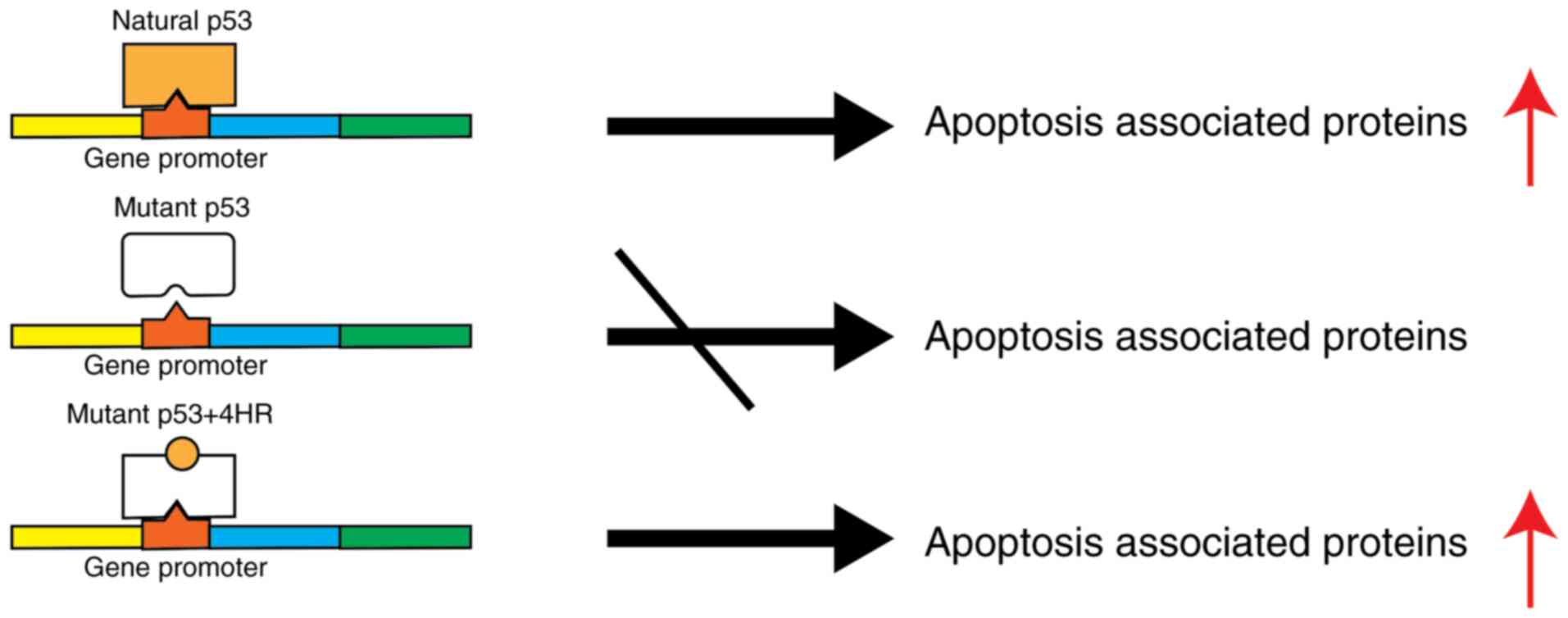
Introduction
p53 is a tumour suppressor and is known as the
‘Guardian of the Genome’ (1). The
function of p53 is the induction of apoptosis, regulation of
cellular proliferation and DNA repair (2). As p53 is a transcription factor, its
function will be dependent on its intranuclear transportation and
its binding ability to the promoter region of target genes. p53 has
6 major domains: The N-terminal transactivation domain, activation
domain 2, proline rich domain, DNA-binding domain, nuclear
localization signalling domain and tetramerization domain (3). The activation of p53 is observed as a
response to cytotoxic stress, such as DNA damage (4). Activated p53 is a tetramer with
acetylation and phosphorylation that translocates into the nucleus
(4–6). Then, p53 binds to the promoter of
genes that transcribe genes associated with apoptosis or DNA damage
repair (4).
The TP53 mutation is a frequent finding in oral
cancer (7,8). Several strategies against mutant TP53
have been developed (8,9). The most frequent site of TP53
mutation is the DNA-binding site (4). Mutant p53 may not bind properly to
DNA, and its transcriptional activity will be lower than that of
wild-type p53 (4,10). As a consequence, the expression
level of genes associated with apoptosis is lowered, and their
protein expression level is also decreased accordingly (4,8).
Tumour cells can undergo uncontrolled cellular proliferation
despite high levels of TP53 or p53 expression (11). Certain types of mutant p53 can act
as gain-of-function mutants (8,12).
In this case, tumour invasion and angiogenesis can be accelerated
with the help of mutant p53 (8,12).
Mutant TP53 can interact with other transcription factors that are
responsible for invasive tumour growth (12). YD-15 cells are a mucoepidermoid
carcinoma cell line in the oral cavity that has a mutation in TP53
(13). The YD-15 cell has an amino
acid change mutation in TP53 from aspartic acid (hydrophilic) to
alanine (hydrophobic) in the DNA binding domain (13). This amino acid change influences
the DNA binding ability of p53. As a consequence, YD-15 cells do
not undergo apoptosis despite a high level of p53 expression. YD-15
cells were selected for the study of mutant p53 function
restoration.
A treatment strategy for carcinoma with the TP53
mutation is restoring the p53 function (9). This strategy can be classified into
the following three categories: i) targeting the degradation or
direct inhibition of mutant p53; ii) binding to mutant p53 and
inducing conformation change to a desirable form; and iii)
targeting the viral enzymes that are responsible for the
degradation of p53 (9). In
particular, small molecules that can bind to mutant p53, inducing
mutant p53 conformational changes, have been a specific focus in
recent studies (9). PRIMA-1,
APR-246 and CP-31398 are chemicals that can bind to p53, inducing
mutant p53 conformational changes (9). Although these chemicals are
interesting in terms of restoring mutant p53 conformation, they
generate reactive oxygen species (ROS) by suppressing antioxidant
enzymes (14). Although ROS may
provide additional toxicity to cancer cells, ROS are also
carcinogenic and toxic to normal cells (15). This is a limitation for the
clinical application of these chemicals (14). Therefore, chemicals that can change
mutant p53 conformation and have antioxidant properties should be
investigated. 4-Hexylresorcinol (4HR) is a chemical chaperone that
has antioxidant properties (16,17)
and is an histone deacetylase (HDAC) inhibitor (18). The administration of 4HR induces
endoplasmic reticulum (ER)-mediated stress potentially by protein
folding changes (16). When 4HR is
administered to HUVECs, the expression level of transforming growth
factor-β1 (TGF-β1) is increased (19). This is a combined action of ER
stress and HDAC inhibition (16,20).
When 4HR is provided to microorganisms with mutant enzymes, the
enzyme activity is recovered by 4HR administration (16). The administration of 4HR to KB
cells shows an anticancer effect, but KB cells have wild-type p53
(21–23). Although 4HR administration has been
shown to have anticancer effects in numerous types of cancer, the
therapeutic effect of 4HR on cancers with TP53 mutations has not
been studied. 4HR may recover p53 DNA binding activity via its
binding to mutant p53 and conformational change. This
conformational change may restore p53-mediated apoptotic gene
expression. In addition, HDAC inhibition by 4HR can increase the
acetylation of p53. The acetylation of p53 is mainly found in its
C-terminal domain and DNA-binding domain (3). The acetylation of p53 in the p53 DNA
binding domain may neutralize its positive charge and improve its
DNA binding ability. HDAC-mediated deacetylation of p53 strongly
inhibits p53-dependent apoptosis (24). As a consequence, tumour cells with
a TP53 mutation may undergo apoptosis by 4HR administration. For
the examination of mutant p53 conformational changes, the peptide
sequence of the p53 DNA-binding domain from YD-15 cells was
acquired and synthesized. The normal peptide sequence of the p53
DNA binding domain was used as a reference. The conformational
changes before and after 4HR administration of the mutant peptide
were examined by Fourier transform infrared (FT-IR) spectra. The
4HR administration capacity to improve the DNA binding ability of
p53 was examined by a p53 transcription activity assay. The
induction of tumour cell apoptosis was evaluated by the examination
of apoptosis markers and cytochrome c leakage. Additionally, the
therapeutic effect of 4HR administration was also studied in an
animal model.
Materials and methods
Cell culture
YD-15 cells originated from the human tongue from
patients with an initial diagnosis of mucoepidermoid carcinoma.
YD-15 Bank (accession no.: CVCL_8930) and YD-9 (accession no.
CVCL_L080) cells were purchased from the Korean Cell Line Bank. The
subsequent culture conditions were in accordance with those
recommended by the Korean Cell Line Bank. Cells were cultured in
six-well culture plates in a humidified 5% CO2 incubator
at 37°C. The freezing media were 52.5% Roswell Park Memorial
Institute-1640 (RPMI-1640) medium supplemented with 40% fetal
bovine serum (FBS) and 7.5% dimethyl sulfoxide (DMSO; all from
Thermo Fisher Scientific, Inc.). Culture media were composed 90% by
RPMI-1640 with L-glutamine (300 mg/l), 25 mM HEPES and 25 mM
NaHCO3, and 10% by heat-inactivated FBS. Two-thirds of
the medium was removed and replaced with fresh medium every 3
days.
Peptide synthesis
Two kinds of artificial peptides were designed from
normal and mutant p53 of YD-15 cells. Glutamic acid at the 258th
amino acid in p53 was replaced by alanine in YD-15 cells (13). The DNA-binding domain of p53 was
from amino acids 102 to 292. Accordingly, mutation in YD-15 cells
was localized in the p53 DNA binding domain (13). The artificial peptides were
fabricated by Peptron. The amino acid sequences were
ILTIITL-[E]-DSSGNLLGRNSF and ILTIITL-[A]-DSSGNLLGRNSF in the
peptide from wild-type p53 and mutant p53 of YD-15 cells,
respectively.
FT-IR spectroscopy
4HR (cat. no. 209495) was purchased from
Sigma-Aldrich; Merck KGaA. A sample was prepared as a mixture with
0.1% ethanol, and the concentrations were p53 Glu-258 (1 mg/ml),
p53 Ala-258 (1 mg/ml), and p53 Ala-258 (1 mg/ml) + 4HR (0.1 mg/ml).
FT-IR absorption spectra were obtained using a Fourier transform
spectrometer (Vertex 80; Bruker Corporation) equipped with an
attenuated total reflectance accessory (MIRacle; PIKE
Technologies). The spectra were recorded in the spectral range of
600 to 4,000 cm−1 at a resolution of 4 cm−1
with a deuterated L-alanine-doped triglycene sulfate detector, and
128 repeated scans were averaged for each spectrum.
Western blot analysis
4HR was solubilized in 0.1% DMSO. When YD-15 and
YD-9 cells were cultured to ~70% confluence, they were treated with
1, 10 and 100 µM 4HR for 2, 8, or 24 h; control cells were treated
with 0.1% DMSO in culture medium. Cultured cells were treated with
0.01% trypsin and 1 mM ethylene-diamine-tetra-acetic acid. Then,
they were harvested with a spatula. Cellular lysis was carried out
with protein lysis buffer (PRO-PREP™; Intron
Biotechnology, Inc.). The protein quantification was performed by
Bradford method as previously described (19). The type of gel used was 15%
SDS-polyacrylamide gel. The amount of protein loaded per lane was
30 µg. The type of membrane used was polyvinylidene difluoride
(cat. no. IPVH00010; Millipore). The membrane was blocked with
TBS-T (25 mM Tris-HCl, 140 mM NaCl, 0.1% Tween-20, pH 7.5) buffer
containing 5% non-fat dry milk for 1 h at room temperature.
Collected lysates underwent western blotting for B-cell lymphoma 2
(BCL2), BCL2 associated X (BAX) and BCL2 associated agonist of cell
death (BAD). Antibodies against BCL2 (cat. no.: sc-7382), BAX (cat.
no. sc-7480), BAD (cat. no. sc-8044) and β-actin (cat. no. sc-8432)
were purchased from Santa Cruz Biotechnology, Inc. Dilution ratio
was 1:1,000. The primary antibodies were incubated at 4°C
overnight. The secondary antibodies used were HRP-conjugated (cat.
nos. sc-2357 and sc-516102, Santa Cruz Biotechnology, Inc.) and its
dilution ratio was 1:1,000. To detect HRP-labelled antibodies,
Immobilon Forte Western HRP substrate (cat. no. WBLUF0500;
MilliporeSigma) was used as chemiluminescence reagent. Blots were
imaged and quantified using a ChemiDoc XRS system and Image Lab 3.0
(both from Bio-Rad Laboratories, Inc.).
p53 transcriptional activity
The p53 transcription factor assay was performed
using a p53 transcription factor assay kit (cat. no. ab207225;
Abcam). All antibodies used for the assay were included in the kit.
For the quantitative measurement of p53 activation in human nuclear
extracts, a specific single-stranded DNA containing the p53
DNA-binding site (5′-GGACATGCCCGGGCATGTCC-3′) was immobilized onto
a 96-well plate. Active p53 present in the nuclear extract
specifically binds to the oligonucleotide. p53 was detected by a
primary antibody that recognizes an epitope of p53 accessible only
when the protein is activated and bound to its target DNA. An
HRP-conjugated secondary antibody provided a sensitive colorimetric
measurement at optical density (OD) of 450 nm. To assess the effect
of 4HR administration on the binding of mutant p53 to the consensus
DNA sequence, 1, 10, and 100 µM 4HR were applied, and nuclear
extracts were collected after 2 h. The subsequent procedures were
performed in accordance with the manufacturer's protocol. Briefly,
samples were added to the appropriate wells and the plate was
incubated for 1 h at room temperature with mild agitation. After
washing each well three times with 200 µl of 1X washing buffer,
primary antibody was added to the wells. After covering the plate,
it was incubated again for 1 h at room temperature without
agitation. After the washing process, an anti-rabbit HRP-conjugated
secondary antibody was applied and its dilution ratio was 1:1,000.
The plate was incubated for 1 h at room temperature without
agitation. After another washing process, a developing solution was
added, and the plate was incubated for 5 min. Then, a stop solution
was added, and the absorbance was measured at 450 nm.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
MTT assays were conducted as follows: YD-15 cells
were cultured as aforementioned. When YD-15 cells were cultured to
~70% confluence, they were treated with 1, 10 and 100 µM 4HR for 24
or 48 h, and control cells were treated with 0.1% DMSO in culture
medium. Then, the cells were incubated with yellow tetrazolium salt
and 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide
(MTT) solution (Cell Proliferation kit I; Roche Molecular
Diagnostics) for 4 h at ambient temperature. Formazan crystals were
solubilized with DMSO overnight, and the products were quantified
spectrophotometrically by measuring the absorbance at 590 nm using
a Victor Multilabel Counter (PerkinElmer, Inc.).
Confocal microscopic examination
When YD-15 cells were cultured on chamber slides at
~70% confluence, they were treated with 1, 10, and 100 µM 4HR for
24 h. After fixation using 4% formaldehyde (diluted in PBS) at 4°C
for overnight, the slides were washed with PBS with Tween-20. Then,
protein blocking was performed with a blocking reagent (cat. no.
X0909; Dako; Agilent Technologies, Inc.) for 30 min at room
temperature. The cytochrome c apoptosis ICC Antibody kit (cat. no.
ab110417; Abcam) was composed of a cytochrome c monoclonal antibody
and ATP synthase V subunit α monoclonal antibody. Cytochrome c and
ATP synthase V are localized in the mitochondria, and only
cytochrome c is released from mitochondria during apoptosis
(25). As anti-cytochrome c
antibody was conjugated with FITC and anti-ATP synthase V antibody
with TxRd, the cells with cytochrome c leakage showed green
fluorescence only. Pre-treated and fixed YD-15 cells were incubated
with anti-cytochrome c antibody and anti-ATP synthase V antibody in
a humidified dark chamber for 1 h. The nuclei were counterstained
with 5 mg/ml DAPI at room temperature for 5 min. After washing,
each slide was mounted. The mounted slides were examined with
Stellaris 5 (Leica Microsystems GmbH) at the Center for Scientific
Instruments, Gangneung-Wonju National University.
Xenograft study
The animal study was approved (approval no.
GWNU-2021-3, approval date 2021.01.15) by the Animal Care and Use
Guidelines of the College of Dentistry at Gangneung-Wonju National
University (Gangneung, Republic of Korea). The approved duration of
the experiment was a year. A total of 20 athymic male nude mice (7
weeks-old, body weight: 21~23.5 g) were approved for the present
study and were introduced from ORIENT BIO, Inc. After receiving the
animals, the adaptation period was allowed for 5 days. The
light/dark cycle was 12/12 h. Mice were kept in individual cage
with ad libitum access to food and water. The cage used was
filter-bonneted individually ventilated cage. Food was sterilized
and water was acidified to pH 2.5-3.0. The temperature in the
specialized husbandry was set at 23–27°C. Cages were changed under
a laminar flow hood. The number of injected YD-15 cells for each
animal was ~3×105 cells. The resuspension solution was
the culture media used for cell culture. They were injected into
the sublingual space through the skin under anaesthetic conditions.
Anaesthesia was induced by inhalation of 3% isoflurane. After
injection, tumour growth was observed daily. In cases of successful
tumour formation, the animals were included in the treatment
groups. The nude mice showing evident tumour formation were
randomly assigned to two groups: the 4HR and control groups. After
the first injection, 12 mice showed tumour formation (Fig. S1). One mouse died. The same number
of mice was assigned to each group. The second tumour injection was
performed for the remaining 7 mice. Another 4 mice showed tumour
formation, and the remaining 3 mice did not show any tumours. A
total of 16 mice were included for drug administration. As the mice
in the control group showed early loss during observation among the
first 12 mice, 9 mice were assigned to the control group, and 7
mice were assigned to the 4HR group. The drug was injected into the
back subcutaneously. 4HR was solubilized by 0.1% β-cyclodextrin,
and the dosage for 4HR was 10 mg/kg of body weight for 28 days,
while the control group received daily injections of the vehicle
(0.1% β-cyclodextrin). The initial body weight and tumour size were
recorded at the time of the first treatment. The mass size and body
weight were recorded every 3 days. The tumour mass size was
calculated using the following formula: Mass volume=a (long
distance) × b (short distance)2/2.
Euthanasia was undertaken when the body weight
decreased by 20%, tumour necrosis was observed, or a back hump was
observed. The procedure was as follows: mice were deeply
anesthetized by 4% isoflurane. Thereafter, mice were quickly
sacrificed by cervical dislocation. The death of mice was verified
by checking loss of heartbeat.
After the animals were sacrificed, tumour masses
were received for histological examination. Briefly, tumour
specimens were fixed using 4% formaldehyde diluted by PBS at 4°C
for overnight. Paraffin-embedded specimens were cut and prepared
for slide. The thickness of cut was 5 µm. Haematoxylin and eosin
(H&E) staining was performed for histological examination. The
staining procedure was conducted at room temperature and duration
was 6 min for haematoxylin and 2 min for eosin.
Immunohistochemistry was performed for BCL-2, BAD and BAX, and the
same antibodies used for western blotting were used. After
hydration, the activity of endogenous peroxidase was blocked by 3%
H2O2 at room temperature for 7 min. The
protein blocking was performed using ready-made reagent (cat. no.
X0909 Dako; Agilent Technologies, Inc.). After colorization by DAB
(cat. no. K5007; Dako Agilent Technologies, Inc.), slides were
examined using a light microscope (BX52; Olympus Corporation).
Statistical analysis
The data from cell and animal experiments were
analysed using Sigma Scan Pro 5.0 and SPSS software ver. 25 (IBM
Corp.). The statistical significance of differences between two
means was examined using unpaired Student's t-test, whereas
multiple values were compared using one-way analysis of variance.
P<0.05 was considered to indicate a statistically significant
differene. The data were presented as the mean ± standard
deviation. The cellular experiments were repeated at least 3 times.
Post hoc analysis was completed using Bonferroni comparisons.
Results
Conformational change of the peptide
from mutant p53 by 4HR administration
Normal p53 has Glu258, but p53 of YD-15 cells has
Ala258. Each peptide has 20 amino acids, including a mutation site.
According to amino acid sequence-based peptide conformation
prediction, this mutation changes the peptide conformation from a
beta-sheet structure to a random coil structure (Fig. 1A) (26,27).
The FT-IR spectrum of the peptide showed a series of absorption
bands at 1,600-1,700 (amide I), 1,480-1,580 (amide II), and
1,230-1,300 cm−1 (amide III) (Fig. 1B) (28–30).
The amide I peak (C=O stretching) position is very sensitive to the
conformation types (β-sheet, α-helix, β-turn, random coil) of the
protein secondary structures (28).
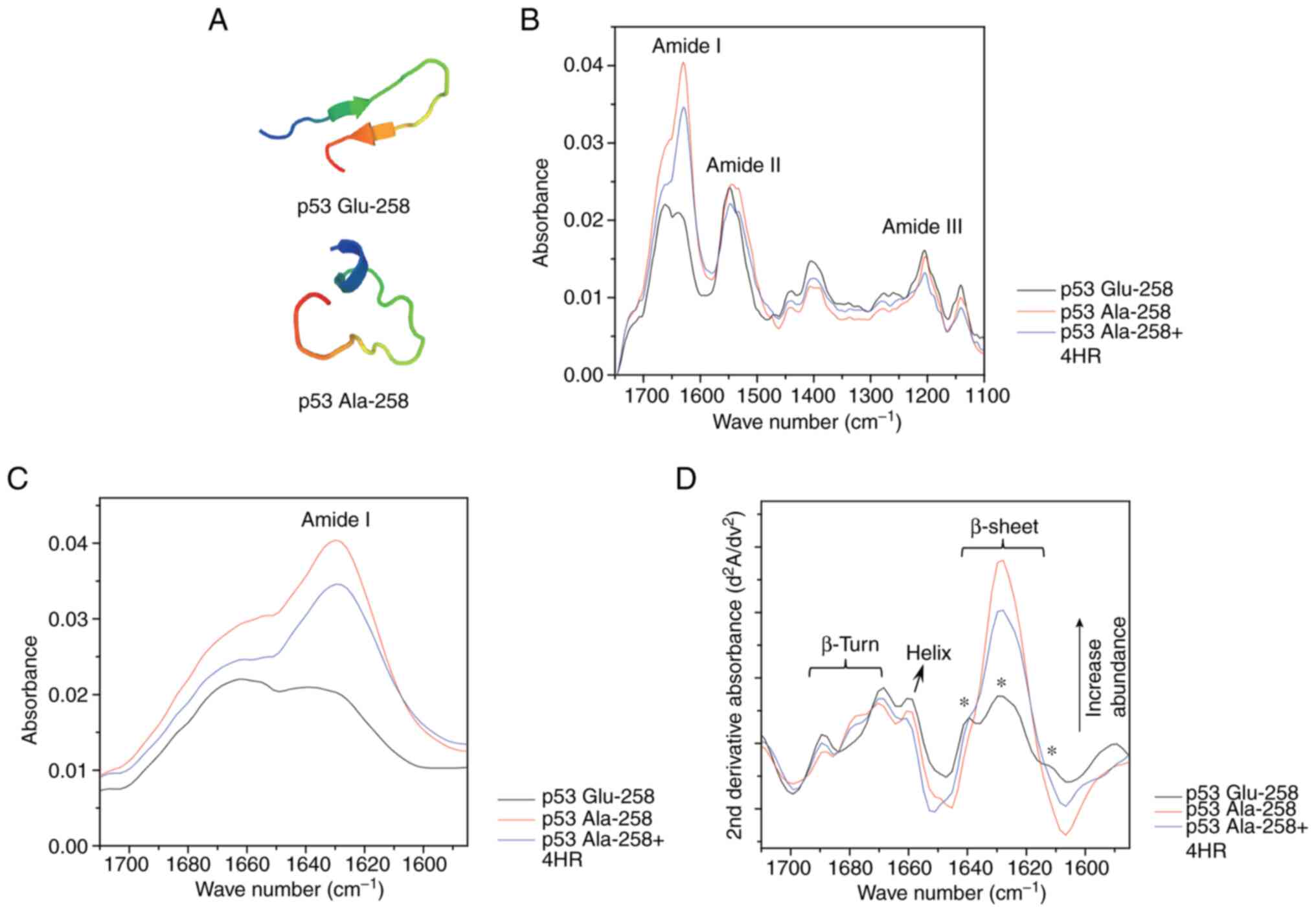 | Figure 1.Conformational change of the peptide
from mutant p53 by 4HR. (A) According to amino acid sequence-based
peptide conformation prediction, the peptide from mutant p53
(p53-Ala-258) has a random coil structure, but the peptide from
wild-type p53 has a beta-sheet structure (p53-Glu-258). (B) The
Fourier transform infrared spectrum of each peptide showed a series
of absorption bands at 1,600-1,700 (amide I), 1,480-1,580 (amide
II) and 1,230-1,300 cm-1 (amide III). (C) The amide I peak (C=O
stretching) of p53-Ala-258 + 4HR was less enhanced than that of
p53-Ala. (D) p53-Glu-258 showed rich β-sheet conformations
detectable at 1,614, 1,628, and 1,639 cm-1 (*), whereas p53 Ala-258
showed a simple conformation mostly detected at 1,628 cm-1. When
p53 Ala-258 was coupled by 4HR, it lost its original conformation
and resembled the conformation of p53 Glu-258. 4HR,
4-Hexylresorcinol. |
Therefore, detailed sub-band assignment of the amide
I band is quite useful for evaluating the relative amount of
protein secondary structures. In addition to the conventional
infrared spectra showing qualitative amounts of protein structures
(Fig. 1C), the second derivative
spectra of the peptides apparently revealed sub-band peaks
assignable to well-known protein secondary structures (Fig. 1D). All examined peptide specimens
showed typical vibrational modes corresponding to β-sheet, β-turn
and helix structures. One notable conformational change was found;
p53 Glu258 showed rich β-sheet conformations detectable at 1,614,
1,628 and 1,639 cm−1, as indicated in Fig. 1D, whereas p53 Ala258 showed a
simple conformation mostly detected at 1628 cm−1. When
p53 Ala-258 was coupled by 4HR, it lost its original conformation
and resembled the conformation of p53 Glu-258.
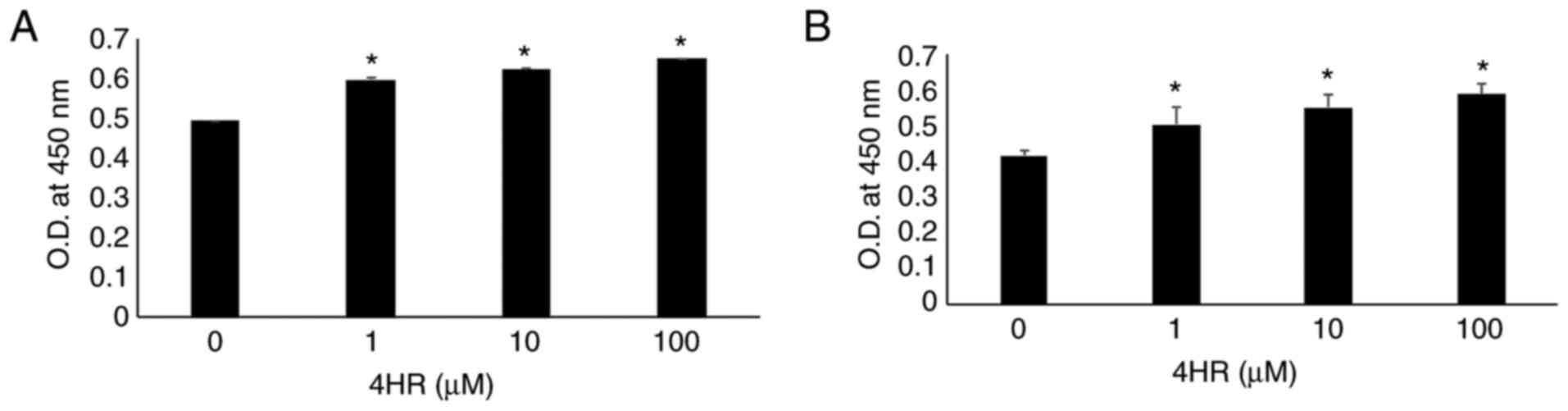
Increase in p53 transcriptional
activity by 4HR administration
p53 transcriptional activity was significantly
increased by 4HR administration (Fig.
2A; P<0.001). The OD value for the untreated control was
0.494±0.001, and the OD values were 0.598±0.007, 0.622±0.008 and
0.649±0.002 for the 1, 10 and 100 µM 4HR treatments, respectively.
In the post hoc comparison, the difference between the untreated
control and 4HR-treated groups was statistically significant
(P<0.001). When comparisons between the 4HR-treated groups were
conducted, a significant difference was identified (P<0.05).
Increased p53-mediated transcriptional activity was also detected
in YD-9 cells (Fig. 2B).
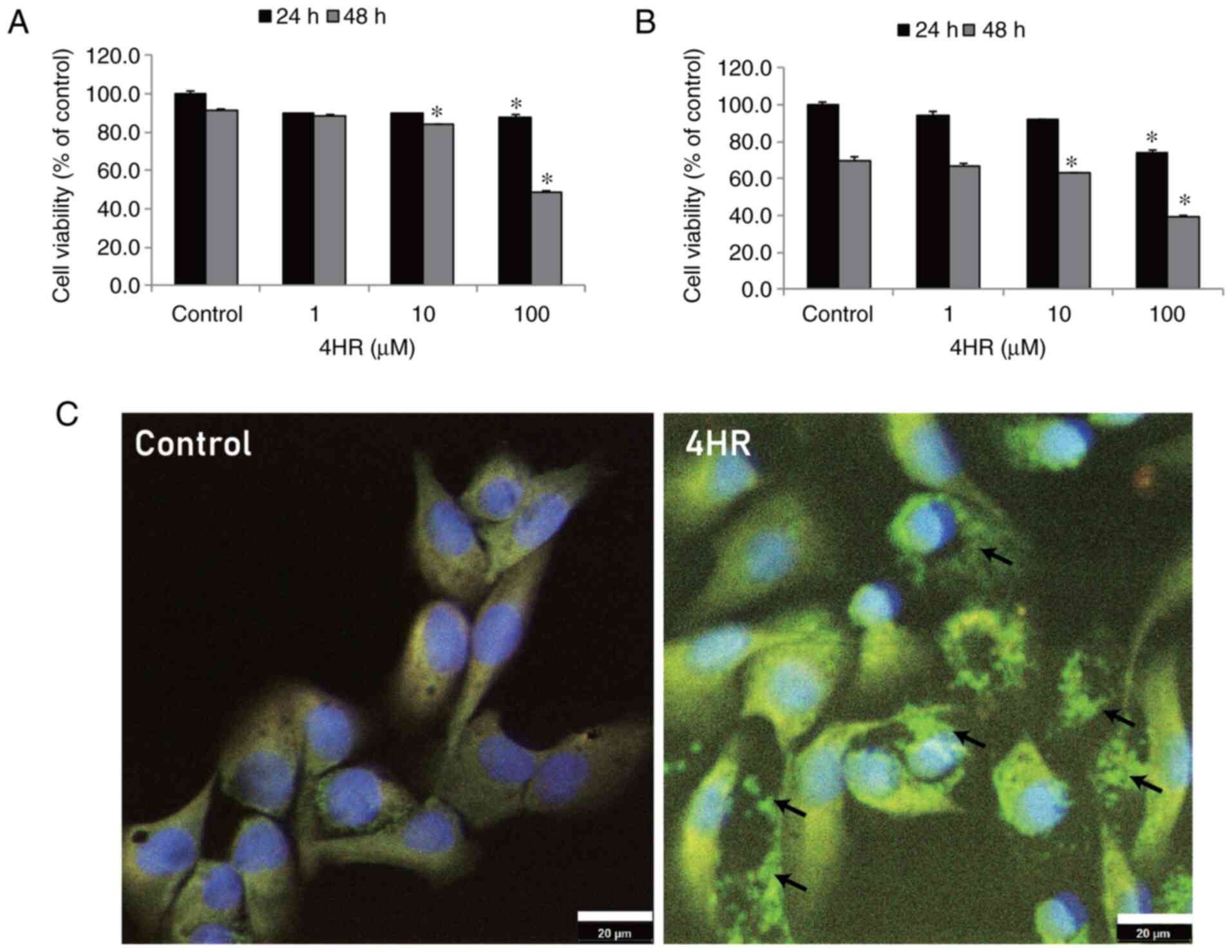
Increase in apoptosis-associated
proteins by 4HR administration
In the MTT assay, the cellular proliferation of
YD-15 cells was suppressed by 4HR administration (Fig. 3A). The suppression of tumour cell
proliferation by 4HR administration was also observed in YD-9 cells
(Fig. 3B). Apoptosis of YD-15
cells increased in a dose-dependent manner as revealed by confocal
microscopic examination (Fig.
3C).
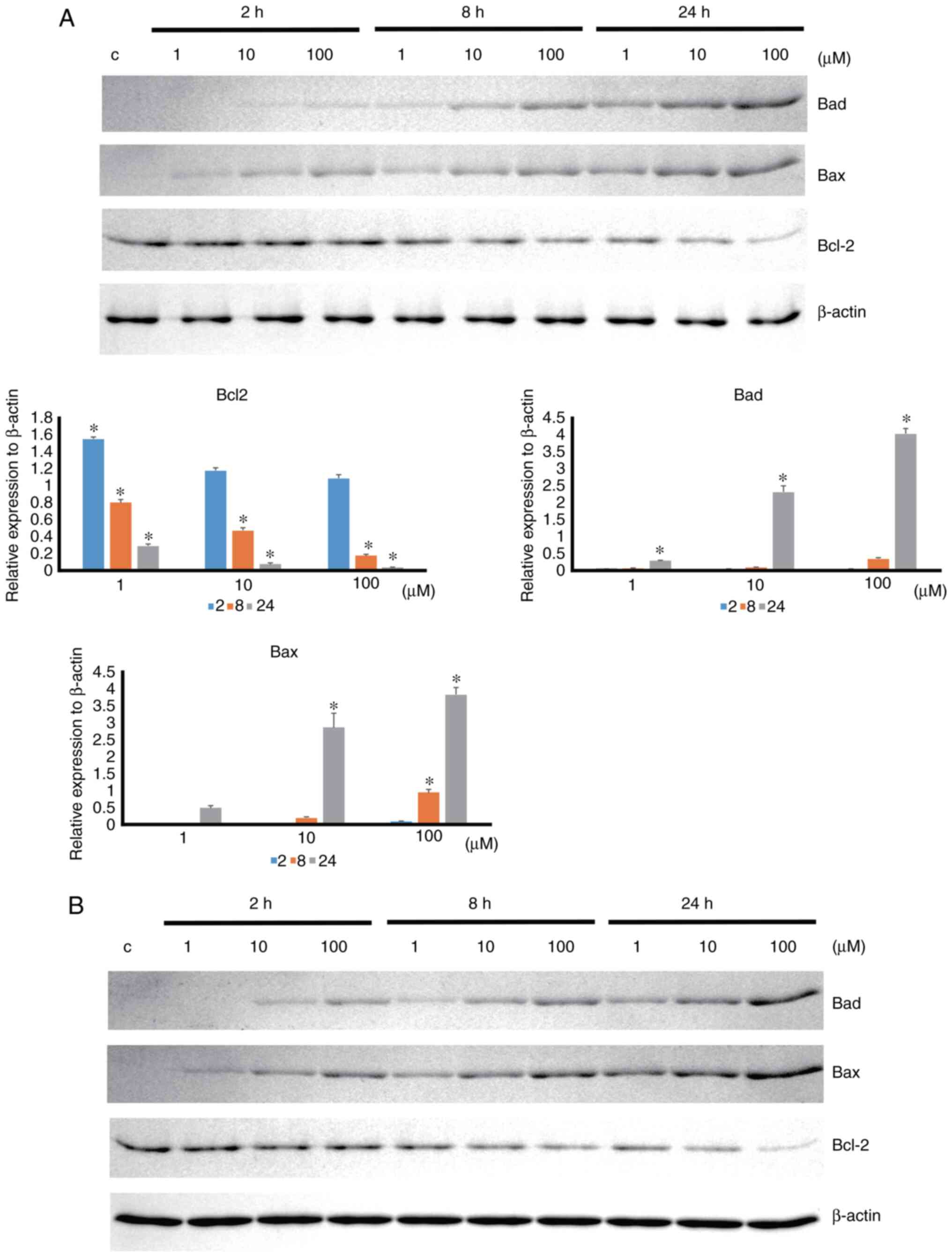
Apoptosis-associated protein expression was
increased by 4HR administration in a time- and dose-dependent
manner (Fig. 4). 4HR
administration significantly decreased the expression level of
BCL-2 anti-apoptotic protein (P<0.001). In the post hoc
comparison, the difference between the untreated control and
4HR-treated groups was statistically significant (P<0.001 for
100 µM 4HR treatment at 2 h and P<0.001 for all groups at 8 and
24 h). The expression levels of BAD and BAX proapoptotic proteins
were significantly increased by 4HR administration (P<0.001). A
post hoc comparison was performed for BAD and BAX. The difference
in BAD expression between the untreated control and 4HR-treated
groups was statistically significant (P=0.003 for 100 µM 4HR
treatment at 8 h, P=0.039 for 1 µM 4HR treatment at 24 h, and
P<0.001 for 10 and 100 µM 4HR treatment at 24 h). The difference
in BAX expression between the untreated control and 4HR-treated
groups was statistically significant (P<0.001 for 100 µM 4HR
treatment at 8 h and P<0.001 for 10 and 100 µM 4HR treatment at
24 h). A similar pattern of expression was also found in YD-9 cells
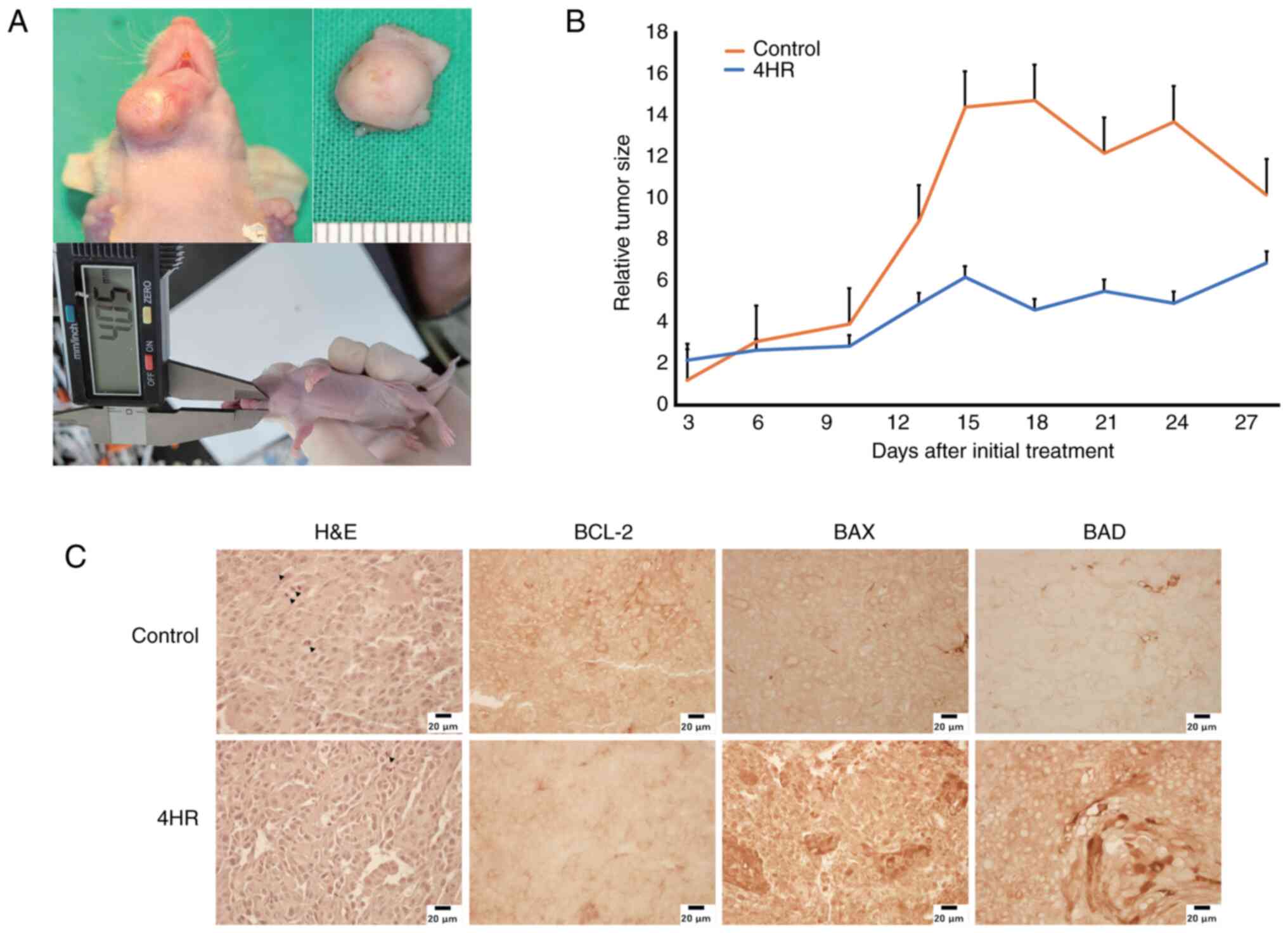
(Fig. 4B). In the xenograft study,
tumours were observed in the sublingual area (Fig. 5A). The 4HR-treated group showed a
smaller increase in tumour size than the control group (Fig. 5B, Table I). However, the difference between
groups was not significant (P>0.05). Mitosis in the tumour mass
was less frequently observed in the 4HR group (Fig. 5C). In the immunohistochemical
study, the expression level of BCL-2 was similar between groups
(Fig. 5C). However, the expression
levels of BAX and BAD were higher in the 4HR group than in the
control group (Fig. 5C).
 | Table I.Initial tumour size and tumour size
at the end of treatment (*Mice were dead within a week and excluded
for plotting in Fig. 5B). |
Table I.
Initial tumour size and tumour size
at the end of treatment (*Mice were dead within a week and excluded
for plotting in Fig. 5B).
| Number | Group | Initial tumour size
(mm3) | Final tumour size
(mm3) |
|---|
| 1 | 4HR | 86.4 | 883.2 |
| 2 | Control | 12.4 | 209.0 |
| 4 | 4HR | 49.1 | 51.4 |
| 5 | Control | 153.3 | 532.9 |
| 6* | Control | 40.7 | 59.7 |
| 7 | 4HR | 55.6 | 158.7 |
| 8* | Control | 36.8 | 43.3 |
| 9 | 4HR | 4.1 | 80.7 |
| 10* | Control | 49.7 | 82.8 |
| 12 | Control | 39.7 | 338.4 |
| 14 | Control | 8.3 | 144.0 |
| 15* | 4HR | 47.7 | 117.2 |
| 16 | Control | 25.3 | 170.7 |
| 17 | Control | 25.1 | 202.1 |
| 19 | 4HR | 82.1 | 49.3 |
| 20 | 4HR | 74.4 | 242.0 |
|
| Mean (control) | 43.5 | 198.1 |
|
| Mean (4HR) | 57.1 | 226.1 |
Discussion
YD-15 cells have a p53 mutation in its DNA binding
domain (13). The peptide from the
p53 DNA binding domain was prepared from both mutant and normal
p53. When 4HR was applied to the mutant peptide, its conformation
changed and was close to that of normal p53. The administration of
4HR increased p53 transcriptional activity in both YD-9 and YD-15
cells. The apoptosis induced by 4HR was confirmed by cytochrome c
leakage in YD-15 cells after 4HR administration. The expression
levels of BAD and BAX were increased by 4HR administration in both
YD-9 and YD-15 cells. The expression levels of BAD and BAX in the
grafted tumour were increased in the 4HR group compared with the
control group. Collectively, 4HR administration to oral cancer
cells with a p53 mutation increased p53 transcriptional activity
via a potential conformational change in mutant p53 (Fig. 6).
Glutamic acid at the 258th amino acid in p53 was
replaced by alanine in YD-15 cells (13). The peptide around the mutant site
was designed and synthesized. The conformation of the peptide from
mutant p53 was predicted to have a random coil structure, but the
conformation of the peptide from normal p53 was a β-sheet structure
(26,27). Notably, the administration of p53
changed the conformation of the peptide from mutant p53 and brought
it close to that of the peptide from normal p53. This
conformational change of mutant p53 by 4HR administration should
improve the transcriptional activity of p53. The transcriptional
activity of p53 was significantly increased by 4HR administration
in both YD-15 and YD-9 cells. Several chemicals have been
identified that can change the conformation of mutant p53 (9). However, most of these increase the
production of ROS (9). Therefore,
while they may be helpful for eliminating tumour cells, they will
also induce toxicity in normal organs by systemic administration
(31). Compared with previous
conformational change chemicals, 4HR has antioxidant activity
(32,33). Thus, its systemic application would
be relatively safe compared with others (33).
Similar to 4HR, numerous chemicals have been
introduced for mutated p53. COTI-2 is a thiosemicarbazone compound.
COTI-2 can interact with both full-length mutated p53 and peptide
of DNA binding domain from mutated p53 (34). The combination of COTI and
cisplatin shows a synergistic effect on head and neck squamous cell
carcinoma (35). Notably, the
combination of 4HR and cisplatin also shows a synergistic effect on
head and neck squamous cell carcinoma (22). When mutated p53 recovers its
function, gene transcription and protein expression should be
subsequently increased. In healthy cells, the administration of
carcinogens activates the p53 signalling pathway in response to DNA
damage. BAX is a gene induced by the p53-dependent DNA damage
response and results in cytochrome c release from the mitochondria
(9). BAD is also a proapoptotic
protein and is activated by p53 (36). In the present study, administration
of 4HR increased the expression levels of BAD and BAX. As a
consequence, the release of cytochrome c was increased with the
inhibition of cellular proliferation.
There are certain limitations to the present study.
First, there may have been limitations in the protein conformation
study using peptides. Peptides are a partial domain of proteins. If
atomic microscopy or X-ray crystallography is used for the study of
mutant p53 conformational changes, more detailed information could
be acquired. Second, numerous compounds that can recover p53
activity in tumours with p53 mutations have p53-independent
effects, and these effects may be beneficial to the treatment of
tumours (12). 4HR is an HDAC
inhibitor (18). The
administration of 4HR decreases the expression level of HDAC4 in
HUVECs (18) and Saos-2 cells
(37). 4HR is a chemical chaperone
(16) that increases the stress of
the ER, which may be induced by changes in protein folding
(38). Thus, investigation of
acetylation and phosphorylation of p53 would be interesting topic.
This will be explored separately in future studies. Third, the
YD-15 cell line has been shown to have poor tumorigenicity in an
animal model (13). In the present
study, some animals showed successful tumour formation, but their
survival rate was poor. Only a few animals survived until the 28th
day after the initial treatment, and a statistically significant
difference between groups was not observed. Thus, confirmation of
antitumor effects in the animal model was not possible in the
current study design. HSC-1, HSC-2 and HSC-4 cells also have
mutations in the DNA-binding domain of p53 (39–41).
Thus, the study of other types of cancer with mutant p53 will be an
interesting topic for future study. In addition, finding essential
mutations via gene sequencing was not the objective of the present
study. This will also be an interesting research subject for future
study. The merit of the current study should be the finding of
recovered transcriptional activity of mutant p53 by the
administration of 4HR.
In conclusion, 4HR is a potential substance for
recovering the loss-of-function in mutant p53 as a chemical
chaperone via protein conformational change.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the ‘Cooperative Research
Program for Agriculture Science and Technology Development (grant
no. PJ01562601)’ of the Rural Development Administration of
Republic of Korea.
Availability of data and materials
Data sharing is not applicable to this article, as
no datasets were generated or analyzed during the current
study.
Authors' contributions
YJK and SGK conceived and designed the study. DWK
and YWG performed the experiments and collected the data. WSC, YJK
and SGK analysed and interpreted the data. YJK wrote the paper. SGK
and HR reviewed the data and information and edited the manuscript.
SGK and HR critically revised the paper for important intellectual
content. YJK, WSC and DWK confirm the authenticity of all the raw
data. All authors read and approved the final version of the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
The animal study was approved (approval no.
GWNU-2021-3; approval date 2021.01.15) by the Animal Care and Use
Guidelines of the College of Dentistry at Gangneung-Wonju National
University (Gangneung, Republic of Korea).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
4HR
|
4-hexylresorcinol
|
|
ROS
|
reactive oxygen species
|
|
HDAC
|
histone deacetylase
|
|
ER
|
endoplasmic reticulum
|
|
TGF-β1
|
transforming growth factor-β1
|
|
FBS
|
fetal bovine serum
|
|
DMSO
|
dimethyl sulfoxide
|
|
FT-IR
|
Fourier transform infrared
|
|
MTT assay
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
assay
|
|
OD
|
optical density
|
|
BAX
|
BCL2-associated X
|
|
BCL-2
|
B-cell lymphoma 2
|
|
BAD
|
BCL2-associated agonist of cell
death
|
References
|
1
|
Lane DP: Cancer. p53, guardian of the
genome. Nature. 358:15–16. 1992. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Levine AJ: p53, the cellular gatekeeper
for growth and division. Cell. 88:323–331. 1997. View Article : Google Scholar
|
|
3
|
Xia Z, Kon N, Gu AP, Tavana O and Gu W:
Deciphering the acetylation code of p53 in transcription regulation
and tumor suppression. Oncogene. 41:3039–3050. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pecorino L: Molecular biology of Cancer.
Oxford University Press; Oxford: 2016
|
|
5
|
Lee JH, Kim HS, Lee SJ and Kim KT:
Stabilization and activation of p53 induced by Cdk5 contributes to
neuronal cell death. J Cell Sci. 120:2259–2271. 2007. View Article : Google Scholar
|
|
6
|
Brooks CL and Gu W: Ubiquitination,
phosphorylation and acetylation: The molecular basis for p53
regulation. Curr Opin Cell Biol. 15:164–171. 2003. View Article : Google Scholar
|
|
7
|
Peltonen JK, Helppi HM, Pääkkö P,
Turpeenniemi-Hujanen T and Vähäkangas KH: p53 in head and neck
cancer: Functional consequences and environmental implications of
TP53 mutations. Head Neck Oncol. 2:362010. View Article : Google Scholar
|
|
8
|
Lindemann A, Takahashi H, Patel A, Osman A
and Myers J: Targeting the DNA damage response in OSCC with TP 53
mutations. J Dent Res. 97:635–644. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
de Bakker T, Journe F, Descamps G, Saussez
S, Dragan T, Ghanem G, Krayem M and Van Gestel D: Restoring p53
function in head and neck squamous cell carcinoma to improve
treatments. Front Oncol. 11:7999932022. View Article : Google Scholar
|
|
10
|
Kaida A and Iwakuma T: Regulation of p53
and cancer signaling by heat shock protein 40/J-domain protein
family members. Int J Mol Sci. 22:135272021. View Article : Google Scholar
|
|
11
|
Kanapathipillai M: Treating p53 mutant
aggregation-associated cancer. Cancers (Basel). 10:1542018.
View Article : Google Scholar
|
|
12
|
Muller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar
|
|
13
|
Lee EJ, Kim J, Lee SA, Kim EJ, Chun YC,
Ryu MH and Yook JI: Characterization of newly established oral
cancer cell lines derived from six squamous cell carcinoma and two
mucoepidermoid carcinoma cells. Exp Mol Med. 37:379–390. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lambert JM, Gorzov P, Veprintsev DB,
Söderqvist M, Segerbäck D, Bergman J, Fersht AR, Hainaut P, Wiman
KG and Bykov VJ: PRIMA-1 reactivates mutant p53 by covalent binding
to the core domain. Cancer Cell. 15:376–388. 2009. View Article : Google Scholar
|
|
15
|
Chen HM, Lee YH and Wang YJ: ROS-triggered
signaling pathways involved in the cytotoxicity and tumor promotion
effects of pentachlorophenol and tetrachlorohydroquinone. Chem Res
Toxicol. 28:339–350. 2015. View Article : Google Scholar
|
|
16
|
Kim SG: 4-Hexylresorcinol: Pharmacologic
chaperone and its application for wound healing. Maxillofac Plast
Reconstr Surg. 44:52022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee IS, Chang JH, Kim DW, Kim SG and Kim
TW: The effect of 4-hexylresorinol administration on NAD+ level and
SIRT activity in Saos-2 cells. Maxillofac Plast Reconstr Surg.
43:392021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim JY, Kweon HY, Kim DW, Choi JY and Kim
SG: 4-Hexylresorcinol inhibits class I histone deacetylases in
human umbilical cord endothelial cells. Appl Sci. 11:34862021.
View Article : Google Scholar
|
|
19
|
Kim DW, Jo YY, Garagiola U, Choi JY, Kang
YJ, Oh JH and Kim SG: Increased level of vascular endothelial
growth factors by 4-hexylresorcinol is mediated by transforming
growth factor-β1 and accelerates capillary regeneration in the
burns in diabetic animals. Int J Mol Sci. 21:34732020. View Article : Google Scholar
|
|
20
|
Jiménez-Uribe AP, Gómez-Sierra T,
Aparicio-Trejo OE, Orozco-Ibarra M and Pedraza-Chaverri J:
Backstage players of fibrosis: NOX4, mTOR, HDAC, and S1P;
companions of TGF-β. Cell Signal. 87:1101232021. View Article : Google Scholar
|
|
21
|
Kim SG, JeonG JH, Park YW, Song JY, Kim
AS, Choi JY and Chae WS: 4-Hexylresorcinol inhibits
transglutaminase-2 activity and has synergistic effects along with
cisplatin in KB cells. Oncol Rep. 25:1597–1602. 2011.PubMed/NCBI
|
|
22
|
Kim SG, Lee SW, Park YW, Jeong JH and Choi
JY: 4-hexylresorcinol inhibits NF-κB phosphorylation and has a
synergistic effect with cisplatin in KB cells. Oncol Rep.
26:1527–1532. 2011.PubMed/NCBI
|
|
23
|
Xue L, Zhou B, Liu X, Qiu W, Jin Z and Yen
Y: Wild-type p53 regulates human ribonucleotide reductase by
protein-protein interaction with p53R2 as well as hRRM2 subunits.
Cancer Res. 63:980–986. 2003.PubMed/NCBI
|
|
24
|
Luo J, Su F, Chen D, Shiloh A and Gu W:
Deacetylation of p53 modulates its effect on cell growth and
apoptosis. Nature. 408:377–381. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Y, Dong Y, Wu X, Lu Y, Xu Z, Knapp
A, Yue Y, Xu T and Xie Z: The mitochondrial pathway of anesthetic
isoflurane-induced apoptosis. J Biol Chem. 285:4025–4037. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shen Y, Maupetit J, Derreumaux P and
Tufféry P: Improved PEP-FOLD approach for peptide and miniprotein
structure prediction. J Chem Theory Comput. 10:4745–4758. 2014.
View Article : Google Scholar
|
|
27
|
Thévenet P, Shen Y, Maupetit J, Guyon F,
Derreumaux P and Tufféry P: PEP-FOLD: An updated de novo structure
prediction server for both linear and disulfide bonded cyclic
peptides. Nucleic Acids Res. 40:(Web Server Issue). W288–W293.
2012. View Article : Google Scholar
|
|
28
|
Kong J and Yu S: Fourier transform
infrared spectroscopic analysis of protein secondary structures.
Acta Biochim Biophys Sin (Shanghai). 39:549–559. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jo YY, Kweon H, Kim DW, Baek K, Kim MK,
Kim SG, Chae WS, Choi JY and Rotaru H: Bone regeneration is
associated with the concentration of tumour necrosis factor-α
induced by sericin released from a silk mat. Sci Rep. 7:155892017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jo YY, Kweon H, Kim DW, Baek K, Chae WS,
Kang YJ, Oh JH, Kim SG and Garagiola U: Silk sericin application
increases bone morphogenic protein-2/4 expression via a toll-like
receptor-mediated pathway. Int J Biol Macromol. 190:607–617. 2021.
View Article : Google Scholar
|
|
31
|
Buonocore G, Perrone S and Tataranno ML:
Oxygen toxicity: Chemistry and biology of reactive oxygen species.
Int J Biol Macromol. 15:186–190. 2010.
|
|
32
|
Yen GC, Duh PD and Lin CW: Effects of
resveratrol and 4-hexylresorcinol on hydrogen peroxide-induced
oxidative DNA damage in human lymphocytes. Free Radic Res.
37:509–514. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guandalini E, Ioppolo A, Mantovani A,
Stacchini P and Giovannini C: 4-Hexylresorcinol as inhibitor of
shrimp melanosis: Efficacy and residues studies; evaluation of
possible toxic effect in a human intestinal in vitro model
(Caco-2); preliminary safety assessment. Food Addit Contam.
15:171–180. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Synnott NC, O'Connell D, Crown J and Duffy
MJ: COTI-2 reactivates mutant p53 and inhibits growth of
triple-negative breast cancer cells. Breast Cancer Res Treat.
179:47–56. 2020. View Article : Google Scholar
|
|
35
|
Lindemann A, Patel AA, Silver NL, Tang L,
Liu Z, Wang L, Tanaka N, Rao X, Takahashi H, Maduka NK, et al:
COTI-2, a novel thiosemicarbazone derivative, exhibits antitumor
activity in HNSCC through p53-dependent and-independent mechanisms.
Clin Cancer Res. 25:5650–5662. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gao L, Yu L, Li CM, Li Y, Jia BL and Zhang
B: Karyopherin α2 induces apoptosis in tongue squamous cell
carcinoma CAL-27 cells through the p53 pathway. Oncol Rep.
35:3357–3362. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee IS, Kim DW, Oh JH, Lee SK, Choi JY,
Kim SG and Kim TW: Effects of 4-hexylresorcinol on craniofacial
growth in rats. Int J Mol Sci. 22:89352021. View Article : Google Scholar
|
|
38
|
Kim JY, Kim DW, Lee SK, Choi JY, Che X,
Kim SG and Garagiola U: Increased expression of TGF-β1 by
4-hexylresorcinol is mediated by endoplasmic reticulum and
mitochondrial stress in human umbilical endothelial vein cells.
Appl Sci. 11:91282021. View Article : Google Scholar
|
|
39
|
Hori M, Suzuki K, Udono MU, Yamauchi M,
Mine M, Watanabe M, Kondo S and Hozumi Y: Establishment of
ponasterone A-inducible the wild-type p53 protein-expressing clones
from HSC-1 cells, cell growth suppression by p53 expression and the
suppression mechanism. Arch Dermatol Res. 301:631–646. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kashiwazaki H: Detectability and
diagnostic criteria of p53 gene mutations in human oral squamous
cell carcinoma using yeast functional assay. Hokkaido Igaku Zasshi.
72:211–224. 1997.(In Japanese).
|
|
41
|
Ichwan SJ, Yamada S, Sumrejkanchanakij P,
Ibrahim-Auerkari E, Eto K and Ikeda MA: Defect in serine 46
phosphorylation of p53 contributes to acquisition of p53 resistance
in oral squamous cell carcinoma cells. Oncogene. 25:1216–1224.
2006. View Article : Google Scholar : PubMed/NCBI
|