Introduction
Dysregulation of the tumor suppressor protein
retinoblastoma (Rb) is commonly found in human cancer.
Hyperphosphorylation rather than mutation of Rb is thought to
promote tumorigenesis (1). The
discovery over 20 years ago that phosphorylation of Rb by CDKs
promotes cellular proliferation led to the development of CDK
inhibitors; however, the early compounds failed due to lack of
specificity or had low affinity for CDK4/6 enzymes (2). Subsequently, three selective
inhibitors of CDK4/6 have been successful in patients. Palbociclib
(Palb), ribociclib and abemaciclib have been approved by the Food
and Drug Administration (FDA). These treatments inhibit CDK4/6 to
reduce Rb phosphorylation, exerting anti-proliferative activity in
breast cancer (3). Furthermore,
studies using preclinical models demonstrate that the efficacy of
these treatments is dependent on Rb (4). These CDK4/6 inhibitors have been
shown to benefit patients with estrogen receptor-positive,
HER2-negative advanced breast cancer (3). Unfortunately, most patients acquire
resistance to treatment with CDK4/6 inhibitors and research into
the mechanisms that trigger resistance is ongoing (5). It is thought that acquired resistance
is due to the activation of alternative growth promoting pathways
in response to the treatment. The PI3K/AKT/mTOR pathway has been
implicated in acquired resistance to targeted therapies in breast
cancer (6), and several
mechanistic studies in breast and pancreatic cancer have described
the activation of this pathway during treatment resistance
(7–13). Not only does the PI3K/AKT/mTOR
pathway stimulate cell growth, survival and proliferation, but it
also enhances tumor aggressiveness by promoting
epithelial-mesenchymal transition (EMT) (14,15).
One downstream effector of this pathway is the enzyme ATP citrate
lyase (ACLY). ACLY is activated by AKT phosphorylation at serine
455 (16). This enzyme connects
glucose and lipid metabolism as it converts excess cytosolic
citrate in cancer cells to acetyl-CoA, which is the precursor to
fatty acid and cholesterol synthesis (17). In keeping with the requirement of
cancer cells for abundant fatty acids needed for membrane
biosynthesis, cancer cells exhibit elevated ACLY activity, and ACLY
gene knockdown suppresses tumor cell growth (18,19).
Recently, lipid reprogramming has been proposed to be essential in
the mechanism of resistance to kinase inhibitors in breast cancer
(20). In 2019, an inhibitor of
ACLY, bempedoic acid (BA), was FDA-approved to reduce levels of
low-density lipoprotein cholesterol in patients (21). Thus, to target ACLY activity in
cancer cells, BA was utilized to investigate the role of ACLY in
tumorigenesis. The present study aimed to determine the combined
effect of ACLY inhibition using BA and CDK4/6 inhibition using Palb
on the proliferation and EMT/invasion of cancer cells.
Materials and methods
Cell culture
Cell culture materials were obtained from Gibco
(Thermo Fisher Scientific, Inc.), unless otherwise indicated. The
MDA-MB-231 (cat. no. HTB-26), T47D (cat. no. HTB-133), MCF7 (cat.
no. HTB-22), Panc1 (cat. no. CRL-1569), MIAPaCa-2 (cat. no.
CRL-1420) and HT-1080 (cat. no. CCL-121) cell lines were obtained
from American Type Culture Collection. All cell lines were handled
using good cell culture practices and utilized within 4 months of
receipt. MCF7 cells were grown in Eagle's Minimum Essential Medium
supplemented with 10% FBS and 1X Penicillin-Streptomycin-Glutamine
(PSG), containing 100 U/ml penicillin, 100 µg/ml streptomycin and 2
mM glutamine. All other cells used in the present study were grown
in DMEM, supplemented with 10% FBS and 1X PSG. Cells were routinely
maintained at 37°C in a humidified, 5% CO2-containing
atmosphere and were split two to three times weekly to maintain
sub-confluent cultures. Cultures were routinely tested for
mycoplasma contamination using the MycoFluor™ Mycoplasma Detection
Kit (Invitrogen; Thermo Fisher Scientific, Inc.). 3D cell culture
was performed either by placement of cells in plates pre-coated
with Matrigel (Corning, Inc.) at room temperature (RT) for 2 h
before cell addition, as previously described (22,23)
or by growing cells in ultra-low attachment plates according to the
manufacturer (Corning, Inc.). A total of 7 days after plating, cell
spheres formed and treatments (0.1% DMSO, 1 µM Palb, 25 µM BA or
the combination of Palb + BA were administered at 37°C for up to 96
h.
Western blotting
Cell extracts were prepared using MDA-MBA-231, Panc1
or HT1080 cells after washing cells in ice-cold TBS [25 mM Tris-HCl
(pH 8.0) and 150 mM NaCl], followed by cell lysis for 15 min at 4°C
in EBC buffer [50 mM Tris-HCl (pH 8.0), 120 mM NaCl and 0.5%
Nonidet P-40] containing 10 µg/ml protease inhibitors (aprotinin
and PMSF). The lysates were cleared by centrifugation at 13,000 × g
for 10 min at 4°C. Protein concentration was determined using the
Bradford protein assay. Electrophoresis was performed using 4–20%
gradient SDS-polyacrylamide gels containing 30 µg total cell
protein in each sample lane. Following electrophoresis, the
proteins were transferred to nitrocellulose membranes. Residual
protein binding sites on the nitrocellulose membranes were blocked
by incubation with TBS-0.5% Tween-20 (TBST) containing 4% non-fat
dry milk for 30–60 min at RT. Subsequently, the nitrocellulose
membranes were incubated in TBST containing 2% non-fat dry milk and
1 µg/ml primary antibody overnight at 4°C. After three washes with
TBST (5 min/wash), the nitrocellulose membranes were probed with
HRP-conjugated anti-IgG antibodies [1:2,000; cat. nos. 1031-05
(anti-mouse) and 4050-05 (anti-rabbit); SouthernBiotech] for 1.5 h
at RT and developed using enhanced chemiluminescence reagent
(Pierce; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. The following primary antibodies were used
in the present study (all 1:1,000 dilution): AKT (cat. no 4691),
phosphorylated (p)-AKT (ser473) (cat. no. 4060), ACLY (cat. no
13390), p-ACLY (ser455) (cat. no. 4331), p-Rb (ser780) (cat. no.
8180), cleaved poly (ADP-ribose) polymerase (cPARP; cat. no. 9541),
p-c-Jun (cat. no. 2361), c-Jun (cat. no. 15683), proliferating cell
nuclear antigen (PCNA; cat. no. 13110), Snail (cat. no. 3879), Slug
(cat. no. 9585), zinc finger E-box-binding homeobox 1 (ZEB1; cat.
no. 3396), E-cadherin (cat. no. 3195), N-cadherin (cat. no. 13116)
and Vimentin (cat. no. 5741) (all from Cell Signaling Technology,
Inc). In addition, β-actin (cat. no. A1978; Sigma-Aldrich; Merck
KGaA), cyclin D3 (cat. no. 610279; BD Biosciences), Rb (cat. no.
sc-102) and minichromosome maintenance complex component 7 (Mcm7;
cat. no. sc-9966) (both from Santa Cruz Biotechnology, Inc.) were
used. Immunoblotting results were quantified using ImageJ software
v0.5.6 (National Institutes of Health).
Transfection
MDA-MB-231 or Panc1 cells were transfected with AKT
small interfering RNA (cat. no. 6211; Cell Signaling Technology,
Inc.) or NT RNA as a control (cat. no. 6568; Cell Signaling
Technology, Inc.) at 100 nM at 37°C for 48 h prior to cell lysis,
using Lipofectamine RNAiMAX transfection reagent (Invitrogen;
Thermo Fisher Scientific, Inc.). Transfections were performed
according to the manufacturer's instructions.
Viability, proliferation and apoptosis
assays
Palb (PD-0332991; cat. no. S1116) and BA (ETC-1002;
cat. no. S7953) were obtained from Selleck Chemicals, and were used
within 3 months of receipt. BA was dissolved in DMSO, while Palb
was dissolved in water. Dose-response curves were generated to
identify the concentration of each drug required to reduce cell
numbers between 20–30% after a 96-h treatment at 37°C compared with
cells treated with water or DMSO (data not shown). The suitable
concentrations determined were 1 µM Palb and 25 µM BA. MCF7 or
MDA-MB-231 cells (6,000/well), T47D cells (12,000/well) and Panc1
or MIAPaCa-2 cells (3,000/well) were plated in 96-well plates and
allowed to proliferate for 24 h. Cells were treated at 37°C for 96
h with full culture medium as control, DMSO (0.1%) or Palb or BA
alone or in combination with n=6. To measure viability, the
following assays were employed (all from Promega Corporation; all
assays performed with n=6 or n=8): CellTiter-Fluor Cell Viability
Assay (cat. no. G6080), utilized on T47D, MCF7, MDA-MBA-231, Panc1
and MiaPaCa2 cells; RealTime-Glo MT Cell Viability assay (cat. no.
G9711), utilized on MDA-MBA-231 and Panc1 cells; or the
CellTiter-Glo 3D Cell Viability Assay (cat. no. G9681), utilized on
MDA-MBA-231 cells. To determine toxicity and apoptosis, additional
Promega assays were employed: the CellTox Green Cytotoxicity Assay
(cat. no. G8741), utilized on MDA-MBA-231 cells; and the
RealTime-Glo Annexin V Apoptosis and Necrosis Assay (cat. no.
JA1000), utilized on T47D and MDA-MBA-231 cells. The Annexin V
assay was utilized to measure apoptosis 48 h after addition of the
aforementioned agents and presented as Annexin V (absorbance)/cell
number (fluorescence). The cell number was determined using the
CellTiter-Fluor Cell Viability Assay. Acetyl-CoA (Sigma-Aldrich;
Merck KGaA) was used to show target validation of BA in MDA-MBA-231
cells in CellTox Green Cytotoxicity assays at a concentration of 1
mM and was administered at the time of BA addition at 37°C.
Proliferation was monitored using the 5-bromo-2-deoxyuridine (BrdU)
Cell Proliferation Assay Kit (cat. no. 6813; Cell Signaling
Technology, Inc.). In the BrdU assay, cells were treated with the
aforementioned agents for 72 h, and cells were fixed using 4%
formaldehyde in PBS for 30 min at RT and a mouse monoclonal
antibody to BrdU (dilution, 1:1,000) was applied, followed by the
use of an anti-mouse HRP-linked antibody (dilution, 1:1,000) that
recognizes the bound detection antibody. The HRP substrate
3,3′,5,5′-tetramethylbenzidine was added to develop color. The
magnitude of the absorbance (450 nm) corresponding to the developed
color is proportional to the quantity of BrdU incorporated into
cells, which is a direct indication of cell proliferation. All
assays were performed as directed by the manufacturer.
Invasion assays
HT1080, Panc1 or MDA-MB-231 cells were treated with
the 0.1% DMSO, 25 µM BA, 1 µM Palb or the combination of BA + Palb
for 24 h, washed in serum-free medium, counted, and 50,000 cells
were plated in 24-well plate Transwell chamber inserts (pore size,
8.0 µm) that were pre-coated with Matrigel at RT for 2 h before
cell addition, while the lower compartment contained medium in the
absence or presence of FBS as a chemoattractant. After incubation
for 24 h at 37°C, cells on the upper surface of the filter were
removed and cells that were motile or invasive on the lower surface
of the filter were quantified by staining with 10 µM Calcein AM
(cat. no. C3100MP; Invitrogen; Thermo Fisher Scientific, Inc.) for
30 min at 37°C followed by measurement of fluorescence using a
microplate reader (GloMax; Promega Corporation).
Statistical analysis
All experiments performed in the present study were
conducted two to three times. Numerical data are presented as the
mean ± standard deviation. All data from each experiment were
analyzed with either unpaired Student's t-test for the comparison
between two groups or with one-way analysis of variance and Tukey's
post hoc test for multiple comparisons using SPSS 19.0 software
(IBM Corp.) as indicated. P<0.05 was considered to indicate a
statistically significant difference.
Results
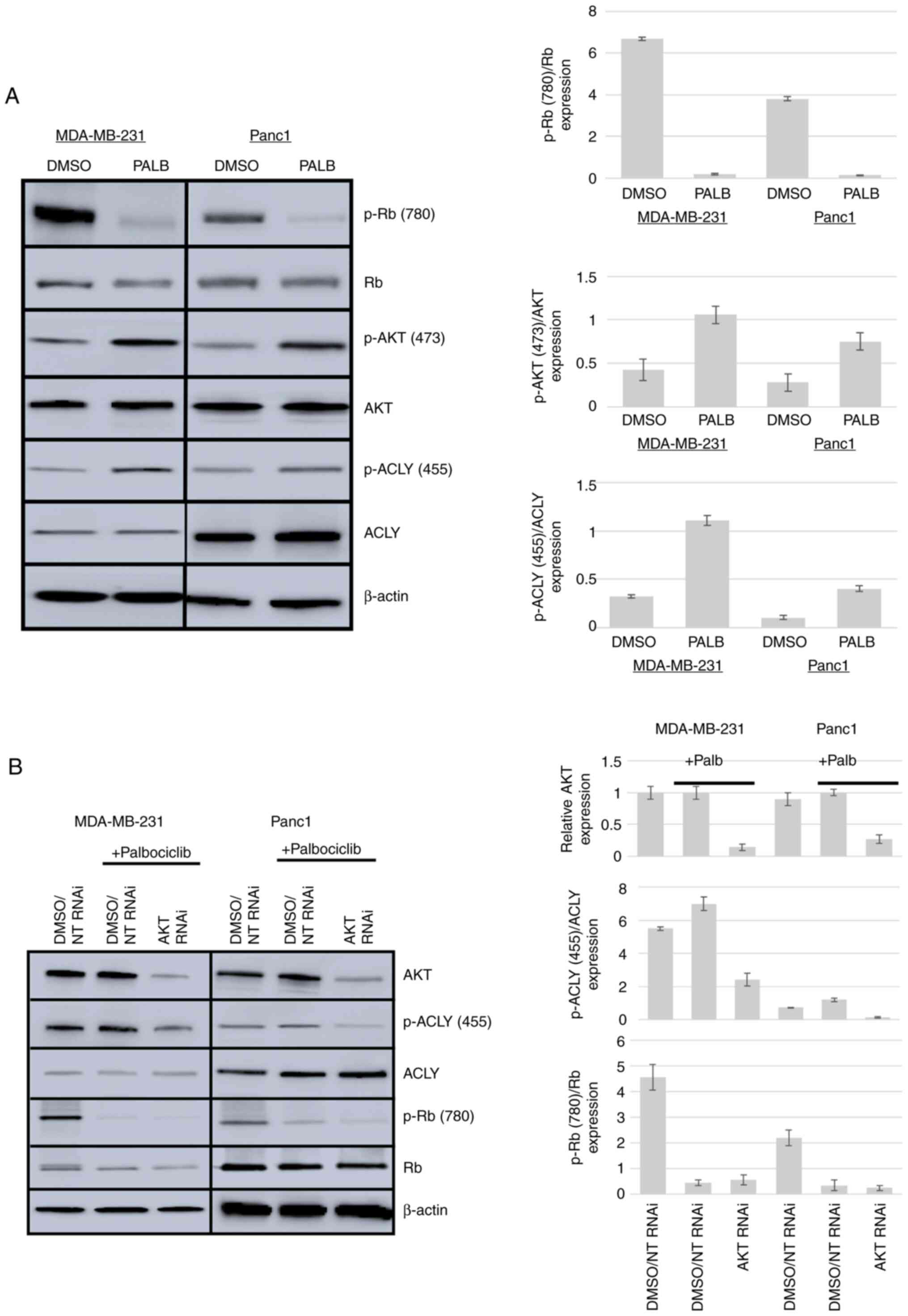
Palb activates ACLY via AKT
To determine whether ACLY, a substrate of AKT, was
activated in response to Palb treatment, MDA-MB-231 cells as a
model of breast cancer and Panc1 cells as a model of pancreatic
cancer were used. These cell types were treated with 1 µM Palb and
CDK4/6 inhibition was verified by measuring the phosphorylation of
Rb. Phosphorylation of AKT at Ser473 and ACLY at Ser455 was
evaluated by western blotting to reveal activation of these
enzymes. Fig. 1A shows that both
AKT and ACLY were activated in MDA-MB-231 and Panc1 cells in
response to CDK4/6 inhibition. In MDA-MB-231 cells, Palb caused a
92% reduction in Rb phosphorylation at amino acid (aa) 780, a
2.6-fold increase in AKT phosphorylation at aa 473 and a 3.6-fold
increase in ACLY phosphorylation at aa 455 relative to pan levels
of AKT and ACLY. In Panc1 cells, Palb caused a 90% reduction in Rb
phosphorylation, a 3-fold increase in AKT phosphorylation at aa 473
and a 4-fold increase in ACLY phosphorylation at aa 455 relative to
the levels of AKT and ACLY, respectively. Furthermore, the
activation of ACLY stimulated by Palb was dependent on AKT
expression in both cell types. In MDA-MB-231 cells, knockdown of
AKT resulted in an 86% reduction of AKT expression and a 55%
decrease in ACLY activation compared with the control. In Panc1
cells, knockdown of AKT resulted in a 73% reduction of AKT
expression and a 68% decrease in ACLY activation compared with the
control (Fig. 1B).
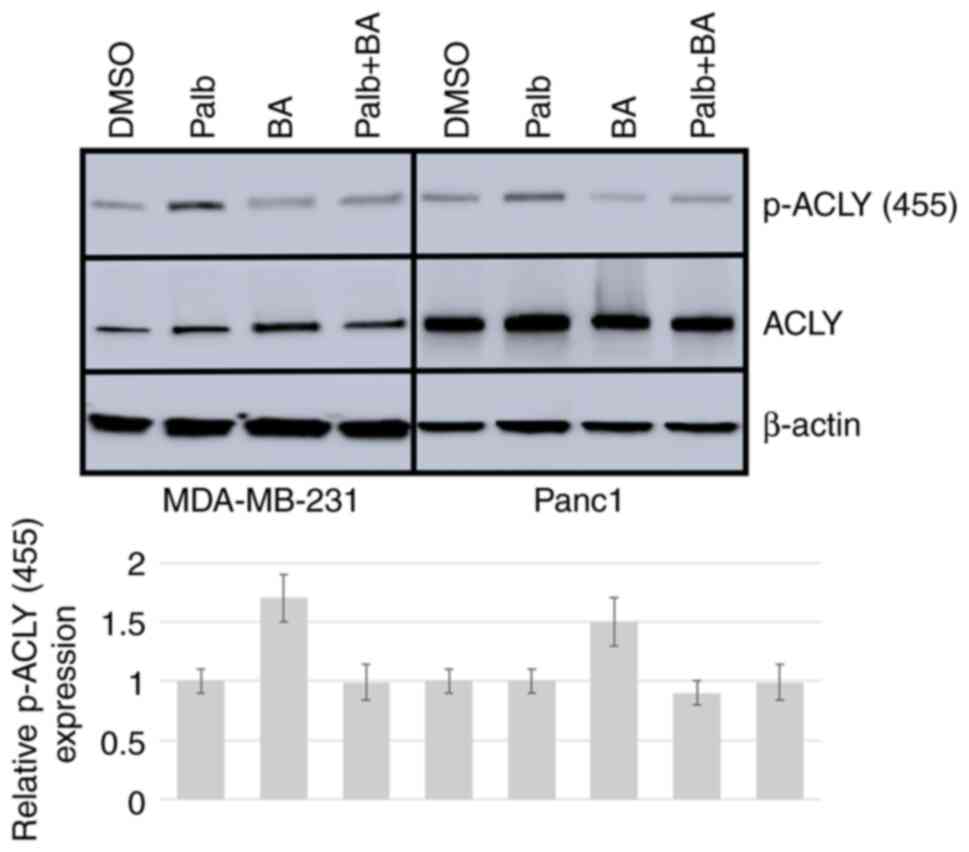
Palb in combination with BA reduces
cancer cell viability
To determine whether ACLY inhibition could block the
Palb-induced activation of ACLY, cells were treated with BA, Palb
or their combination and evaluated the status of ACLY by western
blotting. In both cell types, it was observed that BA reduced the
activation of Palb-induced stimulation of ACLY (Fig. 2). To further investigate the effect
of inhibiting the Palb-mediated ACLY activation in cancer cells,
time course experiments of cell viability measurement using
MDA-MB-231 and Panc1 cells were performed. Fig. 3A and B shows that cells treated
with Palb and BA were significantly less viable that cells treated
with either treatment alone.
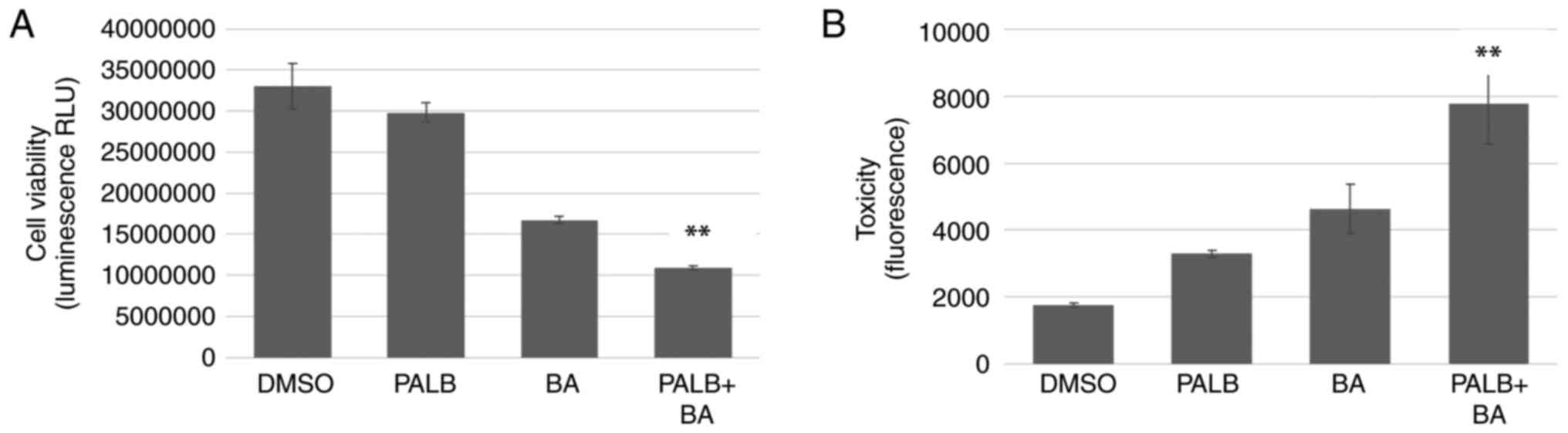
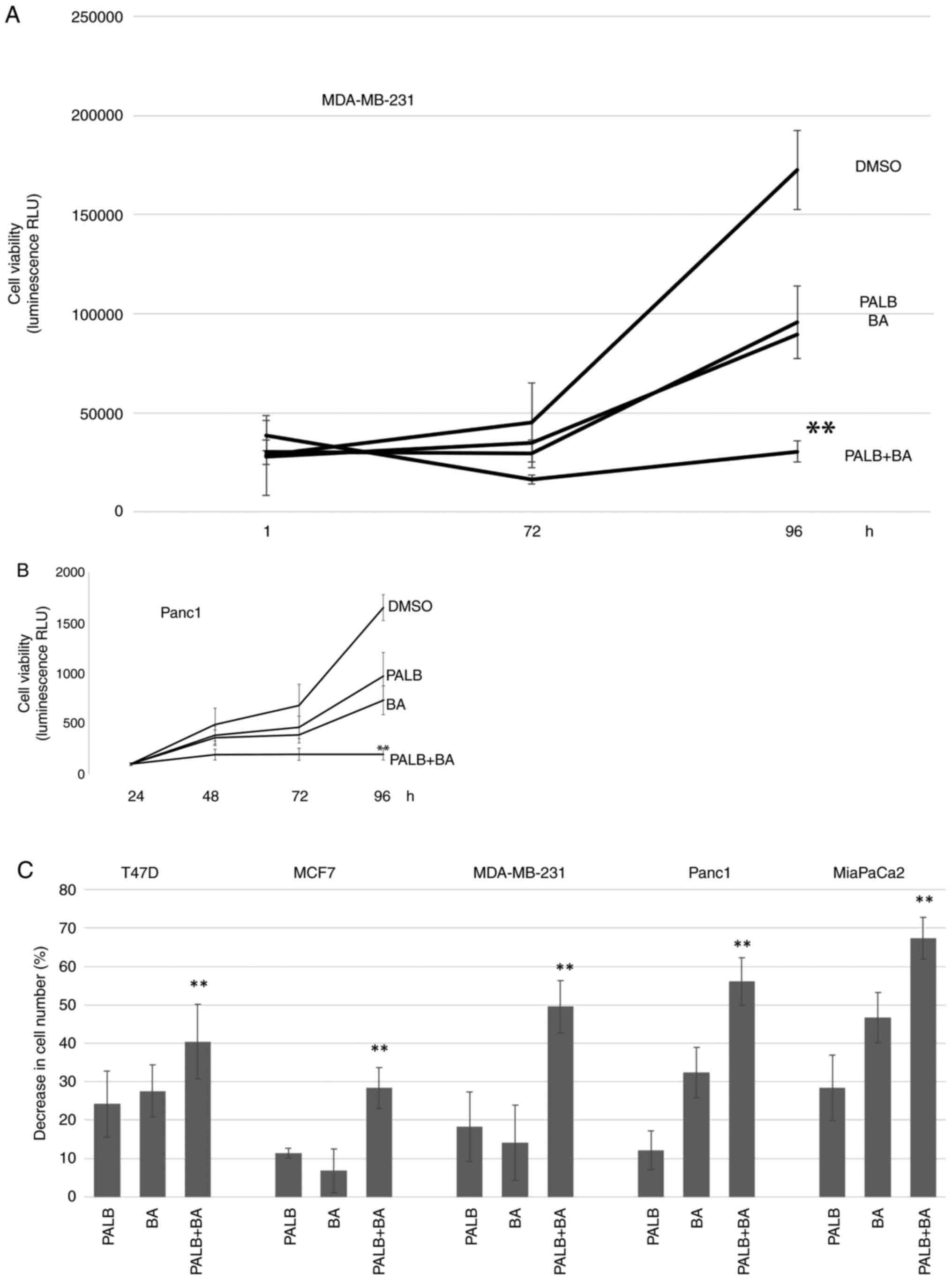 | Figure 3.The combination of Palb and BA
reduces cell viability further than either drug alone. (A)
MDA-MB-231 or (B) Panc1 cells were treated with 0.1% DMSO, 1 µM
Palb, 25 µM BA or the combination of Palb + BA for 96 h. Cell
viability was determined in MDA-MB-231 cells at 1, 72 and 96 h and
in Panc1 cells at 24, 48, 72 and 96 h after drug addition, using
the RealTime-Glo MT Cell Viability assay that measures luminescence
or RLU. Experiments were repeated twice with n=6 Error bars
represent standard deviation of the mean. One-way ANOVA (of 96-h
values) with Tukey's post hoc test was used to determine
statistical significance. **P<0.01 compared with Palb or BA
treatment alone. (C) A panel of breast and pancreatic cancer cell
lines were used in cell viability experiments. The CellTiter-Fluor
Cell Viability Assay was used on cells after treatment with 0.1%
DMSO, 1 µM P or 25 µM BA or the combination P + BA for 96 h. The
average percent decrease in cell number compared with controls is
shown and error bars represent standard deviation of the mean.
Experiments were performed on each cell type three times with n=6.
**P<0.01 compared with Palb or BA treatment alone. Palb,
palbociclib; BA, bempedoic acid; RLU, relative light units. |
The analysis was then expanded to include a panel of
breast and pancreatic cancer cell lines. While Palb and BA had
varied efficacies depending on the cell line, the combination of
Palb and BA reduced the cell number to a greater extent compared
with the effect on the cell number of either treatment alone in all
cell types analyzed. Fig. 3C shows
increased depletion of cell number in breast and pancreatic cell
lines treated with Palb and BA compared with either treatment
alone. These results were extended by using MDA-MB-231 cells grown
in 3D culture. Cells allowed to form 3D structures more closely
recapitulate the tumor environment and represent a good model of
in vivo conditions (24).
Cell viability and toxicity assays performed using MDA-MB-231 cells
grown in 3D culture demonstrated that the combination of Palb and
BA reduced cell viability and increased toxicity compared with
either treatment alone (Fig.
4).
Palb inhibits proliferation, while BA
induces apoptosis
To determine the mechanism of action of Palb and BA
in these experiments, proliferation and apoptosis in cancer cells
treated with Palb or BA were measured. As shown in Fig. 5A, BrdU incorporation assays
indicated that Palb inhibited proliferation while BA had no effect
in both Panc1 and MDA-MBA-231 cells. Conversely, Annexin V assays
revealed that BA induced apoptosis whereas Palb did not (Fig. 5B). To verify the target specificity
of BA, the catalytic product of the ACLY enzyme, acetyl-CoA, was
shown to block toxicity induced by BA in MDA-MBA-231 cells
(Fig. 5C). Finally, the expression
of markers of proliferation and apoptosis were evaluated by western
blotting after cells were treated with either Palb or BA. In
agreement with the results shown in Fig. 5A and B, cyclin D3 expression was
reduced by Palb. In addition, the proliferation markers Mcm7 and
PCNA showed reduced expression in response to Palb, but were
unaffected by BA. After BA treatment, the markers of apoptosis
cPARP and p-c-Jun were induced; however, cPARP was not induced by
Palb (Fig. 5D).
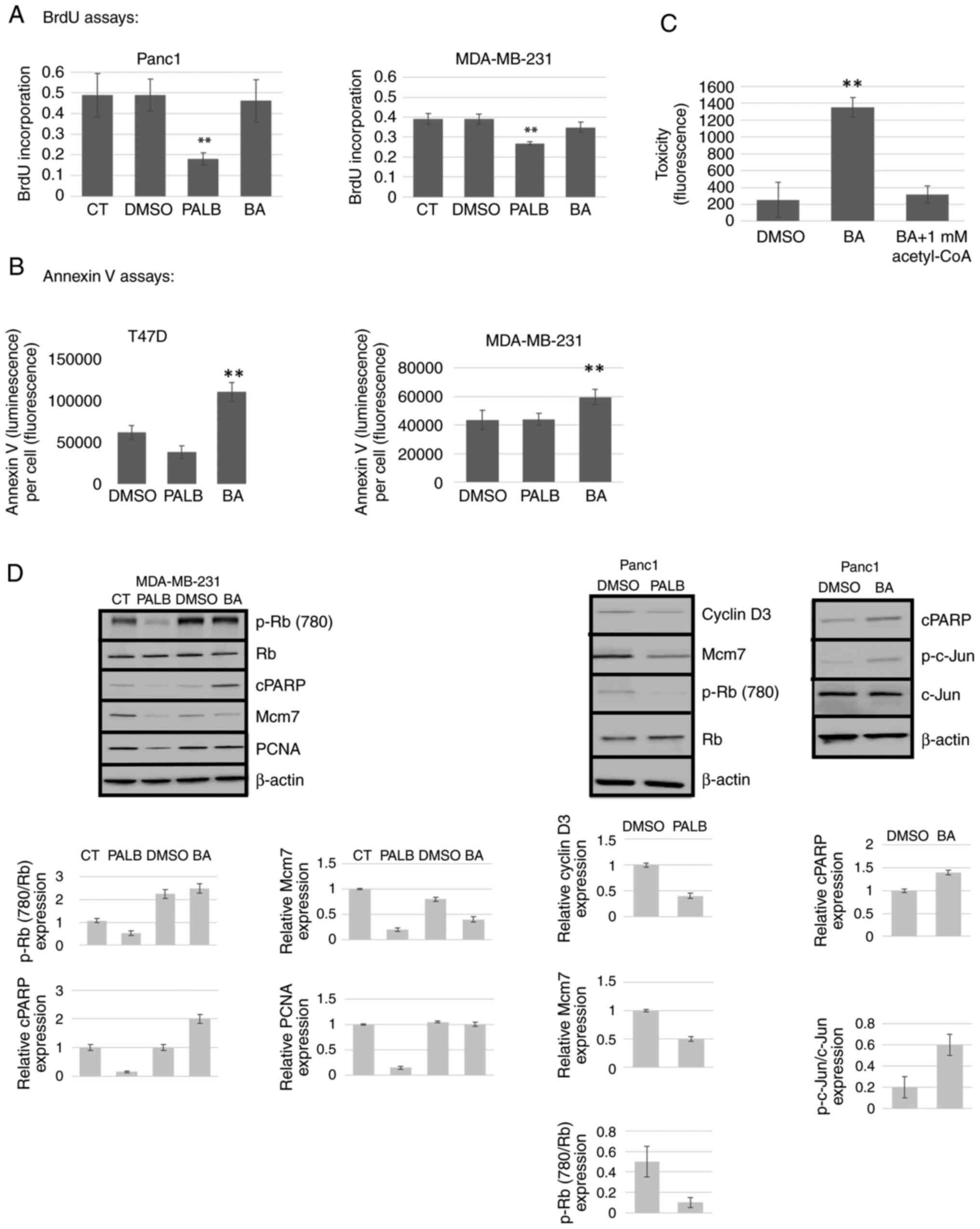 | Figure 5.Palb inhibits proliferation and BA
induces apoptosis. MDA-MB-231, Panc1 and T47D cells were used to
analyze the mechanistic effects of P (1 µM) or BA (25 µM). Culture
medium or 0.1% D were used as controls. (A) The BrdU cell
proliferation assay was performed after drug treatments for 72 h.
(B) The RealTime-Glo Annexin V assay was utilized to measure
apoptosis 48 h after addition of drug and presented as Annexin V
(absorbance)/cell number (fluorescence). Cell number was determined
using the CellTiter-Fluor Cell Viability Assay. (C) Target
validation of BA. The product of the ACLY enzyme (acetyl-CoA) was
shown to reverse toxicity induced by BA using the CellTox Green
Cytotoxicity Assay. Error bars display standard deviation of the
mean of n=8. Statistical analysis was performed using one-way ANOVA
with Tukey's post hoc test. **P<0.01 compared with all other
treatments. (D) Western blotting was performed on cells treated for
96 h as aforementioned using antibodies to p-Rb (780), total Rb,
apoptosis markers cPARP and p-c-Jun and the proliferation markers
Mcm7, cyclin D3 and PCNA. β-actin was used as a loading control.
Results shown were repeated twice. CT, control; p, phosphorylated;
cPARP, cleaved poly (ADP-ribose) polymerase; PCNA, proliferating
cell nuclear antigen; BA, bempedoic acid; Palb, palbociclib; ACLY,
ATP citrate lyase; BrdU, 5-bromo-2-deoxyuridine; Rb,
retinoblastoma; Mcm7, minichromosome maintenance complex component
7. |
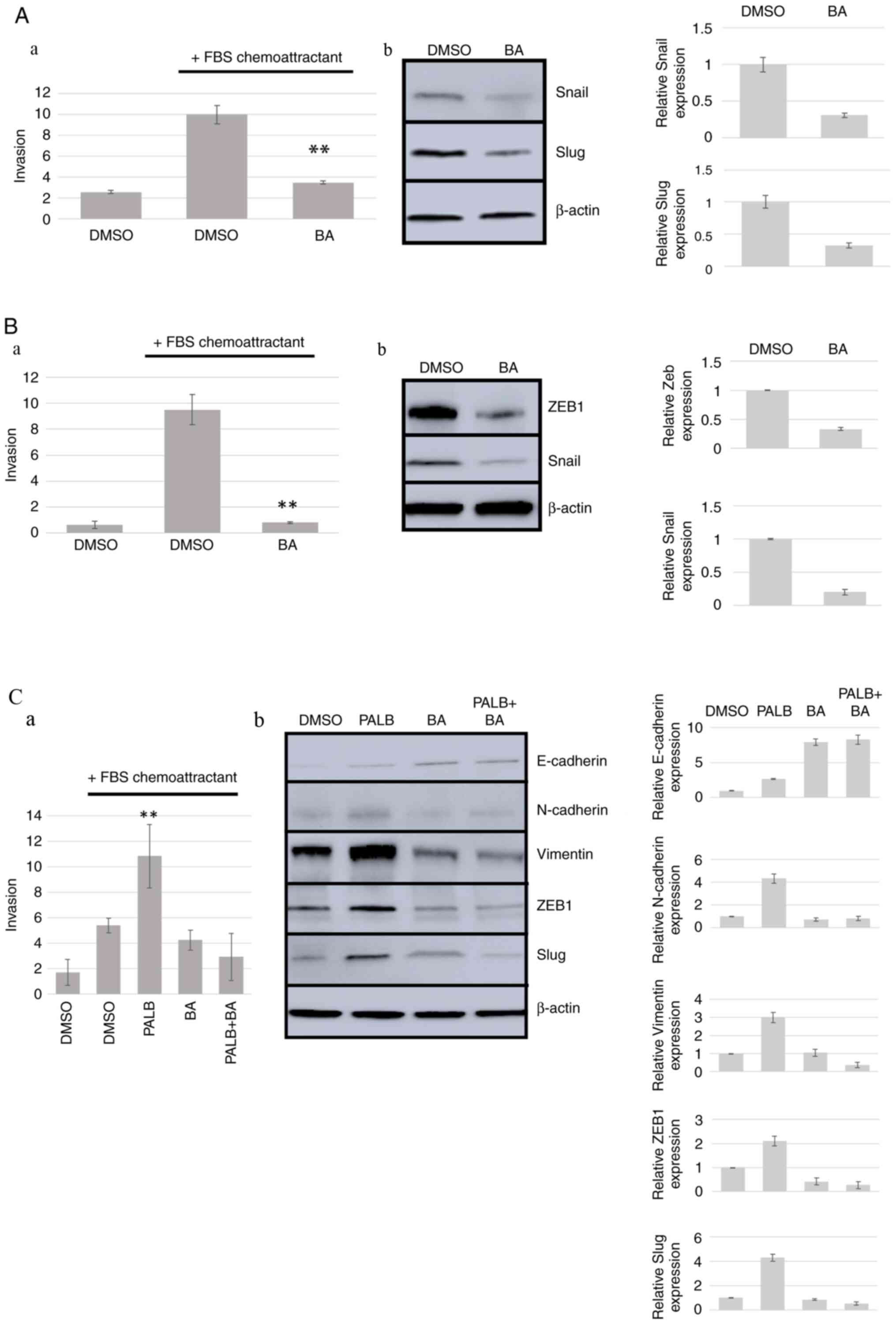
ACLY inhibition reduces cell
invasion
Since ACLY activity is required for fatty acid
synthesis, and fatty acid synthesis is needed for cell membrane
reorganization that occurs in EMT/invasion (25), we hypothesized that ACLY inhibition
may block cancer cell invasion. To evaluate the effect of BA on
cancer cell invasion, the criteria published in 2020 establishing
that conclusions regarding EMT and invasiveness can only be
confirmed by the existence of changes in expression of molecular
markers in addition to changes in cellular properties were followed
(26). Thus, two separate methods
were utilized in the present analysis to evaluate EMT, invasion
assays and western blotting detection of epithelial or mesenchymal
markers. Firstly, the highly invasive fibrosarcoma cell line HT1080
was used. Transwell invasion assays using FBS as a chemoattractant
revealed that treatment of these cells with BA resulted in
significant inhibition of invasion compared with DMSO control and a
decrease in the expression of the mesenchymal markers Snail by 69%
and Slug by 70% compared with DMSO control (Fig. 6A). Similarly, ACLY inhibition in
Panc1 cells blocked invasion and a decrease in the expression of
the mesenchymal markers ZEB1 by 67% and Snail by 80% compared with
control was observed (Fig. 6B).
Finally, the MDA-MB-231 breast cancer cell line exhibited an
increase in invasion when treated with 1 mM Palb that inhibits Rb
phosphorylation in these cells (Figs.
1 and 2) accompanied by
increased expression of the mesenchymal markers N-cadherin (76%),
Vimentin (67%), ZEB1 (52%) and Slug (77%) when compared with DMSO
controls (Fig. 6C). E-cadherin, an
epithelial cell marker, exhibited a 51% increase in expression in
response to BA treatment. Thus, BA treatment reversed the
Palb-induced stimulation of invasion, and ACLY may be a reasonable
target in strategies aimed at reversing AKT-mediated treatment
resistance.
Discussion
Over the last few decades, advances in the
understanding of the crucial molecular mechanisms responsible for
cancer cell signaling have elucidated the importance of the
development of small molecule kinase inhibitors to target specific
oncoproteins that fuel tumorigenesis (27). Kinase inhibition is a useful tool
to specifically focus on a particular biochemical abnormality.
Thus, as of 2022, there are 68 FDA approved kinase inhibitors used
clinically to treat several types of neoplastic disease (27).
As efficacious as kinase inhibitors as a class have
been, a common theme among patients with cancer treated with
small-molecule inhibitors is the development of drug resistance.
There are two mechanisms through which resistance occurs. Lack of
treatment response to therapy is referred to as primary resistance,
whereas resistance that occurs after an initial response to therapy
and while still in treatment is known as acquired resistance. In
the case of acquired resistance, tumors are thought to develop
alternate mechanisms to evade the efficient blockade of cancer
progression (28). The development
of acquired drug resistance remains a major limitation and threat
to the successful management of advanced cancer, and acquired
resistance to the new class of CDK4/6 inhibitors is a significant
clinical hurdle (5). These kinase
inhibitors were developed to target phosphorylation of Rb, as its
inactivation by phosphorylation drives cell cycle progression.
Currently, CDK4/6 inhibitors are widely utilized to treat estrogen
receptor-positive, HER2-negative breast cancer in combination with
hormone therapies, leading to doubling of the progression free
survival compared with hormone therapy plus placebo. In addition,
clinical trials testing the use of CDK4/6 inhibitors in several
other cancer cell types (lung, pancreatic, colorectal, prostate,
glioblastoma and squamous cell carcinoma of the head and neck) are
presently underway (clinicaltrials.gov).
While CDK4/6 inhibition can have favorable effects
in the clinic, most patients will experience disease progression
while on treatment, probably through activation of alternate growth
promoting pathways to acquire resistance (3). Preclinical studies have indicated
that resistance to CDK4/6 inhibition may involve various pathways,
one of which is the PI3K/AKT/mTOR pathway (4). This pathway mediates tumorigenic
processes, such as increased proliferation and alterations in
metabolism (29) Reprogrammed
metabolism is considered a hallmark of cancer (30). Under aerobic conditions, normal
cells convert glucose to pyruvate during glycolysis in the cytosol,
and pyruvate is further metabolized in the mitochondria to generate
most of the cellular ATP.
Several oncogenes have been implicated in
stimulating glycolysis in tumor cells (29,31).
For example, activation of the PI3K/AKT/mTOR pathway is a major
contributor to carcinogenesis, both as a driving factor and as a
mediator of resistance. AKT exerts a diverse collection of
biological effects due to its varied substrates. As such, AKT can
promote cell growth, survival, proliferation and alteration of
metabolic processes that confer an advantage in tumorigenesis. In
fact, the AKT signaling network interconnects oncogenic activity
and cancer metabolism (29), as it
stimulates glucose transport at the plasma membrane to enhance
glucose uptake and glycolysis (32,33).
Thus, AKT and glycolysis collaborate to stimulate the biosynthesis
of lipids that are needed for cell growth and proliferation.
The class of biomolecules called lipids are derived
from fatty acids, and include sterols, mono-, di- and
triglycerides, phospholipids and glycolipids. While somatic cells
obtain lipids from diet or from synthesis in the liver, cancer
cells require an abundance of fatty acids for rapid production of
the cell membrane (34). Lipid
metabolism is often altered in cancer cells, such that de
novo lipogenesis is stimulated making tumor cells impervious to
externally available lipids (35).
Increased lipogenesis has been shown to be required for tumor
growth (34,36). Furthermore, de novo fatty
acid synthesis has been associated with resistance to
chemotherapies (37). In addition,
the metabolic reprogramming of lipids is a newly recognized
mechanism of resistance in breast cancer (20).
In cancer cells, cytoplasmic citrate is cleaved into
acetyl-CoA and oxaloacetate by ACLY (38). Acetyl-CoA is the precursor for the
synthesis of fatty acids, cholesterol and isoprenoids. Not only
does AKT activation result in increased glycolysis and cytosolic
citrate production, AKT directly activates ACLY by phosphorylation
to promote fatty acid synthesis in cancer cells (16,39).
Recently ACLY has been identified as a target with great
therapeutic potential in cancer treatment (17). One of the early studies that
predicted the importance of lipid metabolism in cancer demonstrated
that ACLY exhibited a 160-fold increase in activity in breast
cancer tumors compared with normal cells (40). Abnormally high levels of ACLY
activity have been observed and are correlated with poor prognosis
in several tumor types, such as non-small cell lung, colorectal,
renal, ovarian, prostate, bladder and hepatocellular carcinoma and
glioblastoma (17). Various cancer
cell types, such as A549 lung adenocarcinoma, MCF7 breast cancer,
LNCaP and DU145 prostate adenocarcinoma cells depend on ACLY
activity for proliferation (18,19,39,41).
Thus, we hypothesized that by inhibition of ACLY it may be possible
to reverse the resistance pathway stimulated by AKT in response to
CDK4/6 inhibition. The present study examined the effects of the
combined inhibition of CDK4/6 and ACLY on cell proliferation,
apoptosis and invasiveness of several cancer cell types.
The data described in the present study showed that
CDK4/6 inhibition in cancer cells using Palb caused an increase in
the activation of ACLY, which was mediated by AKT. To the best of
our knowledge, this is the first study showing that CDK4/6
inhibition can lead to activation of this enzyme, and therefore may
be involved in the altered metabolism observed in cells with
reduced CDK4/6 activity (7,42).
Increased ACLY activity due to CDK4/6 inhibition was reversed using
BA. Furthermore, the combination of CDK4/6 inhibition with BA
reduced cell viability more efficaciously than either treatment
alone. This finding applied to several breast and pancreatic cancer
cell types and was observed in breast cancer cells grown in 3D
culture. The reduced cell number was shown to be due to CDK4/6
inhibition reducing proliferation, while ACLY inhibition using BA
caused apoptosis. The combination of reduced proliferation with
increased cell death accounts for the observed decrease in cell
number and viability. It should be noted that these results may be
explained in part by the fact that inhibition of synthesis of
acetyl-CoA will alter the metabolic state of the cell and affect
several processes, such as inflammation, histone acetylation and
gene expression (43,44).
In order to evaluate the invasiveness of cancer
cells treated with a combination of Palb and BA, EMT was analyzed.
In the present study, an increase in the expression of
EMT-transcription factors (TFs) N-cadherin, ZEB1, Vimentin and Slug
in breast cancer cells in response to Palb was observed, suggesting
that CDK4/6 inhibition may affect EMT. These experiments were
undertaken due to the conflicting available information regarding
the effect of CDK4/6 inhibition on invasion. The effect of CDK4/6
inhibition generally and Palb specifically on cancer cell
invasiveness and metastasis is unclear. Interestingly, the phase 3
PALbociclib CoLlaborative Adjuvant Study (PALLAS) clinical trial
measured invasive disease-free survival in patients with estrogen
receptor-positive, HER2-negative breast cancer treated with Palb in
addition to standard adjuvant endocrine therapy. It was reported in
early 2022 that Palb is unlikely to improve patient outcomes over
endocrine therapy alone (45).
With an endpoint of metastatic disease, Palb appears not to improve
patient response. This finding elicits the question of how CDK4/6
inhibition affects cancer cell invasiveness. Preclinical studies,
to date, are conflicting in their findings. In studies that measure
invasiveness or EMT in cancer cells treated with Palb, using short
(<48 h) CDK4/6 inhibitor treatments leads to a decrease in EMT
or invasiveness, while cell treatment for longer time periods (3–8
days) results in a stimulatory effect on invasiveness in response
to Palb treatment (46–48). Interestingly, in a short 24-h Palb
treatment of pancreatic cancer cells, Palb caused inhibition of
EMT; however, this duration of treatment did not lead to activation
of the AKT pathway (47).
Similarly, in MDA-MB-231 breast cancer cells a 48-h treatment of
Palb caused a decrease in ZEB1 protein and inhibition of invasion
(48). However, as observed in the
present study using MDA-MB-231 breast cancer cells, longer
treatment with Palb caused increased expression of the mesenchymal
transcription factor ZEB1 and increased invasiveness.
In agreement with the current results, a study using
treatments that lasted between 72 h and 8 days in pancreatic cancer
cells showed a significant increase in invasion and EMT (46). Thus, it may be that Palb treatment
of a sufficiently longer duration leads to increased invasiveness,
which may be a contributor to Palb-mediated acquired resistance. To
test the effect of ACLY inhibition on cancer cells, HT1080 cells, a
highly invasive fibrosarcoma cell line, was used. It was observed
that ACLY inhibition reduced the invasiveness of HT1080 cells, as
well as that of the pancreatic cell line Panc1. In MDA-MB-231
breast cancer cells, invasion was stimulated by CDK4/6 inhibition,
whereas ACLY inhibition reversed the increase in the observed
invasiveness.
Interestingly, EMT has been implicated as a factor
in the mechanism of resistance. The loss of E-cadherin has been
associated with resistance to growth factor and kinase inhibition.
In non-small cell lung cancer, mesenchymal cell lines (defined as
expressing N-cadherin and Vimentin) exhibited marked resistance to
targeted therapies to the EGFR and the PI3K/AKT pathways compared
with that of epithelial cell lines (expressing high levels of
E-cadherin) (49). The mechanism
of resistance is postulated to occur via EMT-TF-mediated
suppression of apoptosis (50).
Thus, the present results cannot rule out the possibility that an
increase in the expression of EMT-TFs in breast cancer in response
to CDK4/6 inhibition may contribute to acquired resistance. As BA
is a prodrug requiring activation by acyl-CoA synthetase very long
chain family member 1, it is essential to note that this enzyme is
expressed in breast and pancreatic cancer cells (51,52).
In addition, although both Palb and BA are currently utilized to
treat patients with cancer or high cholesterol, the combination of
these treatments has not been tested in animal models to date, to
the best of our knowledge. Thus, additional studies are needed to
further elucidate the usefulness and safety of the combination
strategy utilized in the present study in the clinical setting.
Acknowledgements
Not applicable.
Funding
The present work was supported by the National Cancer Institute
of the National Institutes of Health (grant no. R15CA231372).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
NK initiated the work, performed the apoptosis and
cell proliferation experiments, analyzed data and wrote the
manuscript with BV and CP. BV, KC and RK designed, performed and
analyzed the cell viability experiments. KD designed, performed and
analyzed the AKT knockdown and ACLY activation by western blotting.
CP and DK designed, performed and analyzed the invasion experiments
(Transwell assays and western blotting). NK and BV confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mittnacht S: The retinoblastoma
protein-from bench to bedside. Eur J Cell Biol. 84:97–107. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Asghar U, Witkiewicz AK, Turner NC and
Knudsen ES: The history and future of targeting cyclin-dependent
kinases in cancer therapy. Nat Rev Drug Discov. 14:130–146. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guarducci C, Bonechi M, Boccalini G,
Benelli M, Risi E, Di Leo A, Malorni L and Migliaccio I: Mechanisms
of resistance to CDK4/6 inhibitors in breast cancer and potential
biomarkers of response. Breast Care (Basel). 12:304–308. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hamilton E and Infante JR: Targeting
CDK4/6 in patients with cancer. Cancer Treat Rev. 45:129–138. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McCartney A, Migliaccio I, Bonechi M,
Biagioni C, Romagnoli D, De Luca F, Galardi F, Risi E, De Santo I,
Benelli M, et al: Mechanisms of resistance to CDK4/6 inhibitors:
Potential implications and biomarkers for clinical practice. Front
Oncol. 9:6662019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guerrero-Zotano A, Mayer IA and Arteaga
CL: PI3K/AKT/mTOR: Role in breast cancer progression, drug
resistance, and treatment. Cancer Metastasis Rev. 35:515–524. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Franco J, Balaji U, Freinkman E,
Witkiewicz AK and Knudsen ES: Metabolic reprogramming of pancreatic
cancer mediated by CDK4/6 inhibition elicits unique
vulnerabilities. Cell Rep. 14:979–990. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang J, Xu K, Liu P, Geng Y, Wang B, Gan
W, Guo J, Wu F, Chin YR, Berrios C, et al: Inhibition of Rb
phosphorylation leads to mTORC2-mediated activation of Akt. Mol
Cell. 62:929–942. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jansen VM, Bhola NE, Bauer JA, Formisano
L, Lee KM, Hutchinson KE, Witkiewicz AK, Moore PD, Estrada MV,
Sánchez V, et al: Kinome-wide RNA interference screen reveals a
role for PDK1 in acquired resistance to CDK4/6 inhibition in
ER-positive breast cancer. Cancer Res. 77:2488–2499. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Litchfield LM, Boehnke K, Brahmachary M,
Mur C, Bi C, Stephens JR, Sauder JM, Gutiérrez SM, McNulty AM, Ye
XS, et al: Combined inhibition of PIM and CDK4/6 suppresses both
mTOR signaling and Rb phosphorylation and potentiates PI3K
inhibition in cancer cells. Oncotarget. 11:1478–1492. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Herrera-Abreu MT, Palafox M, Asghar U,
Rivas MA, Cutts RJ, Garcia-Murillas I, Pearson A, Guzman M,
Rodriguez O, Grueso J, et al: Early adaptation and acquired
resistance to CDK4/6 inhibition in estrogen receptor-positive
breast cancer. Cancer Res. 76:2301–2313. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Franco J, Witkiewicz AK and Knudsen ES:
CDK4/6 inhibitors have potent activity in combination with pathway
selective therapeutic agents in models of pancreatic cancer.
Oncotarget. 5:6512–6525. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vora SR, Juric D, Kim N, Mino-Kenudson M,
Huynh T, Costa C, Lockerman EL, Pollack SF, Liu M, Li X, et al: CDK
4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K
inhibitors. Cancer Cell. 26:136–149. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
LoRusso PM: Inhibition of the
PI3K/AKT/mTOR pathway in solid tumors. J Clin Oncol. 34:3803–3815.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu W, Yang Z and Lu N: A new role for the
PI3K/Akt signaling pathway in the epithelial-mesenchymal
transition. Cell Adh Migr. 9:317–324. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Berwick DC, Hers I, Heesom KJ, Moule SK
and Tavare JM: The identification of ATP-citrate lyase as a protein
kinase B (Akt) substrate in primary adipocytes. J Biol Chem.
277:33895–33900. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Granchi C: ATP citrate lyase (ACLY)
inhibitors: An anti-cancer strategy at the crossroads of glucose
and lipid metabolism. Eur J Med Chem. 157:1276–1291. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hatzivassiliou G, Zhao F, Bauer DE,
Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA and
Thompson CB: ATP citrate lyase inhibition can suppress tumor cell
growth. Cancer Cell. 8:311–321. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Migita T, Okabe S, Ikeda K, Igarashi S,
Sugawara S, Tomida A, Taguchi R, Soga T and Seimiya H: Inhibition
of ATP citrate lyase induces an anticancer effect via reactive
oxygen species: AMPK as a predictive biomarker for therapeutic
impact. Am J Pathol. 182:1800–1810. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Feng WW and Kurokawa M: Lipid metabolic
reprogramming as an emerging mechanism of resistance to kinase
inhibitors in breast cancer. Cancer Drug Resist. 3:1–17.
2020.PubMed/NCBI
|
|
21
|
Ray KK, Bays HE, Catapano AL, Lalwani ND,
Bloedon LT, Sterling LR, Robinson PL and Ballantyne CM; CLEAR
Harmony Trial, : Safety and efficacy of bempedoic acid to reduce
LDL cholesterol. N Engl J Med. 380:1022–1032. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Debnath J, Muthuswamy SK and Brugge JS:
Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini
grown in three-dimensional basement membrane cultures. Methods.
30:256–268. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Egger JV, Lane MV, Antonucci LA, Dedi B
and Krucher NA: Dephosphorylation of the retinoblastoma protein
(Rb) inhibits cancer cell EMT via Zeb. Cancer Biol Ther.
17:1197–1205. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Breslin S and O'Driscoll L: The relevance
of using 3D cell cultures, in addition to 2D monolayer cultures,
when evaluating breast cancer drug sensitivity and resistance.
Oncotarget. 7:45745–45756. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Luo X, Cheng C, Tan Z, Li N, Tang M, Yang
L and Cao Y: Emerging roles of lipid metabolism in cancer
metastasis. Mol Cancer. 16:762017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang J, Antin P, Berx G, Blanpain C,
Brabletz T, Bronner M, Campbell K, Cano A, Casanova J, Christofori
G, et al: Guidelines and definitions for research on
epithelial-mesenchymal transition. Nat Rev Mol Cell Biol.
21:341–352. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Roskoski R Jr: Properties of FDA-approved
small molecule protein kinase inhibitors: A 2022 update. Pharmacol
Res. 175:1060372022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lovly CM and Shaw AT: Molecular pathways:
Resistance to kinase inhibitors and implications for therapeutic
strategies. Clin Cancer Res. 20:2249–2256. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hoxhaj G and Manning BD: The PI3K-AKT
network at the interface of oncogenic signalling and cancer
metabolism. Nat Rev Cancer. 20:74–88. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hanahan D: Hallmarks of cancer: New
dimensions. Cancer Discov. 12:31–46. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
DeBerardinis RJ and Chandel NS:
Fundamentals of cancer metabolism. Sci Adv. 2:e16002002016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kohn AD, Summers SA, Birnbaum MJ and Roth
RA: Expression of a constitutively active Akt Ser/Thr kinase in
3T3-L1 adipocytes stimulates glucose uptake and glucose transporter
4 translocation. J Biol Chem. 271:31372–31378. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Roberts DJ, Tan-Sah VP, Smith JM and
Miyamoto S: Akt phosphorylates HK-II at Thr-473 and increases
mitochondrial HK-II association to protect cardiomyocytes. J Biol
Chem. 288:23798–23806. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Currie E, Schulze A, Zechner R, Walther TC
and Farese RV Jr: Cellular fatty acid metabolism and cancer. Cell
Metab. 18:153–161. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Menendez JA and Lupu R: Fatty acid
synthase and the lipogenic phenotype in cancer pathogenesis. Nat
Rev Cancer. 7:763–777. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Santos CR and Schulze A: Lipid metabolism
in cancer. FEBS J. 279:2610–2623. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rysman E, Brusselmans K, Scheys K,
Timmermans L, Derua R, Munck S, Van Veldhoven PP, Waltregny D,
Daniëls VW, Machiels J, et al: De novo lipogenesis protects cancer
cells from free radicals and chemotherapeutics by promoting
membrane lipid saturation. Cancer Res. 70:8117–8126. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Srere PA: The citrate cleavage enzyme. I.
Distribution and purification. J Biol Chem. 234:2544–2547. 1959.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bauer DE, Hatzivassiliou G, Zhao F,
Andreadis C and Thompson CB: ATP citrate lyase is an important
component of cell growth and transformation. Oncogene.
24:6314–6322. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Szutowicz A, Kwiatkowski J and Angielski
S: Lipogenetic and glycolytic enzyme activities in carcinoma and
nonmalignant diseases of the human breast. Br J Cancer. 39:681–687.
1979. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang C, Liu J, Huang G, Zhao Y, Yue X, Wu
H, Li J, Zhu J, Shen Z, Haffty BG, et al: Cullin3-KLHL25 ubiquitin
ligase targets ACLY for degradation to inhibit lipid synthesis and
tumor progression. Genes Dev. 30:1956–1970. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lorito N, Bacci M, Smiriglia A, Mannelli
M, Parri M, Comito G, Ippolito L, Giannoni E, Bonechi M, Benelli M,
et al: Glucose metabolic reprogramming of ER breast cancer in
acquired resistance to the CDK4/6 inhibitor palbociclib. Cells.
9:6682020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Santarsiero A, Convertini P, Todisco S,
Pierri CL, De Grassi A, Williams NC, Iacobazzi D, De Stefano G,
O'Neill LAJ and Infantino V: ACLY nuclear translocation in human
macrophages drives proinflammatory gene expression by NF-κB
acetylation. Cells. 10:29622021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shi L and Tu BP: Acetyl-CoA and the
regulation of metabolism: Mechanisms and consequences. Curr Opin
Cell Biol. 33:125–131. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gnant M, Dueck AC, Frantal S, Martin M,
Burstein HJ, Greil R, Fox P, Wolff AC, Chan A, Winer EP, et al:
Adjuvant palbociclib for early breast cancer: The PALLAS trial
results (ABCSG-42/AFT-05/BIG-14-03). J Clin Oncol. 40:282–293.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu F and Korc M: Cdk4/6 inhibition
induces epithelial-mesenchymal transition and enhances invasiveness
in pancreatic cancer cells. Mol Cancer Ther. 11:2138–2148. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rencuzogulları O, Yerlikaya PO, Gürkan AÇ,
Arısan ED and Telci D: Palbociclib, a selective CDK4/6 inhibitor,
restricts cell survival and epithelial-mesenchymal transition in
Panc-1 and MiaPaCa-2 pancreatic cancer cells. J Cell Biochem.
121:508–523. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang Z, Li J, Ou Y, Yang G, Deng K, Wang
Q, Wang Z, Wang W, Zhang Q, Wang H, et al: CDK4/6 inhibition blocks
cancer metastasis through a USP51-ZEB1-dependent deubiquitination
mechanism. Signal Transduct Target Ther. 5:252020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Witta SE, Gemmill RM, Hirsch FR, Coldren
CD, Hedman K, Ravdel L, Helfrich B, Dziadziuszko R, Chan DC, Sugita
M, et al: Restoring E-cadherin expression increases sensitivity to
epidermal growth factor receptor inhibitors in lung cancer cell
lines. Cancer Res. 66:944–950. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Du B and Shim JS: Targeting
epithelial-mesenchymal transition (EMT) to overcome drug resistance
in cancer. Molecules. 21:9652016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Monaco ME: Fatty acid metabolism in breast
cancer subtypes. Oncotarget. 8:29487–29500. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hansel DE, Rahman A, House M, Ashfaq R,
Berg K, Yeo CJ and Maitra A: Met proto-oncogene and insulin-like
growth factor binding protein 3 overexpression correlates with
metastatic ability in well-differentiated pancreatic endocrine
neoplasms. Clin Cancer Res. 10:6152–6158. 2004. View Article : Google Scholar : PubMed/NCBI
|