Introduction
Genomic alterations play a significant role in human
carcinogenesis, and the genomic instability of cancer cells is
frequently antagonized by activation of the sentinel cellular
stress protein TP53 (referred to as p53) gene. This gene has the
ability to respond to a number of high-stress situations, including
DNA damage, heat shock, hypoxia and oncogene overexpression; thus,
it is involved in monitoring DNA damage to induce growth arrest,
DNA repair or apoptosis (1,2).
Primarily, p53 plays a vital role in the maintenance of
genome stability and integrity by regulating DNA repair and a
diverse range of biological responses (3). p53 is a key initiator of DNA
damage response signaling and activates numerous DNA damage repair
genes, depending on the specific type of DNA damage. Functional
loss of p53 is therefore associated with increased levels of
genomic alterations that are often associated with oncogenic
progression (4). In addition,
p53-mutated tumors exert selective pressure, leading to the
clonal expansion of mutator phenotypes with increased survival
capabilities and metastatic potential (5).
p53 is the most mutated gene across all
cancer types, and is genetically altered in >50% of all tumors
(6). The majority of mutations in
p53 are missense mutations that give rise to single amino
acid substitutions. These disrupt p53 DNA binding found in the
central DNA-binding domain (DBD), which spans 200 amino acids, and
account for 80% of all p53 mutations identified (7). Almost all DBD mutations lead to the
loss of wild-type p53 (wtp53) functions (8), and loss of p53 activity predisposes
cells to the acquisition of oncogenic mutations and favour genetic
instability (9). A heterozygous
p53 mutation not only inactivates wtp53 function, but may
also confer gain of function (GOF) activities to mutant p53
(mutp53) that promotes tumorigenesis. A number of GOF activities
have previously been reported, and the most important GOF activity
is to inhibit DNA repair which stimulates genome instability.
Moreover, it has previously been hypothesized that treatment of
p53-mutated tumors with chemotherapeutic agents which induce
double-stranded breaks (DSBs), such as cisplatin, doxorubicin and
etoposide, results in enhanced genomic instability (10,11).
Frequent mutations, high levels of missense mutp53
expression and the existence of tumor-promoting GOF activities make
p53 a relevant and promising target for therapeutic
intervention. Over the last decade, significant efforts have been
employed in the identification and characterization of small
molecules that can restore wild-type activity to mutp53. Of these,
APR-246 (PRIMA-1met) has demonstrated a high level of
potential in a clinical setting. Preclinical studies have
demonstrated that APR-246 modifies mutp53 protein by
sequence-specific DNA-binding, leading to antitumor activity
(12,13). APR-246 is currently employed in
phase I/II clinical trials for ovarian cancer, haematological
malignancies and prostate cancer (14,15).
Similarly, another small molecule, CP-31398, interacts with the DBD
of mutp53 proteins, thereby promoting the correct folding of the
mutant protein; thus, restoring its function (16). Results of our previous study
demonstrated that mutant-p53 reactivation by CP31398 promotes
natural killer cell-mediated lysis in breast cancer cell lines
through an autophagy-dependent mechanism (17).
The present study aimed to investigate the efficacy
of small molecule APR-246 in regulating the DNA repair functions
that are downstream of p53. The functional efficacy of p53
reactivation was analyzed by measuring the mutational frequency in
p53-mutated breast cancer cell lines using whole exome
sequencing (WES). Results of the present study demonstrated that
APR-246 controls genomic instability by enhancing the DNA damage
response in mutated p53 cells by upregulating DNA repair genes.
Materials and methods
Cell culture, cell lines and mutation
status
Human breast cancer cell lines MDA-MB-231
(p53mut) and MCF-7 (p53wt), used in the
present study, were obtained from INSERM U1186, Gustave Roussy.
MDA-MB-231 cells have gain of function mutations specific to the
genomic region c.839G>A leading to a change in protein
sequence-R248Q (18). MDA-MB-231
cells were cultured in DMEM (Gibco; Thermo Fisher Scientific,
Inc.), supplemented with 10% FBS (Gibco; Thermo Fisher Scientific,
Inc.) and 1% penicillin/streptomycin (Sigma-Aldrich; Merck KGaA).
The human breast cancer cell line MCF-7 was maintained in RPMI
medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with
10% FBS, 1% penicillin/streptomycin and 20 mM L-glutamine
(Sigma-Aldrich; Merck KGaA). Cell lines were incubated under
standard conditions at 37°C in a humidified 5% CO2
atmosphere. The mutation status of the cell lines was confirmed
using direct Sanger sequencing and specific p53 primers (Table SI).
Drug treatments for measuring the
extent of DNA damage
APR246/PRIMA-1Met was purchased from
Sigma-Aldrich; Merck KGaA and a 10 mmol/l stock was prepared by
dissolving in DMSO. MDA-MB-231 and MCF-7 cell lines were treated
with 50 µM APR-246 for 24 h for monotherapy. The synergistic effect
of APR-246 with cisplatin for DNA damage response was evaluated by
treating the cells with APR-246 and cisplatin (10 µM) for 24 h. The
final concentration of DMSO was kept constant and did not exceed
0.05% (vol/vol).
Long term treatment for genomic
instability analysis
Briefly, long term treatment for analyzing genomic
instability was performed following the procedures used by Hansen
et al (18) with minor
modifications. MDA-MB-231 and MCF-7 cell lines were cultured in
medium alone or medium containing 50 µM APR-246 for 15 passages
[from passage 1 (p5) until passage 15 (p20)] with splitting (ratio,
1:10) every fifth day. Cells cultured in medium alone were used as
the control. Mutations arising after 15 passages were analyzed as a
measure of genomic instability. Genomic instability was analyzed in
APR-246-p20 cells and control-p20 cells with vehicle DMSO, and
these were compared with control cells at p5 using WES
analysis.
Immunofluorescence
Following the aforementioned drug treatment, cells
were collected and washed with PBS, and fixed with 4%
paraformaldehyde for 30 min. Cells were subsequently permeabilized
with 100 mM Tris, 50 mM EDTA and 0.5% Triton X-100 for 30 min, and
blocked in 3% bovine serum albumin in PBS. Between each of these
steps, cells were washed with washing buffer (5% FBS in PBS) and
stained overnight with the primary antibody. Following incubation
with the primary antibody, cells were incubated with the Alexa
fluor-conjugated secondary antibody for 1 h at room temperature.
Cells were subsequently stained with DAPI (cat. no. D1306; Thermo
Fisher Scientific, Inc.) and mounted on a slide with ProLong Gold
Antifade reagent (cat. no. P10144; Thermo Fisher Scientific, Inc.).
The slides were visualized using a confocal microscope (Zeiss LSM
800 with Airyscan; Carl Zeiss AG) and analyzed for the presence of
foci. The antibodies used were as follows: Anti-p53 (1:1,000; cat.
no. 2527; Cell Signaling Technology, Inc.), anti-53BP1 (1:1,000;
cat. no. PA1-46147; Thermo Fisher Scientific, Inc.) and
anti-phosphorylated histone H2A variant H2AX (γH2AX; 1:2,000; cat.
no. 05-636; MilliporeSigma).
Alkaline comet assay
The alkaline comet assay was performed following the
protocol described by Olive and Banath (19). The second layer of agarose gel was
prepared using ~5,000 cells per slide. The slides were immersed
overnight in lysis buffer, followed by alkaline unwinding for 30
min and electrophoresis at 0.65 volts/cm for 35 min. The slides
were visualized and imaged using a confocal microscope
(magnification, ×20). A total of 100 randomly captured nuclei were
scored and the assay was repeated in triplicate. The comet images
were analyzed for the presence of comets using the OpenComet tool
(20) on ImageJ (version 1.51;
National Institutes of Health). Hydrogen peroxide-treated cells
(200 µM for 30 min) were used as the positive control. All three
parameters of the comet assay were presented: tail length (TL),
tail intensity (TI) and tail moment (TM). Data are presented as the
mean ± standard deviation.
Reverse transcription-quantitative
(RT-q)PCR assay
Total RNA was extracted from the control and treated
cells using a RNeasy kit (cat. no. 74104; Qiagen, Inc.), and
reverse transcribed into cDNA using the High Cap cDNA Reverse
Transcription kit (cat. no. 4374966; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. qPCR was
performed using the SYBR Green PCR Master Mix (cat. no. 4368577;
Applied Biosystems; Thermo Fisher Scientific, Inc.) and relative
mRNA expression was normalized to GAPDH/ß-actin. PCR primers used
in the present study are described in Table SII.
WES analysis
WES analysis was performed using MDA MB-231 and
MCF-7 cell lines treated with APR-246 (50 µM) for 15 passages. MDA
MB-231 and MCF-7 cell lines at p5 were used before the initiation
of treatment, and cell lines at p20 treated with DMSO were used as
controls. DNA was isolated using the QiaAmp DNA mini kit (Qiagen,
Inc.) following the manufacturer's protocols. The isolated DNA was
quantified using Qubit 2.0 (Thermo Fisher Scientific, Inc.). The
exome libraries were prepared using Ampliseq Exome library panel of
12 primer pools (cat. no. A38264; Thermo Fisher Scientific, Inc.)
which was designed to cover ~300,000 amplicons. Generated amplicons
of each sample were pooled and barcoded using Ion Express barcode
adapter. Thus, the generated exome library was quantified using the
Ion Library TaqMan Quantitation kit (cat. no. 4468802; Thermo
Fisher Scientific, Inc.), and the libraries were further diluted to
100 pM and pooled equally with two individual samples per pool. The
pooled libraries were amplified using emulsion PCR using ion chef
reagents (cat. no. A30011; Thermo Fisher Scientific, Inc.)
following the manufacturer's protocol. Prepared template libraries
were subsequently sequenced using the Ion S5 XL Semiconductor
sequencer and an Ion 540 Chip.
Bioinformatic analysis of WES
data
The raw data was aligned with the hg38 version of
the genome using Ion Torrent Suite software and the bam files were
processed. Briefly, the pileup of treated and non-treated pairs of
samples was obtained using samtools mpileup followed by Varscan2
somatic variant and indel calling (21), where non-treated control p5 was
considered as a reference sample, and APR-246 p20 and non-treated
control p20 were considered as case samples. Varscan2 revealed
variants only in the regions where coverage was >10 reads. Only
high confidence (hc), as marked by Varscan2 in its default
settings, were used in subsequent analyses. Obtained vcf files were
processed to filter out variants with strand bias. The ratio of
variants supporting reads from different strands were maintained
within a range of 0.5-2, and all remaining variants were filtered
out. Subsequently, the maf object was created using the vcf2maf
tool. Variants from the vcf files were annotated using the Variant
Effect Predictor (VEP) (22). MAF
objects were created separately for MCF-7 and MDA-MB-231 cells. MAF
objects were subsequently analyzed using maftools (23).
Statistical analysis
For all statistical analyses associated with comet
and immunofluorescence assays, a one-way ANOVA followed by
Bonferroni's post-hoc test was performed using GraphPad Prism
(version 8.0; GraphPad Software, Inc.). P<0.05 was considered to
indicate a statistically significant difference. All results are
presented as the mean ± standard error of the mean for three
independent experiments.
Results
APR-246 increases the DNA damage
response and repair mechanisms in mutated p53 cell line
MDA-MB-231
To determine whether the efficiency of p53-dependent
DNA repair is modified following pharmacological reactivation of
mutp53, the breast carcinoma cell line MDA-MB-231
(p53R280K) was used, along with MCF-7 cells
(p53WT) as a control. The presence of c.839G>A
variation in MDA-MB-231 cells was verified by sanger sequencing
(Fig. S1). Cells were treated
with 10 µM cisplatin to inflict DNA damage for 24 h, followed by
the incubation of cells with medium alone, or medium containing
APR-246 for a further 24 h. Although there was clear activation of
DNA damage response, the viability of cells remained at 95% after
24 h (Fig. S2). The actual values
were plotted, after counting 500 cells from a Trypan blue assay,
and the axis as percent viability. Subsequently, the ability of
APR-246 in mitigating the genomic instability was analyzed by
measuring the residual DNA damage using an alkaline comet assay
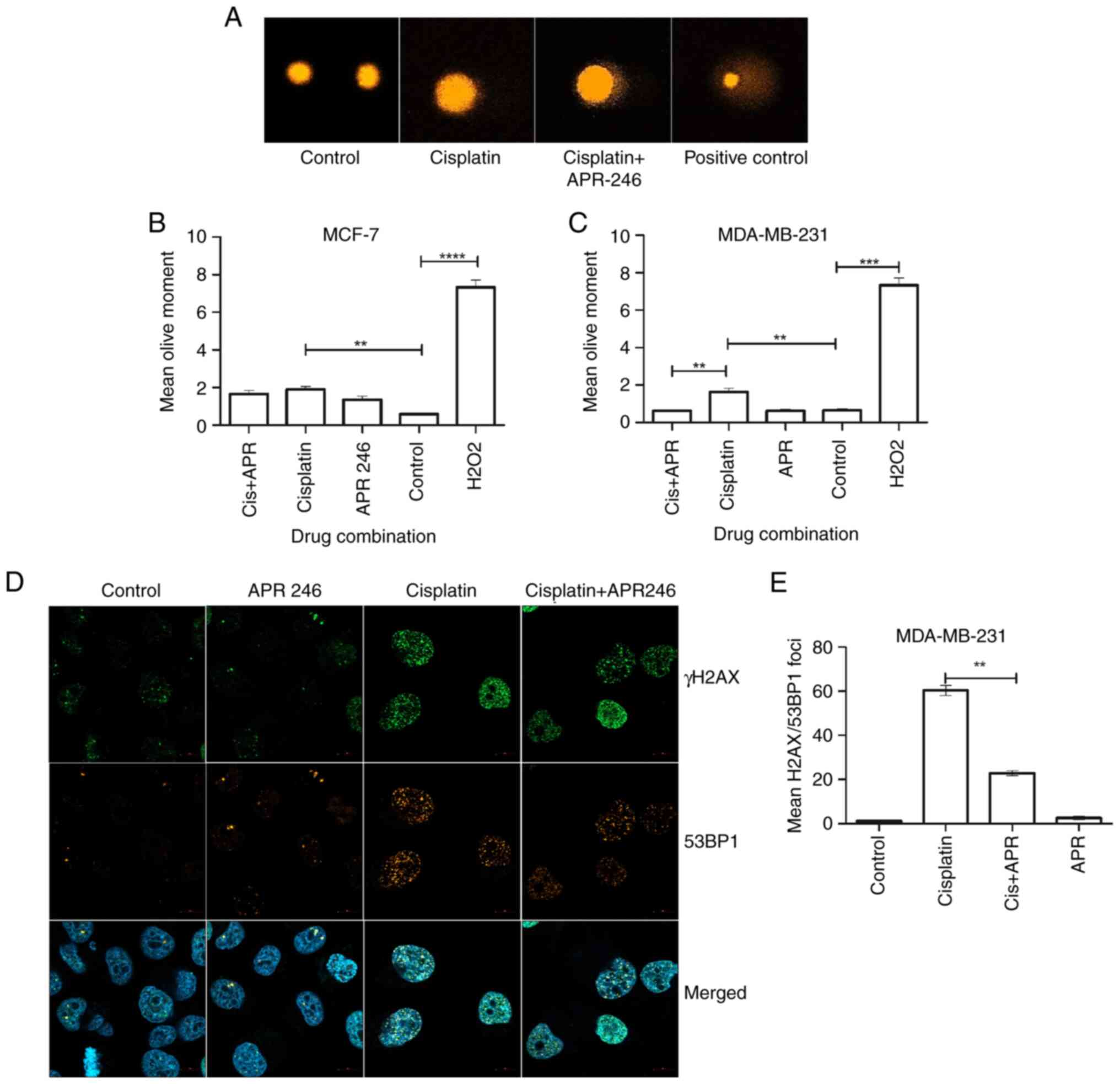
(Fig. 1A). The results
demonstrated that the extent of DNA damage was significantly
reduced in MDA-MB-231 cells that had been treated with a
combination of cisplatin and APR-246 (Cis+APR) compared with cells
that had been treated with cisplatin alone (Fig. 1C). Conversely, no significant
difference in residual DNA damage was observed in the p53 wild-type
cell line MCF-7, following both cisplatin and cisplatin/APR-246
treatment (Fig. 1B).
To analyze the activation of the DNA damage
response, the extent of phosphorylation of γ-H2AX at Serine
(Ser)139 was investigated, which is widely abundant and has
previously been used as a sensitive marker for DNA damage,
including DSBs (24). Results of
the present study demonstrated an increase in phosphorylated
(phospho)-γ-H2AX/53BP1-positive foci in cisplatin-treated cells
using confocal microscopy, which reflected the presence of DSBs
(Fig. 1D). We also noted that the
cells treated with either cisplatin alone or the combination of
cisplatin + APR-246 were sensitive to washing steps and almost
30-40% of the cells were washed out after the
fixing/permeabilization steps of immunocytochemistry. Thus, the
number of cells were lower in the cisplatin and combination
treatment groups. Similarly, phospho-γ-H2AX/53BP1 foci were
significantly reduced in the MDA-MB-231 cells co-treated with
cisplatin + APR-246, compared with control cells treated with
cisplatin alone (Fig. 1E).
Collectively, these data suggest that mutp53 reactivation using
APR-246 may induce DNA repair mechanisms in breast cancer
cells.
APR-246 increases the expression of
p53-dependent DNA repair genes
As the results of the present study revealed a
reduction in the levels of DNA damage following mutp53
reactivation, the impact of APR-246 on p53-dependent DNA repair
gene expression was explored. MDA-MB231 cells were treated with 10
µM cisplatin alone, and/or in the presence of 50 µM APR-246 for 24
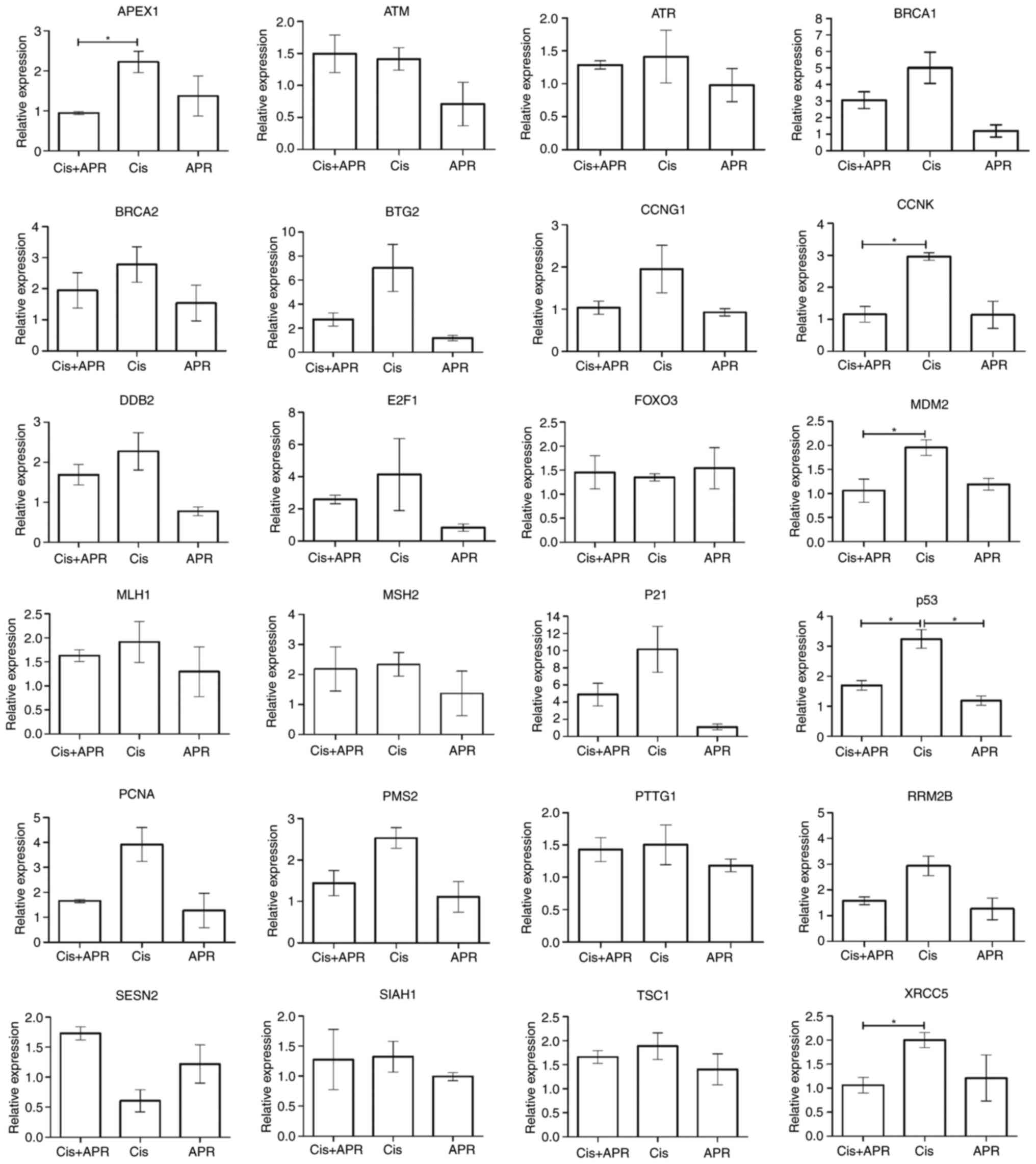
h. Subsequently, RT-qPCR analysis was performed using 24 genes
associated with DNA repair to establish potential associations with
the p53 gene (Fig. 2). Results of
the present study demonstrated a notable increase in DNA
repair-associated gene expression following treatment with
cisplatin. Moreover, treatment with cisplatin in combination with
APR-246 (Cis+APR) led to an increase in the level of gene
expression, compared with treatment with APR-246 alone. Notably,
the results showed an increase in the expression of p21, which is
downstream of p53 (Fig. 2). In
addition, a significant increase in the expression levels of cyclin
K (CCNK), E3 ubiquitin-protein ligase MDM2 and X-ray
repair cross-complementing protein 5 (XRCC5) was observed.
Increased expression levels of the aforementioned genes denotes the
activation of p53-dependent DNA repair enzymes contributing to the
DNA repair process, thereby aiding in the control of genomic
instability.
Pharmacological reactivation by
APR-246 controls the genomic instability of MDA MB-231 cells
The impact of the pharmacological reactivation of
p53 using APR-246 on the regulation of genomic instability was
investigated. WES analysis was performed using MDA-MB-231
(p53R280K) and MCF-7 (p53WT) cells that had
been subcultured for 15 passages with or without 50 µM APR-246. All
mutational analyses were normalized to control cells (p0). Results
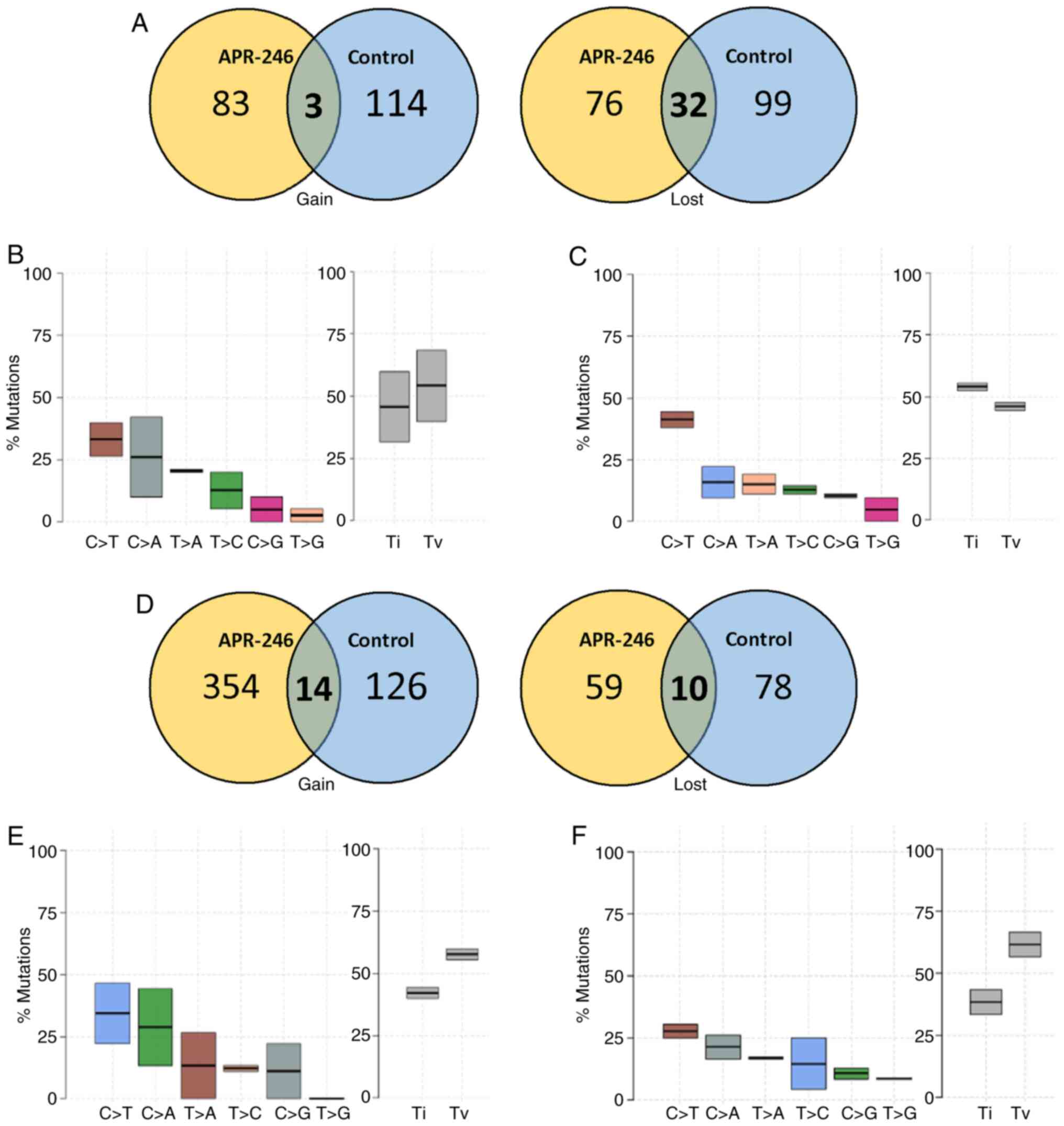
of the WES analysis demonstrated a high level of heterogeneous DNA
variations throughout the exome under all conditions Notably,
results of the present study revealed a gain of 99 somatic
variations in APR-246-treated p53-mutated MDA-MB231 cells, compared
with 129 somatic variations in untreated cells (data not shown). In
addition, mutations may also arise as a function of loss or
deletion of a base in the genomic regions. Novel genomic changes
that arise in the p20 passage when compared with the original p5
passage (starting point) are characterized as mutations that have
been gained, whereas the nucleotides originally recognized in p5,
but are subsequently lost in p20, are characterized as mutations
that have been lost. Results of the present study demonstrated that
there were a significant number of deletions and a loss of
mutations in the control group, compared with cells grown in
APR-246 (Fig. 3A). Further
analysis of the transitions and transversions demonstrated that
there was little or no variation in the pattern of mutational types
(Fig. 3B and C). However, the
transversions were significantly reduced in the APR-246-treated
groups, compared with the control.
Notably, WES analysis of MCF7 cells revealed a
higher mutational rate following treatment with APR-246. A total of
274 and 500 variations were observed in the control-p20 and
APR-246-p20 groups, respectively (Fig.
3D). A total of 354 mutations were gained in the APR-246-p20
group, compared with only 126 mutations in the control-p20 group
(Fig. 3E and F). However, APR-246
treatment did not affect the pattern of transitions and
transversions in MCF-7 cells. These results highlight the ability
of APR-246 in controlling genomic instability in p53-mutated
cells.
Discussion
As previous studies have demonstrated that >50%
of all human cancers exhibit mutations in p53, targeting mutp53 is
an attractive research focus at present. Previous evidence suggests
that the biological mechanisms underlying p53 are complex,
and p53 is involved in the support of cell cycle arrest,
senescence, apoptosis, autophagy, DNA repair and the maintenance of
genomic integrity. As a key player in the cellular stress response,
p53 is activated by numerous stress signals, including DNA damage,
oncogene activation, hypoxia and nutrient deprivation (4,25).
Once activated, p53 functions as a transcription factor, regulating
both genes and microRNAs (25).
Moreover, mutp53 confers gain of function (GOF) activities compared
with non-functional p53, which promotes tumorigenesis. The
GOF/mutp53 inhibits DNA repair which in turn increases genomic
instability, and this increases the levels of oncogenic progression
and therapeutic resistance (26).
Several small molecules have been claimed to
reactivate mutp53, including CP-31398, PRIMA-1,
PRIMA-1MET/APR-246, WR-1065 and MIRA-1 (27). However, all previous studies
analyzed the effect of these molecules on apoptosis and cell cycle
arrest. APR-246 is of great interest, as there are currently seven
clinical trials, three with acute myeloid leukemia (NCT03931291;
NCT03072043; NCT03588078); two with ovarian cancer (NCT03268382;
NCT02098343); mutant myelodysplastic syndromes (NCT03745716) and
one with prostate cancer (NCT00900614) (14). However, in breast cancer, APR-246
has been tested in combination with 2aG4, a monoclonal antibody
that disrupts tumor vasculature with increased effectiveness in
tumor control (28), and reduced
metastasis of p53-mutated triple-negative breast cancer
cells to the lungs (29). Further
mechanistic in vivo studies are required. APR-246 gets
converted to a reactive electrophile-methylene quinuclidinone (MQ),
covalently binding to the core domain of mutp53, thus reactivating
the protein (30). A previous
study revealed that cysteine (Cys)124 and Cys277 are required for
APR-246-mediated functional restoration as a prime binding target
for MQ in p53 (15). Liang et
al (28) demonstrated that
APR-246 effectively reduced in vitro cell viability in
mutant breast cancer cell lines (BT-474 and T47-D). They also found
that APR-246 was effective in inducing apoptosis and in
significantly reducing proliferation in tumor xenografts (29). In colorectal cancer cells, APR-246
induced robust cell death by apoptosis through activation of
pro-apoptotic molecules, Noxa and PUMA (31), and through induction of autophagy
by upregulating the mTOR/AMPK-ULK1-Vps34 signaling cascade
(32). However, to the best of our
knowledge, studies demonstrating p53 reactivation in DNA damage
response and genomic instability are limited. Moreover, assessing
changes at the genetic level following chemotherapeutic treatment
is important to understand the clonal evolution in tumors, and also
in the context of sensitivity or resistance to chemotherapy. To the
best of our knowledge, the present study is the first to use WES
analysis for further investigation into the impact of the
mutp53-targeting drug APR-246 on the genomic instability of breast
cancer cells.
DNA damage is a primary cause of genome instability.
DNA damage can manifest from single or double-stranded DNA breaks,
DNA cross-links, replication stress/replication fork collapse,
telomere attrition and nucleotide depletion (26). Cisplatin, a metal-based anticancer
drug widely used in a number of cancers, causes DNA damage by
forming bivalent adducts at the 1,2 intrastrand crosslink, thus
activating various signaling pathways (26). p53 is a critical sensor and
mediator in DNA damage signaling and upregulates various DNA repair
mechanisms. Chemotherapeutic drugs, such as cisplatin and
doxorubicin induce DNA damage, triggering wild-type p53 activation
and p53-induced apoptosis, resulting in tumor control. Thus, the
present study aimed to analyze the DNA damage and repair patterns
in wild-type MCF-7 and mutp53 MDA-MB-231 cell lines following
cisplatin treatment. Results generated using a comet assay for DNA
damage response revealed an increase in DNA damage in
cisplatin-treated cells, compared with cells treated with a
combination of cisplatin and APR-246. Moreover, p53 expression was
upregulated following DNA damage caused by cisplatin, and p53
reactivation by APR-246 induced a DNA repair response; thus, DNA
damage was significantly reduced following treatment with both
APR-246 and cisplatin. These results suggested that APR-246 was
effective in activating mutp53 in MDA-MB-231 cells. Similarly,
γ-H2AX foci analysis further demonstrated a significant reduction
in the number of foci in cells following treatment with APR-246 and
cisplatin in combination. A previous report by Makhale et al
(33) analyzed proteins associated
with p53 signaling, and observed the levels of phosphorylation in
DNA damage and replication stress markers, such as ataxia
telangiectasia mutated, ataxia telangiectasia and rad3-related
protein, replication protein a and H2AX (33). Moreover, Synnott et al
(34) demonstrated an increase in
gene expression that may regulate intracellular reactive oxygen
species (ROS) following APR-246 treatment in breast cancer cell
lines (34). APR-246 has
previously been shown to enhance the levels of ROS in cancer cells
by targeting thioredoxin reductase 1 without affecting mutp53
expression (35). An increase in
intracellular ROS plays a significant role in the induction of DNA
damage in cells (36). However, an
increase in ROS or the subsequent DNA damage was not directly
measured in the aforementioned study. Furthermore, the results of
the present study demonstrated an increase in the expression of
p53-dependent DNA repair genes using RT-qPCR.
Due to its intrinsic nature, genomic DNA is highly
unstable, and >1 million DNA lesions per cell per day may arise
under certain physiological conditions. Although cellular DNA
repair systems are robust, even a single unrepaired event may
result in the accumulation of mutations, leading to genomic
instability (37,38). Inactivation of the mismatch repair
system is known to cause mutations not only in microsatellites, but
also in random sequences (39). As
a result, patients with cancer undergoing chemotherapy should be
screened for genomic instability, as the level of
chemotherapy-induced genomic instability under the influence of
mutp53 can lead to resistant phenotypes. A recent review reported
that mutp53 facilitates cancer progression through the induction of
genomic instability, mainly in the form of interchromosomal
translocations, aneuploidy and destabilization of DNA replication
mechanisms (40). However, to the
best of our knowledge, there are no recent studies that have
reported on single nucleotide changes. In the present study,
results of the WES analysis demonstrated that in MDA-MB-231
(mutp53) cells, treatment with APR-246 significantly reduced the
mutational gain and loss activities (99 and 154), compared with
control-p20 cells (129 and 180). MDA-MB-231 cells exhibit
aggressive tumor phenotypes and inadequate DNA repair systems,
which may contribute to the formation of mutations. Collectively,
results of the present study revealed that treatment with APR-246
adequately reactivated p53, and controls genomic instability in
MDA-MB-231 cells. Notably, MCF7 control cells demonstrated an
increased mutational load following treatment with APR-246 (500 and
480, respectively), compared with the control. In both cell lines
used in the present study, frameshift variations were notably
frequent. The results of the present study are in accordance with
those by Watanabe et al (41), who demonstrated that MCF-7 and
MDA-MB-231 cells exhibited a 2.9-12-fold increase in the mutation
rate of the hprt gene per each cell division (41). Additionally, both cell lines
exhibited mutations in various oncogenes, and enrichment analysis
in MCF-7 cells revealed that various pathways were affected by
these mutations, including microtubule cytoskeleton organization,
structure homeostasis, DNA metabolic processes and DNA repair.
However, an increased dose of APR-246 treatment alone or in
combination with docetaxel demonstrated cell death through ROS
induction and apoptosis. The DNA repair capacity of cell lines
analyzed in this study were not available (32). At present, the exact mechanisms
underlying APR-246-induced maintenance of genomic stability and DNA
repair in tumors remain to be fully elucidated, as the effect is
dose-, time- and cell line-dependent (12).
There are a few limitations to this study. The genes
for qPCR experiments were basically selected from the RNA
sequencing experiments (data not shown as only two independent
experiments were performed). Nevertheless, the data indicated that
DNA repair genes (Fig. 2) were
highly upregulated when the cells were treated for 24 h with
APR-246 (25 µM). However, we did see some inconsistency in qPCR
data. Also, for the whole exome sequencing, the inconsistency in
results may be attributed to smaller sample size. The
phosphorylation of ATM, ATR, CHK1 and H2AX are the key steps
downstream of p53 activation. We have not verified the effect of
cisplatin and APR-246 on the phosphorylation status of the key
proteins downstream of p53. We have not analyzed the effect of
long-term treatment of APR-246 in this study. It will be
interesting to understand the proliferation and survival to get a
clear picture of the final metabolic effect of APR-246 on breast
cancer cells.
In conclusion, the pharmacological reactivation of
mutp53 may be a promising strategy for the treatment of tumors with
inactive p53, and APR-246 plays an important role in regulating the
DNA repair mechanisms, thus maintaining genomic integrity. Further
research into the influence of APR-246 on the mutational spectrum
in p53-mutated cancer is required.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by Gulf Medical University (GMU)
(UAE) (TRIPM) grants.
Availability of data and materials
The data generated using whole exome sequencing is
available at TRIPM, GMU and can be provided upon reasonable
request.
Authors' contributions
FA, JT and SC conceived and designed the study. FA,
GHV, HHN, ASM, ZNN, MSK performed the experiments, collected and
analyzed the data and confirm the accuracy of the data. BW
performed the bioinformatic analysis. FA and GHV wrote the paper.
FA, GHV, JT and SC reviewed and edited the manuscript. All authors
read and approved the manuscript and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
No samples were used in the study. Thus, ethical
approval was not necessary.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rieber M and Strasberg-Rieber M: Hypoxia,
Mn-SOD and H2O2 regulate p53 reactivation and
PRIMA-1 toxicity irrespective of p53 status in human breast cancer
cells. Biochem Pharmacol. 84:1563–1570. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brady CA, Jiang D, Mello SS, Johnson TM,
Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ,
McLaughlin ME, et al: Distinct p53 transcriptional programs dictate
acute DNA-damage responses and tumor suppression. Cell.
145:571–583. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gupta A, Shah K, Oza MJ and Behl T:
Reactivation of p53 gene by MDM2 inhibitors: A novel therapy for
cancer treatment. Biomed Pharmacother. 109:484–492. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Labuschagne CF, Zani F and Vousden KH:
Control of metabolism by p53-Cancer and beyond. Biochim Biophys
Acta Rev Cancer. 1870:32–42. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nakayama M, Hong CP, Oshima H, Sakai E,
Kim SJ and Oshima M: Loss of wild-type p53 promotes mutant
p53-driven metastasis through acquisition of survival and
tumor-initiating properties. Nat Commun. 11:23332020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Forbes SA, Beare D, Boutselakis H, Bamford
S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, et al:
COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids
Res. 45((D1)): D777–D783. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bouaoun L, Sonkin D, Ardin M, Hollstein M,
Byrnes G, Zavadil J and Olivier M: TP53 Variations in human
cancers: New lessons from the IARC TP53 database and genomics data.
Hum Mutat. 37:865–876. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hwang LA, Phang BH, Liew OW, Iqbal J, Koh
XH, Koh XY, Othman R, Xue Y, Richards AM, Lane DP and Sabapathy K:
Monoclonal antibodies against specific p53 hotspot mutants as
potential tools for precision medicine. Cell Rep. 22:299–312. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
van Oijen MG and Slootweg PJ:
Gain-of-function mutations in the tumor suppressor gene p53. Clin
Cancer Res. 6:2138–2145. 2000.PubMed/NCBI
|
|
10
|
Sears CR and Turchi JJ: Complex
cisplatin-double strand break (DSB) lesions directly impair
cellular non-homologous end-joining (NHEJ) independent of
downstream damage response (DDR) pathways. J Biol Chem.
287:24263–24272. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bykov VJ and Wiman KG: Mutant p53
reactivation by small molecules makes its way to the clinic. FEBS
Lett. 588:2622–2627. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu DS, Read M, Cullinane C, Azar WJ,
Fennell CM, Montgomery KG, Haupt S, Haupt Y, Wiman KG, Duong CP, et
al: APR-246 potently inhibits tumour growth and overcomes
chemoresistance in preclinical models of oesophageal
adenocarcinoma. Gut. 64:1506–1516. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Russo D, Ottaggio L, Foggetti G, Masini M,
Masiello P, Fronza G and Menichini P: PRIMA-1 induces autophagy in
cancer cells carrying mutant or wild type p53. Biochim Biophys
Acta. 1833:1904–1913. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Perdrix A, Najem A, Saussez S, Awada A,
Journe F, Ghanem G and Krayem M: PRIMA-1 and PRIMA-1Met
(APR-246): From mutant/wild type p53 reactivation to unexpected
mechanisms underlying their potent anti-tumor effect in
combinatorial therapies. Cancers (Basel). 9:1722017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Q, Bykov VJN, Wiman KG and
Zawacka-Pankau J: APR-246 reactivates mutant p53 by targeting
cysteines 124 and 277. Cell Death Dis. 9:4392018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Muller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chollat-Namy M, Ben Safta-Saadoun T,
Haferssas D, Meurice G, Chouaib S and Thiery J: The pharmalogical
reactivation of p53 function improves breast tumor cell lysis by
granzyme B and NK cells through induction of autophagy. Cell Death
Dis. 10:6952019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hansen SN, Ehlers NS, Zhu S, Thomsen MB,
Nielsen RL, Liu D, Wang G, Hou Y, Zhang X, Xu X, et al: The
stepwise evolution of the exome during acquisition of docetaxel
resistance in breast cancer cells. BMC Genomics. 17:4422016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Olive PL and Banath JP: The comet assay: A
method to measure DNA damage in individual cells. Nat Protoc.
1:23–29. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gyori BM, Venkatachalam G, Thiagarajan PS,
Hsu D and Clement MV: OpenComet: An automated tool for comet assay
image analysis. Redox Biol. 2:457–465. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Koboldt DC, Zhang Q, Larson DE, Shen D,
McLellan MD, Lin L, Miller CA, Mardis ER, Ding L and Wilson RK:
VarScan 2: Somatic mutation and copy number alteration discovery in
cancer by exome sequencing. Genome Res. 22:568–576. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McLaren W, Gil L, Hunt SE, Riat HS,
Ritchie GR, Thormann A, Flicek P and Cunningham F: The ensembl
variant effect predictor. Genome Biol. 17:1222016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mayakonda A, Lin DC, Assenov Y, Plass C
and Koeffler HP: Maftools: Efficient and comprehensive analysis of
somatic variants in cancer. Genome Res. 28:1747–1756. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pilch DR, Sedelnikova OA, Redon C, Celeste
A, Nussenzweig A and Bonner WM: Characteristics of gamma-H2AX foci
at DNA double-strand breaks sites. Biochem Cell Biol. 81:123–129.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Berkers CR, Maddocks OD, Cheung EC, Mor I
and Vousden KH: Metabolic regulation by p53 family members. Cell
Metab. 18:617–633. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Williams AB and Schumacher B: p53 in the
DNA-damage-repair process. Cold Spring Harb Perspect Med.
6:a0260702016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu X, Vazquez A, Levine AJ and Carpizo DR:
Allele-specific p53 mutant reactivation. Cancer Cell. 21:614–625.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liang Y, Besch-Williford C, Benakanakere
I, Thorpe PE and Hyder SM: Targeting mutant p53 protein and the
tumor vasculature: An effective combination therapy for advanced
breast tumors. Breast Cancer Res Treat. 125:407–420. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liang Y, Besch-Williford C, Cook MT,
Belenchia A, Brekken RA and Hyder SM: APR-246 alone and in
combination with a phosphatidylserine-targeting antibody inhibits
lung metastasis of human triple-negative breast cancer cells in
nude mice. Breast Cancer (Dove Med Press). 11:249–259.
2019.PubMed/NCBI
|
|
30
|
Lambert JM, Gorzov P, Veprintsev DB,
Söderqvist M, Segerbäck D, Bergman J, Fersht AR, Hainaut P, Wiman
KG and Bykov VJ: PRIMA-1 reactivates mutant p53 by covalent binding
to the core domain. Cancer Cell. 15:376–388. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li XL, Zhou J, Chan ZL, Chooi JY, Chen ZR
and Chng WJ: PRIMA-1met (APR-246) inhibits growth of colorectal
cancer cells with different p53 status through distinct mechanisms.
Oncotarget. 6:36689–36699. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li XL, Zhou J, Xia CJ, Min H, Lu ZK and
Chen ZR: PRIMA-1met induces autophagy in colorectal cancer cells
through upregulation of the mTOR/AMPK-ULK1-Vps34 signaling cascade.
Oncol Rep. 45:862021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Makhale A, Nanayakkara D, Raninga P,
Khanna KK and Kalimutho M: CX-5461 enhances the efficacy of APR-246
via induction of DNA damage and replication stress in
triple-negative breast cancer. Int J Mol Sci. 22:57822021.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Synnott NC, Madden SF, Bykov VJN, Crown J,
Wiman KG and Duffy MJ: The mutant p53-targeting compound APR-246
Induces ROS-modulating genes in breast cancer cells. Transl Oncol.
11:1343–1349. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mantovani F, Collavin L and Del Sal G:
Mutant p53 as a guardian of the cancer cell. Cell Death Differ.
26:199–212. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Srinivas US, Tan BWQ, Vellayappan BA and
Jeyasekharan AD: ROS and the DNA damage response in cancer. Redox
Biol. 25:1010842019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Khanna A: DNA damage in cancer
therapeutics: A boon or a curse? Cancer Res. 75:2133–2138. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Garcia-de Teresa B, Hernandez-Gomez M and
Frias S: DNA Damage as a driver for growth delay: Chromosome
instability syndromes with intrauterine growth retardation. Biomed
Res Int. 2017:81938922017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Eischen CM: Genome stability requires p53.
Cold Spring Harb Perspect Med. 6:a0260962016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhu G, Pan C, Bei JX, Li B, Liang C, Xu Y
and Fu X: Mutant p53 in Cancer progression and targeted therapies.
Front Oncol. 10:5951872020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Watanabe N, Okochi E, Mochizuki M,
Sugimura T and Ushijima T: The presence of single nucleotide
instability in human breast cancer cell lines. Cancer Res.
61:7739–7742. 2001.PubMed/NCBI
|