Introduction
Lung cancer is the leading cause of cancer mortality
among men and women worldwide and causes 1.59 million deaths each
year (1,2). Histopathologically, lung cancer is
divided into non-small cell lung cancer (NSCLC) and small-cell lung
cancer and is usually diagnosed at a late stage because it often
has no symptoms until it has spread (3). Furthermore, ~85% of lung cancers are
NSCLC, of which >50% are advanced at the time of diagnosis and
the 5-year survival rate for all stages of NSCLC is <15%
(4,5). In the late stage, the most common
symptoms include cough, dyspnea and hemoptysis and the cancer has
metastasized beyond the lungs and into other areas of the body,
such as the lymph nodes, brain or other organs (6). At present, surgery and paclitaxel
(PTX)- or platinum-based combination chemotherapy are the most
common applications in the clinical treatment of NSCLC (7). PTX is a tubulin-disrupting agent and
has demonstrated antitumor efficacy against a broad variety of
tumors, such as lung, breast and ovarian cancer (8,9). PTX
is a first-line chemotherapy drug in the treatment of advanced
NSCLC (10). The initial response
to PTX in the treatment of NSCLC is favorable; however, the
patients often develop drug resistance to PTX leading to treatment
failure (11). Therefore, it is
urgent to investigate the mechanism underlying the development of
PTX resistance and to develop novel therapeutic strategies for
overcoming PTX resistance.
The chloride voltage-gated channel-3 (ClC-3) is a
member of the ClC voltage-gated Cl− channel family.
Accumulating studies have suggested that ClC-3 is expressed in a
number of cancer cells and serves a well-defined role in cell
proliferation, apoptosis and metastasis (12–15).
Furthermore, abnormality of ClC-3 expression has been demonstrated
to be associated with the development of drug resistance, including
PTX, cisplatin and etoposide resistance, in various tumor cells
(16–19). However, the potential regulatory
mechanism of ClC-3 in PTX resistance of NSCLC remains largely
unknown.
Cancer stem cells (CSCs) are a small subpopulation
of cancer cells with characteristics that are associated with stem
cells (20). CSCs are considered
to be the main cause of chemotherapy resistance (21,22).
SRY-box transcription factor 2 (SOX2) is not only a pluripotent
stem cell-related factor but also a key transcription factor and
serves a role in maintaining stem cell properties and determining
the fate of cells (23).
Researchers have revealed that SOX2 is aberrantly expressed in
different types of cancer and that SOX2 expression is positively
associated with cancer cell stemness and multi-drug resistance
(24–26). Therefore, SOX2 may be an attractive
therapeutic target for overcoming chemotherapy resistance.
In the present study, SOX2 and ClC-3 were highly
expressed in PTX-resistant A549 NSCLC cells and SOX2 increased the
sensitivity of A549 NSCLC cells to PTX treatment via downregulation
of the levels of ClC-3. The molecular mechanism between SOX2 and
ClC-3 was further explored using cleavage under targets and
tagmentation (CUT&Tag) sequencing prediction results. Taken
together, the present study provided novel insights into targeting
the SOX2/ClC-3 axis as a potential therapeutic strategy for
patients with NSCLC with PTX resistance.
Materials and methods
Cell lines and cell culture
Human A549 NSCLC cell lines were obtained from
American Type Culture Collection. The PTX-resistant A549 NSCLC
(A549-PTX) cells were established by gradual exposure of A549 cells
to increasing concentrations of PTX, as previously described
(27). In order to maintain the
PTX-resistant phenotype of A549-PTX cells, 0.1 µM PTX was added
into the culture medium. The A549 cells used in the present study
were cultured in parallel during the establishment of A549-PTX
cells. All cells were cultured in DMEM (Corning, Inc.) supplemented
with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.)
and 1X penicillin/streptomycin (HyClone; Cytiva). All cultures were
maintained in a humidified tissue culture incubator at 37°C with 5%
CO2.
Cell transfection
For RNA interference, SOX2 small interfering RNA
(siRNA/si) and ClC-3 siRNA were purchased from Guangzhou RiboBio
Co., Ltd. 2×105 cells per well were seeded in 6-well
plates for 24 h before transfection. Subsequently, cells were
transfected with SOX2 siRNA (25 nM), ClC-3 siRNA (25 nM) or their
negative control (siNC, (25 nM)) using Lipofectamine®
RNAiMAX Transfection Reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. Following
transfection for 12 h in a humidified tissue culture incubator at
37°C with 5% CO2, the fresh medium with 10% FBS was
replaced and incubated for 48 h. The siRNA target sequences used
are presented in Table I.
 | Table I.siRNA target sequences. |
Table I.
siRNA target sequences.
| siRNA | Target sequences
(5′-3′) |
|---|
| siClC-3-1 |
CCTGGTTCTTATATCATGA |
| siClC-3-2 |
GATGGCTAGTAGTAACACT |
| siClC-3-3 |
GCCTTAGTGCGTTGTGGTA |
| siSOX2-1 |
CCAAGACGCTCATGAAGAA |
| siSOX2-2 |
GGAGCACCCGGATTATAAA |
| siSOX2-3 |
GCTCGCAGACCTACATGAA |
Lentiviral infection
Human SOX2 and ClC-3 were subcloned into the
lentiviral plasmids pCMV-3XFlag-Puro vector and the plasmids and
lentivirus particles were generated by OBiO Technology (Shanghai)
Corp., Ltd. Lentivirus were produced in 293T cells using a third
generation lentiviral system (Shanghai OBiO Technology Co., Ltd.).
Briefly, 5×106 293T cells were seeded in a 100-mm
culture dish at 24 h before transfection. 5 µg lentiviral construct
and 5 µg lentiviral envelope and packaging plasmids (both from
Shanghai OBiO Technology Co., Ltd.) were co-transfected into 293T
cells (the mixed ratio was lentiviral construct: lentiviral
envelope and packaging plasmids, 1:1) by using Lentiviral Packaging
Transfection kit (Shanghai OBiO Technology Co., Ltd.). Following
transfection for 8 h at 37°C in a CO2 incubator, the
medium was replaced with fresh culture medium. After 48 h, the
lentivirus-containing supernatants were harvested, centrifuged at
2,000 × g for 10 min at room temperature and filtered by using 0.22
µm filter. The cells were infected with the 10 MOI lentivirus of
empty vector pCMV-3XFlag-Puro, pCMV-ClC-3-3Xflag-Puro or
pCMV-SOX2-3Xflag-Puro construct to create SOX2- or
ClC-3-overexpressing stable cell lines. Cells were infected with 10
MOI lentivirus and then selected in medium containing 1 µg/ml
puromycin for 1 week. Finally, cells were maintained in medium
containing 0.1 µg/ml puromycin medium. SOX2 or ClC-3 expression was
confirmed by western blot analysis.
Cell counting kit-8 (CCK-8)
assays
Cell viability was evaluated using CCK-8 (Dojindo
Molecular Technologies, Inc.) according to the manufacturer's
protocol. Briefly, 5,000 cells per well were seeded in 96-well
plates and allowed to adhere overnight. Cells were then treated
with different concentrations of PTX or DIDS for 48 h. Next, DMEM
containing 10% CCK-8 solution was supplemented into each well and
the cells were incubated in a 37°C incubator for 2 h. The optical
density of each well was measured using a microplate reader
(Synergy H1; BioTeke Corporation) at a wavelength of 450 nm. The
cytotoxicity of PTX to cell lines was evaluated.
Cell colony formation assays
A total of 1×103 cells per well were
seeded in 6-well plates and incubated for 24 h, following treatment
with PTX (50 or 100 µM) or DMSO as a control in a humidified tissue
culture incubator at 37°C with 5% CO2 for 48 h. Media
were replaced every 3 days. After 2 weeks of growth, the medium was
discarded and the cells were fixed with 4% formaldehyde, following
Please give temperature and duration of staining. according to the
manufacturer's instructions. Images were captured using a digital
scanner (Canon, Inc.). Colonies were counted using ImageJ 1.80
software (National Institutes of Health).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cultured cells at 80%
confluence using TRIzol® (Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. cDNA synthesis was
carried out using SuperScript II Reverse Transcriptase and random
hexanucleotide primers (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocols. qPCR was performed using
synthesized primers (Tsingke Biological Technology) and SYBR green
master mix (Tiangen Biotech Co., Ltd.) to detect the mRNA levels.
PCR conditions were as follows: Pre-denaturation at 95°C for 1 min;
followed by 40 cycles of denaturation at 95°C for 20 sec, annealing
at 60°C for 20 sec and elongation at 72°C for 30 sec. The reaction
was performed using an Applied Biosystems 7500 Fast Sequence
Detection system (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The expression levels of the target genes were quantitated
using the 2−ΔΔCq method and β actin (ACTB) was
used as the internal control to normalize the qPCR data (28). The primer sequences are presented
in Table II. All samples were
examined at least three times.
| Table II.Primer sequences. |
Table II.
Primer sequences.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| CLCN3 |
CCTCTTTCCAAAGTATAGCAC |
TTACTGGCATTCATGTCATTTC |
| SOX2 |
TACAGCATGTCCTACTCGCAG |
GAGGAAGAGGTAACCACAGGG |
| OCT4 |
GCAGCGACTATGCACAACGA |
CCAGAGTGGTGACGGAGACA |
| KLF4 |
ATCTTTCTCCACGTTCGCGTCTG |
AAGCACTGGGGGAAGTCGCTTC |
| Nanog |
AAGGTCCCGGTCAAGAAACAG |
CTTCTGCGTCACACCATTGC |
| P-gp |
GCTGTCAAGGAAGCCAATGCCT |
TGCAATGGCGATCCTCTGCTTC |
| MDR1 |
CCCATCATTGCAATAGCAGG |
TGTTCAAACTTCTGCTCCTGA |
| ABCC2 |
GCCAACTTGTGGCTGTGATAGG |
ATCCAGGACTGCTGTGGGACAT |
| ABCC10 |
CCTAGTGCTGACCGTGTTGT |
TAGGTTGGCTGCAGTCTGTG |
| ACTB |
CACCATTGGCAATGAGCGGTTC |
AGGTCTTTGCGGATGTCCACGT |
Western blotting and
immunoprecipitation (IP) assays
Protein was extracted from cells using M-PER (Thermo
Fisher Scientific, Inc.) and the protein concentration was
determined using a BCA Protein Assay Kit (Thermo Fisher Scientific,
Inc.). The protein extracts (20 µg per lane) were separated by
using 10% SDS-PAGE and then transferred to a polyvinylidene
fluoride (PVDF) membrane (MilliporeSigma), followed by blocking
with 5% skimmed milk powder in room temperature for 1 h and
incubation with primary antibodies overnight at 4°C. The primary
antibodies used were: ClC-3 (1:1,000; cat. no. 13359; Cell
Signaling Technology, Inc.), SOX2 (1:1,000; cat. no. 23064; Cell
Signaling Technology, Inc.), octamer-binding transcription factor 4
(OCT4; 1:1,000; cat. no. 2750; Cell Signaling Technology, Inc.),
NANOG (1:1,000; cat. no. 4903; Cell Signaling Technology, Inc.),
KLF transcription factor 4 (KLF4; 1:1,000; cat. no. 4038; Cell
Signaling Technology, Inc.), multidrug resistance mutation 1 (MDR1;
1:1,000; cat. no. 13342; Cell Signaling Technology, Inc.), ATP
binding cassette subfamily C member 2 (ABCC2; 1:1,000; cat. no.
4446; Cell Signaling Technology, Inc.), ATP binding cassette
subfamily C member 10 (ABCC10; 1:1,000; cat. no. ab69296; Abcam),
GAPDH (1:10,000; cat. no. ARG65680; Arigo Biolaboratories Corp.)
and tubulin (1:10,000; cat. no. ARG65693; Arigo Biolaboratories
Corp.). Subsequently, the membranes were incubated with
peroxidase-conjugated secondary antibody. The secondary antibodies
used were Goat anti-Rabbit IgG (1:10,000; cat. no. ARG65351; Arigo
Biolaboratories Corp.) and Goat anti-Mouse IgG (1:10,000; cat. no.
ARG65350; Arigo Biolaboratories Corp.). The protein signals were
determined using the ChemiDoc XRS+ System (Bio-Rad Laboratories,
Inc.) and the ECL detection kit (MilliporeSigma). The gray value of
the protein bands was analyzed by ImageJ software (version: 1.53;
National Institutes of Health).
For IP analysis, the cells were treated with 30 µM
MG132 for 6 h in a tissue culture incubator at 37°C with 5%
CO2 before collection and then the cells were lysed in
IP lysis buffer (Beyotime Biotechnology Inc.). Next, the lysates
were immunoprecipitated with antibody of SOX2 (1:100; cat. no.
23064; Cell Signaling Technology, Inc.) or ClC-3 (1:50; cat. no.
13359; Cell Signaling Technology, Inc.) together with Protein A/G
magnetic beads at 4°C overnight. The samples were boiled in 5X
loading buffer for 10 min and then separated from the beads using
magnetic separator. The samples were detected by western blot
analysis according to the aforementioned procedure.
CUT&Tag assays and CUT&Tag
qPCR
The CUT&Tag assay was performed using a NovoNGS
CUT&Tag 3.0 HighSensitivity kit (Novoprotein Scientific Inc.)
according to the manufacturer's instructions. Briefly, NovoNGS ConA
Beads were washed using ConA Binding Buffer. A total of
1×105 A549 cells were harvested and washed using 1X wash
buffer. The cells with beads were incubated with the SOX2 antibody
(1:50; cat. no. 23064; Cell Signaling Technology, Inc.) overnight
at 4°C, followed by incubation with a secondary antibody at room
temperature for 1 h. The secondary antibody used was Goat
anti-Rabbit IgG H&L (1:100, cat. no. N269-01A; Novoprotein
Scientific Inc.). After washing away the unbounded secondary
antibody, the cells were incubated with NovoNGS ChiTag pA-Tn5 for 1
h at room temperature. Next, the cells were washed by ChiTag
Buffer, followed by tagmentation using Tagmentation Buffer for 1 h
at 37°C. The tagmentation reaction was stopped by addition of 10%
SDS at 55°C for 10 min. DNA was isolated using Tagment DNA Extract
Beads (Novoprotein Scientific Inc.) and dissolved in TE Buffer. DNA
was amplified with N5 and N7 primers and purified with NovoNGS DNA
Clean Beads for sequencing and qPCR assays. For CUT&Tag
sequencing, the libraries were sequenced and analyzed by Guangzhou
Epibiotek Co., Ltd. Briefly, the reads were aligned using Bowtie2
(version: 2.2.9; http://bowtie-bio.sourceforge.net/bowtie2/index.shtml).
Peak calling was performed with MACS2 (version: 2.1.1; https://pypi.org/project/MACS2/2.1.1.20160309/) and
annotated using HOMER (http://homer.ucsd.edu/homer/). The heatmap was
generated using deepTools (version: 2.4.1; http://deeptools.ie-freiburg.mpg.de/). The peaks
visualization in the genome was shown by IGV software (version:
2.13.2; http://software.broadinstitute.org/software/igv).
Functional Gene Ontology (GO) enrichment analysis were performed
using GENEONTOLOGY database (http://geneontology.org/). The purified DNA from the
CUT&Tag assay was quantified by qPCR using SuperReal PreMix
SYBR Green on an Applied Biosystems 7500 Fast Sequence Detection
system. The ClC-3 binding sites of SOX2 at the gene promoter
regions were predicted in CUT&Tag sequencing and primers were
designed by Primer software. The primers of the CLCN3
promoter used were as follows: Forward, 5′-AACCTCCGCCTTCCA-3′;
Reverse, 5′-AAACCAGCCTGAGCAAC-3′.
Luciferase reporter assays
The luciferase reporter plasmid containing the
putative ClC-3 promoter in pGL4 basic vector were purchased by OBiO
Technology Corp., Ltd. Luciferase reporter assays were carried out
in A549-ClC-3 and empty vector stably transfected cells. Cells were
transfected with ClC-3 promoter and Renilla luciferase
plasmids in 6-well plates using Lipofectamine® 3000
according to the manufacturer's instructions (Invitrogen; Thermo
Fisher Scientific, Inc.). After 48 h of transfection, the
luciferase activity was measured using the Dual-Luciferase Reporter
Assay System (Promega Corporation) according to the manufacturer's
instructions and the cell lysates were analyzed by western blot
analysis according to the aforementioned procedure.
Graphic scheme of study
methodology
The stages of study methodologies are shown in
Fig. S1.
Statistical analysis
All statistical analyses were performed using
GraphPad Prism (8.0; GraphPad Software, Inc.) and all data were
repeated at least three times from three independent experiments
for analysis and are presented as the mean ± standard deviation.
The normality of the data was tested by Shapiro-Wilk test and all
of the datasets are homogeneity of variance. Unpaired two-tail
Student's t-test was used to analyze the difference between the two
groups (Figs. 1A-D, 2B, C and F, 3B, D, E and H). The difference between
the three groups of data were analyzed by one-way ANOVA. Dunnett
test was used for multiple comparisons of cell viability (Figs. 1E, 2E, 3G,
5B and D). Tukey's HSD test was
used for multiple comparisons of protein expression (Figs. 4H and I, 5A and C). P<0.05 was considered to
indicate a statistically significant difference.
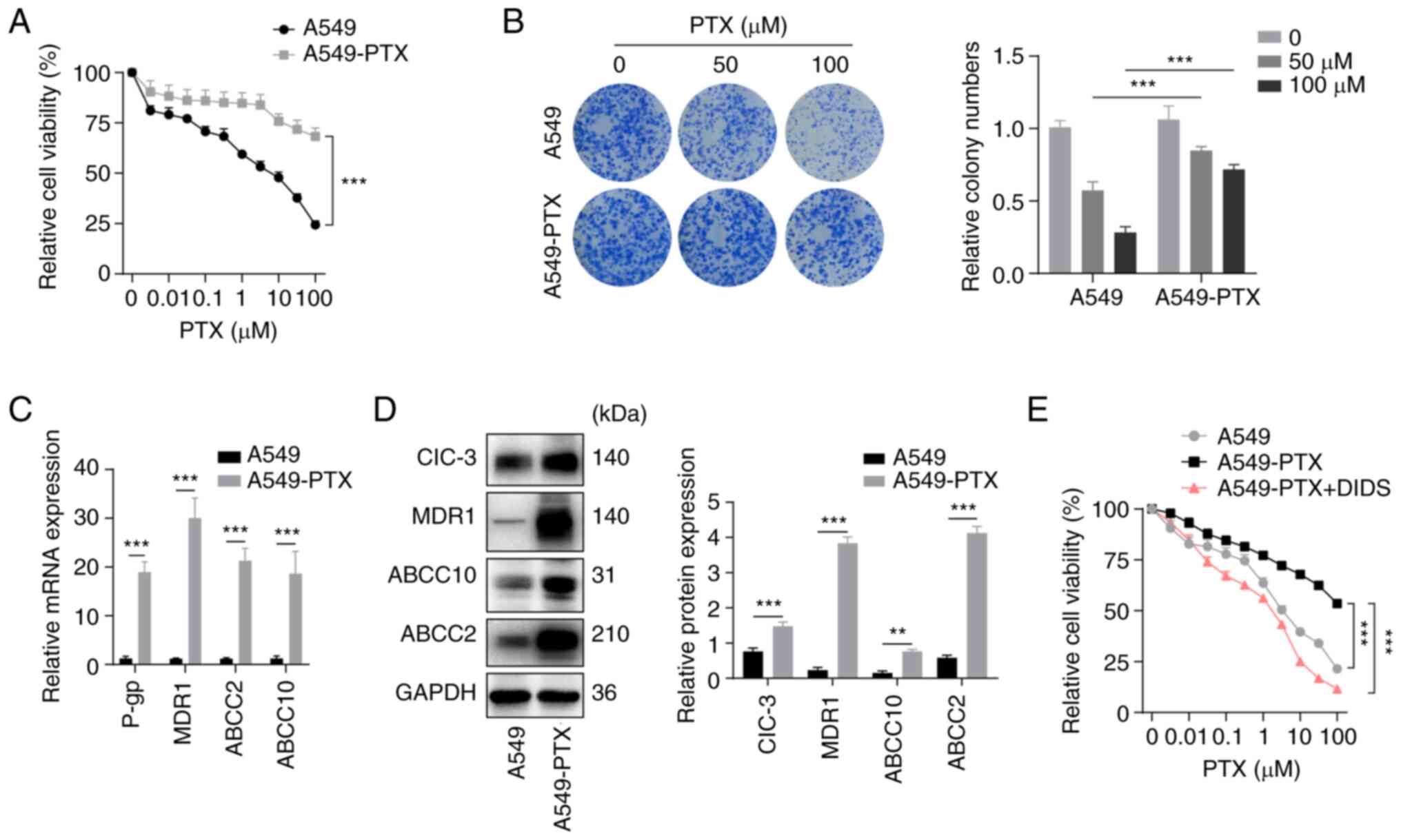 | Figure 1.Establishment of PTX-resistant A549
non-small cell lung cancer cells. (A) Viability was assessed using
CCK-8 assays in A549 and A549-PTX cells treated with PTX for 48 h.
(B) (Left) Cell proliferation was examined by colony formation
assays in A549 and A549-PTX cells treated with PTX. (Right) Colony
numbers as quantified using ImageJ. (C) mRNA expression levels of
drug resistance-related genes were measured by reverse
transcription-quantitative PCR in A549 and A549-PTX cells. (D)
(Left) Protein expression levels of ClC-3, MDR1, ABCC10, ABCC2 and
GAPDH were examined by western blotting in A549 and A549-PTX cells.
(Right) Protein expression was semi-quantified using ImageJ. (E)
Viability was assessed using CCK-8 assays in A549, A549-PTX or
DIDS-treated A549-PTX cells treated with PTX for 48 h. DIDS, 10 µM
for 48 h. GAPDH was used as a loading control in western blotting.
All data are presented as the mean ± standard deviation.
**P<0.01, ***P<0.001. Relative, vs. respective control. PTX,
paclitaxel; CCK-8, Cell Counting Kit-8; A549-PTX cells,
PTX-resistant A549 NSCLC cells; CIC-3, chloride voltage-gated
channel 3; MDR1, multidrug resistance mutation 1; ABCC10, ATP
binding cassette subfamily C member 10; ABCC2, ATP binding cassette
subfamily C member 2; DIDS,
4,4-diisothiocyanatostilbene-2,2-disulfonate. |
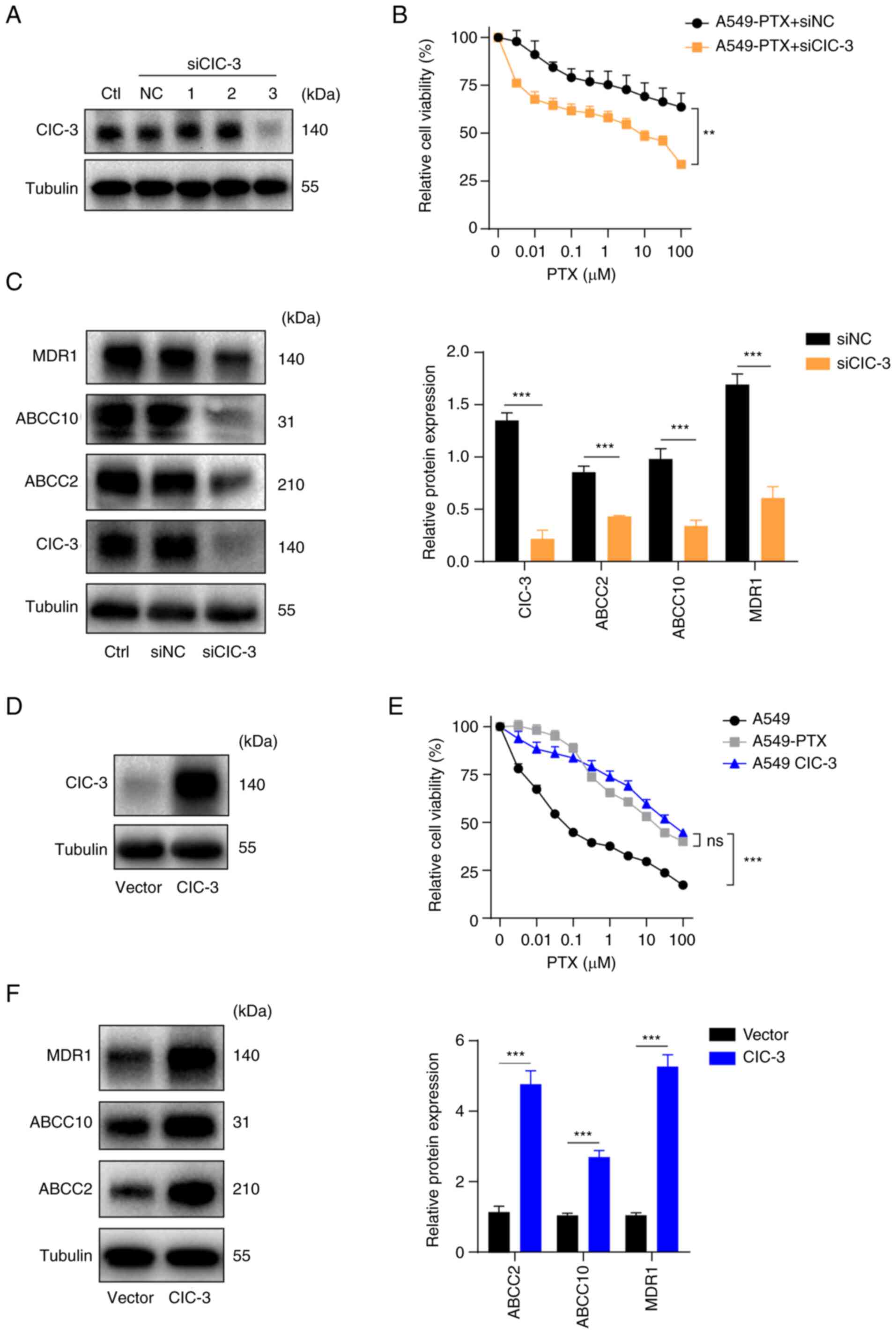 | Figure 2.ClC-3 is upregulated in PTX-resistant
A549 cells. (A) ClC-3 protein expression was measured by western
blotting in A549-PTX cells transfected with siNC or ClC-3 siRNA
(siClC-3-1, siClC-3-2 and siClC-3-3). (B) Viability was examined by
CCK-8 assays in A549-PTX cells transfected with siClC-3 or control
treated with PTX for 48 h. (C) (Left) Protein expression levels of
MDR1, ABCC10, ABCC2 and ClC-3 were examined by western blotting in
A549-PTX cells transfected with siClC-3 or control. (Right) Protein
expression was semi-quantified using ImageJ. (D) Protein expression
levels of ClC-3 were examined by western blotting in A549 cells
overexpressing ClC-3 or its vector control. (E) Viability was
examined by CCK-8 assays in A549, A549-PTX and ClC-3-overexpressing
A549 cells treated with PTX for 48 h. (F) (Left) Protein expression
levels of MDR1, ABCC10 and ABCC2 were examined by western blotting
in A549 cells overexpressing ClC-3 or its vector control. (Right)
Protein expression was semi-quantified using ImageJ. Tubulin was
used as a loading control in western blotting. All data are
presented as the mean ± standard deviation. **P<0.01,
***P<0.001. ns, not significant. Relative, vs. respective
control. ClC-3, chloride voltage-gated channel 3; PTX, paclitaxel;
A549-PTX cells, PTX-resistant A549 non-small cell lung cancer
cells; siRNA/si, small interfering RNA; siNC, control siRNA; ClC-3,
chloride voltage-gated channel 3; MDR1, multidrug resistance
mutation 1; ABCC10, ATP binding cassette subfamily C member 10;
ABCC2, ATP binding cassette subfamily C member 2; CCK-8, Cell
Counting Kit-8; Ctl, control. |
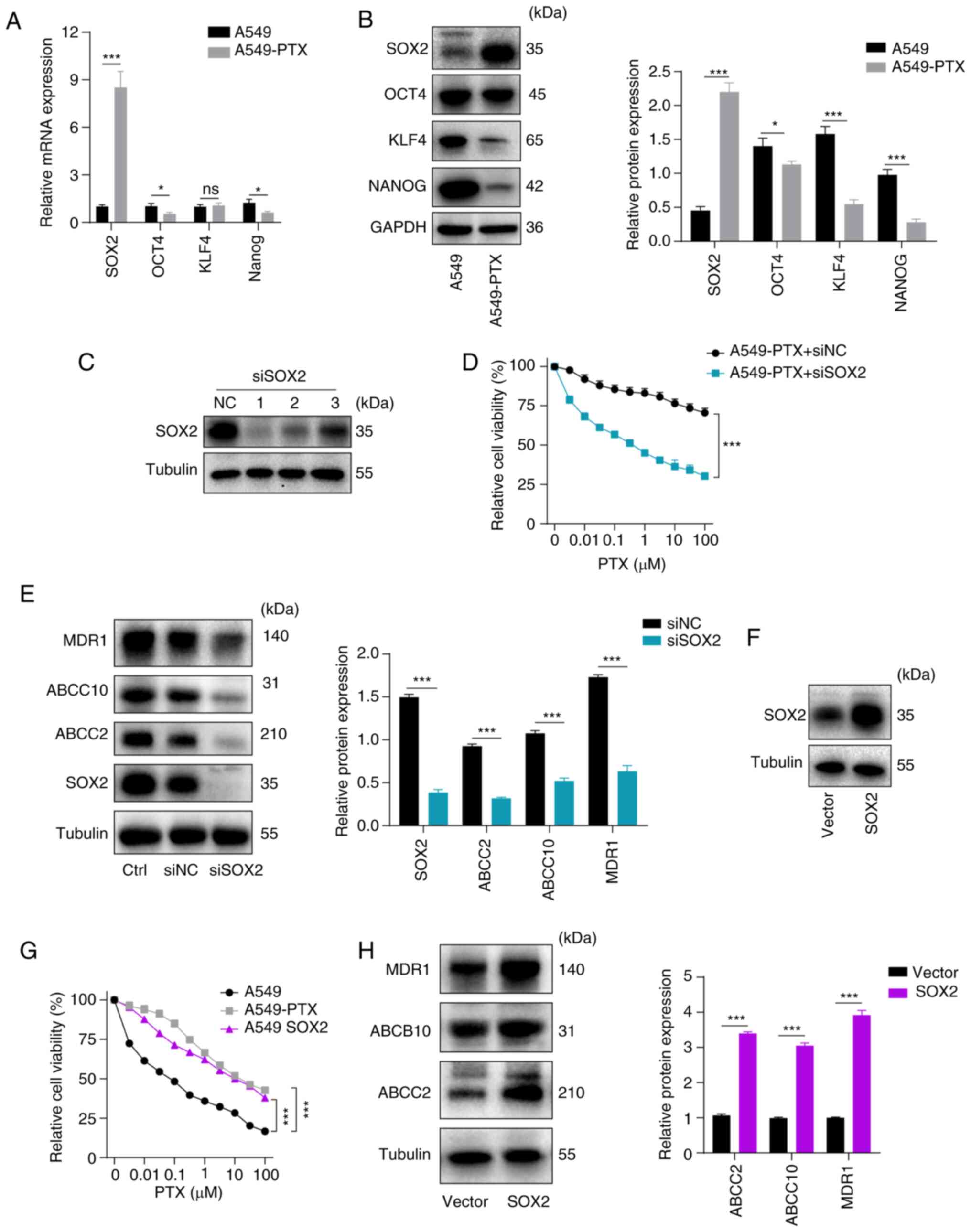 | Figure 3.Higher levels of SOX2 confer PTX
resistance in A549 non-small cell lung cancer cells. (A) mRNA
expression levels of stemness-related genes were measured by
reverse transcription-quantitative PCR in A549 and A549-PTX cells.
(B) (Left) Protein expression levels of NANOG, KLF4, OCT4 and SOX2
were examined by western blotting in A549 and A549-PTX cells.
(Right) Protein expression was semi-quantified using ImageJ. (C)
Protein expression levels of SOX2 were examined by western blotting
in A549-PTX cells transfected with siNC or SOX2 siRNA (siSOX2-1,
siSOX2-2 and siSOX2-3). (D) Viability was examined using CCK-8
assays in A549-PTX cells transfected with siSOX2 or its control and
treated with PTX for 48 h. (E) (Left) Protein expression levels of
MDR1, ABCC10, ABCC2 and SOX2 were examined by western blotting in
A549-PTX cells transfected with siSOX2 or its control. (Right)
Protein expression was semi-quantified using ImageJ. (F) Protein
expression levels of SOX2 were examined by western blotting in A549
cells overexpressing SOX2 or its vector control. (G) Viability was
examined using CCK-8 assays in A549, A549-PTX and
SOX2-overexpressing A549 cells treated with PTX for 48 h. (H)
(Left) Protein expression levels of MDR1, ABCC10 and ABCC2 were
examined by western blotting in A549 cells overexpressing SOX2 or
its vector control. (Right) Protein expression was semi-quantified
using ImageJ. GAPDH or Tubulin were used as a loading control in
western blotting. All data are presented as the mean ± standard
deviation. *P<0.05, ***P<0.001. ns, not significant.
Relative, vs. respective control. SOX2, SRY-box transcription
factor 2; PTX, paclitaxel; A549-PTX cells, PTX-resistant A549
non-small cell lung cancer cells; KLF4, Krüppel like factor 4;
OCT4, octamer-binding transcription factor 4; siRNA/si, small
interfering RNA; siNC, control siRNA; CCK-8, Cell Counting Kit-8;
MDR1, multidrug resistance mutation 1; ABCC10, ATP binding cassette
subfamily C member 10; ABCC2, ATP binding cassette subfamily C
member 2; Ctl, control. |
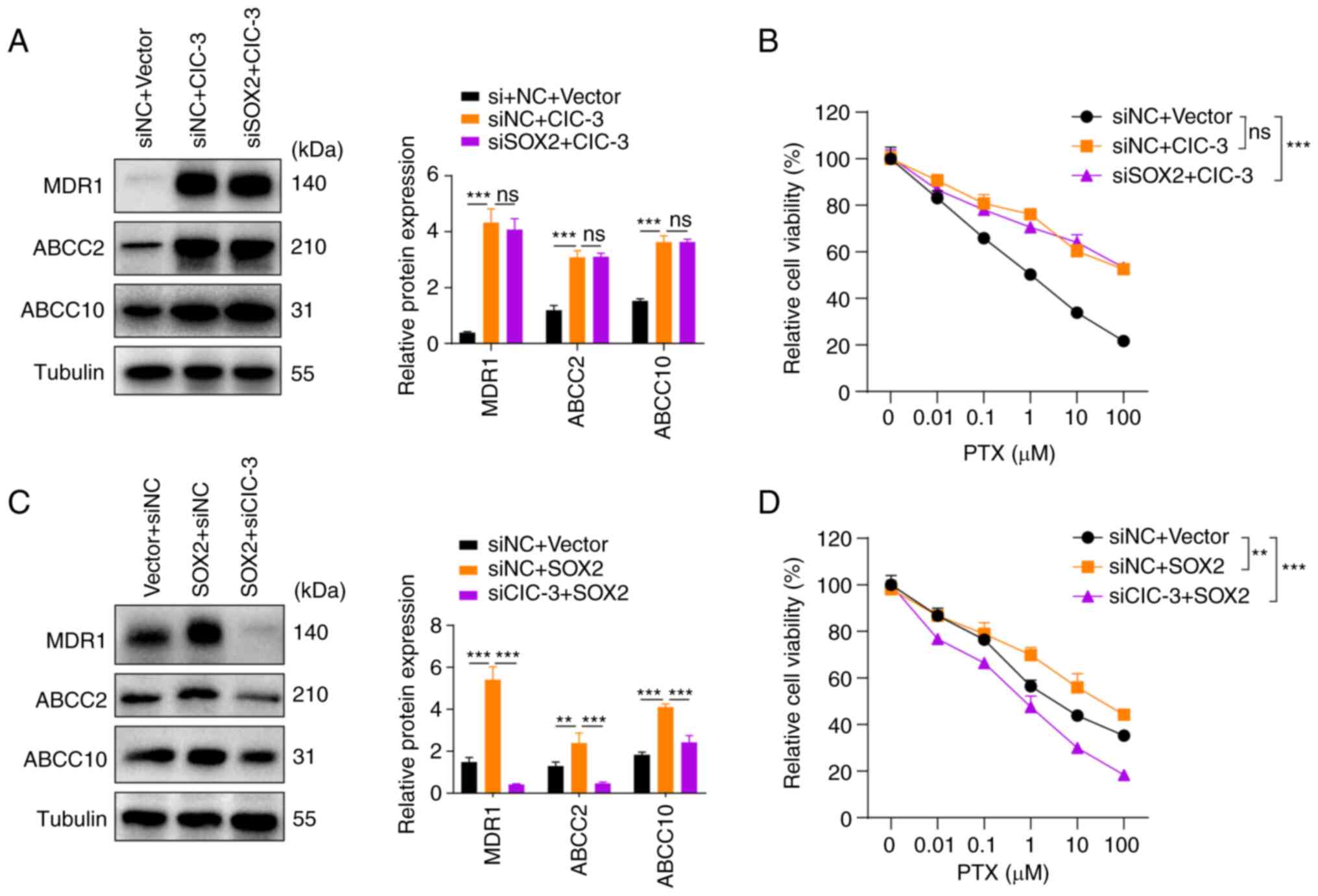 | Figure 5.Knockdown of ClC-3 expression in A549
cells prevents PTX resistance induced by upregulation of SOX2
expression. (A) (Left) Protein expression levels of MDR1, ABCC2,
ABCC10 and tubulin in A549 cells transfected with ClC-3 plasmid or
siSOX2 and its control vector or siNC. (Right) Protein expression
was semi-quantified using ImageJ. (B) Viability was examined using
a CCK-8 assay in A549 cells transfected with ClC-3 plasmid or
siSOX2 and its control vector or siNC. Cells were treated with PTX
for 48 h. (C) (Left) Protein expression levels of MDR1, ABCC2,
ABCC10 and tubulin in A549 cells transfected with SOX2 plasmid or
siClC-3 and its control vector or siNC. (Right) Protein expression
was semi-quantified using ImageJ. (D) Viability was examined using
a CCK-8 assay in A549 cells transfected with SOX2 plasmid or
siClC-3 and its control vector or siNC. Cells were treated with PTX
for 48 h. Tubulin were used as a loading control in western
blotting. All data are presented as the mean ± standard deviation.
**P<0.005, ***P<0.001. ns, not
significant. Relative, vs. respective control. ClC-3, chloride
voltage-gated channel 3; A549-PTX cells, paclitaxel-resistant A549
non-small cell lung cancer cells; PTX, paclitaxel; MDR1, multidrug
resistance mutation 1; ABCC2, ATP binding cassette subfamily C
member 2; ABCC10, ATP binding cassette subfamily C member 10;
siRNA/si, small interfering RNA; CCK-8, Cell Counting Kit-8. |
Results
Establishment of PTX-resistant A549
NSCLC cells
A549 cells were cultured with 1 µM PTX for >6
months to establish the PTX-resistant A549 subline as described in
a previous study (27). In order
to verify whether the established A549 cells were resistant to PTX,
the proliferation of A549-PTX cells and the parental A549 cells
treated with PTX was compared in CCK-8 and colony formation assays.
The results of the CCK-8 assay revealed that the viability of
A549-PTX cells was higher compared with that of A549 cells
(Fig. 1A). Additionally, the
results of the colony formation assays indicated that the
proliferation capacity was strongly increased in A549-PTX cells at
the same concentration of PTX compared with A549 cells (Fig. 1B). Subsequently, the levels of drug
resistance markers in A549 and A549-PTX cells were compared using
RT-qPCR and western blotting. RT-qPCR and western blotting revealed
increased expression levels of P-glycoprotein (P-gp), MDR1, ABCC2
and ABCC10 in A549-PTX cells (Fig. 1C
and D). Overall, the data indicated that the establishment of
PTX-resistant A549 cells was successful.
ClC-3 promotes PTX resistance in A549
NSCLC cells
It has been reported that ClC-3 contributes to PTX
resistance in A549 NSCLC cells (17). ClC-3 was significantly upregulated
in A549-PTX cells consistent with the results of previous studies
(16,17). To verify the role of ClC-3 in PTX
resistance, 4,4-diisothiocyanatostilbene-2,2-disulfonate (DIDS), a
specific chloride channel inhibitor, was used. CCK-8 assays
revealed that DIDS increased the sensitivity of A549-PTX cells to
PTX (Fig. 1E). Subsequently, the
present study examined whether ClC-3 directly modulated PTX
resistance. The expression levels of ClC-3 in A549-PTX cells were
knocked down by exogenous introduction of ClC-3 siRNAs (siClC-3-1,
siClC-3-2 and siClC-3-3). Western blotting was conducted to detect
the knockdown efficiency and the results demonstrated that
transfection with siClC-3-3 led to a reduction of ClC-3 expression
in A549-PTX cells and used in subsequent assays (Fig. 2A). CCK-8 assays revealed that ClC-3
silencing significantly increased the sensitivity of A549-PTX cells
to PTX (Fig. 2B). Western blotting
demonstrated that knockdown of ClC-3 downregulated the expression
levels of MDR1, ABCC2 and ABCC10 in A549-PTX cells (Fig. 2C). Next, ClC-3 was overexpressed in
A549 cells by infection with lentiviral vector and the
overexpression efficiency of ClC-3 was verified by western blotting
(Fig. 2D). The CCK-8 assay results
revealed that ClC-3 overexpression decreased the sensitivity of
A549 cells to PTX (Fig. 2E) and
western blot analysis revealed that ClC-3 overexpression
upregulated the expression levels of MDR1, ABCC2 and ABCC10 in A549
cells (Fig. 2F). Taken together,
these results indicated that ClC-3 was upregulated in A549-PTX
cells and that ClC-3 is required for sustaining PTX resistance in
A549 NSCLC cells. Western blotting demonstrated that knockdown of
ClC-3 downregulated the expression levels of MDR1, ABCC2 and ABCC10
in A549-PTX cells.
Higher levels of SOX2 confer PTX
resistance in A549 NSCLC cells
Previous reports have demonstrated that stemness
factors are involved in the development of multi-drug resistance
(20,21,29).
Initially, the expression levels of stemness factors were examined
by RT-qPCR and it was observed that SOX2, OCT4 and
NANOG were downregulated and KLF4 expression was not
significantly altered in A549-PTX cells compared with A549 cells
(Fig. 3A). The results of western
blotting demonstrated that SOX2 was upregulated but OCT4, KLF4 and
NANOG were downregulated in A549-PTX cells compared with A549 cells
(Fig. 3B). The present study next
examined whether SOX2 is required for PTX resistance in A549 NSCLC
cells. The expression levels of SOX2 were knocked down in A549-PTX
cells by exogenous introduction of SOX2 siRNAs (siSOX2-1, siSOX2-2
and siSOX2-3). Western blot analysis was conducted to detect the
knockdown efficiency and the results indicated that siSOX2-1 led to
a reduction of SOX2 expression in A549-PTX cells and used in
subsequent assays (Fig. 3C). CCK-8
assays demonstrated that SOX2 silencing significantly increased the
sensitivity of A549-PTX cells to PTX (Fig. 3D). Western blot analysis
demonstrated that knockdown of SOX2 downregulated the expression
levels of MDR1, ABCC2 and ABCC10 in A549-PTX cells (Fig. 3E). Next, SOX2 was overexpressed in
A549 cells by infection with lentiviral vector and the
overexpression efficiency of SOX2 was verified by western blotting
(Fig. 3F). The CCK-8 assay results
revealed that SOX2 overexpression decreased the sensitivity of A549
cells to PTX (Fig. 3G) and western
blot analysis demonstrated that SOX2 overexpression upregulated the
expression levels of MDR1, ABCC2 and ABCC10 in A549 cells (Fig. 3H). Taken together, the data
suggested that SOX2 mediated the PTX resistance of NSCLC cells.
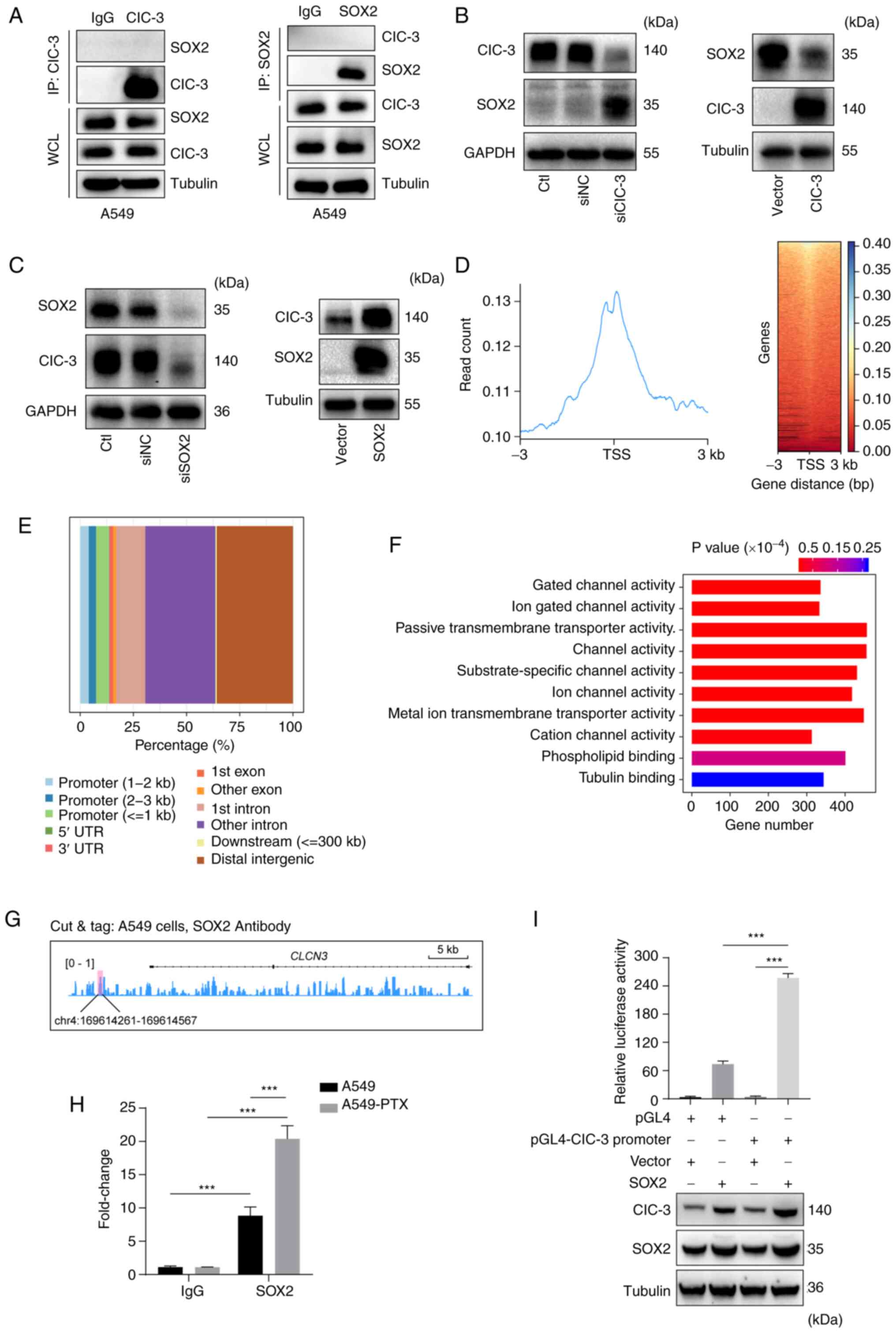
SOX2 promotes ClC-3 transcription
Our previous study revealed that SOX2 interacts with
ClC-3 in DU145 prostatic carcinoma cells and contributes to
tumorigenesis (30). To further
examine whether there was a potential interaction between ClC-3 and
SOX2 in PTX-resistant A549 NSCLC cells, the interaction of SOX2 and
ClC-3 was detected using an IP assay. The IP assay demonstrated
that there was no interaction between ClC-3 and SOX2 in A549 cells
(Fig. 4A). To further examine the
potential regulation between ClC-3 and SOX2, western blotting was
performed and revealed that SOX2 expression was increased after
transfection with siClC-3 in A549 cells and ClC-3 overexpression
downregulated SOX2 expression in A549 cells (Fig. 4B). However, knockdown of SOX2
downregulated the levels of ClC-3 and SOX2 overexpression
upregulated the expression levels of ClC-3 in A549 cells (Fig. 4C). These results revealed that
ClC-3 is a downstream effector of SOX2. Given that SOX2 is a
transcription factor (31), the
present study demonstrated that SOX2 could bind to the promoter
region of ClC-3 and examined the binding sites using CUT&Tag in
A549 cells. CUT&Tag using antibodies against SOX2 and analysis
with deepTools revealed clear enrichment of SOX2 peaks and SOX2
peaks were localized in the transcription start site of gene
promoters (±3 kb) in A549 cells (Fig.
4D). The wide genomic distribution of SOX2 in A549 cells is
shown in Fig. 4E. Next, to
investigate the attendant epigenetic modulatory impacts of SOX2 in
A549 cells, the target genes of different SOX2 binding peaks at the
promoter were classified into different GO pathways. These GO
pathways included ‘Gated channel activity’, ‘Ion gated channel
activity’, ‘Passive transmembrane transporter activity’, ‘Channel
activity’ and ‘Cation channel activity’ (Fig. 4F). Specifically, the binding of
SOX2 on ClC-3 (ClCN3) loci is shown in Fig. 4G and the potential SOX2 binding
site in the promoter of ClCN3 was in
chr4:169614261-169614567. CUT&Tag-qPCR analysis indicated that
the SOX2 levels on ClC-3 promoters were significantly elevated in
A549-PTX cells compared with A549 cells (Fig. 4H). A dual-luciferase reporter gene
assay revealed that the luciferase activity in cells infected with
the SOX2 vector was increased compared with that in cells infected
with the promoter vector (Fig.
4I). These results suggested that SOX2 could promote gene
transcription of ClC-3.
SOX2 promotes PTX resistance of A549
cells via ClC-3 expression
The aforementioned results indicated that SOX2
promoted the transcriptional expression of ClC-3 in PTX-resistant
A549 NSCLC cells. The present study subsequently explored whether
SOX2 mediates PTX resistance via ClC-3 expression. Western blotting
and cell viability assay results revealed that silencing of SOX2
could not reverse the PTX resistance induced by ClC-3
overexpression (Fig. 5A and B);
however, knockdown of ClC-3 expression in A549 cells prevented PTX
resistance induced by upregulation of SOX2 expression (Fig. 5C and D). Collectively, these
results provided additional evidence suggesting that SOX2 modulates
PTX resistance via ClC-3.
Discussion
NSCLC is the main type of lung cancer with a high
incidence and mortality (2). PTX
is a broad-spectrum anticancer drug; however, development of
resistance to PTX remains an important clinical problem (32,33).
Previous research has demonstrated that CSCs have the ability of
self-renewal and multi-directional differentiation, which is
related to tumor progression, metastasis, drug resistance and tumor
recurrence (34). CSCs are
considered to be the main cause of chemotherapy resistance and
tumor recurrence (21,22). SOX2 is a key transcription factor
maintaining the pluripotency of stem cells and serves a key role in
maintaining stemness and conferring chemotherapy resistance. Piva
et al (35) revealed that
the tamoxifen resistance of breast cancer cells is related to
SOX2-dependent activation of Wnt signaling. MLN4924 inhibits stem
cell properties and makes cancer cells sensitive to chemotherapy by
inactivating the F-box and WD repeat domain containing 2/msh
homeobox 2/SOX2 axis in human lung cancer (36). Silencing of SOX2 increases the
sensitivity to cisplatin in NSCLC by regulating
apurinic/apyrimidinic endonuclease 1 signaling (26). The present study revealed that the
mRNA and protein expression levels of SOX2 were increased in
A549-PTX cells. SOX2 silencing in A549-PTX cells decreased
viability by increasing the sensitivity to PTX; however,
overexpression of SOX2 reversed these effects.
Chloride channels serve an important role in tumor
drug resistance and have attracted wide attention. For example,
ClC-3 increases the proportion of the free form of β-tubulin and
decreases the proportion of the polymerized form of β-tubulin and
finally decreases ovarian cancer cell sensitivity to PTX by
interacting with SOX2 (16). ClC-3
participates in PTX resistance in A549 lung cancer cells through
NF-κB signaling-dependent P-gp expression (17). Chloride voltage-gated channel 5
(ClC-5) induces multiple myeloma cell drug resistance to bortezomib
by increasing pro-survival autophagy by inhibiting the AKT-mTOR
signaling pathway (37). In the
present study, PTX-resistant A549 cells were established according
to a previous protocol (27). The
protein and mRNA expression levels of drug resistance-related genes
were increased in A549-PTX cells. ClC-3 was upregulated in A549-PTX
cells. Treatment with DIDS, a chloride channel inhibitor, increased
the sensitivity to PTX and downregulation of ClC-3 expression in
PTX-resistant A549 NSCLC cells could significantly increase the
sensitivity to PTX. However, the relationship between SOX2 and
ClC-3 in PTX-resistant NSCLC cells is unclear.
Our previous study showed that SOX2 regulates the
progression of prostate cancer cells by interacting with ClC-3
(30); however, there was no
interaction between SOX2 and ClC-3 in PTX-resistant A549 NSCLC
cells. Furthermore, ClC-3 is a downstream molecule of SOX2;
however, SOX2 expression was increased following transfection with
siClC-3 and SOX2 expression was decreased by ClC-3 overexpression.
It has been previously reported that microRNA (miR)-103 increases
PC-12 cell viability and reduced cell apoptosis via upregulation of
SOX2 (38). Low miR-1181
expression increases pancreatic cancer cell viability and reduces
cell apoptosis via upregulation of SOX2 and STAT3 (39). PTX has been reported to induce cell
apoptosis in various tumors (40).
It was hypothesized that SOX2 may protect against apoptosis and
SOX2 expression was upregulated after transfection with siClC-3.
Additionally, SOX2 combined with the promoter of ClC-3 and
increased the transcriptional activity of ClC-3 in PTX-resistant
A549 NSCLC cells. Recent studies have indicated that SOX2 combined
with β-catenin and increased the transcriptional activity of ABCC2
to promote chemoresistance in colorectal cancer (41,42).
Furthermore, SOX2 cooperates with Nup153 to control transcriptional
programs in neural progenitor cells (NeuPCs) to enable bimodal gene
regulation and maintenance of NeuPCs (43). Therefore, it was hypothesized that
the transcriptional regulation of ClC-3 by SOX2 is an indirect
regulation. SOX2 may interact with other factors to regulate the
transcription of ClC-3. The factors which combine with SOX2 warrant
further exploration.
Researchers have found that a variety of ion
channels, such as voltage-gated K+, Na+,
Ca2+ and transient receptor potential channels, as well
as epithelial Na+/degenerin family ion channels except
chloride channels, are abnormally expressed in various cancer types
and are involved in the growth, migration, invasion and drug
resistance of cancer cells (44–46).
Notably, GO enrichment of CUT&Tag analysis showed that SOX2
also regulated the transcription of other ion channel genes,
including ‘Gated channel activity’, ‘Ion gated channel activity’,
‘Passive transmembrane transporter activity’, ‘Channel activity’
and ‘Cation channel activity’.
Weinstein (47)
indicated that gene addiction is a potential Achilles' heel of
cancer, that is, the expression of oncogenes is necessary not only
to initiate tumorigenesis, but also to maintain malignant
phenotypes, such as tumor invasion, metastasis and drug resistance
(48–49). In order to survive, tumor cells
would promote the emergence of new tumor clones or develop gene
mutations that make tumors insensitivity to drug treatment
(50). For example, targeting
BCR-ABL fusion gene with the small molecule inhibitor
serine/threonine kinase inhibitor Gleevec could cure chronic
myelogenous leukemia patients. However, despite the great clinical
success of Gleevec, the drug resistance of Gleevec has also
developed, which is caused by obtaining mutations in the Gleevec
binding site (51). It is evident
that combined therapies are required to cure cancer. Given the
important role of SOX2/ClC-3 axis in CSCs and PTX-resistance in
NSCLC, it is hypothesized that SOX2/ClC-3-based therapy combined
with PTX may be a promising path to pursue.
The current study only confirmed these results in
A549 cells. NSCLC is a group of lung cancers with several subtypes
such as squamous cell carcinoma, adenocarcinoma and large cell
carcinoma (52). Other PTX NSCLC
cells such H1299, PC9 and SPC-A1 will be generated to explore the
consistency of SOX2/ClC-3 involved PTX resistance in different
subtypes NSCLS cells. In addition, whether SOX2/ClC-3 axis is
involved in PTX or other drug resistance in human lung cancer
specimens will also be explored in future studies. In summary, the
present study revealed a novel mechanism whereby the SOX2/ClC-3
axis regulates NSCLC PTX resistance. The present findings may
contribute to the development of novel therapeutic candidates for
NSCLC PTX resistance and provides potentially useful experimental
evidence for PTX-resistant cancer therapy.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Pearl River S&T Nova
Program of Guangzhou (grant no. 201906010069 to HZ).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YH designed the study, analyzed the data and wrote
the manuscript. XW and RH performed the study and data analysis. GP
analyzed the data and constructed the graphs. XL wrote the
manuscript and revised the manuscript. YH, XW and RH confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Herbst RS, Morgensztern D and Boshoff C:
The biology and management of non-small cell lung cancer. Nature.
553:446–454. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gildea TR, DaCosta Byfield S, Hogarth DK,
Wilson DS and Quinn CC: A retrospective analysis of delays in the
diagnosis of lung cancer and associated costs. Clinicoecon Outcomes
Res. 9:261–269. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Travis WD: Lung cancer pathology: Current
concepts. Clin Chest Med. 41:67–85. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Travis WD, Brambilla E, Nicholson AG,
Yatabe Y, Austin JHM, Beasley MB, Chirieac LR, Dacic S, Duhig E,
Flieder DB, et al: The 2015 World Health Organization
classification of lung tumors: Impact of genetic, clinical and
radiologic advances since the 2004 classification. J Thorac Oncol.
10:1243–1260. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Latimer KM: Lung cancer: Clinical
presentation and diagnosis. FP Essent. 464:23–26. 2018.PubMed/NCBI
|
|
7
|
Huang LT, Cao R, Wang YR, Sun L, Zhang XY,
Guo YJ, Zhao JZ, Zhang SL, Jing W, Song J, et al: Clinical option
of pemetrexed-based versus paclitaxel-based first-line
chemotherapeutic regimens in combination with bevacizumab for
advanced non-squamous non-small-cell lung cancer and optimal
maintenance therapy: Evidence from a meta-analysis of randomized
control trials. BMC Cancer. 21:4262021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jordan MA and Wilson L: Microtubules as a
target for anticancer drugs. Nat Rev Cancer. 4:253–265. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schiff PB, Fant J and Horwitz SB:
Promotion of microtubule assembly in vitro by taxol. Nature.
277:665–667. 1979. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Weaver BA: How taxol/paclitaxel kills
cancer cells. Mol Biol Cell. 25:2677–2681. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cui H, Arnst K, Miller DD and Li W: Recent
advances in elucidating paclitaxel resistance mechanisms in
non-small cell lung cancer and strategies to overcome drug
resistance. Curr Med Chem. 27:6573–6595. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hong S, Bi M, Wang L, Kang Z, Ling L and
Zhao C: CLC-3 channels in cancer (review). Oncol Rep. 33:507–514.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mu H, Mu L and Gao J: Suppression of CLC-3
reduces the proliferation, invasion and migration of colorectal
cancer through Wnt/β-catenin signaling pathway. Biochem Biophys Res
Commun. 533:1240–1246. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Du S and Yang L: ClC-3 chloride channel
modulates the proliferation and migration of osteosarcoma cells via
AKT/GSK3β signaling pathway. Int J Clin Exp Pathol. 8:1622–1630.
2015.PubMed/NCBI
|
|
15
|
Ye D, Luo H, Lai Z, Zou L, Zhu L, Mao J,
Jacob T, Ye W, Wang L and Chen L: ClC-3 chloride channel proteins
regulate the cell cycle by up-regulating cyclin D1-CDK4/6 through
suppressing p21/p27 expression in nasopharyngeal carcinoma cells.
Sci Rep. 6:302762016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Feng J, Peng Z, Gao L, Yang X, Sun Z, Hou
X, Li E, Zhu L and Yang H: ClC-3 promotes paclitaxel resistance via
modulating tubulins polymerization in ovarian cancer cells. Biomed
Pharmacother. 138:1114072021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen Q, Liu X, Luo Z, Wang S, Lin J, Xie
Z, Li M, Li C, Cao H, Huang Q, et al: Chloride channel-3 mediates
multidrug resistance of cancer by upregulating P-glycoprotein
expression. J Cell Physiol. 234:6611–6623. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weylandt KH, Nebrig M, Jansen-Rosseck N,
Amey JS, Carmena D, Wiedenmann B, Higgins CF and Sardini A: ClC-3
expression enhances etoposide resistance by increasing
acidification of the late endocytic compartment. Mol Cancer Ther.
6:979–986. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Han Y, Zhou Y, Zhou L, Jia X, Yu X, An X
and Shi Z: Blockade of chloride channel-3 enhances cisplatin
sensitivity of cholangiocarcinoma cells though inhibiting
autophagy. Can J Physiol Pharmacol. 100:584–593. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ajani JA, Song S, Hochster HS and
Steinberg IB: Cancer stem cells: The promise and the potential.
Semin Oncol. 42 (Suppl 1):S3–S17. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Donnenberg VS and Donnenberg AD: Multiple
drug resistance in cancer revisited: The cancer stem cell
hypothesis. J Clin Pharmacol. 45:872–877. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Phi LTH, Sari IN, Yang YG, Lee SH, Jun N,
Kim KS, Lee YK and Kwon HY: Cancer stem cells (CSCs) in drug
resistance and their therapeutic implications in cancer treatment.
Stem Cells Int. 2018:54169232018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou C, Yang X, Sun Y, Yu H, Zhang Y and
Jin Y: Comprehensive profiling reveals mechanisms of SOX2-mediated
cell fate specification in human ESCs and NPCs. Cell Res.
26:171–189. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu K, Lin B, Zhao M, Yang X, Chen M, Gao
A, Liu F, Que J and Lan X: The multiple roles for Sox2 in stem cell
maintenance and tumorigenesis. Cell Signal. 25:1264–1271. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Song WS, Yang YP, Huang CS, Lu KH, Liu WH,
Wu WW, Lee YY, Lo WL, Lee SD, Chen YW, et al: Sox2, a stemness
gene, regulates tumor-initiating and drug-resistant properties in
CD133-positive glioblastoma stem cells. J Chin Med Assoc.
79:538–545. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen TY, Zhou J, Li PC, Tang CH, Xu K, Li
T and Ren T: SOX2 knockdown with siRNA reverses cisplatin
resistance in NSCLC by regulating APE1 signaling. Med Oncol.
39:362022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang C, Zhang X, Jiang L, Zhang L, Xiang
M and Ren H: FoxM1 induced paclitaxel resistance via activation of
the FoxM1/PHB1/RAF-MEK-ERK pathway and enhancement of the ABCA2
transporter. Mol Ther Oncolytics. 14:196–212. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Prieto-Vila M, Takahashi RU, Usuba W,
Kohama I and Ochiya T: Drug resistance driven by cancer stem cells
and their niche. Int J Mol Sci. 18:25742017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen J, Wang F, Lu Y, Yang S, Chen X,
Huang Y and Lin X: CLC-3 and SOX2 regulate the cell cycle in DU145
cells. Oncol Lett. 20:3722020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu Q, Zhang L, Su P, Lei X, Liu X, Wang H,
Lu L, Bai Y, Xiong T, Li D, et al: MSX2 mediates entry of human
pluripotent stem cells into mesendoderm by simultaneously
suppressing SOX2 and activating NODAL signaling. Cell Res.
25:1314–1332. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Adrianzen Herrera D, Ashai N, Perez-Soler
R and Cheng H: Nanoparticle albumin bound-paclitaxel for treatment
of advanced non-small cell lung cancer: An evaluation of the
clinical evidence. Expert Opin Pharmacother. 20:95–102. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Scripture CD, Figg WD and Sparreboom A:
Paclitaxel chemotherapy: From empiricism to a mechanism-based
formulation strategy. Ther Clin Risk Manag. 1:107–114. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sullivan JP, Minna JD and Shay JW:
Evidence for self-renewing lung cancer stem cells and their
implications in tumor initiation, progression, and targeted
therapy. Cancer Metastasis Rev. 29:61–72. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Piva M, Domenici G, Iriondo O, Rábano M,
Simões BM, Comaills V, Barredo I, López-Ruiz JA, Zabalza I, Kypta R
and Vivanco MD: Sox2 promotes tamoxifen resistance in breast cancer
cells. EMBO Mol Med. 6:66–79. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yin Y, Xie CM, Li H, Tan M, Chen G, Schiff
R, Xiong X and Sun Y: The FBXW2-MSX2-SOX2 axis regulates stem cell
property and drug resistance of cancer cells. Proc Natl Acad Sci
USA. 116:20528–20538. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang H, Pang Y, Ma C, Li J, Wang H and
Shao Z: ClC5 decreases the sensitivity of multiple myeloma cells to
bortezomib via promoting prosurvival autophagy. Oncol Res.
26:421–429. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li G, Chen T, Zhu Y, Xiao X, Bu J and
Huang Z: MiR-103 alleviates autophagy and apoptosis by regulating
SOX2 in LPS-injured PC12 cells and SCI rats. Iran J Basic Med Sci.
21:292–300. 2018.PubMed/NCBI
|
|
39
|
Jiang J, Li Z, Yu C, Chen M, Tian S and
Sun C: MiR-1181 inhibits stem cell-like phenotypes and suppresses
SOX2 and STAT3 in human pancreatic cancer. Cancer Lett.
356:962–970. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Khing TM, Choi WS, Kim DM, Po WW, Thein W,
Shin CY and Sohn UD: The effect of paclitaxel on apoptosis,
autophagy and mitotic catastrophe in AGS cells. Sci Rep.
11:234902021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhu Y, Huang S, Chen S, Chen J, Wang Z,
Wang Y and Zheng H: SOX2 promotes chemoresistance, cancer stem
cells properties, and epithelial-mesenchymal transition by
β-catenin and Beclin1/autophagy signaling in colorectal cancer.
Cell Death Dis. 12:4492021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kim BH, Oh HK, Kim DW, Kang SB, Choi Y and
Shin E: Clinical implications of cancer stem cell markers and ABC
transporters as a predictor of prognosis in colorectal cancer
patients. Anticancer Res. 40:4481–4489. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Toda T, Hsu JY, Linker SB, Hu L, Schafer
ST, Mertens J, Jacinto FV, Hetzer MW and Gage FH: Nup153 interacts
with Sox2 to enable bimodal gene regulation and maintenance of
neural progenitor cells. Cell Stem Cell. 21:618–634.e7. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wulff H, Castle NA and Pardo LA:
Voltage-gated potassium channels as therapeutic targets. Nat Rev
Drug Discov. 8:982–1001. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yamashita N, Hamada H, Tsuruo T and Ogata
E: Enhancement of voltage-gated Na+ channel current associated with
multidrug resistance in human leukemia cells. Cancer Res.
47:3736–3741. 1987.PubMed/NCBI
|
|
46
|
Catterall WA and Swanson TM: Structural
basis for pharmacology of voltage-gated sodium and calcium
channels. Mol Pharmacol. 88:141–150. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Weinstein IB: Cancer. Addiction to
oncogenes-the Achilles heal of cancer. Science. 297:63–64. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yan W, Zhang W and Jiang T: Oncogene
addiction in gliomas: Implications for molecular targeted therapy.
J Exp Clin Cancer Res. 30:582011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Nagel R, Semenova EA and Berns A: Drugging
the addict: Non-oncogene addiction as a target for cancer therapy.
EMBO Rep. 17:1516–1531. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sosa Iglesias V, Giuranno L, Dubois LJ,
Theys J and Vooijs M: Drug resistance in non-small cell lung
cancer: A potential for NOTCH targeting? Front Oncol. 8:2672018.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gorre ME, Mohammed M, Ellwood K, Hsu N,
Paquette R, Rao PN and Sawyers CL: Clinical resistance to STI-571
cancer therapy caused by BCR-ABL gene mutation or amplification.
Science. 293:876–880. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Pikor LA, Ramnarine VR, Lam S and Lam WL:
Genetic alterations defining NSCLC subtypes and their therapeutic
implications. Lung Cancer. 82:179–189. 2013. View Article : Google Scholar : PubMed/NCBI
|