; Fig. 5A). There was no significant
difference in tumor growth between the AZD6738 alone group and the
control group (Fig. S2).
In addition, immunohistochemical staining of γH2AX
revealed a significant increase in the percentage of γH2AX-positive
cells in the AZD6738 combination (9.82%) group compared with the
5-FU monotherapy group (3.55%) (P<0.05; Fig. 5C-E). Thus, in vivo, the
combination of 5-FU and AZD6738 increased DNA damage in cancer
cells and enhanced tumor growth inhibition compared with 5-FU
alone.
Discussion
The increasing resistance to chemotherapy or
radiotherapy, or both, in malignant tumors causes major
difficulties in their treatment and management. It has been
reported that cancer cells may acquire therapeutic resistance by
activating specific DNA repair pathways (35), and focusing on this process is
important for predicting the treatment response and developing
novel therapeutic strategies to prevent the emergence of treatment
resistance. Therefore, the combination of cytotoxic agents with
chemosensitizing agents, such as inhibitors of cell cycle
checkpoints or of DNA repair pathways, is likely to result in
synthetic lethality in specific types of cancer cells.
A number of G2 checkpoint inhibitors have
been developed, and several of these have been proposed as
promising candidates for treating p53-deficient cancer cells
because survival of these cells after DNA damage is dependent on
the ATR/Chk1-mediated G2 checkpoint (36,37).
In this context, ATR has been considered a promising therapeutic
target because it is an essential kinase for G2 arrest
in response to various genotoxic stresses (38). However, a number of checkpoint
kinases also serve an important role in normal cell survival and
genome maintenance, and their inhibition may have unexpected
adverse effects on normal cell function (38). To be used as a clinical therapeutic
agent, specificity to the target kinase is more important (39). The first ATR inhibitor, Schisandrin
B, has been reported to abrogate the UV-induced G2
checkpoint, but its efficacy against ATR is insufficient for
clinical use (40). Since then,
ATR-selective inhibitors, such as VE-821 (41,42)
and AZ20 (43), have been
developed. AZD6738, used in the present study, is an ATR-selective
agent with superior solubility, bioavailability and pharmacokinetic
properties compared with other agents (18,44).
Also known as ceralasetrib, AZD6738 is currently undergoing phase I
and II trials (24); however, to
the best of our knowledge, no studies have evaluated its effect in
combination with 5-FU. One of our previous studies elucidated the
mechanism of action of CBP-93872 (6), a G2 checkpoint inhibitor,
and another one of our previous studies reported its efficacy in
combination with radiotherapy and multiple anticancer agents
(34). Based on this, the present
study focused on AZD6738.
5-FU serves a central role in the treatment of
colorectal cancer in the first and second chemotherapy lines. It
has three mechanisms of action: DNA damage by its uptake into DNA
(1), RNA damage by its uptake into
RNA (45) and the inhibition of
DNA de novo synthesis by inhibition of thymidylate synthase
(TS) (46). In the case of DNA
damage, 5-FU taken into cells is converted into
5-fluoro-2′-deoxyuridine 5′-monophosphate (FdUMP). After that it is
converted into 5-fluoro-2′-deoxyuridine 5′-triphosphate and taken
into DNA (47,48). Such replication stress causes DNA
damage and activates ATR, which phosphorylates multiple downstream
substrates responsible for the DDR (6). Cells can stabilize their DNA and
survive replication stress by preventing firing of replication
origins, stabilizing arrested replication forks, and promoting DNA
repair and cell cycle checkpoints (6). The simultaneous use of AZD6738 and
5-FU is likely to inhibit these processes. Therefore,
AZD6738-induced cell death may be mediated by destabilization of
one or more of the aforementioned pathways.
Another effect of 5-FU on the cell cycle is arrest
in the S phase (49). This cell
cycle arrest is due to inhibition of DNA synthesis. By forming a
ternary complex with TS and 5,10-methylenetetrahydrofolate, FdUMP
inhibits TS, which in turn causes a reduction in deoxythymidine
monophosphate, which is required for deoxythymidine triphosphate
synthesis, ultimately leading to thymidine depletion and S phase
arrest (49). In the present
study, although 5-FU induced S-phase arrest when combined with
AZD6738, this was followed by inhibition of the G2
checkpoint, which enhanced the effect of 5-FU both in vitro
and in vivo.
AZD6738 specifically suppresses the G2
checkpoint by inhibiting DNA damage-dependent activation of ATR
(39). Importantly, AZD6738 has
little effect on normal cells because normal cells have a
functioning ATM/p53/p21 signaling pathway (6). The present results suggest that
AZD6738, when used in combination with 5-FU, has the potential to
enhance the therapeutic effect of chemotherapy on colorectal cancer
by efficiently acting from the first line and preventing
resistance.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Ms Seiko Inumaru and
Ms Ryoko Hara for handling tumor samples in the present study
(Department of Gastroenterological Surgery, Nagoya City University
Graduate School of Medical Sciences, Nagoya, Japan).
Funding
The present study was supported by Grants-in-Aid for Scientific
Research (Japan Society for the Promotion of Science; grant no.
19K18158).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
TS and TH contributed to the conception and design
of the study, analyzed and interpreted the data, and wrote and
reviewed the manuscript. AMa, KW, TY, NN, YMae, KS, RO, AMi, MK,
YMae, HT and ST designed the study. AMa, SH, KW, HU, NN, YMat and
HT analyszd and interpreted data. TS, SH, HU, AMa, AMi and MK
acquired the data. TS, TH, AMi, KW and MK confirm the authenticity
of all the raw data. TS, TH, SH, KW and HT wrote the manuscript.
TH, YMat, AMi and ST supervised the study. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
In vivo mouse experiments were performed
according to the Guidelines for Animal Experiments of the Nagoya
City University Graduate School of Medical Sciences and approved by
the Animal Care and Use Committee of the Nagoya City University
Graduate School of Medical Sciences (reference number, 20-011;
Nagoya, Japan).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Saito K, Nagashima H, Noguchi K, Yoshisue
K, Yokogawa T, Matsushima E, Tahara T and Takagi S: First-in-human,
phase I dose-escalation study of single and multiple doses of a
first-in-class enhancer of fluoropyrimidines, a dUTPase inhibitor
(TAS-114) in healthy male volunteers. Cancer Chemother Pharmacol.
73:577–583. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ghosh S: Cisplatin: The first metal based
anticancer drug. Bioorg Chem. 88:1029252019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ward JF: DNA damage produced by ionizing
radiation in mammalian cells: Identities, mechanisms of formation,
and reparability. Prog Nucleic Acid Res Mol Biol. 35:95–125. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Blondy S, David V, Verdier M, Mathonnet M,
Perraud A and Christou N: 5-Fluorouracil resistance mechanisms in
colorectal cancer: From classical pathways to promising processes.
Cancer Sci. 111:3142–3154. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Harper JW and Elledge SJ: The DNA damage
response: Ten years after. Mol Cell. 28:739–745. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hirokawa T, Shiotani B, Shimada M, Murata
K, Johmura Y, Haruta M, Tahara H, Takeyama H and Nakanishi M:
CBP-93872 inhibits NBS1-mediated ATR activation, abrogating
maintenance of the DNA double-strand break-specific G2 checkpoint.
Cancer Res. 74:3880–3889. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shimada M and Nakanishi M: DNA damage
checkpoints and cancer. J Mol Histol. 37:253–260. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Niida H, Katsuno Y, Banerjee B, Hande MP
and Nakanishi M: Specific role of Chk1 phosphorylations in cell
survival and checkpoint activation. Mol Cell Biol. 27:2572–2581.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shimada M, Niida H, Zineldeen DH, Tagami
H, Tanaka M, Saito H and Nakanishi M: Chk1 is a histone H3
threonine 11 kinase that regulates DNA damage-induced
transcriptional repression. Cell. 132:221–232. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shimada M and Nakanishi M: Checkpoints
meet the transcription at a novel histone milestone (H3-T11). Cell
Cycle. 7:1555–1559. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu Q, Guntuku S, Cui XS, Matsuoka S,
Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A,
et al: Chk1 is an essential kinase that is regulated by Atr and
required for the G(2)/M DNA damage checkpoint. Genes Dev.
14:1448–1459. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Iacopetta B: TP53 mutation in colorectal
cancer. Hum Mutat. 21:271–276. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kandoth C, McLellan MD, Vandin F, Ye K,
Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al:
Mutational landscape and significance across 12 major cancer types.
Nature. 502:333–339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Song Y, Li L, Ou Y, Gao Z, Li E, Li X,
Zhang W, Wang J, Xu L, Zhou Y, et al: Identification of genomic
alterations in oesophageal squamous cell cancer. Nature. 509:91–95.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fan S, Smith ML, Rivet DJ II, Duba D, Zhan
Q, Kohn KW, Fornace AJ Jr and O'Connor PM: Disruption of p53
function sensitizes breast cancer MCF-7 cells to cisplatin and
pentoxifylline. Cancer Res. 55:1649–1654. 1995.PubMed/NCBI
|
|
16
|
Goto H, Izawa I, Li P and Inagaki M: Novel
regulation of checkpoint kinase 1: Is checkpoint kinase 1 a good
candidate for anti-cancer therapy? Cancer Sci. 103:1195–1200. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ma CX, Janetka JW and Piwnica-Worms H:
Death by releasing the breaks: CHK1 inhibitors as cancer
therapeutics. Trends Mol Med. 17:88–96. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sundar R, Brown J, Ingles Russo A and Yap
TA: Targeting ATR in cancer medicine. Curr Probl Cancer.
41:302–315. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vendetti FP, Lau A, Schamus S, Conrads TP,
O'Connor MJ and Bakkenist CJ: The orally active and bioavailable
ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of
cisplatin to resolve ATM-deficient non-small cell lung cancer in
vivo. Oncotarget. 6:44289–44305. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Foote KM, Nissink JWM, McGuire T, Turner
P, Guichard S, Yates JW, Lau A, Blades K, Heathcote D, Odedra R, et
al: Discovery and characterization of AZD6738, a potent inhibitor
of ataxia telangiectasia mutated and Rad3 related (ATR) kinase with
application as an anticancer agent. J Med Chem. 61:9889–9907. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wallez Y, Dunlop CR, Johnson TI, Koh SB,
Fornari C, Yates JWT, Bernaldo de Quirós Fernández S, Lau A,
Richards FM and Jodrell DI: The ATR inhibitor AZD6738 synergizes
with gemcitabine in vitro and in vivo to induce pancreatic ductal
adenocarcinoma regression. Mol Cancer Ther. 17:1670–1682. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dok R, Glorieux M, Bamps M and Nuyts S:
Effect of ATR Inhibition in RT response of HPV-negative and
HPV-positive head and neck cancers. Int J Mol Sci. 22:15042021.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dillon MT, Barker HE, Pedersen M, Hafsi H,
Bhide SA, Newbold KL, Nutting CM, McLaughlin M and Harrington KJ:
Radiosensitization by the ATR inhibitor AZD6738 through generation
of acentric micronuclei. Mol Cancer Ther. 16:25–34. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gorecki L, Andrs M, Rezacova M and
Korabecny J: Discovery of ATR kinase inhibitor berzosertib (VX-970,
M6620): Clinical candidate for cancer therapy. Pharmacol Ther.
210:1075182020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim R, Kwon M, An M, Kim ST, Smith SA,
Loembé AB, Mortimer PGS, Armenia J, Lukashchuk N, Shah N, et al:
Phase II study of ceralasertib (AZD6738) in combination with
durvalumab in patients with advanced/metastatic melanoma who have
failed prior anti-PD-1 therapy. Ann Oncol. 33:193–203. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shah PD, Wethington SL, Pagan C, Latif N,
Tanyi J, Martin LP, Morgan M, Burger RA, Haggerty A, Zarrin H, et
al: Combination ATR and PARP Inhibitor (CAPRI): A phase 2 study of
ceralasertib plus olaparib in patients with recurrent,
platinum-resistant epithelial ovarian cancer. Gynecol Oncol.
163:246–253. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dillon MT, Boylan Z, Smith D, Guevara J,
Mohammed K, Peckitt C, Saunders M, Banerji U, Clack G, Smith SA, et
al: PATRIOT: A phase I study to assess the tolerability, safety and
biological effects of a specific ataxia telangiectasia and
Rad3-related (ATR) inhibitor (AZD6738) as a single agent and in
combination with palliative radiation therapy in patients with
solid tumours. Clin Transl Radiat Oncol. 12:16–20. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yap TA, Krebs MG, Postel-Vinay S, Bang YJ,
El-Khoueiry A, Abida W, Harrington K, Sundar R, Carter L,
Castanon-Alvarez E, et al: Phase I modular study of AZD6738, a
novel oral, potent and selective ataxia telangiectasia Rad3-related
(ATR) inhibitor in combination (combo) with carboplatin, olaparib
or durvalumab in patients (pts) with advanced cancers. Eur J
Cancer. 69 (Suppl 1):S22016. View Article : Google Scholar
|
|
29
|
Young LA, O'Connor LO, de Renty C,
Veldman-Jones MH, Dorval T, Wilson Z, Jones DR, Lawson D, Odedra R,
Maya-Mendoza A, et al: Differential activity of ATR and WEE1
inhibitors in a highly sensitive subpopulation of DLBCL linked to
replication stress. Cancer Res. 79:3762–3775. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nam AR, Jin MH, Bang JH, Oh KS, Seo HR, Oh
DY and Bang YJ: Inhibition of ATR increases the sensitivity to WEE1
inhibitor in biliary tract cancer. Cancer Res Treat. 52:945–956.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Min A, Im SA, Jang H, Kim S, Lee M, Kim
DK, Yang Y, Kim HJ, Lee KH, Kim JW, et al: AZD6738, A novel oral
inhibitor of ATR, induces synthetic lethality with ATM deficiency
in gastric cancer cells. Mol Cancer Ther. 16:566–577. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shimabukuro M, Hayashi K, Kishida R,
Tsuchiya A and Ishikawa K: No-observed-effect level of silver
phosphate in carbonate apatite artificial bone on initial bone
regeneration. ACS Infect Dis. 8:159–169. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rodrigues NR, Rowan A, Smith ME, Kerr IB,
Bodmer WF, Gannon JV and Lane DP: p53 mutations in colorectal
cancer. Proc Natl Acad Sci USA. 87:7555–7559. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Iwata T, Uchino T, Koyama A, Johmura Y,
Koyama K, Saito T, Ishiguro S, Arikawa T, Komatsu S, Miyachi M, et
al: The G2 checkpoint inhibitor CBP-93872 increases the sensitivity
of colorectal and pancreatic cancer cells to chemotherapy. PLoS
One. 12:e01782212017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stover EH, Konstantinopoulos PA, Matulonis
UA and Swisher EM: Biomarkers of response and resistance to DNA
repair targeted therapies. Clin Cancer Res. 22:5651–5660. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Landau HJ, McNeely SC, Nair JS, Comenzo
RL, Asai T, Friedman H, Jhanwar SC, Nimer SD and Schwartz GK: The
checkpoint kinase inhibitor AZD7762 potentiates
chemotherapy-induced apoptosis of p53-mutated multiple myeloma
cells. Mol Cancer Ther. 11:1781–1788. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Meng X, Laidler LL, Kosmacek EA, Yang S,
Xiong Z, Zhu D, Wang X, Dai D, Zhang Y, Wang X, et al: Induction of
mitotic cell death by overriding G2/M checkpoint in endometrial
cancer cells with non-functional p53. Gynecol Oncol. 128:461–469.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu S, Shiotani B, Lahiri M, Maréchal A,
Tse A, Leung CC, Glover JN, Yang XH and Zou L: ATR
autophosphorylation as a molecular switch for checkpoint
activation. Mol Cell. 43:192–202. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Foote KM, Lau A and Nissink JW: Drugging
ATR: Progress in the development of specific inhibitors for the
treatment of cancer. Future Med Chem. 7:873–891. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nishida H, Tatewaki N, Nakajima Y, Magara
T, Ko KM, Hamamori Y and Konishi T: Inhibition of ATR protein
kinase activity by schisandrin B in DNA damage response. Nucleic
Acids Res. 37:5678–5689. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Charrier JD, Durrant SJ, Golec JM, Kay DP,
Knegtel RM, MacCormick S, Mortimore M, O'Donnell ME, Pinder JL,
Reaper PM, et al: Discovery of potent and selective inhibitors of
ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase
as potential anticancer agents. J Med Chem. 54:2320–2330. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Šalovská B, Fabrik I, Ďurišová K, Link M,
Vávrová J, Řezáčová M and Tichý A: Radiosensitization of human
leukemic HL-60 cells by ATR kinase inhibitor (VE-821):
Phosphoproteomic analysis. Int J Mol Sci. 15:12007–12026. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Foote KM, Blades K, Cronin A, Fillery S,
Guichard SS, Hassall L, Hickson I, Jacq X, Jewsbury PJ, McGuire TM,
et al: Discovery of
4-{4-[(3R)-3-Methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-yl}-1H-indole
(AZ20): A potent and selective inhibitor of ATR protein kinase with
monotherapy in vivo antitumor activity. J Med Chem. 56:2125–2138.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jones CD, Blades K, Foote KM, Guichard SM,
Jewsbury PJ, McGuire T, Nissink JW, Odedra R, Tam K, Thommes P, et
al: Abstract 2348: Discovery of AZD6738, a potent and selective
inhibitor with the potential to test the clinical efficacy of ATR
kinase inhibition in cancer patients. Cancer Res. 73 (Suppl
8):S23482013.
|
|
45
|
Chalabi-Dchar M, Fenouil T, Machon C,
Vincent A, Catez F, Marcel V, Mertani HC, Saurin JC, Bouvet P,
Guitton J, et al: A novel view on an old drug, 5-fluorouracil: An
unexpected RNA modifier with intriguing impact on cancer cell fate.
NAR Cancer. 3:zcab0322021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Very N, Hardivillé S, Decourcelle A,
Thévenet J, Djouina M, Page A, Vergoten G, Schulz C, Kerr-Conte J,
Lefebvre T, et al: Thymidylate synthase O-GlcNAcylation: A
molecular mechanism of 5-FU sensitization in colorectal cancer.
Oncogene. 41:745–756. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Longley DB, Harkin DP and Johnston PG:
5-fluorouracil: Mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mori R, Yoshida K, Futamura M, Suetsugu T,
Shizu K, Tanahashi T, Tanaka Y, Matsuhashi N and Yamaguchi K: The
inhibition of thymidine phosphorylase can reverse acquired
5FU-resistance in gastric cancer cells. Gastric Cancer. 22:497–505.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hagenkort A, Paulin CBJ, Desroses M, Sarno
A, Wiita E, Mortusewicz O, Koolmeister T, Loseva O, Jemth AS,
Almlöf I, et al: dUTPase inhibition augments replication defects of
5-fluorouracil. Oncotarget. 8:23713–23726. 2017. View Article : Google Scholar : PubMed/NCBI
|