Introduction
Metformin is a biguanide developed from galegine, a
guanidine derivative; it is a hydrophilic base, present as a
cationic species at physiological pH. Metformin is absorbed mainly
in the small intestine, and then taken up by the liver to play an
antihyperglycemic role. Metformin is excreted unchanged in the
urine (1). As the primary drug for
treating type 2 diabetes, it controls blood glucose by reducing
glycogen decomposition, inhibiting gluconeogenesis, inhibiting
intestinal glucose absorption and increasing insulin sensitivity in
the surrounding tissues (2,3). The
molecular target of metformin is presenilin enhancer protein 2
(PEN2). Metformin binds PEN2 and initiates a signaling pathway that
interacts with the glucose-sensing pathway via V-type proton ATPase
subunit S1 to activate the lysosomal 5′-adenosine
monophosphate-activated protein kinase (AMPK) pathway, which is an
indispensable mechanism that enables metformin to inhibit hepatic
gluconeogenesis and improve insulin sensitivity (4,5).
Recent studies have found that metformin can inhibit the occurrence
and development of lung cancer in addition to its hypoglycemic
effect (Fig. 1).
An extensive cohort study published in 2009
discovered lower cancer morbidity rates (6), while another study discovered lower
mortality rates (7), including
those for lung cancer, in patients with type 2 diabetes treated
with metformin compared with patients who had never used metformin.
Cancer was diagnosed in 7.3% of 4,085 metformin users compared with
11.6% of 4,085 comparators (6). In
patients taking metformin compared with patients not taking
metformin at baseline, the adjusted hazard ratio (HR) for cancer
mortality was 0.43 (95% CI, 0.23-0.80) (7). Several studies have indicated that
metformin inhibits tumor growth (6,8–10). A
meta-analysis found that metformin increased the survival time of
patients with lung cancer plus type 2 diabetes, suggesting that
metformin may improve the prognosis of these patients (11). A study using Surveillance,
Epidemiology, and End Results public data included 750 mergers of
patients with type 2 diabetes plus stage IV non-small cell lung
cancer (NSCLC), with 61% patients on metformin. After controlling
for social-demographic characteristics, the types of lung cancer
and treatment, the results showed that patients with advanced lung
cancer undergoing metformin treatment had higher survival rates
compared with patients not treated with metformin (12). Therefore, certain researchers began
to study metformin in lung cancer treatment and its mechanism,
using animal tests and clinical studies. Studies have shown that
metformin has cytotoxic effects on human lung cancer cell lines
(including squamous cell carcinoma (13), adenocarcinoma (14), large cell carcinoma (15), small cell carcinoma and
non-transformed cell lines (16).
Tan et al (17) conducted
clinical trials to compare the efficacy of metformin with insulin
and other hypoglycemic drugs in the treatment of NSCLC, and
discovered that patients treated with metformin had longer overall
survival (OS) (P=0.007) and progression-free survival (PFS)
(P=0.002) times. In a meta-analysis of 14 randomized controlled
trials (RCTs), Stevens et al (18) discovered no significant effect of
metformin on cancer mortality. Therefore, the effect of metformin
on the treatment and prognosis of lung cancer remains
controversial, requiring confirmation from further studies.
Mechanisms of metformin in the treatment of
lung cancer
Antitumor effects through liver kinase
B1 (LKB1)-dependent AMPK kinase pathways
Metformin can produce antitumor effects through the
LKB1-AMPK kinase pathway. LKB1 is a tumor suppressor gene; its
encoding product, LKB1 protein, is a serine/threonine kinase that
can regulate various cell physiological and pathological processes.
Somatic LKB1 gene mutations exist in numerous malignant tumors,
including lung cancer, colon cancer, breast cancer and
Peutz-Jeghers syndrome (a cancer susceptibility disease) (19,20).
Gene mutations in the somatic LKB1-AMPK pathway increase the risk
of precancerous lesions (21). In
NSCLC, 13% of adenocarcinomas and 5% of squamous cell carcinomas
have LKB1 mutations. However, the LKB1-AMPK pathway can still be
activated by metformin and inhibit tumor growth (22). These results suggest that metformin
may have other mechanisms to inhibit tumor genesis and development.
Mammalian target of rapamycin (mTOR), the downstream target of the
LKB1-AMPK pathway, is an important target of metformin in tumor
inhibition. mTOR is the catalytic subunit of two multi-protein
complexes, mTOR complex 1 (mTORC1) and mTORC2, regulating cell
growth and integrating input signals from various hormonal and
energy-sensing pathways (23).
These signals include insulin and insulin-like growth factor 1
(IGF-1), IGF-2 and AMPK (24).
AMPK activates tumor suppressor gene binding sclerosis complex 1
(TSC1) and TSC2/mTORC1 to form mTOR inhibitory complex, leading to
mTORC1 downregulation. AMPK can also directly inhibit the positive
regulator of mTOR, namely regulatory-associated protein of mTOR,
leading to its downregulation (25). Metformin can also inhibit the IGF-1
insulin signaling pathway through the AMPK-dependent insulin
receptor substrate 1 (IRS-1) phosphorylation pathway, inhibiting
the phosphatidylinositol 3-kinase (PI3K)/AKT pathway and
downregulating the mTOR signaling pathway to interfere with protein
synthesis, affecting tumor cell proliferation (26). As the target molecule of
PI3K/AKT/mTOR signaling, mTOR contributes to the control of protein
synthesis. There is a positive correlation between protein
synthesis rates and proliferation rates. In turn, the production of
mitochondrial ATP is needed to fuel protein synthesis and
proliferation. The production of mitochondrial energy, protein
synthesis and proliferation are co-regulated processes, and mTORC1
stimulates the synthesis of numerous nuclear-encoded mitochondrial
regulators, such as TFAM, mitochondrial ribosomal proteins and
complex I and V components via the upregulated translation of
corresponding mRNAs (27). mTOR
plays a major part in coupling mitochondrial functions and
translation. As well as regulating nuclear-encoded mitochondrial
regulator synthesis, mTOR regulates the translation of mRNAs
encoding proteins that promote proliferation, including cyclins,
ornithine decarboxylase (ODC) and Myc. Through the aforementioned
mechanisms, the synthesis of cyclins, ODC and Myc, which can
promote tumor cell proliferation, are inhibited by the
downregulation of PI3K/AKT/mTOR signaling (28). Therefore, tumor cell proliferation
is inhibited.
Metformin in the treatment of type 2 diabetes can
improve type 1 diabetes insulin resistance and the inflammatory
response through the p53/RAP2A pathway, and regulate the p53/RAP2A
pathway to improve insulin resistance (29). Similarly, AMPK-dependent p53
activation has been associated with the antitumor effects of
metformin. p53, a tumor suppressor protein and one of the
downstream targets of AMPK, achieves its antitumor function by
increasing transcriptional expression of proteins involved in DNA
repair, apoptosis and the prevention of cell proliferation,
alteration and senescence. When DNA suffers oxidative damage,
intracellular damage recognition signals activate p53 and its
transcriptional targets, protecting genomic integrity and
regulating cell metabolism and the cell cycle. p53 inhibits tumors
by activating multiple genes that inhibit the AKT and mTORC1
pathways; it can be phosphorylated by serine and activated by AMPK
(30–33). According to a previous study,
metformin-treated p53 mutant cells had significantly higher
apoptosis rates than wild-type colon cancer cells (34). However, another study has shown
that metformin can enhance the efficacy of radiotherapy,
independent of AMPK and p53 status (35). Metformin can inhibit mTOR and slow
cell cycle progression through regulated in DNA damage and
development 1 independently of AMPK (36). In addition, metformin selectively
inhibits tumor growth and triggers apoptosis in p53-deficient
HCT116 ×enografts (34).
Therefore, metformin can act through AMPK or p53, but the specific
pathway is complex. In addition, AMPK can induce endoribonuclease
DICER (DICER) expression, an enzyme involved in microRNA
(miRNA/miR) synthesis, whose change and loss of function can lead
to complex tumor syndrome, suggesting that metformin-induced DICER
expression may be one of the antitumor mechanisms (37). DICER belongs to the double-stranded
RNA-specific endonuclease family, which are able to convert the
miRNA precursor forms into their mature forms through a stepwise
process. The methylation levels of DICER are significantly higher
in patients with lung cancer. Methylation analysis of the first
region of the DICER can distinguish patients with NSCLC from
healthy individuals (38). In one
study, DICER was regulated by MDA-7/interleukin (IL)-24 through the
downregulation of microphthalmia-associated transcription factor in
lung cancer cells (39). Dicer
expression of NSCLC was significantly increased in stage II tumors
compared with that in stage I tumors, and in stage III tumors
compared with that in stage II and I tumors (40). Dicer contributes to the resistance
to gefitinib in lung cancer (41),
and can promote autophagy and cisplatin resistance in NSCLC by
downregulating let-7i-5p, as well as inhibiting the activation of
the PI3K/AKT/mTOR pathway (42).
Activated AMPK can also trigger UNC-51-like kinase 1 (a
serine/threonine kinase that is an autophagy promoter in mammals),
inducing apoptosis, cell cycle arrest and autophagy to exert
antitumor effects (43). Metformin
also inhibits proto-oncogene c-Myc and hypoxia-inducible factor 1α
(HIF-1α) via AMPK (44). HIF-1α is
a transcription factor that promotes the expression of glycolysis
enzyme glucose transporter 1 (GLUT1) and monocarboxylic acid
transporter 4, both of which play a key role in the metabolic
transformation of cancer. There is no expression of HIF-1α in human
normal lung tissues, while HIF-1α is highly expressed in lung
cancer tissues. HIF-1α is mainly expressed in the nucleus and
cytoplasm of lung cancer cells, presenting with obvious
heterogeneity (45). In a previous
study, the expression of HIF-1α around the tumor necrosis area and
the infiltrating edge of the tumor was significantly increased, and
the expression intensity in SCLC with a high degree of malignancy,
strong invasion and early metastasis was significantly higher than
that of squamous cell carcinoma and adenocarcinoma (46). The expression intensity of HIF-1α
was also closely associated with the degree of differentiation and
postoperative survival of lung cancer cells. HIF-1α promotes the
increase in vascular endothelial growth factor (VEGF) production,
angiogenesis and permeability, provides more oxygen and nutrients
for tumor cells, and ensures the proliferation of tumor cells
(47). On the other hand, HIF-1α
can also induce anti-apoptotic factors to make cells resist
apoptosis, or increase the transcription of other enzymes
associated with glycolysis and glycogen generation, in order to
increase the proliferation activity of lung cancer cells, enable
invasion and metastasis, and shorten the survival period of
affected patients (48). These
studies indicate that the AMPK signaling pathway is a promising new
target for tumor therapy. However, another study has found that
AMPK may lose its regulatory role in cancer cells due to
mutations/deletions of its upstream regulatory kinase Lkb1/Stk11 or
ubiquitination of the MAGE-A3/6-TRIM28 E3 ligase complex. This
results in autophagy inhibition, mTOR signaling pathway activation
and metformin hypersensitivity (49). Therefore, further animal
experiments and clinical studies are required to explore the role
of this pathway in the antitumor effect of metformin.
Downregulating the
GRB/IRS-1/PI3K/AKT/mTOR pathway
IGFs are multifunctional cell proliferation
regulators that promote cell differentiation, proliferation and
individual growth and development. IGFs include IGF-1 and IGF-2.
Type 1 IGF receptor (IGF-1R) belongs to the receptor tyrosine
kinases (RTKs) family. It can be activated by IGF-1 or IGF-2,
causing phosphorylation of its tyrosine kinase domain and
initiating intracellular signal transduction, thereby regulating
cell growth and differentiation, development, senescence and other
life activities (50). Studies
have revealed that IGF-1R can regulate blood glucose and stimulate
the growth of NSCLC cell lines, promoting carcinogenic
transformation, growth and survival of tumor cells (51,52).
In addition, IGF can activate the Ras/Raf/ERK signaling pathway
through growth factor receptor-bound protein 2 and promote tumor
cell proliferation (53).
Downstream activation of IGF-1R upregulates the PI3K/AKT/mTOR
pathway and the mitogen-activated protein kinase (MAPK)/ERK pathway
(also known as the KRAS-Raf-MEK-ERK pathway) to enhance cell
proliferation. IGFs are associated with the occurrence, development
and metastasis of tumors. Metformin can downregulate IGF-1 by
inhibiting the PI3K/AKT and MEK/ERK signaling pathways, regulating
lung cancer cell metabolism and inhibiting cell proliferation
(54,55). In NSCLC, activating the
PI3K/AKT/mTOR signaling pathway leads to more aggressive lung
cancer and a worse prognosis, especially in squamous cell lung
cancer. Metformin can block the IGF-1-insulin signaling pathway by
phosphorylating IRS-1, inhibiting the IRS-1/PI3K/AKT and
PI3K/AKT/mTOR signaling pathways, thereby preventing mTOR
activation and inhibiting NSCLC (55). Other studies have demonstrated that
abnormal activation of the PI3K/AKT/mTOR signaling pathway is one
of the mechanisms of acquired imported epidermal growth factor
receptor (EGFR)-tyrosine kinase inhibitor (TKI) resistance in
patients with adenocarcinoma and EGFR activation mutation (56). Inhibiting the PI3K/AKT/mTOR
signaling pathway may also overcome radiotherapy and chemotherapy
resistance, as well as immune escape in NSCLC (57). Animal experiments have indicated
that nicotine derivatives can reduce the number of tumor-related
regulatory T cells (Tregs) by inhibiting mTOR in tumor cells
(58) and creating a favorable
environment for the occurrence and development of nicotine-induced
lung tumors (59). Rapamycin
activation by metformin can prevent the occurrence of lung tumors.
Metformin mildly inhibits the mTOR pathway in tumors, which can
reduce the occurrence of lung tumors by 40–50%. One study
discovered that mTOR inhibition in lung tissue is associated with
lower circulating IGF-1 and insulin levels rather than lower AMPK
(60).
A variety of anti-IGF-1R monoclonal antibody drugs
have been developed. A study has disclosed that metformin alone or
combined with figitumumab (anti-IGF-1R monoclonal antibody) can
achieve antitumor effects on NSCLC by inhibiting the PI3K/AKT and
MEK/ERK signaling pathways, and downregulating the IGF-1R signaling
pathway. It has been suggested that metformin combined with
figitumumab may have a good therapeutic value in treating NSCLC
(54).
Inhibiting mTORC1 to regulate glucose
and amino acid concentration
Metformin can inhibit the mTORC1 signal by
inhibiting and regulating the activity of the Rag GTPases complex.
A number of environmental signaling factors, including nutrition
and growth factors, activate the mTORC1 pathway to regulate the
growth of organisms (61).
Cell-based studies showed that mTORC1 senses amino acids through
the RagA-D family of GTPases (also known as RRAGA, B, C and D)
(62). However, their importance
in mammalian physiology is unclear. In animal experiments, mice
expressed the active form of RagA (RagAGTP) by inserting
endogenous promoters through a gene knock-in method. Fasted
RagAGTP/GTP newborn mice could not trigger autophagy and
produce amino acids used to convert glucose, resulting in an
imbalance in glucose homeostasis. However, severe hypoglycemia did
not inhibit mTORC1 in RagAGTP/GTP newborn mice (62). We hypothesize that the Rag pathway
may signal the availability of glucose and amino acids to mTORC1,
inhibiting mTORC1 (63). In
RagAGTP/GTP fibroblasts, mTORC1 is resistant to glucose
deprivation, and glucose, like amino acids, controls its
recruitment on the lysosomal surface, where mTORC1 is activated
(64). Therefore, Rag GTPases
transmit glucose and amino acid concentration signals to mTORC1,
playing a key role in autophagy induction, nutritional homeostasis
and the survival ability of newborn mice (62).
Metformin directly affects glucose metabolism and
inhibits tumor growth. Glucose metabolism in lung cancer mainly
includes the glycolysis, aerobic oxidation and pentose phosphate
pathways. The glycolysis pathway produces less energy (ATP) per
mole of glucose than the aerobic oxidation pathway, but glycolysis
pathway can provide energy more quickly. Under aerobic conditions,
tumor cells also preferentially utilize glucose glycolysis capacity
as their primary energy source (Warburg effect) (55). In NSCLC, adenocarcinoma uses
glycolysis for energy under normal oxygen conditions. Squamous cell
carcinoma is more likely to have a high rate of anaerobic
glycolysis due to hypoxia in the tumor microenvironment, slow
diffusion and metastasis. Metformin can promote glycolysis by
changing the activity of certain glucose metabolism enzymes
(including fructose-2,6-biphosphatase) and can promote the
conversion of glucose metabolism to glycolysis in NSCLC cells
(65). This may stimulate the
growth of the NSCLC cells. However, due to less energy per unit
provided by glycolysis, reduced ATP production leads to increased
AMP levels, which results in an increased intracellular ratio of
AMP to ATP and an imbalance of energy metabolism, thereby realizing
the antitumor effect of metformin (66).
A study has revealed that squamous cell carcinoma
has a higher uptake rate of 18F-fluorodeoxyglucose (FDG) than
adenocarcinoma, but adenocarcinoma has a higher metastasis
potential and poorer disease-free survival time (67). Another phase II study in advanced
NSCLC cancer randomized patients to receive metformin (1,000 mg
twice daily) combined with platinum-containing chemotherapy in a
controlled diet with or without metformin. It discovered that the
uptake of 18F-FDG on baseline positron emission tomography (PET)
images was significantly higher in squamous cell carcinoma than
that in non-squamous NSCLC. Metformin significantly reduced the
risk of tumor progression and death in lung squamous cell carcinoma
with high uptake of 18F-FDG, indicating that the antitumor effect
of metformin is highly dependent on glucose metabolism (68). In a recent single-blind phase II
trial (69), metformin was used to
treat inoperable early-stage NSCLC. PET scans were performed at the
beginning of treatment, in the middle of treatment (after 2 weeks
of metformin or placebo administration) and after 6 months. The
results revealed that most metformin-treated subjects had PET
Response Criteria in Solid Tumors (PERCIST) (70) metabolic responses on PET imaging,
leading to increased glucose metabolic activity in most tumors,
demonstrating that the effect of metformin on tumor growth may be
influenced by glucose metabolism in the tumor environment. In
addition, insulin is a growth hormone that promotes division, while
metformin can directly or indirectly inhibit tumor growth by
reducing serum insulin levels and improving glucose metabolism in
hyperinsulinemia (71,72).
Inhibiting complex I of the
mitochondrial respiratory chain
Studies have found that metformin can cross the
plasma (1,55) and mitochondrial (73) membranes to affect tumor metabolism
by reaching the mitochondria. Metformin gets positively charged at
physiological pH, and organic cation transporters mediate the
movement of metformin across the cell membrane in NSCLC. Metformin
targets mitochondrial complex I, inhibits mitochondrial complex I
and attenuates the oxidation of nicotinamide adenine dinucleotide,
reducing the proton gradient across the mitochondrial intima and
proton-driven ATP synthesis. Metformin directly inhibits adenosine
deaminase to increase AMP, leading to an increase in the ratio of
AMP to ATP in cells, catalyzing the conversion of ATP to cyclic AMP
(cAMP), resulting in imbalanced cell energy metabolism and
inhibiting tumor cell growth (74). Therefore, metformin increases the
cAMP level, which activates 5′-AMPK and its downstream signaling
pathway, inhibiting tumor growth and proliferation (55). Simultaneously, metformin reduces
reactive oxygen species production, oxidative stress and DNA damage
by inhibiting mitochondrial complex I, thus reducing the risk of
mutation (75). In addition,
interactions between biguanides and mitochondrial copper ions are
critical for metformin metabolism, with copper chelators inhibiting
metformin-activated 5′-AMPK-dependent signaling and S6 protein
phosphorylation (76). Extensive
P-electron delocalization can stabilize the binding of the
biguanides to mitochondrial copper, enabling the biguanides to
regulate AMPK, glucose production, gluconeogenesis gene expression
and mitochondrial respiration (77).
Regulating lung miRNA
An animal study revealed that metformin regulated 42
out of 1,281 pulmonary miRNAs in smoke-free mice through multiple
mechanisms, including AMPK, stress response, inflammation, NF-κB,
Tlr9, TGF, p53, cell cycle, apoptosis and antioxidant pathways,
Ras, Myc, Dicer, angiogenesis, stem cell recruitment and
angiogenesis. In smoke-exposed mice, metformin significantly
reduced DNA adduct levels and oxidative DNA damage, normalized the
expression of certain miRNAs, and thus protected mouse lungs from
smoke-induced changes in DNA and miRNA, thereby inhibiting pretumor
lesions of the lungs and kidneys (78). Among the miRNAs involved, miR-148b
and miR-30b are known miRNA families that can regulate AMPK
(79) and are associated with the
activation of AMPK by metformin. In addition, metformin regulates
the expression of a number of miRNAs involved in cell cycle
regulation, such as let-7f, miR-30b, miR-362, miR-376c, miR-466h,
miR-490 and miR-574. They are also important mediators of the
antitumor activity of metformin through the AMPK pathway (80). miR-137 targeting SLC22A18 has been
revealed to significantly inhibit the proliferation, invasion and
migration of NSCLC cells (81).
miR-7 regulates the occurrence and development of lung cancer
through PI3K regulatory subunit γ/AKT, Bcl-2, IGF-1R and other
signaling pathways. In NSCLC, metformin regulation of the AMPK
pathway is also associated with the upregulation of miR-7 (82,83).
Dong et al (84) also found
that metformin significantly upregulates miR-7 in a time and
dose-dependent manner through the AMPK pathway, and that the
upregulation of miR-7 reduces growth, migration and invasion of
NSCLC cells. Recently, Jin et al (85) found that metformin reduced the
growth, migration, invasion and epithelial-mesenchymal transition
(EMT) of NSCLC cells by regulating miR-381/yes-associated protein
(YAP) activity.
Affecting the tumor and its
microenvironment
Peripheral immune cells and angiogenesis are two
major components of the interaction of a tumor with its
microenvironment. Altering the tumor microenvironment can
significantly affect tumor growth and the therapeutic effect. A
study on tumor cell lines, including those for NSCLC, found that
metformin enhanced CD8+ T cell memory by altering fatty
acid metabolism, and promoted rejection of solid tumors in control
mice, but did not exert this effect on T cell-deficient mice
(86). Metformin treatment of
tumor tissue significantly increased the number and activity of
CD8+ tumor-infiltrating lymphocytes (TILs), and
protected them from apoptosis and exhaustion. Metformin-mediated
effects were significantly reduced when AMPK was knocked out
(87). Furthermore, metformin
treatment reduced the expression of Ki67 (a proliferation signal)
and caspase-3 (an apoptosis signal), but this effect was attenuated
when CD8+ T cells were deficient. These results
suggested that metformin reduces Ki67 and caspase-3 expression
through CD8+ TILs in the tumor microenvironment
(80). Similarly, when
CD4+ T cells were depleted in the tumor
microenvironment, the antitumor effect of metformin was
significantly weakened (88).
In addition, the antitumor effect of metformin is
closely associated with the adaptive immune response to the tumor
microenvironment. Studies on animals have confirmed that metformin
treatment can reduce lung cancer-associated Foxp3+ Tregs
by 65% and tumor-associated Tregs by 50% (88). Foxp3+ Tregs are
necessary for KRAS-mediated lung tumorigenesis in the tumor
microenvironment (58). In
addition, due to changes in blood supply and the energy imbalance
of tumor cells, the concentration of glucose and other metabolites
in the tumor microenvironment is low, resulting in an acidic
interstitium and low oxygen content (89), and a lack of energy supply for
locally infiltrating tumor T cells. Recent study has revealed that
metformin inhibits tumor cell oxidative metabolism and oxygen
levels in the tumor microenvironment. Metformin increases oxygen
supply to TILs, rescues T cells from an anoxic environment and
enhances their role, and may have potential for the immune
treatment of patients (90).
Downregulation of silent information
regulator T1 (SIRT1) can enhance the antitumor effect of cells
SIRT1 is involved in the development of a variety of
tumors through the deacetylation of histones and non-histones. A
study found that 62% of NSCLC tissues overexpressed SIRT1,
significantly reducing the OS rate of affected patients (91). In NSCLC cell lines with different
LKB1 expression states, metformin combined with SIRT1 inhibitor
Tenovin 6 could synergically inhibit SIRT1 expression in NSCLC
cells regardless of LKB1 status. Even in LKB1-deficient A549 cells,
the combination of metformin and Tenovin 6 significantly reduced
SIRT1 expression, increased the acetylation of p53 at lysine 382
and enhanced the stability of p53 (91). Metformin inhibited SIRT1 promoter
activity by upregulating the hypermethylation binding of
hypermethylated in cancer 1 protein on the SIRT1 promoter and
synergistically induced caspase 3-dependent apoptosis. The research
has confirmed that metformin combined with Tenovin 6 enhances the
antitumor effect by downregulating SIRT1 expression independently
of LKB1 (91).
In a previous study, the activation of protein
phosphatase 2 (PP2A) by metformin inhibited the growth, invasion
and activity of A549 and H1651 tumor cells and promoted apoptosis
(92). PP2A is a tumor suppressor
in a number of cancer types; it inhibits the carcinogenic activity
of AKT and Myc by catalyzing serine dephosphorylation. PP2A
inhibitor α4 is often overexpressed in cancer cells (92). Metformin activates PP2A by
preventing PP2A inhibitors (PP2A regulatory subunit α4 and E3
ubiquitin ligase midline 1) from interacting with their catalytic
subunits, resulting in increased BAX expression, decreased Myc
expression and AKT inactivation (92).
Inhibiting YAP expression in lung
cancer cells
YAP is a carcinogenic protein whose overexpression
and activation are associated with lung, liver, colon, ovarian and
breast cancer; it has been linked to a poor prognosis, metastasis
and progression of lung cancer due to its ability to promote cell
cycle progression and inhibit apoptosis (93). Jin et al (94) found that YAP mRNA and protein
levels in NSCLC tissues were higher than those in normal lung
tissues. Metformin treatment significantly reduced YAP mRNA and
protein levels and their downstream targets. Metformin was shown to
interfere with the binding of the transcription factor interferon
regulatory factor-1 to YAP promoter. Thus, YAP expression in lung
cancer cells was decreased. Inhibition of YAP promoter activity
reduced cell proliferation, migration, invasion and EMT, and
increased cell senescence and apoptosis. In mice with lung cancer,
250 mg/kg/day metformin reduced tumor volume, increased survival
rate and decreased YAP expression level in transplanted tumors
(94).
Metformin promotes survivin degradation, induces
apoptosis and inhibits NSCLC cell proliferation through the
AMPK-dependent protein kinase A (PKA)/glycogen synthase kinase 3β
(GSK-3β) pathway. Survivin is an anti-apoptotic protein that is
often overexpressed in malignant cells (95). Luo et al (95) found that metformin downregulated
survivin level, without changing its mRNA level, enhancing its
proteasome degradation by inhibiting PKA activity through
downstream GSK-3β activation. PKA activator (8-Br-camp and
forskolin) and GSK-3β inhibitor (LiCl and small interfering RNA)
can increase survivin activity and enhance lung cancer cell
proliferation (94).
Salani et al (96) demonstrated that microcystin 1 in
NSCLC can inhibit the effect of metformin on the IGF-1 pathway and
that microcystin is necessary for tumor inhibition by metformin.
The study also proposed that metformin can significantly upregulate
the transcription levels of intracellular matrix metalloproteinase
2 (MMP2) and MMP9, enhancing the migratory rate and invasive
ability of human lung adenocarcinoma A549 cells in vitro
(96).
Others
Ataxia telangiectasia mutated (ATM) encodes a tumor
suppressor protein, a key component of the DNA damage response
network system, and is required for DNA repair and cell cycle
control. As a cellular stressor, metformin participates in
ATM-mediated repair through AMPK-dependent and AMPK-independent
mechanisms and activates the cell repair process, which may have a
protective effect on the malignant transformation of cells
(97).
Metformin promotes apoptosis through the MAPK
signaling pathway and upregulation of GADD153. MAPKs,
serine/threonine proteases, regulate various cell physiological
processes and play an important role in apoptosis. Metformin can
induce lung cancer cell cycle arrest through the MAPK signal
transduction pathway, thus playing an anti-proliferative and
pro-apoptotic role in lung cancer cells (98).
In summary, the antitumor mechanisms of metformin
are not completely clear. However, certain pathways have been
established as aforementioned. One of these pathways acts to block
protein synthesis by inhibiting mTORC1. This effect can be achieved
through AMPK-dependent and AMPK-independent pathways. Anabolic
events in the plasma membrane, cytoplasm and mitochondria of tumor
cells are tightly regulated by the LKB1-AMPK pathway. AMPK is
indicated to regulate the PI3K/AKT/mTOR pathway, which stimulates
gene expression, cellular growth and survival. Activation of the
LKB1-AMPK pathway by metformin leads to downregulation of the
downstream target mTOR. Inhibition of mTORC1 blocks protein
synthesis. The inhibition of mTOR can also be achieved by
inhibiting IGFs and their downstream targets. In addition,
metformin can inhibit the mTORC1 signal by inhibiting and
regulating the activity of the Rag GTPases complex. This process is
mediated by regulating glucose and amino acid concentration.
Metformin also inhibits tumor growth by affecting tumor energy
metabolism; it suppresses tumor progression by inhibiting
glycolysis and the mitochondrial respiratory chain. The immune
microenvironment is critical for tumor growth. The effects of
metformin on the tumor microenvironment are closely associated with
the decrease in Foxp3+ Tregs and the increase in
CD8+ T cells. Metformin regulates pulmonary miRNAs
associated with DNA damage or the cell cycle. The mechanisms of
metformin in the treatment of lung cancer are summarized in
Fig. 2.
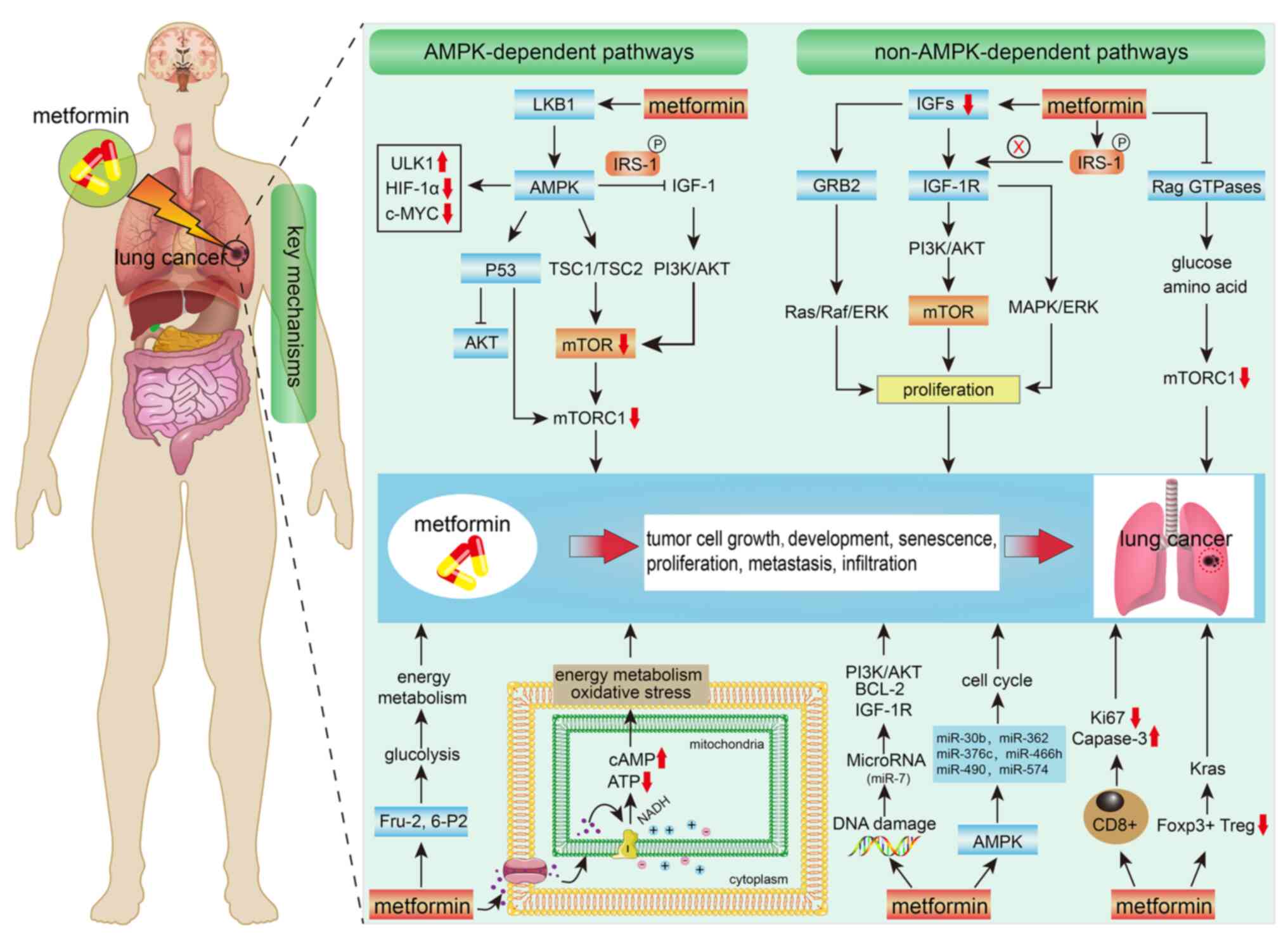 | Figure 2.Mechanisms of metformin in the
treatment of lung cancer. AMPK, adenosine monophosphate-activated
protein kinase; LKB1, liver kinase B1; ULK1, UNC-51-like kinase 1;
HIF-1α, hypoxia-inducible factor 1α; IRS-1, insulin receptor
substrate 1; IGF-1, insulin-like growth factor 1; TSC, tumor
suppressor gene binding sclerosis complex; mTOR, mammalian target
of rapamycin; mTORC1, mTOR complex 1; IGF-1R, type 1 IGF receptor;
GRB2, growth factor receptor-bound protein 2; IRS-1, insulin
receptor substrate 1; PI3K, phosphatidylinositol 3-kinase; AKT,
serine/threonine kinase; c-MYC, myelocytomatosis oncogene; RAS,
renin-angiotensin system; MAPK, mitogen-activated protein kinase;
Raf, v-raf-leukemia oncogene; ERK, extracellular signal-regulated
kinase; Bcl-2, B-cell lymphoma 2; miR, microRNA; KRAS, Kirsten rat
sarcoma viral oncogene homolog; Foxp3, forkhead box P3; ATP,
adenosine triphosphate; cAMP, adenosine 3′5′-cyclic monophosphate;
NADH, nicotinamide adenine dinucleotide phosphate; Treg, regulatory
T cell; Rag, recombination activation gene; GTPases, guanosine
triphosphatases. |
Application of metformin in lung cancer
treatment
Metformin and chemotherapy in lung
cancer
Chemotherapy is one of the main treatments for lung
cancer, but most patients will develop drug resistance as the
treatment progresses, resulting in tumor recurrence and
progression. Several metformin trials have proved that metformin
can increase chemotherapy sensitivity, reverse the resistance of
chemotherapy drugs and improve the therapeutic effect of tumor
chemotherapy.
Metformin increases the sensitivity of
chemotherapy in lung cancer
The interaction between metformin and chemotherapy
drugs has been studied using a mouse lung adenocarcinoma
transplanted tumor model. Metformin and doxorubicin were combined
to treat lung adenocarcinoma in mice. Compared with the doxorubicin
alone group, the metformin treatment group did not exhibit an
increased tumor recurrence rate, even in the lower doxorubicin dose
group, suggesting that metformin can increase the chemotherapy
sensitivity of doxorubicin to lung adenocarcinoma (99). Iliopoulos et al (99) used mice injected with A549 lung
cancer cells in the right flank as a research model to explore the
antitumor effect of metformin combined with chemotherapy drugs and
found that the tumor volume decreased more significantly in the
metformin + chemotherapy group than that in the chemotherapy group
alone, and no tumor recurrence was found in the metformin group.
However, tumor recurrence occurred in the group with chemotherapy
alone (99). Tseng et al
(52) found that metformin at 0.1
mmol/l combined with paclitaxel had a stronger cytotoxic effect on
lung cancer cell lines than paclitaxel chemotherapy alone,
suggesting that metformin could improve the therapeutic effect of
paclitaxel on lung cancer (52).
One RCT included 99 patients with stage IV NSCLC who received
platinum-based chemotherapy (without radiotherapy) from five
hospitals (including 19 patients whose lung cancer recurred after
treatment), although there was no statistical difference in OS time
between the groups. However, the metformin chemotherapy group
(n=39) was superior to the insulin (n=35) and other hypoglycemic
drug (n=25) groups in terms of PFS time (17). In addition, another study has
revealed that metformin can increase the antitumor effect of
cisplatin or etoposide on large cell lung cancer (15).
To date, two clinical trials of metformin combined
with chemotherapy in NSCLC have been conducted, including a phase
II trial in which all patients received metformin in combination
with chemotherapy [carboplatin (area under the curve (AUC)=5) +
pemetrexed (500 mg/m2) intravenously every 21 days for 4
cycles]. Subjects maintained pemetrexed treatment until disease
progression or until they could no longer tolerate treatment. Oral
metformin (500 mg), administered twice daily for 1 week, starting
from the 1st day of chemotherapy cycle 1 (C1D1), and increased by
500 mg/day at C1D8 and C1D15, eventually reaching 1,000 mg twice
daily, was continued as oral metformin treatment until disease
progression or intolerance (100). In another open-label phase II
trial, patients were randomized 3:1 to receive chemotherapy with or
without metformin. The chemotherapy regimen in this study was
carboplatin (AUC 6) + paclitaxel (200 mg/m2) +
bevacizumab (15 mg/kg), in an intravenous infusion every 21 days
for 1 day, for a total of 46 cycles (101). However, the two trials remain in
progress, and the results are expected to guide the therapeutic
dose and course of metformin in lung cancer, and evaluate the
adverse reactions and therapeutic effects of metformin combined
with chemotherapy. A pooled analysis of individualized data from
two phase II trials evaluated metformin in combination with
platinum-based chemotherapy with or without bevacizumab in
untreated non-diabetic patients with advanced NSCLC (102). A total of 33 patients were
included in the pooled analysis, and the combined median PFS and
combined median OS times for all patients were 6.0 and 14.8 months,
respectively. PFS and combined median OS were 6.6 and 13.3 months,
respectively, in patients with EGFR mutation, and 17.5 and 13.3
months, respectively, in patients with KRAS mutation. This study
confirmed the efficacy and tolerability of metformin combined with
chemotherapy, suggesting that KRAS or EGFR mutations may be key
molecules affecting the difference in efficacy of metformin
combined with standard chemotherapy (102).
An open-label randomized controlled study of
gemcitabine plus cisplatin + metformin in patients with stage IV
NSCLC showed no improvement in objective response rate (ORR) or OS
compared with gemcitabine plus cisplatin (P=0.109 and P=0.119)
(103). Another prospective study
of the pemetrexed + carboplatin + metformin regimen in the
treatment of advanced NSCLC also yielded negative results (100). In summary, most current studies
suggest that metformin can enhance the efficacy of chemotherapy
drugs for lung cancer. However, some clinical trials have shown no
further benefit, so more in vitro experiments and clinical
studies are needed to verify the efficacy of metformin combined
with chemotherapy for lung cancer. Metformin can reduce the
chemotherapeutic drug resistance in lung cancer. At present,
platinum-based chemotherapy is the first-line treatment for
advanced NSCLC, but drug resistance is inevitable at the late stage
of treatment. A study has found that cisplatin resistance is
related to signal transducer and activator of transcription 3
(STAT3) phosphorylation, reactive oxygen species (ROS) production
and IL-6 secretion, while metformin can inhibit cisplatin-induced
ROS generation, STAT3 phosphorylation and autocrine IL-6 secretion,
thus improving the chemical sensitivity of NSCLC to cisplatin
(104). STAT3 promotes tumor
proliferation, tumor cell survival and angiogenesis through
overexpression of anti-apoptotic proteins (Bcl-2-like protein 1 and
myeloid cell leukemia 1), cell cycle regulating proteins (cyclin D1
and c-Myc) and VEGF in NSCLC (105). The STAT3 signaling pathway
activates various cytokines and growth factors, which are critical
in tumor cell growth and apoptosis. Metformin improves the
cisplatin resistance of lung cancer cells by inhibiting STAT3
activity through the LKB1-AMPK pathway and mTOR pathway-dependent
mechanisms (106). Studies have
revealed that metformin can improve cisplatin cytotoxicity and
improve the cisplatin resistance of tumor cells (107,108).
Metformin and radiotherapy for lung
cancer
Radiotherapy is another important treatment for lung
cancer, especially for patients who lose the opportunity for
surgery at a later stage or cannot tolerate chemotherapy. However,
radiotherapy may cause radiation-related side effects, such as
insensitivity to bone marrow suppression pneumonia secondary to
lung infection. Koritzinsky suggested that radiosensitivity was
related to the efficiency of the insulin receptor, including the
repair of tumor cell DNA damage, cell redistribution in the cell
cycle, re-replication (tumor cell replication) and the
reoxygenation ability (the degree of hypoxia in the tumor)
(109). The study by Storozhuk
et al (30) confirmed that
metformin combined with radiotherapy could continuously activate
the ATM/AMPK/p53/p21cip1 signaling pathway and inhibit
the AKT/mTOR/eukaryotic initiation factor (eIF) 4E-binding protein
1 (4EBP1) signaling pathway, thus improving the sensitivity of
radiotherapy. EIF4E and 4EBP1 have been found to be overexpressed
in cancer tissues, such as lung cancer, breast cancer and
colorectal cancer (110). This
leads to a significant increase in the activity of the eIF4F
complex, which further promotes the translation initiation process
of various proteins, such as c-myc, cyclin D1, VEGF and ODC, and
induces tumor resistance to radiation. Inhibiting the 4EBP1 pathway
would enhance sensitivity to radiotherapy (111). DNA damage after irradiation
(mainly DNA double-strand breaks) can activate
serine/threonine-protein kinase Chk2 through ATM to block the cell
cycle for DNA repair, thereby activating AMPK and p53, and
p53-mediated apoptosis is one of the main mechanisms of cell death
after irradiation (112). ATM
inhibition has been revealed to enhance the sensitivity of
radiotherapy combined with cisplatin in NSCLC cell lines (112). Notably, a study found that
metformin can enhance the effect of radiotherapy in the absence of
AMPK, suggesting that there are other mechanisms to enhance the
sensitivity of radiotherapy (113). The interaction between the tumor
and its microenvironment (including immune cells) is also
considered to impact radiotherapy response significantly (114,115). Irradiation increases the number
of TILs, induces upregulation of programmed death ligand 1 (PD-L1)
on tumor cells and diversifies T-cell receptor libraries (116,117). In a retrospective analysis of 74
patients with NSCLC who received concurrent chemoradiotherapy,
CD8+ TIL density was associated with good survival
(118). The effect of irradiation
and metformin on TILs is one of the mechanisms of increasing
radiotherapy sensitivity. In addition, metformin has been shown to
be a good radiosensitization agent by inhibiting the G1
phase of the cell cycle, angiogenesis and the AMPK/AKT/mTOR/4EBP1
pathways in different NSCLC cell lines (30). Storozhuk et al (30) studied NCI-H1299, A549, SK-MES and
other lung cancer cells, and found that metformin combined with
radiotherapy could significantly reduce the tumor proliferation
capacity and survival coefficient of cells. In an A549 transplanted
mouse tumor model, the tumor inhibition in the metformin combined
with radiotherapy group was more prominent than that in the
radiotherapy group. Metformin activated the
ATM/AMPK/p53/p21cip1 pathway, inhibited the
AKT/mTOR/4EBP1 pathway, induced G1 phase cell cycle
arrest and enhanced apoptosis. Metformin or irradiation inhibited
xenograft growth, while the combination treatment enhanced it more
than each treatment used alone. Ionising radiation and metformin
induced sustained activation of the ATM/AMPK/p53/p21cip1
pathway and inhibition of the AKT/mTOR/4EBP1 pathway in the tumors,
reduced expression of angiogenesis and enhanced expression of
apoptotic markers (30). The
cytotoxicity of metformin in cancer stem cells, a rare cell pool,
could theoretically also partially improve the efficacy of
irradiation, but further studies are needed to confirm this
(119,120).
Metformin synchronous
chemoradiotherapy for lung cancer
Since 2013, three studies have involved metformin
combined with radiotherapy for NSCLC. In a retrospective
multicenter study of all patients with stage III NSCLC plus type 2
diabetes who received platinum-based chemotherapy and chest
irradiation (mean total dose, 66.1 Gy), metformin improved
radiotherapy response in reoxygenated tumors. The results suggested
that metformin could improve PFS during concurrent
chemoradiotherapy in diabetic patients with locally advanced
(LA)-NSCLC (121).
Simultaneously, metformin combined with concurrent radiotherapy and
chemotherapy for treating LA-NSCLC was studied, and it was found
that this type of treatment in LA-NSCLC could effectively improve
the short-term efficacy and prolong the survival time of patients,
without increasing the adverse reactions. Although metformin has
enhanced radiotherapeutic effects on NSCLC in vitro, these
effects have not been proven in the clinic. The combination of
metformin and radiotherapy in NSCLC treatment shows an antagonistic
effect. Therefore, the design of future clinical studies of
metformin and radiotherapy in NSCLC treatment should be
cautious.
Effect of metformin on targeted drug
therapy for lung cancer
Metformin combined with targeted drug therapy
exhibits a synergistic effect. Targeted drugs for lung cancer have
been widely used in clinical practice. The main medications used
are monoclonal antibodies and small-molecule TKIs. In human lung
squamous cell carcinoma cells, gefitinib downregulated the
expression of DNA mismatch repair protein MSH2 through the p38/MAPK
pathway, and enhanced the cytotoxic and growth inhibitory effects
of gefitinib on lung cancer cells (122). Retrospective analysis showed that
metformin and EGFR-TKIs have a synergistic therapeutic effect on
patients with NSCLC plus type 2 diabetes with EGFR mutation
(123,124). In addition, metformin combined
with EGFR-TKIs has been reported to significantly improve the
clinical efficacy in patients with NSCLC plus type 2 diabetes
(125). In a study targeting the
treatment of LKB1 wild-type NSCLC cells, the addition of gefitinib
to metformin inhibited EGFR phosphorylation and its downstream
signaling. Increased c-Raf/B-Raf isomerization induced MAPK
activation, thereby inducing significant apoptosis in vitro
and in vivo, which suggests a synergic effect of metformin
combined with EGFR-TKIs on LKB1 wild-type NSCLC cells (126).
The main studies on metformin combined with TKIs in
lung cancer treatment include the trial NCT03071705 (127), which evaluated the efficacy and
safety of various TKIs (erlitinib, afatinib or gefitinib) ±
metformin as a second-line treatment for diabetic patients with
advanced NSCLC and EGFR mutation. The results have been published.
The median PFS time was significantly longer in the EGFR-TKI plus
metformin group (13.1 months; 95% CI, 9.8-16.3) compared with the
EGFR-TKI group (9.9 months; 95% CI, 7.5-12.2) (hazard ratio, 0.60;
95% CI, 0.40-0.94; P=0.03). The median OS time was also
significantly longer for patients receiving the combination therapy
(31.7 months; 95% CI, 20.5-42.8; vs. 17.5 months; 95% CI,
11.4-23.7; P=0.02) (127). Two
ongoing studies to determine whether metformin and EGFR-TKIs have a
synergistic effect in patients with non-diabetic lung cancer are
the CGMT (NCT01864681) and METLUNG (NCT05445791) trials. The CGMT
trial consists of >200 patients in a multicenter, phase II,
randomized, double-blind, placebo-controlled study and aims to
assess the safety and efficacy of treatment with metformin as
first-line therapy for stage IIIb-IV NSCLC with EGFR mutation; the
main purpose of this experiment is a comparative study of the
1-year PFS rate. The secondary objective of this trial was to
compare the 2-year OS rate, the 2-year PFS rate, the ORR and the
DCR between the two treatments and evaluate their relative
therapeutic safety. The METLUNG trial was designed to evaluate the
efficacy and safety of metformin + erlotinib as a treatment for
patients with EGFR mutant-type stage IIIB-IV NSCLC.
Metformin combined with targeted drugs can overcome
the resistance to targeted drugs. Applying targeted drugs in lung
cancer has significantly improved the prognosis of patients with
lung cancer, but almost all targeted drugs will cause resistance in
the treatment process and affect the therapeutic effect. TKI drug
resistance is a common and intractable problem in the clinical
treatment of lung cancer, resulting in poor treatment effects and
shortened survival times for patients. There are multiple
mechanisms of drug resistance, which can be divided into primary
resistance and acquired resistance. Currently, the identified
acquired drug resistance of first-generation EGFR-TKIs is mainly
caused by the mutation of EGFR-T790M and the gene amplification of
c-Met, which account for ~50 and 20% of cases, respectively
(128). Other possible drug
resistance mechanisms include the occurrence of phenotypic EMT of
tumor cells, the interaction between IGF-1R and the EGFR receptor
signaling pathway, and the activation of P13K/AKT/mTOR signaling by
the loss of the PTEN gene (129).
Metformin can reverse EGFR-TKI resistance by
inhibiting the PI3K/AKT/mTOR signaling pathway (56). Li et al (130) reported that metformin combined
with EGFR TKI blockers (gefitinib or erlotinib) in vivo and
in vitro inhibited the IL-6/STAT3 signaling pathway,
reversed EMT and overcame drug resistance in NSCLC cells. In a
study of lung cancer cell lines with KRAS/LKB1 mutation, EGFR-TKIs
induced apoptosis and drug resistance through the PI3K/AKT/mTOR
signaling pathway. The addition of metformin and mTOR inhibitor
MLN0128 induced a significant therapeutic response. The
adenocarcinoma cells showed a higher therapeutic response than
squamous cell carcinoma cells. Furthermore, the addition of an AKT
inhibitor (MK2206) in squamous cell lung cancer cells also reversed
the drug resistance of KRAS/LKB1 mutant cell lines and led to
growth inhibition of lung squamous cell tumors (131).
Similarly, in combination with MEK inhibitors,
metformin has shown anti-proliferative/pro-apoptotic effects and
reduced EMT in LKB1 wild-type human NSCLC cell lines independent of
KRAS mutation status (132). In
addition, metformin can overcome IL-6-induced EGFR-TKI resistance
in lung cancer cells by inhibiting STAT3 and AKT phosphorylation,
and by enhancing AMPK activation (133). Pan et al (134) investigated whether metformin
sensitized primary resistant NSCLC cells to gefitinib and found
that primary resistance was more dependent on the IGF-1R pathway
than acquired resistance. The IGF-1R pathway is more highly
activated in primary EGFR-TKI resistant cells than in EGFR-TKI
sensitive cells or those with acquired resistance. Compared with
gefitinib alone, combined metformin treatment can lead to growth
inhibition, IGF-1R signaling pathway inhibition and increased
apoptosis via the inhibition of AKT and the upregulation of
Bcl2-like protein 11, resulting in increased sensitivity of primary
drug-resistant cells to gefitinib (134). A study has shown that metformin
can restore the sensitivity of drug-resistant NSCLC cells to the
anaplastic lymphoma kinase (ALK) inhibitor crizotinib by inhibiting
the IGF-1R pathway (135).
However, a study comparing metformin alone with metformin in
combination treatment with crizotinib in a xenograft mouse model of
ALK-positive lung cancer found that metformin alone (100 mg/kg per
day for 14 days) had a statistically significant effect on tumor
growth inhibition. When combined with metformin, treatment with
crizotinib (25 mg/kg) did not produce a stronger tumor-suppressive
effect than crizotinib alone (136).
Furthermore, increased expression of hepatocyte
growth factor (HGF) and its RTK c-Met has been observed in certain
ALK-positive NSCLC tumor tissues, associated with acquired
resistance to various TKIs (137,138). Alectinib, a second-generation ALK
inhibitor, has become an important drug in the first-line treatment
of advanced ALK-positive NSCLC. It was found that HGF level in the
supernatant of ALK-positive cell lines increased over time. Neither
exogenous nor endogenous HGF showed resistance to crizotinib, an
ALK/MET dual-targeted small molecule inhibitor, but it was an
important cause of alectinib resistance (137). GRB2-associated binding protein 1
(Gab1) is a key effector of the HGF/MET signal transduction pathway
mediating alectinib resistance. Metformin combined with alectinib
overcomes HGF by destroying the complex between MET and Gab1,
inhibiting Gab1 phosphorylation and activating downstream signal
transduction pathways, suggesting that metformin combined with
alectinib may help overcome the alectinib resistance caused by the
activation of the HGF/MET signaling pathway and improve the
efficacy of alectinib (137).
Third-generation EGFR-TKIs, including lochitinib,
have been used to treat patients with T790M mutations selectively,
but resistance to third-generation EGFR-TKIs can still emerge
during the treatment. Pan et al (139) investigated the effect of
metformin on rociletinib sensitivity in drug-resistant NSCLC cells.
The drug-resistant cells showed higher expression of p50/p65
heterodimer, phosphorylated (p)-AKT, IKK and IKBα, as well as
higher phosphorylation levels of IKBα and IKK, compared with the
parental control cells. Drug-resistant cells mediated NF-κB
activation through the PI3K/AKT pathway leading to increased p-AKT
level. Adding NF-κB inhibitor TPCA-1 to the rociletinib treatment
decreased cell viability, increased proliferation inhibition and
apoptosis, and significantly reduced p-AKT, p50/p65, p-IKK and
p-IKBα levels. These results suggested that inhibition of NF-κB may
sensitize the drug-resistant cells to rociletinib. A combination of
metformin and rociletinib had a similar effect. Metformin inhibited
NF-κB activity, resulting in increased sensitivity to rociletinib,
decreased p-AKT, p-IKBα, p-IKK, p50 and p65 levels, and reduced
nuclear translocation of p50/p65. Compared with treatment alone,
combination therapy significantly reduced the proliferation,
viability and invasion of NSCLC cells. Therefore, metformin and
rociletinib synergistically inhibited the NF-κB signaling pathway
and overcame EGFR-TKI resistance in T790M mutant NSCLC cells
(139). These findings suggest
that metformin may delay the emergence of EGFR-TKI resistance in
patients with NSCLC.
Metformin and immunotherapy of lung
cancer
Overexpression of PD-L1 often occurs in NSCLC,
resulting in a poor prognosis for patients with lung cancer
(140,141). Immunotherapy is currently
approved as the standard first- and second-line treatment for
advanced NSCLC and has achieved marked results in the treatment of
NSCLC. Animal experiments found that LKB1 and PD-L1 expression in
NSCLC tissues were significantly correlated (142). Downregulated LKB1 reduced the
PD-L1 level in TC-1 cells cells, while overexpressed LKB1 increased
the PD-L1 level in A549 cells, further confirming that AMPK
mediates PD-L1 upregulation through LKB1. The inhibition of AMPK
significantly reduced PD-L1 levels in NSCLC cells with intact LKB1.
The combination of metformin and anti-programmed cell death protein
1 (PD-1) antibody effectively inhibited the growth of tumors
expressing LKB1. LKB1 upregulated the expression of PD-L1 in NSCLC
by activating the AMPK and KEAP1/NRF2 signaling pathways, and
improved the therapeutic effect of PD-1 inhibitors on LKB1
wild-type NSCLC (142). In
addition, to study the association between AMPK activation and NK
cells in PD-1 therapy, metformin was used as an AMPK activator to
induce AMPK activation. The results showed that metformin-induced
AMPK activation combined with NK cell-mediated killing of tumor
cells could significantly inhibit tumor growth in mice (143). Studies showed that NSCLC with
KRAS mutation was more sensitive to PD-1/PD-L1 inhibitor therapy
(144–147), and LKB1 loss was detected in
one-third of KRAS-mutant NSCLC (148). However, NSCLC with this mutant
subtype was more aggressive and resistant to immunotherapy
(149,150). Another study showed that the
efficacy of PD-1 inhibitors was reduced in patients with lung
adenocarcinoma with LKB1 mutation, but not in patients with KRAS
mutation (151). In addition,
patients with lung adenocarcinoma without concurrent LKB1 or EGFR
mutations and TP53 mutations had prolonged PFS times when treated
with anti-PD-1 inhibitors, suggesting that LKB1 plays a key role in
NSCLC response to PD-1/PD-L1 inhibitors (149). Afzal et al (152) studied the clinical efficacy of
metformin in combination with immune checkpoint inhibitors (ICIs)
in patients with NSCLC. A total of 50 patients with NSCLC received
ICIs plus metformin or no metformin. The results revealed that the
total response rate, DCR, median OS and PFS times were higher in
the combined metformin group than those in the non-combined
metformin group, and the same results were obtained in the subgroup
analysis (second-line/third-line ICIs) (152). These results suggest that the
prognosis of patients with NSCLC is better for those who receive
both metformin and ICI therapy. A study reported a unique case of a
patient with SCLC who received nivolumab monotherapy for 2 years
until disease progression, and then metformin plus nivolumab, which
resulted in a sustained partial response for >6 months. These
results suggest that metformin may help overcome the acquired
resistance to PD-1 inhibitors (153). However, further clinical studies
are required to confirm this. At present, studies have indicated
that metformin has anticancer effects, and numerous clinical
studies have demonstrated that metformin significantly improves
anticancer activity in patients with NSCLC. The therapeutic
efficacies of metformin on lung cancer are summarized in Table I. Notably, most of the studies show
that metformin provides better outcomes when used in addition to
existing treatments, including chemotherapy, targeted therapy and
immunotherapy. There were three studies investigating the efficacy
of metformin combined with EGFR-TKI, and all three confirmed that
EGFR-TKI combined with metformin could achieve longer PFS and OS
times compared with EGFR-TKI treatment alone. Yendamuri et
al (154) found that ICIs
combined with metformin could result in better OS time. This effect
was limited by body mass index. Notably, these results may provide
a new strategy to strengthen the therapeutic effects of EGFR-TKI
and ICIs. At present, most of the studies support the antitumor
effects of metformin. However, some clinical trials show no further
benefits on NSCLC. There may be several reasons for the different
conclusions: i) A high degree of clinical heterogeneity among
different trials, as studies were conducted in different
ethnicities and different regions; ii) the doses of metformin and
the combination treatments were different; and iii) some studies
included individuals with diabetes, while others did not. This also
affected the conclusions. Therefore, the effect of metformin on the
treatment and prognosis of lung cancer remains controversial,
requiring confirmation with further tests and studies. Further
studies are needed to evaluate the effect of metformin on the
outcome of patients with NSCLC. The ClinicalTrials.gov website
indicates that a number of prospective clinical trials (Table II) are currently assessing the
effects of metformin on lung cancer. Notably, the clinical trial
NCT05445791 is committed to recruit 312 participants with NSCLC
(stage IIIB-IV) to evaluate the PFS time in patients with NSCLC and
EGFR mutations undergoing treatment with TKIs plus placebo vs. TKIs
plus metformin. The clinical trial NCT01864681 also focuses on the
effect of metformin on EGFR-TKI therapy. These trials may provide a
new strategy for overcoming EGFR-TKI resistance. The clinical trial
NCT02115464 showed that the addition of metformin to
chemoradiotherapy was associated with worse treatment efficacy and
increased toxic effects compared with combined modality therapy
alone. Metformin was not recommended for patients with LA-NSCLC who
are candidates for chemoradiotherapy (155). Despite low accrual rates, the
clinical trial NCT02285855 showed that the majority of patients
treated with metformin exhibited metabolic responses according to
PERCIST criteria on PET imaging. In contrast to the effect of
metformin on the majority of physiological tissues, most tumors had
increased metabolic activity in response to metformin (69). The clinical trial NCT01578551
showed that there was a significant benefit in terms of PFS with
the use of metformin in advanced NSCLC (101). No published results are available
for other completed or terminated trials.
 | Table I.Therapeutic efficacies of metformin
in lung cancer. |
Table I.
Therapeutic efficacies of metformin
in lung cancer.
| First author,
year | Number of
subjects | Combination
treatment | Effect of
treatment/conclusions | (Refs.) |
|---|
| Tan et al,
2011 | 99 | Chemotherapy | Improved PFS
time | (17) |
| Marrone et
al, 2018 | 25 | Carboplatin,
paclitaxel, bevacizumab | Improved PFS rate
at 1 year | (101) |
| Parikh et
al, 2019 | 33 | Platinum-based
chemotherapy | Effect of metformin
on PFS and OS time is related to the mutation status of KRAS and
EGFR. | (102) |
| Sayed et al,
2015 | 30 | Gemcitabine and
cisplatin | Metformin
administration reduced occurrence of chemotherapy-induced nausea.
Metformin had no effect on ORR, PFS time and OS time. | (103) |
| Wink et al,
2016 | 682 | Concurrent
chemoradiotherapy | Metformin use was
associated with an improved distant metastasis-free survival (DMFS)
rate at 2 years and PFS time. | (121) |
| Tsakiridis et
al, 2021 | 96 |
Chemoradiotherapy | Metformin is not
recommended in patients with locally advanced NSCLC who are
candidates for. chemoradiotherapy | (155) |
| Arrieta et
al, 2022 | 70 | EGFR-TKI | Improved PFS and OS
time of patients with a BMI of ≥24 | (165) |
| Chen et al,
2015 | 90 | EGFR-TKI | Improved PFS time,
OS time, ORR and DCR | (123) |
| Arrieta et
al, 2019 | 139 | EGFR-TKI | Improved PFS and OS
time. | (127) |
| Afzal et al,
2019 | 50 | ICIs | Improved ORR, DCR,
median OS time and PFS time | (152) |
| Yendamuri et
al, 2019 | 434 | ICIs | A tendency to
improved OS in metformin users only in patients with a BMI >25
kg/m2, and the strength of the association was higher in
patients with a BMI >30 kg/m2 | (154) |
| Table II.Active clinical trials with metformin
in lung cancer (www.ClinicalTrials.gov; accessed June 2022). |
Table II.
Active clinical trials with metformin
in lung cancer (www.ClinicalTrials.gov; accessed June 2022).
| Clinical trial
number | Trial phase | Title |
Region/institute | Metformin dose | Combination
treatment | Stage | Investigation
purpose | Status | Study type |
|---|
| NCT02115464 | Phase 2 | Advanced lung
cancer treatment with metformin and chemoradiotherapy | Canada | 2,000 mg, daily,
for 12 months | Chemotherapy and
radiotherapy | Stage IIIa or stage
IIIb | To determine the
effect of metformin on the proportion of patients free of disease
progression at 12 months after initiation of drug treatment | Terminated | Interventional |
| NCT02285855 | Phase 2 | Metformin in
non-small cell lung cancer (NSCLC) | United States | 2,000 mg, daily,
for 3 weeks | Radiotherapy | Stage I–II | To determine the
effect of metformin on the response of patients with NSCLC treated
with hypofractionated radiation therapy | Terminated | Interventional |
| NCT02019979 | Phase 2 | Metformin and
carbohydrate restriction with platinum-based chemotherapy in stage
IIIB/IV non-squamous non-small cell lung cancer (NS-NSCLC) | United States | 1,000 mg, bid, for
3 weeks | Carbohydrate
restricted diet, platinum based chemotherapy | Stage IIIB or
IV | To determine the
effect of metformin and carbohydrate restriction on the response of
patients with non-squamous NSCLC treated with platinum-based
chemotherapy | Terminated | Interventional |
| NCT04931017 | Phase 2 | Metformin for
chemoprevention of lung cancer in overweight or obese individuals
at high risk for lung cancer | United States,
Canada | Unknown | - | - | To examine whether
metformin extended release as a preventative treatment may lower
the chance of developing lung cancer, and whether it may help the
patients' immune system learn to lower a certain type of immune
cell (regulatory T cells) that are linked to tumor development | Recruiting | Interventional |
| NCT03086733 | Phase 2 | Phase II lung
metcore preoperative metformin for lung cancer | United States | 850 mg, bid | Surgery | Stage I–IIIa | To determine the
effect of metformin on Ki67 apoptosis in patients with NSCLC | Completed | Interventional |
| NCT01717482 | Phase 2 | Metformin as a
chemoprevention agent in non-small cell lung cancer | United States | 850 mg, bid | Standard of Care
Observation | Stage Ia-IIIa | To investigate
whether it is better to receive the drug metformin with standard of
care for lung cancer or just standard of care | Terminated | Interventional |
| NCT01997775 | Phase 2 | Metformin in stage
IV lung adenocarcinoma | Taiwan | 500 mg, tid | Chemotherapy
combining cisplatin and pemetrexed or targeted therapy | Stage IV | To determine
whether metformin is effective in lowering plasma IL-6 level and
improving the treatment response in patients with NSCLC | Terminated | Interventional |
| NCT03874000 | Phase 2 | Sintilimab combined
with metformin in first-line chemotherapy refractory advanced NSCLC
patients | China | 500 mg, bid | Sintilimab | Stage IV | To determine the
effect of metformin on objective response rate in patients with
first-line chemotherapy refractory advanced NSCLC | Unknown | Interventional |
| NCT02109549 | - | Influence of the
use of the diabetic drug metformin on the OS and treatment-related
toxicity in advanced stage non-small cell lung cancer patients | Netherlands | Unknown | Concurrent
radiochemo-therapy | Advanced stage | To test whether
patients suffering from non-insulin-dependent diabetes mellitus,
treated with metformin, have improved local tumor control | Completed | Observational |
| NCT03048500 | Phase 2 | Nivolumab and
metformin hydrochloride in treating patients with stage III–IV
non-small cell lung cancer that cannot be removed by surgery | United States | Unknown | Nivolumab | Stage IV or
non-resectable stage III | To assess antitumor
activity of the combination treatment of metformin hydrochloride
(metformin) with nivolumab in patients with NSCLC with and without
prior exposure to PD-1/PD-L1 inhibitors | Active, not
recruiting | Interventional |
| NCT03994744 | Phase 2 | Assessing safety
and efficacy of sintilimab and metformin combination therapy in
SCLC | China | 1,000 mg, bid | Sintilimab | Extended disease
stage of SCLC | To assess safety
and efficacy of sintilimab and metformin combination therapy in
SCLC | Recruiting | Interventional |
| NCT03071705 | Not Applicable | Metformin plus TKI
use in patients with non-small cell lung carcinoma | Instituto National
de Cancerologia | 500 mg, bid | EGFR-TKI | Unknown | To assess the PFS
period in patients with advanced NSCLC in treatment with TKIs and
metformin vs. TKI alone | Unknown | Interventional |
| NCT02186847 | Phase 2 | Chemotherapy and
radiation therapy with or without metformin hydrochloride in
treating patients with stage III non-small cell lung cancer | Multicenter | 500 mg, bid (1–7
days); 500 mg, tid (8–14 days); 2,000 mg daily (15–126 days) | Radiation and
chemotherapy can improve PFS in patients with locally advanced
NSCLC | Stage III | To determine
whether metformin hydrochloride added to chemoradiotherapy | Active, not
recruiting | Interventional |
| NCT04170959 | Phase 2 | The addition of
metformin to definitive radiotherapy in patients with stage III
NSCLC (RADFORMIN) | Belgium | 500 mg, daily (1–7
days); 500 mg bid (7–14 days) | Standard of
care | Stage III | To identify subsets
of patients who derive maximum benefit of adding metformin to
radiotherapy using innovative biomarkers | Terminated | Interventional |
| NCT01864681 | Phase 2 | Combination of
metformin with gefitinib to treat NSCLC | China | 500 mg, bid | Gefitinib | Stage IV | To determine
whether metformin in combination with gefitinib is effective in
patients with previously untreated advanced or metastatic NSCLC
with EGFR mutations | Completed | Interventional |
| NCT01578551 | Phase 2 | Study of metformin
plus paclitaxel/carboplatin/bevacizumab in patients with
adenocarcinoma | United States | 500 mg, bid-1,000
mg, bid | Paclitaxel,
carboplatin, bevacizumab | Stage IV | To determine the
1-year PFS rate of the combination of metformin and standard
chemotherapy in patients with previously untreated advanced or
metastatic pulmonary adenocarcinoma | Terminated | Interventional |
| NCT05445791 | Phase 3 | Metformin plus
tyrosine kinase inhibitors for treatment of patients with non-small
cell lung cancer with EGFR mutations (METLUNG) | Mexico | 500 mg, bid | Tyrosine kinase
inhibitors | IIIB-IV | To evaluate the PFS
in patients with NSCLC with EGFR mutations under going treatment
with TKIs plus placebo vs. TKIs plus metformin | Recruiting | Interventional |
Limitations and challenges of using
metformin in lung cancer
Dose of metformin for lung cancer
Although several studies support the efficacy and
feasibility of metformin in cancer treatment, metformin is mainly
used in patients with diabetes. In clinical application, for
non-diabetic patients, the feasibility of treating lung cancer with
metformin alone or in combination with other anti-lung cancer
treatment schemes may be limited due to its possible adverse
reactions leading to intolerance in some patients (156). The most common toxicity of
metformin is gastrointestinal toxicity, including mild anorexia, a
metallic taste in the mouth, nausea, diarrhea and abdominal pain
(157). Although the symptoms are
typically mild, transient and reversible with dose reduction or
withdrawal, the side effects of metformin may increase when
combined with chemotherapy (especially platinum-based) and
radiotherapy (158). However, a
small prospective randomized phase II study that included 15
patients with stage IV NSCLC who received both metformin and
chemotherapy, found that metformin combined with chemotherapy
reduced gastrointestinal reactions to chemotherapy. The metformin
group was found to have a lower incidence of nausea compared with
the combined treatment group (26.7 vs. 66.7%; P=0.03) (103). Metformin can accumulate in the
body and cause a rare but severe form of lactic acidosis. In
addition, the main risks of metformin treatment are kidney damage,
sepsis, dehydration, liver damage and acute congestive heart
failure (157). Based on the
experience of Wink et al (121), a conventional dose of metformin
combined with concurrent chemoradiotherapy is safe and feasible.
However, in this study, the dose of metformin was not reported, and
only a cautious, gradual increase in the dose during the first few
weeks of administration was recommended (121). The antitumor effects of metformin
seem to increase with increasing dose. In addition, an important
limitation of a number of experimental studies is that the
metformin concentrations used in numerous experiments are greater
than the conventional doses applied for diabetes treatment
(72). However, these high doses
are inappropriate for practical clinical use due to the potential
drug toxicity. Most current retrospective studies and corresponding
meta-analyses were conducted on diabetic patients, and metformin
was used at a conventional treatment dose. In the in vitro
tests on the effect of metformin on lung cancer, the concentration
of metformin was significantly higher than the blood concentration
of the treatment dose of diabetic patients and showed a
concentration dependence (130).
Therefore, whether the clinical metformin dose has such an effect
on the tumor is debatable.
In addition, a number of factors affect the
effectiveness and reactivity of metformin in tissues. For example,
tissue expression of transporters that mediate metformin uptake
differs between normal and tumor cells, and may be affected by
various drugs, such as antibiotics and proton pump inhibitors
(159). Malabsorption of
metformin in target cells may limit its potential to treat cancer.
Overall, the metabolic state of the patient and the interactions
between tumor molecules add to the complexity of the impact of
metformin on the tumor (160). It
remains to be further confirmed whether the anti-lung cancer effect
of a conventional metformin dose can be achieved in in vitro
trials.
Administration route of metformin for
lung cancer
Metformin has been used orally in clinical studies
of diabetes mellitus and lung cancer, but the study by Memmott
et al (60) has shown that
intraperitoneal injection of metformin can produce higher plasma
metformin levels, resulting in tissue-specific regulation of the
AMPK and mTOR pathways. Intraperitoneal injection of metformin
inhibits the mTOR pathway in lung tissue by reducing the response
to insulin or IGF-1, independent of AMPK. In this research,
metformin administration through intraperitoneal injection was
unexpectedly well tolerated throughout the study and did not
significantly affect body weight in the mice (data were not shown).
Metformin reduced tumor diversity by 66%, mean tumor volume by 50%
and total tumor load by 72%. S6 phosphorylation in the tumor was
reduced by 40%. Therefore, the intraperitoneal injection of
metformin was more effective than oral injection in preventing
NNK-induced lung tumorigenesis and inhibiting mTOR (60). A recent in vitro study
evaluating metformin sterol liposomes as an inhaled treatment for
lung cancer was conducted by mixing stearin and cholesterol.
Metformin liposome showed a significant inhibitory effect on A549
cells (P<0.05), and it increased significantly with the increase
of dose and exposure time. The feasibility of liposomes for aerosol
delivery provides a new strategy for metformin inhalation
administration, which may be an effective inhalation therapy for
lung cancer (161). Therefore,
the administration route of metformin in treating tumors needs to
be further explored in clinical trials.
Population selection of combined
metformin therapy for lung cancer
A number of clinical trials have evaluated the
anticancer activity of metformin, but the results have been
inconsistent. There are numerous factors influencing the trial
results, such as the use of different subgroups of patients with
lung cancer. Further studies that screen for metformin-sensitive
subgroups of lung cancer to improve the efficacy and reduce adverse
drug reactions in patients with lung cancer are awaited. A
randomized phase II study reported that EGFR-TKI combined with
metformin showed better PFS and OS times than EGFR-TKI alone in
patients with EGFR-mutated lung adenocarcinoma (127). Some case-control studies have
suggested that squamous cell carcinoma may have more metabolic
characteristics than adenocarcinoma. There is a markedly elevated
expression of the glucose transporter 1 (GLUT1) in lung squamous
cell carcinoma, which augments glucose uptake and glycolytic flux.
Elevated GLUT1-mediated glycolysis in lung squamous cell carcinoma
strongly correlates with high 18F-FDG uptake and poor
prognosis (162). Compared with
lung adenocarcinoma, lung squamous cell carcinoma has a higher
level of glucose metabolism to maintain the metabolism required for
rapid tumor growth. The expression of HK2 (the rate-limit enzyme
and the first committed step in glucose metabolism) in lung
squamous cell carcinoma was significantly higher than that in lung
adenocarcinoma and normal tissues (13). A recent study showed that metformin
significantly improved the prognosis of patients with squamous cell
carcinoma and high FDG uptake (68). These results suggest that metformin
may be more effective in patients with squamous cell lung cancer
with high FDG uptake. The aforementioned research also found that
within the subgroup with TP53 mutations (n=33), the metformin group
exhibited better OS than the control group (HR, 0.23; 95% CI,
0.17-0.38; P=0.021). By contrast, within the subgroup with
wild-type TP53 (n=80), the metformin group exhibited worse OS (HR,
1.61; 95% CI, 0.97-2.81; P=0.091). The results suggest that
metformin may have a better antitumor effect on patients with lung
cancer and TP53 mutation (39).
However, all the aforementioned studies are small sample trials,
and larger studies are required to explore and find a suitable
population for lung cancer combined with metformin to improve the
effective rate of lung cancer treatment and prolong the PFS and OS
times of affected patients.
Recently, the effects of metformin on lung cancer
have gained the attention of a number of researchers, and numerous
reviews have already been previously published. Li et al
(163) and Chen et al
(164) reviewed the effects of
metformin on lung cancer. While the association between metformin
and lung cancer was complex, the effects of metformin on lung
cancer remained controversial and the mechanisms were intricate.
Therefore, new studies were instigated. The present review gives a
more comprehensive overview of the mechanisms, in addition to the
involvement of the AMPK signaling pathway and the AMPK-independent
signaling pathway. In contrast to the review by Chen et al
(164), the present review
summarized other mechanisms. The mechanisms of metformin inhibiting
complex I of the mitochondrial respiratory chain, regulating lung
miRNAs, and affecting the tumor and its microenvironment were
preliminarily reviewed. The present review described the
administration route of metformin for lung cancer and the
population selection of combined metformin therapy for lung cancer.
This will help further research select an appropriate
administration route and population. In contrast to the review by
Li et al (163), the
present review illustrated the fact that metformin can enhance the
effect of immunotherapy, and more comprehensive and updated studies
were included.
Conclusion
Numerous studies have revealed that metformin plays
a direct or indirect antitumor role in regulating the lung cancer
cell cycle, inhibiting cell proliferation and promoting apoptosis.
Metformin alone or in combination with chemoradiotherapy, targeted
drug therapy and immunotherapy has therapeutic effects on lung
cancer and is expected to improve the prognosis of patients with
lung cancer. However, more randomized, prospective, standardized
and quantitative trial results are needed to further explore the
selection of metformin, the effective treatment dose and the
treatment approaches, to improve the survival time and prognosis of
patients with lung cancer.
Acknowledgements
Not applicable.
Funding
This study was supported by the Wanzhou District Science and
Technology Joint Medical Research Project, Chongqing (grant no.
wzstc-kw2021011), the Chongqing Medical Scientific Research Project
(grant no. 2021MSXM042) and the General Program of Chongqing
Natural Science Foundation (grant no. cstc2020jcyj-msxmX0713).
Availability of data and materials
Not applicable.
Authors' contributions
PH, JZ, QL and KS were responsible for gathering
the associated research and designing the review. PH was
responsible for creating the figures. JX, QL and KS contributed to
the study design, interpretation of the research articles, editing
of the manuscript and critical revision of the manuscript. All
authors read and approved the final manuscript. Data authentication
is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Graham GG, Punt J, Arora M, Day RO, Doogue
MP, Duong JK, Furlong TJ, Greenfield JR, Greenup LC, Kirkpatrick
CM, et al: Clinical pharmacokinetics of metformin. Clin
Pharmacokinet. 50:81–98. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Apostolova N, Iannantuoni F, Gruevska A,
Muntane J, Rocha M and Victor VM: Mechanisms of action of metformin
in type 2 diabetes: Effects on mitochondria and
leukocyte-endothelium interactions. Redox Biol. 34:1015172020.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rena G, Hardie DG and Pearson ER: The
mechanisms of action of metformin. Diabetologia. 60:1577–1585.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ma T, Tian X, Zhang B, Li M, Wang Y, Yang
C, Wu J, Wei X, Qu Q, Yu Y, et al: Low-dose metformin targets the
lysosomal AMPK pathway through PEN2. Nature. 603:159–165. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Viollet B, Guigas B, Sanz Garcia N,
Leclerc J, Foretz M and Andreelli F: Cellular and molecular
mechanisms of metformin: An overview. Clin Sci (Lond). 122:253–270.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Libby G, Donnelly LA, Donnan PT, Alessi
DR, Morris AD and Evans JM: New users of metformin are at low risk
of incident cancer: A cohort study among people with type 2
diabetes. Diabetes Care. 32:1620–1625. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Landman GW, Kleefstra N, van Hateren KJ,
Groenier KH, Gans RO and Bilo HJ: Metformin associated with lower
cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care.
33:322–326. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shaw RJ, Bardeesy N, Manning BD, Lopez L,
Kosmatka M, DePinho RA and Cantley LC: The LKB1 tumor suppressor
negatively regulates mTOR signaling. Cancer Cell. 6:91–99. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tomic T, Botton T, Cerezo M, Robert G,
Luciano F, Puissant A, Gounon P, Allegra M, Bertolotto C, Bereder
JM, et al: Metformin inhibits melanoma development through
autophagy and apoptosis mechanisms. Cell Death Dis. 2:e1992011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Taubes G: Cancer research. Cancer
prevention with a diabetes pill? Science. 335:292012.PubMed/NCBI
|
|
11
|
Tian RH, Zhang YG, Wu Z, Liu X, Yang JW
and Ji HL: Effects of metformin on survival outcomes of lung cancer
patients with type 2 diabetes mellitus: A meta-analysis. Clin
Transl Oncol. 18:641–649. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin JJ, Gallagher EJ, Sigel K, Mhango G,
Galsky MD, Smith CB, LeRoith D and Wisnivesky JP: Survival of
patients with stage IV lung cancer with diabetes treated with
metformin. Am J Respir Crit Care Med. 191:448–454. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guo W, Kuang Y, Wu J, Wen D, Zhou A, Liao
Y, Song H, Xu D, Wang T, Jing B, et al: Hexokinase 2 depletion
confers sensitization to metformin and inhibits glycolysis in lung
squamous cell carcinoma. Front Oncol. 10:522020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang J, Gao Q, Wang D, Wang Z and Hu C:
Metformin inhibits growth of lung adenocarcinoma cells by inducing
apoptosis via the mitochondria-mediated pathway. Oncol Lett.
10:1343–1349. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Teixeira SF, Guimarães Idos S, Madeira KP,
Daltoé RD, Silva IV and Rangel LB: Metformin synergistically
enhances antiproliferative effects of cisplatin and etoposide in
NCI-H460 human lung cancer cells. J Bras Pneumol. 39:644–649. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ashinuma H, Takiguchi Y, Kitazono S,
Kitazono-Saitoh M, Kitamura A, Chiba T, Tada Y, Kurosu K, Sakaida
E, Sekine I, et al: Antiproliferative action of metformin in human
lung cancer cell lines. Oncol Rep. 28:8–14. 2012.PubMed/NCBI
|
|
17
|
Tan BX, Yao WX, Ge J, Peng XC, Du XB,
Zhang R, Yao B, Xie K, Li LH, Dong H, et al: Prognostic influence
of metformin as first-line chemotherapy for advanced nonsmall cell
lung cancer in patients with type 2 diabetes. Cancer.
117:5103–5111. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stevens RJ, Ali R, Bankhead CR, Bethel MA,
Cairns BJ, Camisasca RP, Crowe FL, Farmer AJ, Harrison S, Hirst JA,
et al: Cancer outcomes and all-cause mortality in adults allocated
to metformin: Systematic review and collaborative meta-analysis of
randomised clinical trials. Diabetologia. 55:2593–2603. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ece H, Cigdem E, Yuksel K, Ahmet D, Hakan
E and Oktay TM: Use of oral antidiabetic drugs (metformin and
pioglitazone) in diabetic patients with breast cancer: How does it
effect serum Hif-1 alpha and 8Ohdg levels? Asian Pac J Cancer Prev.
13:5143–5148. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Berbée JF, Boon MR, Khedoe PP, Bartelt A,
Schlein C, Worthmann A, Kooijman S, Hoeke G, Mol IM, John C, et al:
Brown fat activation reduces hypercholesterolaemia and protects
from atherosclerosis development. Nat Commun. 6:63562015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hardie DG and Alessi DR: LKB1 and AMPK and
the cancer-metabolism link-ten years after. BMC Biol. 11:362013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Koivunen JP, Kim J, Lee J, Rogers AM, Park
JO, Zhao X, Naoki K, Okamoto I, Nakagawa K, Yeap BY, et al:
Mutations in the LKB1 tumour suppressor are frequently detected in
tumours from Caucasian but not Asian lung cancer patients. Br J
Cancer. 99:245–252. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Han D, Li SJ, Zhu YT, Liu L and Li MX:
LKB1/AMPK/mTOR signaling pathway in non-small-cell lung cancer.
Asian Pac J Cancer Prev. 14:4033–4039. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shaw RJ and Cantley LC: Ras, PI(3)K and
mTOR signalling controls tumour cell growth. Nature. 441:424–430.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gwinn DM, Shackelford DB, Egan DF,
Mihaylova MM, Mery A, Vasquez DS, Turk BE and Shaw RJ: AMPK
phosphorylation of raptor mediates a metabolic checkpoint. Mol
Cell. 30:214–226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rabiee A, Krüger M, Ardenkjær-Larsen J,
Kahn CR and Emanuelli B: Distinct signalling properties of insulin
receptor substrate (IRS)-1 and IRS-2 in mediating insulin/IGF-1
action. Cell Signal. 47:1–15. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang X and Proud CG: The mTOR pathway in
the control of protein synthesis. Physiology (Bethesda).
21:362–369. 2006.PubMed/NCBI
|
|
28
|
Morita M, Gravel SP, Hulea L, Larsson O,
Pollak M, St-Pierre J and Topisirovic I: mTOR coordinates protein
synthesis, mitochondrial activity and proliferation. Cell Cycle.
14:473–480. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ren GF, Xiao LL, Ma XJ, Yan YS and Jiao
PF: Metformin decreases insulin resistance in type 1 diabetes
through regulating p53 and RAP2A in vitro and in vivo. Drug Des
Devel Ther. 14:2381–2392. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Storozhuk Y, Hopmans SN, Sanli T, Barron
C, Tsiani E, Cutz JC, Pond G, Wright J, Singh G and Tsakiridis T:
Metformin inhibits growth and enhances radiation response of
non-small cell lung cancer (NSCLC) through ATM and AMPK. Br J
Cancer. 108:2021–2032. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Blagih J, Buck MD and Vousden KH: p53,
cancer and the immune response. J Cell Sci. 133:jcs2374532020.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu G, Pei F, Yang F, Li L, Amin AD, Liu
S, Buchan JR and Cho WC: Role of autophagy and apoptosis in
non-small-cell lung cancer. Int J Mol Sci. 18:3672017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bykov VJN, Eriksson SE, Bianchi J and
Wiman KG: Targeting mutant p53 for efficient cancer therapy. Nat
Rev Cancer. 18:89–102. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Buzzai M, Jones RG, Amaravadi RK, Lum JJ,
DeBerardinis RJ, Zhao F, Viollet B and Thompson CB: Systemic
treatment with the antidiabetic drug metformin selectively impairs
p53-deficient tumor cell growth. Cancer Res. 67:6745–6752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dogan Turacli I, Candar T, Yuksel BE and
Demirtas S: Role of metformin on base excision repair pathway in
p53 wild-type H2009 and HepG2 cancer cells. Hum Exp Toxicol.
37:909–919. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ben Sahra I, Regazzetti C, Robert G,
Laurent K, Le Marchand-Brustel Y, Auberger P, Tanti JF,
Giorgetti-Peraldi S and Bost F: Metformin, independent of AMPK,
induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer
Res. 71:4366–4372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Blandino G, Valerio M, Cioce M, Mori F,
Casadei L, Pulito C, Sacconi A, Biagioni F, Cortese G, Galanti S,
et al: Metformin elicits anticancer effects through the sequential
modulation of DICER and c-MYC. Nat Commun. 3:8652012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Szczyrek M, Grenda A, Kuźnar-Kamińska B,
Krawczyk P, Sawicki M, Batura-Gabryel H, Mlak R, Szudy-Szczyrek A,
Krajka T, Krajka A, et al: Methylation of DROSHA and DICER as a
biomarker for the detection of lung cancer. Cancers (Basel).
13:61392021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pradhan AK, Bhoopathi P, Talukdar S,
Scheunemann D, Sarkar D, Cavenee WK, Das SK, Emdad L and Fisher PB:
MDA-7/IL-24 regulates the miRNA processing enzyme DICER through
downregulation of MITF. Proc Natl Acad Sci USA. 116:5687–5692.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Prodromaki E, Korpetinou A, Giannopoulou
E, Vlotinou E, Chatziathanasiadou Μ, Papachristou NI, Scopa CD,
Papadaki H, Kalofonos HP and Papachristou DJ: Expression of the
microRNA regulators drosha, dicer and Ago2 in non-small cell lung
carcinomas. Cell Oncol (Dordr). 38:307–317. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen JC, Su YH, Chiu CF, Chang YW, Yu YH,
Tseng CF, Chen HA and Su JL: Suppression of Dicer increases
sensitivity to gefitinib in human lung cancer cells. Ann Surg
Oncol. 21 (Suppl 4):S555–563. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li C, Chen L, Song W, Peng B, Zhu J and
Fang L: DICER activates autophagy and promotes cisplatin resistance
in non-small cell lung cancer by binding with let-7i-5p. Acta
Histochem. 123:1517882021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu M, Zhang Z, Wang H, Chen X and Jin C:
Activation of AMPK by metformin promotes renal cancer cell
proliferation under glucose deprivation through its interaction
with PKM2. Int J Biol Sci. 15:617–627. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Volm M and Koomägi R: Hypoxia-inducible
factor (HIF-1) and its relationship to apoptosis and proliferation
in lung cancer. Anticancer Res. 20:1527–1533. 2000.PubMed/NCBI
|
|
46
|
Yang SL, Ren QG, Wen L and Hu JL:
Clinicopathological and prognostic significance of
hypoxia-inducible factor-1 alpha in lung cancer: A systematic
review with meta-analysis. J Huazhong Univ Sci Technolog Med Sci.
36:321–327. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang J, Wang Q, Wang Q, Guo P, Wang Y,
Xing Y, Zhang M, Liu F and Zeng Q: Chrysophanol exhibits
anti-cancer activities in lung cancer cell through regulating
ROS/HIF-1a/VEGF signaling pathway. Naunyn Schmiedebergs Arch
Pharmacol. 393:469–480. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Akakura N, Kobayashi M, Horiuchi I, Suzuki
A, Wang J, Chen J, Niizeki H, Kawamura K, Hosokawa M and Asaka M:
Constitutive expression of hypoxia-inducible factor-1alpha renders
pancreatic cancer cells resistant to apoptosis induced by hypoxia
and nutrient deprivation. Cancer Res. 61:6548–6554. 2001.PubMed/NCBI
|
|
49
|
Pineda CT, Ramanathan S, Fon Tacer K, Weon
JL, Potts MB, Ou YH, White MA and Potts PR: Degradation of AMPK by
a cancer-specific ubiquitin ligase. Cell. 160:715–728. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Fürstenberger G and Senn HJ: Insulin-like
growth factors and cancer. Lancet Oncol. 3:298–302. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Favoni RE, de Cupis A, Ravera F, Cantoni
C, Pirani P, Ardizzoni A, Noonan D and Biassoni R: Expression and
function of the insulin-like growth factor I system in human
non-small-cell lung cancer and normal lung cell lines. Int J
Cancer. 56:858–866. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tseng SC, Huang YC, Chen HJ, Chiu HC,
Huang YJ, Wo TY, Weng SH and Lin YW: Metformin-mediated
downregulation of p38 mitogen-activated protein kinase-dependent
excision repair cross-complementing 1 decreases DNA repair capacity
and sensitizes human lung cancer cells to paclitaxel. Biochem
Pharmacol. 85:583–594. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kuribayashi A, Kataoka K, Kurabayashi T
and Miura M: Evidence that basal activity, but not transactivation,
of the epidermal growth factor receptor tyrosine kinase is required
for Insulin-like growth factor I-induced activation of
extracellular signal-regulated kinase in oral carcinoma cells.
Endocrinology. 145:4976–4984. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Cao H, Dong W, Qu X, Shen H, Xu J, Zhu L,
Liu Q and Du J: Metformin enhances the therapy effects of
Anti-IGF-1R mAb figitumumab to NSCLC. Sci Rep. 6:310722016.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Pernicova I and Korbonits M:
Metformin-mode of action and clinical implications for diabetes and
cancer. Nat Rev Endocrinol. 10:143–156. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fumarola C, Bonelli MA, Petronini PG and
Alfieri RR: Targeting PI3K/AKT/mTOR pathway in non small cell lung
cancer. Biochem Pharmacol. 90:197–207. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Heavey S, O'Byrne KJ and Gately K:
Strategies for co-targeting the PI3K/AKT/mTOR pathway in NSCLC.
Cancer Treat Rev. 40:445–456. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Granville CA, Memmott RM, Balogh A,
Mariotti J, Kawabata S, Han W, Lopiccolo J, Foley J, Liewehr DJ,
Steinberg SM, et al: A central role for Foxp3+ regulatory T cells
in K-Ras-driven lung tumorigenesis. PLoS One. 4:e50612009.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zou W: Regulatory T cells, tumour immunity
and immunotherapy. Nat Rev Immunol. 6:295–307. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Memmott RM, Mercado JR, Maier CR, Kawabata
S, Fox SD and Dennis PA: Metformin prevents tobacco
carcinogen-induced lung tumorigenesis. Cancer Prev Res (Phila).
3:1066–1076. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kalender A, Selvaraj A, Kim SY, Gulati P,
Brûlé S, Viollet B, Kemp BE, Bardeesy N, Dennis P, Schlager JJ, et
al: Metformin, independent of AMPK, inhibits mTORC1 in a rag
GTPase-dependent manner. Cell Metab. 11:390–401. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Efeyan A, Zoncu R, Chang S, Gumper I,
Snitkin H, Wolfson RL, Kirak O, Sabatini DD and Sabatini DM:
Regulation of mTORC1 by the Rag GTPases is necessary for neonatal
autophagy and survival. Nature. 493:679–683. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Bar-Peled L, Schweitzer LD, Zoncu R and
Sabatini DM: Ragulator is a GEF for the rag GTPases that signal
amino acid levels to mTORC1. Cell. 150:1196–1208. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kim J and Kim E: Rag GTPase in amino acid
signaling. Amino Acids. 48:915–928. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Liu Y, He C and Huang X: Metformin
partially reverses the carboplatin-resistance in NSCLC by
inhibiting glucose metabolism. Oncotarget. 8:75206–75216. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Salani B, Del Rio A, Marini C, Sambuceti
G, Cordera R and Maggi D: Metformin, cancer and glucose metabolism.
Endocr Relat Cancer. 21:R461–R471. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Schuurbiers OC, Meijer TW, Kaanders JH,
Looijen-Salamon MG, de Geus-Oei LF, van der Drift MA, van der
Heijden EH, Oyen WJ, Visser EP, Span PN, et al: Glucose metabolism
in NSCLC is histology-specific and diverges the prognostic
potential of 18FDG-PET for adenocarcinoma and squamous cell
carcinoma. J Thorac Oncol. 9:1485–1493. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lee Y, Joo J, Lee YJ, Lee EK, Park S, Kim
TS, Lee SH, Kim SY, Wie GA, Park M, et al: Randomized phase II
study of Platinum-based chemotherapy plus controlled diet with or
without metformin in patients with advanced Non-small cell lung
cancer. Lung Cancer. 151:8–15. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Chun SG, Liao Z, Jeter MD, Chang JY, Lin
SH, Komaki RU, Guerrero TM, Mayo RC, Korah BM, Koshy SM, et al:
Metabolic responses to metformin in inoperable early-stage
non-small cell lung cancer treated with stereotactic radiotherapy:
Results of a randomized Phase II clinical trial. Am J Clin Oncol.
43:231–235. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
O JH, Lodge MA and Wahl RL: Practical
PERCIST: A Simplified Guide to PET response criteria in solid
tumors 1.0. Radiology. 280:576–584. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Goodwin PJ, Ligibel JA and Stambolic V:
Metformin in breast cancer: Time for action. J Clin Oncol.
27:3271–3273. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Pollak MN: Investigating metformin for
cancer prevention and treatment: The end of the beginning. Cancer
Discov. 2:778–790. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wheaton WW, Weinberg SE, Hamanaka RB,
Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM,
Budigner GS, et al: Metformin inhibits mitochondrial complex I of
cancer cells to reduce tumorigenesis. Elife. 3:e022422014.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Andrzejewski S, Gravel SP, Pollak M and
St-Pierre J: Metformin directly acts on mitochondria to alter
cellular bioenergetics. Cancer Metab. 2:122014. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Algire C, Moiseeva O, Deschênes-Simard X,
Amrein L, Petruccelli L, Birman E, Viollet B, Ferbeyre G and Pollak
MN: Metformin reduces endogenous reactive oxygen species and
associated DNA damage. Cancer Prev Res (Phila). 5:536–543. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Boyle KA, Van Wickle J, Hill RB, Marchese
A, Kalyanaraman B and Dwinell MB: Mitochondria-targeted drugs
stimulate mitophagy and abrogate colon cancer cell proliferation. J
Biol Chem. 293:14891–14904. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Logie L, Harthill J, Patel K, Bacon S,
Hamilton DL, Macrae K, McDougall G, Wang HH, Xue L, Jiang H, et al:
Cellular responses to the metal-binding properties of metformin.
Diabetes. 61:1423–1433. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Izzotti A, Balansky R, D'Agostini F,
Longobardi M, Cartiglia C, Micale RT, La Maestra S, Camoirano A,
Ganchev G, Iltcheva M, et al: Modulation by metformin of molecular
and histopathological alterations in the lung of cigarette
Smoke-exposed mice. Cancer Med. 3:719–730. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Dweep H, Sticht C, Pandey P and Gretz N:
miRWalk-database: Prediction of possible miRNA binding sites by
‘Walking’ the genes of three genomes. J Biomed Inform. 44:839–847.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Bost F, Ben-Sahra I and Tanti JF:
Prevention of mutagenesis: New potential mechanisms of metformin
action in neoplastic cells. Cancer Prev Res (Phila). 5:503–506.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zhang B, Liu T, Wu T, Wang Z, Rao Z and
Gao J: microRNA-137 functions as a tumor suppressor in human
non-small cell lung cancer by targeting SLC22A18. Int J Biol
Macromol. 74:111–118. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Kefas B, Godlewski J, Comeau L, Li Y,
Abounader R, Hawkinson M, Lee J, Fine H, Chiocca EA, Lawler S, et
al: microRNA-7 inhibits the epidermal growth factor receptor and
the Akt pathway and is down-regulated in glioblastoma. Cancer Res.
68:3566–3572. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhao X, Dou W, He L, Liang S, Tie J, Liu
C, Li T, Lu Y, Mo P, Shi Y, et al: MicroRNA-7 functions as an
anti-metastatic microRNA in gastric cancer by targeting
Insulin-like growth Factor-1 receptor. Oncogene. 32:1363–1372.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Dong J, Peng H, Yang X, Wu W, Zhao Y, Chen
D, Chen L and Liu J: Metformin mediated microRNA-7 upregulation
inhibits growth, migration, and invasion of non-small cell lung
cancer A549 cells. Anticancer Drugs. 31:345–352. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Jin D, Guo J, Wu Y, Chen W, Du J, Yang L,
Wang X, Gong K, Dai J, Miao S, et al: Metformin-repressed
miR-381-YAP-snail axis activity disrupts NSCLC growth and
metastasis. J Exp Clin Cancer Res. 39:62020. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Pearce EL, Walsh MC, Cejas PJ, Harms GM,
Shen H, Wang LS, Jones RG and Choi Y: Enhancing CD8 T-cell memory
by modulating fatty acid metabolism. Nature. 460:103–107. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Eikawa S, Nishida M, Mizukami S, Yamazaki
C, Nakayama E and Udono H: Immune-mediated antitumor effect by type
2 diabetes drug, metformin. Proc Natl Acad Sci USA. 112:1809–1814.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Zhao D, Long XD, Lu TF, Wang T, Zhang WW,
Liu YX, Cui XL, Dai HJ, Xue F and Xia Q: Metformin decreases IL-22
secretion to suppress tumor growth in an orthotopic mouse model of
hepatocellular carcinoma. Int J Cancer. 136:2556–2565. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Justus CR, Sanderlin EJ and Yang LV:
Molecular connections between cancer cell metabolism and the tumor
microenvironment. Int J Mol Sci. 16:11055–11086. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Scharping NE, Menk AV, Whetstone RD, Zeng
X and Delgoffe GM: Efficacy of PD-1 blockade is potentiated by
metformin-induced reduction of tumor hypoxia. Cancer Immunol Res.
5:9–16. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Lee BB, Kim Y, Kim D, Cho EY, Han J, Kim
HK, Shim YM and Kim DH: Metformin and tenovin-6 synergistically
induces apoptosis through LKB1-independent SIRT1 Down-regulation in
non-small cell lung cancer cells. J Cell Mol Med. 23:2872–2889.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zhou X, Liu S, Lin X, Xu L, Mao X, Liu J,
Zhang Z, Jiang W and Zhou H: Metformin inhibit lung cancer cell
growth and invasion in vitro as well as tumor formation in vivo
partially by activating PP2A. Med Sci Monit. 25:836–846. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Moroishi T, Hansen CG and Guan KL: The
emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 15:73–79.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Jin D, Guo J, Wang D, Wu Y, Wang X, Gao Y,
Shao C, Xu X and Tan S: The antineoplastic drug metformin
downregulates YAP by interfering with IRF-1 binding to the YAP
promoter in NSCLC. EBioMedicine. 37:188–204. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Luo Z, Chen W, Wu W, Luo W, Zhu T, Guo G,
Zhang L, Wang C, Li M and Shi S: Metformin promotes survivin
degradation through AMPK/PKA/GSK-3β-axis in non-small cell lung
cancer. J Cell Biochem. Feb 21–2019.doi: 10.1002/jcb.28470 (Epub
ahead of print). View Article : Google Scholar
|
|
96
|
Salani B, Maffioli S, Hamoudane M, Parodi
A, Ravera S, Passalacqua M, Alama A, Nhiri M, Cordera R and Maggi
D: Caveolin-1 is essential for metformin inhibitory effect on IGF1
action in Non-small-cell lung cancer cells. FASEB J. 26:788–798.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Vazquez-Martin A, Oliveras-Ferraros C,
Cufí S, Martin-Castillo B and Menendez JA: Metformin activates an
ataxia telangiectasia mutated (ATM)/Chk2-regulated DNA Damage-like
response. Cell Cycle. 10:1499–1501. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Wu N, Gu C, Gu H, Hu H, Han Y and Li Q:
Metformin induces apoptosis of lung cancer cells through activating
JNK/p38 MAPK pathway and GADD153. Neoplasma. 58:482–490. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Iliopoulos D, Hirsch HA and Struhl K:
Metformin decreases the dose of chemotherapy for prolonging tumor
remission in mouse xenografts involving multiple cancer cell types.
Cancer Res. 71:3196–3201. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Parikh AB, Kozuch P, Rohs N, Becker DJ and
Levy BP: Metformin as a repurposed therapy in advanced Non-small
cell lung cancer (NSCLC): Results of a phase II trial. Invest New
Drugs. 35:813–819. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Marrone KA, Zhou X, Forde PM, Purtell M,
Brahmer JR, Hann CL, Kelly RJ, Coleman B, Gabrielson E, Rosner GL,
et al: A Randomized Phase II study of metformin plus
Paclitaxel/Carboplatin/Bevacizumab in patients with
Chemotherapy-Naïve advanced or metastatic nonsquamous non-small
cell lung cancer. Oncologist. 23:859–865. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Parikh AB, Marrone KA, Becker DJ, Brahmer
JR, Ettinger DS and Levy BP: A pooled analysis of two phase II
trials evaluating metformin plus platinum-based chemotherapy in
advanced non-small cell lung cancer. Cancer Treat Res Commun.
20:1001502019. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Sayed R, Saad AS, El Wakeel L, Elkholy E
and Badary O: Metformin addition to chemotherapy in stage IV
non-small cell lung cancer: An open label randomized controlled
study. Asian Pac J Cancer Prev. 16:6621–6626. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Lin CC, Yeh HH, Huang WL, Yan JJ, Lai WW,
Su WP, Chen HH and Su WC: Metformin enhances cisplatin cytotoxicity
by suppressing signal transducer and activator of transcription-3
activity independently of the liver kinase B1-AMP-activated protein
kinase pathway. Am J Respir Cell Mol Biol. 49:241–250. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Weerasinghe P, Garcia GE, Zhu Q, Yuan P,
Feng L, Mao L and Jing N: Inhibition of Stat3 activation and tumor
growth suppression of non-small cell lung cancer by G-quartet
oligonucleotides. Int J Oncol. 31:129–136. 2007.PubMed/NCBI
|
|
106
|
Chiang GG and Abraham RT: Targeting the
mTOR signaling network in cancer. Trends Mol Med. 13:433–442. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Wang Y, Lin B, Wu J, Zhang H and Wu B:
Metformin inhibits the proliferation of A549/CDDP cells by
activating p38 mitogen-activated protein kinase. Oncol Lett.
8:1269–1274. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Wang J, Xia S and Zhu Z: Synergistic
effect of phenformin in non-small cell lung cancer (NSCLC) ionizing
radiation treatment. Cell Biochem Biophys. 71:513–518. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Koritzinsky M: Metformin: A novel
biological modifier of tumor response to radiation therapy. Int J
Radiat Oncol Biol Phys. 93:454–464. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Bitterman PB and Polunovsky VA:
eIF4E-mediated translational control of cancer incidence. Biochim
Biophys Acta. 1849:774–780. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Chen B, Zhang B, Xia L, Zhang J, Chen Y,
Hu Q and Zhu C: Knockdown of eukaryotic translation initiation
factor 4E suppresses cell growth and invasion, and induces
apoptosis and cell cycle arrest in a human lung adenocarcinoma cell
line. Mol Med Rep. 12:7971–7978. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Sanli T, Rashid A, Liu C, Harding S,
Bristow RG, Cutz JC, Singh G, Wright J and Tsakiridis T: Ionizing
radiation activates AMP-activated kinase (AMPK): A target for
radiosensitization of human cancer cells. Int J Radiat Oncol Biol
Phys. 78:221–229. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Muaddi H, Chowdhury S, Vellanki R, Zamiara
P and Koritzinsky M: Contributions of AMPK and p53 dependent
signaling to radiation response in the presence of metformin.
Radiother Oncol. 108:446–450. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Barker HE, Paget JT, Khan AA and
Harrington KJ: The tumour microenvironment after radiotherapy:
Mechanisms of resistance and recurrence. Nat Rev Cancer.
15:409–425. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Levy A, Nigro G, Sansonetti PJ and Deutsch
E: Candidate immune biomarkers for radioimmunotherapy. Biochim
Biophys Acta Rev Cancer. 1868:58–68. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Levy A, Bardet E, Lacas B, Pignon JP, Adam
J, Lacroix L, Artignan X, Verrelle P and Le Péchoux C: A phase II
Open-label multicenter study of gefitinib in combination with
irradiation followed by chemotherapy in patients with inoperable
stage III non-small cell lung cancer. Oncotarget. 8:15924–15933.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Twyman-Saint Victor C, Rech AJ, Maity A,
Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi
PM, et al: Radiation and dual checkpoint blockade activate
non-redundant immune mechanisms in cancer. Nature. 520:373–377.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Tokito T, Azuma K, Kawahara A, Ishii H,
Yamada K, Matsuo N, Kinoshita T, Mizukami N, Ono H, Kage M, et al:
Predictive relevance of PD-L1 expression combined with CD8+ TIL
density in stage III non-small cell lung cancer patients receiving
concurrent chemoradiotherapy. Eur J Cancer. 55:7–14. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Song CW, Lee H, Dings RP, Williams B,
Powers J, Santos TD, Choi BH and Park HJ: Metformin kills and
radiosensitizes cancer cells and preferentially kills cancer stem
cells. Sci Rep. 2:3622012. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Moncharmont C, Levy A, Gilormini M,
Bertrand G, Chargari C, Alphonse G, Ardail D, Rodriguez-Lafrasse C
and Magné N: Targeting a cornerstone of radiation resistance:
Cancer stem cell. Cancer Lett. 322:139–147. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Wink KC, Belderbos JS, Dieleman EM, Rossi
M, Rasch CR, Damhuis RA, Houben RM and Troost EG: Improved
progression free survival for patients with diabetes and locally
advanced non-small cell lung cancer (NSCLC) using metformin during
concurrent chemoradiotherapy. Radiother Oncol. 118:453–459. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Ko JC, Chiu HC, Wo TY, Huang YJ, Tseng SC,
Huang YC, Chen HJ, Syu JJ, Chen CY, Jian YT, et al: Inhibition of
p38 MAPK-dependent MutS homologue-2 (MSH2) expression by metformin
enhances gefitinib-induced cytotoxicity in human squamous lung
cancer cells. Lung Cancer. 82:397–406. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Chen H, Yao W, Chu Q, Han R, Wang Y, Sun
J, Wang D, Wang Y, Cao M and He Y: Synergistic effects of metformin
in combination with EGFR-TKI in the treatment of patients with
advanced non-small cell lung cancer and type 2 diabetes. Cancer
Lett. 369:97–102. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Li KL, Li L, Zhang P, Kang J, Wang YB,
Chen HY and He Y: A multicenter Double-blind Phase II study of
metformin with gefitinib as First-Line therapy of locally advanced
non-small-cell lung cancer. Clin Lung Cancer. 18:340–343. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Nguyen GH, Murph MM and Chang JY: Cancer
stem cell radioresistance and enrichment: Where frontline radiation
therapy may fail in lung and esophageal cancers. Cancers (Basel).
3:1232–1252. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Morgillo F, Sasso FC, Della Corte CM,
Vitagliano D, D'Aiuto E, Troiani T, Martinelli E, De Vita F,
Orditura M, De Palma R, et al: Synergistic effects of metformin
treatment in combination with gefitinib, a selective EGFR tyrosine
kinase inhibitor, in LKB1 wild-type NSCLC cell lines. Clin Cancer
Res. 19:3508–3519. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Arrieta O, Barrón F, Padilla MS,
Avilés-Salas A, Ramírez-Tirado LA, Arguelles Jiménez MJ, Vergara E,
Zatarain-Barrón ZL, Hernández-Pedro N, Cardona AF, et al: Effect of
metformin plus tyrosine kinase inhibitors compared with tyrosine
kinase inhibitors alone in patients with epidermal growth factor
receptor-mutated lung adenocarcinoma: A phase 2 randomized clinical
trial. JAMA Oncol. 5:e1925532019. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Chung C: Tyrosine kinase inhibitors for
epidermal growth factor receptor gene mutation-positive non-small
cell lung cancers: An update for recent advances in therapeutics. J
Oncol Pharm Pract. 22:461–476. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Bidkhori G, Moeini A and Masoudi-Nejad A:
Modeling of tumor progression in NSCLC and intrinsic resistance to
TKI in loss of PTEN expression. PLoS One. 7:e480042012. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Li L, Han R, Xiao H, Lin C, Wang Y, Liu H,
Li K, Chen H, Sun F, Yang Z, et al: Metformin sensitizes
EGFR-TKI-resistant human lung cancer cells in vitro and in vivo
through inhibition of IL-6 signaling and EMT reversal. Clin Cancer
Res. 20:2714–2726. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Momcilovic M, McMickle R, Abt E, Seki A,
Simko SA, Magyar C, Stout DB, Fishbein MC, Walser TC, Dubinett SM,
et al: Heightening energetic stress selectively targets
LKB1-deficient non-small cell lung cancers. Cancer Res.
75:4910–4922. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Della Corte CM, Ciaramella V, Di Mauro C,
Castellone MD, Papaccio F, Fasano M, Sasso FC, Martinelli E,
Troiani T, De Vita F, et al: Metformin increases antitumor activity
of MEK inhibitors through GLI1 downregulation in LKB1 positive
human NSCLC cancer cells. Oncotarget. 7:4265–4278. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Yeh HH, Lai WW, Chen HH, Liu HS and Su WC:
Autocrine IL-6-induced Stat3 activation contributes to the
pathogenesis of lung adenocarcinoma and malignant pleural effusion.
Oncogene. 25:4300–4309. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Pan YH, Jiao L, Lin CY, Lu CH, Li L, Chen
HY, Wang YB and He Y: Combined treatment with metformin and
gefitinib overcomes primary resistance to EGFR-TKIs with EGFR
mutation via targeting IGF-1R signaling pathway. Biologics.
12:75–86. 2018.PubMed/NCBI
|
|
135
|
Li L, Wang Y, Peng T, Zhang K, Lin C, Han
R, Lu C and He Y: Metformin restores crizotinib sensitivity in
crizotinib-resistant human lung cancer cells through inhibition of
IGF1-R signaling pathway. Oncotarget. 7:34442–34452. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Bland AR, Shrestha N, Bower RL, Rosengren
RJ and Ashton JC: The effect of metformin in EML(4)-ALK+ lung
cancer alone and in combination with crizotinib in cell and rodent
models. Biochem Pharmacol. 183:1143452021. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Chen H, Lin C, Peng T, Hu C, Lu C, Li L,
Wang Y, Han R, Feng M, Sun F, et al: Metformin reduces HGF-induced
resistance to alectinib via the inhibition of Gab1. Cell Death Dis.
11:1112020. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Isozaki H, Ichihara E, Takigawa N, Ohashi
K, Ochi N, Yasugi M, Ninomiya T, Yamane H, Hotta K, Sakai K, et al:
Non-small cell lung cancer cells acquire resistance to the ALK
inhibitor alectinib by activating alternative receptor tyrosine
kinases. Cancer Res. 76:1506–1516. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Pan YH, Lin CY, Lu CH, Li L, Wang YB, Chen
HY and He Y: Metformin synergistically enhances the antitumor
activity of the third-generation EGFR-TKI CO-1686 in lung cancer
cells through suppressing NF-κB signaling. Clin Respir J.
12:2642–2652. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Mu CY, Huang JA, Chen Y, Chen C and Zhang
XG: High expression of PD-L1 in lung cancer may contribute to poor
prognosis and tumor cells immune escape through suppressing tumor
infiltrating dendritic cells maturation. Med Oncol. 28:682–688.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Miyazawa T, Marushima H, Saji H, Kojima K,
Hoshikawa M, Takagi M and Nakamura H: PD-L1 expression in
non-small-cell lung cancer including various adenocarcinoma
subtypes. Ann Thorac Cardiovasc Surg. 25:1–9. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Shen X, Zhao Y, Liu G, Zhou HL, Fan J,
Zhang L, Li YL, Wang Y, Liang J and Xu ZX: Upregulation of
programmed death ligand 1 by liver kinase B1 and its implication in
programmed death 1 blockade therapy in non-small cell lung cancer.
Life Sci. 256:1179232020. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Xia W, Qi X, Li M, Wu Y, Sun L, Fan X,
Yuan Y and Li J: Metformin promotes anticancer activity of NK cells
in a p38 MAPK dependent manner. Oncoimmunology. 10:19959992021.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Lee CK, Man J, Lord S, Cooper W, Links M,
Gebski V, Herbst RS, Gralla RJ, Mok T and Yang JC: Clinical and
molecular characteristics associated with survival among patients
treated with checkpoint inhibitors for advanced Non-small cell lung
carcinoma: A systematic review and Meta-analysis. JAMA Oncol.
4:210–216. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Kauffmann-Guerrero D, Tufman A, Kahnert K,
Bollmann BA, Reu S, Syunyaeva Z, Schneider C, Manapov F, Huber RM
and Golpon H: Response to checkpoint inhibition in Non-small cell
lung cancer with molecular driver alterations. Oncol Res Treat.
43:289–298. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Cinausero M, Laprovitera N, De Maglio G,
Gerratana L, Riefolo M, Macerelli M, Fiorentino M, Porcellini E,
Buoro V, Gelsomino F, et al: KRAS and ERBB-family genetic
alterations affect response to PD-1 inhibitors in metastatic
nonsquamous NSCLC. Ther Adv Med Oncol. 11:17588359198855402019.
View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Liu C, Zheng S, Jin R, Wang X, Wang F,
Zang R, Xu H, Lu Z, Huang J, Lei Y, et al: The superior efficacy of
anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung
cancer that correlates with an inflammatory phenotype and increased
immunogenicity. Cancer Lett. 470:95–105. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Calles A, Sholl LM, Rodig SJ, Pelton AK,
Hornick JL, Butaney M, Lydon C, Dahlberg SE, Oxnard GR, Jackman DM,
et al: Immunohistochemical Loss of LKB1 is a biomarker for more
aggressive biology in KRAS-mutant lung adenocarcinoma. Clin Cancer
Res. 21:2851–2860. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Biton J, Mansuet-Lupo A, Pécuchet N,
Alifano M, Ouakrim H, Arrondeau J, Boudou-Rouquette P, Goldwasser
F, Leroy K, Goc J, et al: TP53, STK11, and EGFR mutations predict
tumor immune profile and the response to Anti-PD-1 in lung
adenocarcinoma. Clin Cancer Res. 24:5710–5723. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Rizvi H, Sanchez-Vega F, La K, Chatila W,
Jonsson P, Halpenny D, Plodkowski A, Long N, Sauter JL, Rekhtman N,
et al: Molecular determinants of response to anti-programmed cell
death (PD)-1 and Anti-programmed Death-ligand 1 (PD-L1) blockade in
patients with Non-small-cell lung cancer profiled with targeted
Next-generation sequencing. J Clin Oncol. 36:633–641. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Skoulidis F, Goldberg ME, Greenawalt DM,
Hellmann MD, Awad MM, Gainor JF, Schrock AB, Hartmaier RJ, Trabucco
SE, Gay L, et al: STK11/LKB1 mutations and PD-1 inhibitor
resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov.
8:822–835. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Afzal MZ, Dragnev K, Sarwar T and Shirai
K: Clinical outcomes in Non-small-cell lung cancer patients
receiving concurrent metformin and immune checkpoint inhibitors.
Lung Cancer Manag. 8:LMT112019. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Kim Y, Vagia E, Viveiros P, Kang CY, Lee
JY, Gim G, Cho S, Choi H, Kim L, Park I, et al: Overcoming acquired
resistance to PD-1 inhibitor with the addition of metformin in
small cell lung cancer (SCLC). Cancer Immunol Immunother.
70:961–965. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Yendamuri S, Barbi J, Pabla S, Petrucci C,
Punnanitinont A, Nesline M, Glenn ST, Depietro P,
Papanicalou-Sengos A, Morrison C, et al: Body mass index influences
the salutary effects of metformin on survival after lobectomy for
stage I NSCLC. J Thorac Oncol. 14:2181–2187. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Tsakiridis T, Pond GR, Wright J, Ellis PM,
Ahmed N, Abdulkarim B, Roa W, Robinson A, Swaminath A, Okawara G,
et al: Metformin in combination with chemoradiotherapy in locally
advanced Non-small cell lung cancer: The OCOG-ALMERA randomized
clinical trial. JAMA Oncol. 7:1333–1341. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Syngelaki A, Nicolaides KH, Balani J, Hyer
S, Akolekar R, Kotecha R, Pastides A and Shehata H: Metformin
versus placebo in obese pregnant women without diabetes mellitus. N
Engl J Med. 374:434–443. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Okayasu S, Kitaichi K, Hori A, Suwa T,
Horikawa Y, Yamamoto M, Takeda J and Itoh Y: The evaluation of risk
factors associated with adverse drug reactions by metformin in type
2 diabetes mellitus. Biol Pharm Bull. 35:933–937. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Zhang HH and Guo XL: Combinational
strategies of metformin and chemotherapy in cancers. Cancer
Chemother Pharmacol. 78:13–26. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Mallik R and Chowdhury TA: Metformin in
cancer. Diabetes Res Clin Pract. 143:409–419. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Skuli SJ, Alomari S, Gaitsch H, Bakayoko
A, Skuli N and Tyler BM: Metformin and cancer, an ambiguanidous
relationship. Pharmaceuticals (Basel). 15:6262022. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Osama H, Sayed OM, Hussein RRS, Abdelrahim
M and A Elberry A: Design, optimization, characterization, and in
vivo evaluation of sterosomes as a carrier of metformin for
treatment of lung cancer. J Liposome Res. 30:150–162. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Goodwin J, Neugent ML, Lee SY, Choe JH,
Choi H, Jenkins DMR, Ruthenborg RJ, Robinson MW, Jeong JY, Wake M,
et al: The distinct metabolic phenotype of lung squamous cell
carcinoma defines selective vulnerability to glycolytic inhibition.
Nat Commun. 8:155032017. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Li C, Xue Y, Xi YR and Xie K: Progress in
the application and mechanism of metformin in treating Non-small
cell lung cancer. Oncol Lett. 13:2873–2880. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Chen N, Zhou YS, Wang LC and Huang JB:
Advances in metformin-based metabolic therapy for non-small cell
lung cancer (Review). Oncol Rep. 47:552022. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Arrieta O, Zatarain-Barrón ZL, Turcott JG,
Barrón F, Yendamuri S, Cardona AF and Rosell R: Association of BMI
with benefit of metformin plus epidermal growth factor
Receptor-tyrosine kinase inhibitors in patients with advanced lung
adenocarcinoma: A secondary analysis of a phase 2 randomized
clinical trial. JAMA Oncol. 8:477–479. 2022. View Article : Google Scholar : PubMed/NCBI
|