Introduction
Radical prostatectomy or radiotherapy with or
without androgen deprivation therapy are the current treatments for
localized prostate cancer (1).
However, disease recurrence and the emergence of
castration-resistant prostate cancer (CRPC) are frequent
occurrences following therapy, and treatment options for these
remain insufficient; in addition, the survival rate for patients
with advanced disease remains low (2), highlighting the importance of basic
research required for this disease.
The overexpression of galectins (GALs), carbohydrate
binding proteins, with affinity for N-acetylglucosamine, their
roles in cancer progression (3) and
their potential as therapeutic targets have been demonstrated in
various tumor types over the past 10 years (4). The role of GAL in prostate cancer has
previously been described (5,6). These
previous studies have mainly focused on GAL-1 and GAL-3, although
the importance of GAL-4, GAL-7, GAL-8 and GAL-9 has also been
highlighted in this disease (7).
The expression of GAL-3 at the mRNA and protein level in the tumor
decreases during prostate cancer progression (8–17).
However, cytoplasmic overexpression in tumors cells has also been
shown to be positively associated with disease progression
(11), suggesting the dual role of
GAL-3 in prostate cancer cells, depending on its subcellular
localization (18).
In vitro studies have revealed that GAL-3
inhibits the apoptosis of prostate cancer cells (19–21),
and induces T-cell apoptosis (22)
and tumor cell adhesion to endothelial cells (23,24).
It has also been demonstrated that in androgen-independent (PC-3
cells) and androgen-dependent prostate cancer cells (LNCaP)
transfected with GAL-3 (LNCaP-GAL-3 cells), GAL-3 induces
proliferation, migration and invasion (13,25).
These findings were corroborated in vivo using tumor
xenograft mouse models, in which GAL-3 inhibition with
pharmacological or RNA interference (RNAi) strategies impaired
tumor growth (13,25) angiogenesis (22) and metastasis (21,22,26).
LNCaP cells do not express GAL-3, whereas the
androgen-independent prostate cancer cells, DU-145 and PC-3, highly
express GAL-3 (21,25). As previously demonstrated, the
overexpression of GAL-3 in LNCaP cells (21,25) or
the knockdown of GAL-3 in PC-3 cells does not alter the expression
level of the androgen receptor (AR) (21); similarly, it has been demonstrated
that the overexpression of AR in PC-3 cells has no regulatory
effect on the expression of GAL-3 (21).
The molecular regulatory mechanisms responsible for
the expression of GAL-3 in tumor cells are not yet clear (27,28).
The expression of GAL-3, at both the transcriptional and
translational level, can be regulated by various stimuli (27,28).
It has been suggested that promoter methylation is not the only
factor regulating the expression of GAL-3 (29). The expression of GAL-3 is increased
by transcription factors, such as the RUNX protein family,
homeodomain-interacting protein kinase 2, cAMP-response
element-binding protein, the NF-κB transcription factor,
hypoxia-inducible factor-1α, and inflammatory cytokines and the
Ras/MAPK pathway (27,28). Several of these transcription
factors and signaling pathways are activated by estrogen receptors
(ERs) or interact with ERs (30,31).
The regulatory effects of ERs on the expression of GAL-3 remain to
be explored in prostate cancer cells.
The authors have previously demonstrated the
presence of the ERs, ERα (ESR1) and ERβ (ESR2), in the
androgen-independent PC-3 and DU-145 prostate cancer cells, and
these receptors are mostly located outside the cell nucleus
(32,33). The activation of ERα and ERβ can
activate rapid cell signaling pathways in these cells, including an
increase in the phosphorylation of ERK1/2 in PC-3 and DU-145 cells
(32,33), and SRC and AKT in PC-3 cells
(34,35). It is noteworthy that the expression
of ERα (unpublished data, Fig. S1)
and ERβ (33) is higher in DU-145
than in PNT1A and PC-3 cells, so the present study focused on
DU-145 cells.
The present study aimed to examine the roles of ERα,
ERβ and GAL-3 in the migration and invasion of DU-145 cells, and to
determine the regulatory effects of the activation of these
receptors on the expression of the GAL-3.
Materials and methods
Cells and cell culture
The human post-pubertal prostate epithelial cell
line, PNT1A, was obtained from Public Health England Culture
Collections (lot 11B010; cat. no. 95012614). The DU-145 (derived
from brain metastasis) and PC-3 (derived from bone metastasis) cell
lines were obtained from ATCC (DU-145 cells, lot7000 9869, cat. no.
HTB-81; and PC-3 cells, lot BCRJ:0269, cat. no. CRL-1435; deposited
at the Rio de Janeiro Cell Bank). Mycoplasma testing was carried
out for all cell lines used. The PNT1A, DU-145 and PC-3 cells were
cultured as previously described (32–36).
The culture medium was replaced by serum free medium for 24 h
before the assays. All experimental procedures (cell culture,
western blot analysis, immunofluorescence, wound healing, cell
invasion and cell viability analyses, and statistical analysis)
were described, submitted, analyzed and approved by the Research
Ethics Committee at the Paulista School of Medicine (EPM), Federal
University of São Paulo (UNIFESP; no. 3527220917).
Western blot analysis for the
detection of GAL-3 and ERα (ESR1)
The PNT1A and PC-3 (used in some experiments) and
the DU-145 cells were incubated in the absence (control, untreated
cells) or presence of 17β-estradiol (E2, 10 nM; MilliporeSigma),
the ERα-selective agonist,
4,4′,4”-(4-propyl-(1H)-pyrazole-1,3,5-triyl)trisphenol (PPT; 10 nM,
MilliporeSigma) or the ERβ-selective agonist,
2,3-bis(4-hydroxyphenyl)-propionitrile (diarylprepionitrile; DPN;
10 nM, MilliporeSigma) for 30 min, 1, 2 and 4 h at 37°C. At these
concentrations, the agonists are highly selective, as previously
reported (32,37,38).
Total cell lysates (20 or 50 µg of protein/lane),
SDS/PAGE and western blot analysis were performed as previously
described (33,39). The protein concentration was
determined using the Bio-Rad protein assay, using bovine serum
albumin as standard (Bio Rad Laboratories, Inc.). Briefly, rabbit
polyclonal antibody raised against a peptide mapping at the
carboxyterminal of ERα of mouse origin, similar to human ERα
[MC-20, sc-542, Santa Cruz Biotechnology, Inc.; diluted at 1:200 in
Tris-buffered saline containing 0.2% Tween-20 (TBS-T) (Sigma
Chemical Co.) and 10% non-fat dry milk (Nestle), pH 7.6, overnight
at 4°C] and anti-GAL-3 [hybridoma M3/38.1.2.8 HL.2, TIB-166™, ATCC;
donated by Professor Roger Chammas, Center for Translational
Research in Oncology, Instituto do Cancer do Estado de São Paulo,
São Paulo, SP, Brazil; diluted at 1:100 in phosphate-buffered
saline containing 0.1% Tween-20 (PBS-T) (Sigma Chemical Co.) and 5%
non-fat dry milk (Nestle), pH 7.2, for 1 h at room temperature],
were used. Proteins were visualized by enhanced chemiluminescence
reagent (ECL, GE Healthcare), after incubation for 1 h at room
temperature, with the appropriate HRP-conjugated secondary antibody
(GE Healthcare) diluted in TBS-T at 1:3,000 or in PBS-T 1:3,500.
The band intensities of ERα, GAL-3, β-tubulin and GAPDH from
individual experiments were quantified using the densitometric
analysis of linear-range autoradiograms, using an Epson Expression
1680 scanner (Epson America, Inc.) and the quick Scan 2000 WIN
software (Helena Laboratories Co.). β-tubulin or GAPDH were used as
protein loading controls. The results were normalized to the
respective expression of β-tubulin or GAPDH, expressed in relation
to the control (C=1) or in arbitrary unit and plotted (mean ± SEM)
from three to six independent experiments. The blots are
representative of three to six independent experiments.
Immunofluorescence staining for the
detection of the GAL-3
The DU-145 cells were incubated in the absence
(control) or presence of E2, 10 nM); PPT (10 nM) or DPN (10 nM) for
2, 4 and 24 h at 37°C. The cells were also untreated or pre-treated
with the ERα-selective antagonist, 1,3-bis(4-hydrox-
yphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole
dihydrochloride (MPP; 10 nM, MilliporeSigma) and the ERβ-selective
antagonist,
4-[2-phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]phenol
(PHTPP; 10 nM, Tocris Bioscience) for 30 min at 37°C. Incubation
was continued in the absence or presence of E2 (10 nM), PPT (10 nM)
or DPN (10 nM) for 2 and 4 h at 37°C, as previously described
(33). Subsequently, the DU-145
cells were washed with ice-cold PBS, fixed in 2% formalin
(formaldehyde EM grade, Electron Microscopy Sciences) for 20 min at
room temperature, and washed with PBS containing 0.1 M glycine
(Sigma Chemical Co). The immunofluorescence assays were performed
as previously described (32,33).
Briefly, rat monoclonal antibody raised against GAL-3, at 1:50
dilution, in PBS containing 0.01% saponin (Sigma Chemical Co.) and
1% BSA (Sigma Chemical Co.), for 1 h at room temperature. The cells
were also incubated with Alexa Fluor 594-labeled secondary antibody
(anti-rat; 1:300; Molecular Probes®, Invitrogen; Thermo
Fisher Scientific, Inc.). Nuclear staining was performed with DAPI
(4′,6-diamidino-2-phenylindole, Sigma Chemical Co.). Negative
controls were performed in the absence of primary antibodies. The
immunostaining of GAL-3 was visualized under a confocal microscope
Leica Microsystems TCSSP8 (Leica Microsystems GmbH). Images of five
random microscope fields containing ~20 cells were captured, in
duplicate, in each assay (three independent experiments) and
analyzed using LAS-X software version: 3.7.0.20979 (Leica
Microsystems CMS GmbH). Images are representative of two to four
independent experiments performed in duplicate. The fluorescence
intensity of whole cell was obtained and analyzed using ImageJ
software 1.53t (National Institutes of Health) from the control and
treated cells and expressed in arbitrary units.
Wound healing assay
The DU-145 cells in culture medium without serum
containing a blocking DNA replication mitomycin C (10 µg/l;
MilliporeSigma) to avoid cell proliferation, were wounded using 200
µl sterile pipette tips as previously described (40,41).
The DU-145 cells were incubated in the absence (control, basal
level of cellular function) or presence of E2 (10 nM), PPT (10 nM)
DPN (10 nM) for 24 h at 37°C. The cells were also untreated or
pre-treated with MPP (10 nM), PHTTP (10 nM), simultaneously with
MPP (10 nM) and PHTPP (10 nM), or with the inhibitor of GAL-3,
1,2,3-triazole-galactosyl arylsulfadimethoxine [VA03; donated by
Professor Vanessa Leiria Campo Barão de Mauá University Center
(CBM), Ribeirão Preto, SP, Brazil. 200 µM] (42) for 30 min at 37°C. Incubation was
continued in the absence or presence of E2 (10 nM), PPT (10 nM) or
DPN (10 nM) for 24 h at 37°C. Wound healing analysis was performed
as previously described (40,41).
For measuring the closure of the wound in the control and treated
cells, images of the same area of the wound were obtained at 0 and
24 h. Images were captured using an inverted optical microscope
Axio Observer Z1 (Zeiss Nikon Eclipse, Zeiss GmbH) and ZEN 3.3 blue
edition software (eiss Nikon Eclipse, Zeiss GmbH). The areas that
were occupied by migrating cells after 24 h of incubation (control
and treated cells) were calculated by subtracting the background
levels at 0 h. The experiments were quantified using ImageJ
software 1.53t (National Institutes of Health). The results were
expressed in relation to the control (C=100%) and plotted (mean ±
SEM) from three to five independent experiments, in duplicate.
Images are representative of three to five independent experiments
performed in duplicate.
Cell invasion assay
The DU-145 cells in culture medium without serum
were seeded in Thincert® chambers (Greiner Bio-One) with
polyethylene terephthalate membranes (8 µm pore size) pre-coated
with 50 µl of phenol red-free Matrigel (1:10, Corning, Inc.). These
chambers were placed in 24-well plates containing culture medium
with 10% of fetal bovine serum in the lower chamber. The DU-145
cells in the upper chamber were incubated in the absence (control)
or presence of E2 (10 nM), PPT (10 nM) or DPN (10 nM) for 24 h at
37°C. The cells were also untreated or pre-treated with MPP (10
nM), PHTPP (10 nM), simultaneously with MPP (10 nM) and PHTPP (10
nM), or VA03 (200 µM) for 30 min at 37°C (42). Incubation was continued in the
absence or presence of E2 (10 nM), PPT (10 nM) or DPN (10 nM), for
24 h at 37°C. Cell invasion analysis was performed as previously
described (35,41). Briefly, the membranes were washed
thoroughly with 10 mM PBS (Sigma Chemical Co.), fixed in 4%
paraformaldehyde (Electron Microscopy Science) for 30 min, and
stained with 0.2% crystal violet (Merck KGaA) for 10 min (35,41).
Non-invading cells from the membrane upper surface were removed
using a sterile cotton swab. The membranes containing the invaded
cells (under the surface of membrane), were photographed. Images of
three random microscope fields, in duplicate, were captured using
an inverted optical microscope (Nikon Eclipse, Nikon Corporation).
The areas of invaded cells were analyzed using Micrometrics SE
Premium 4 software (Nikon Eclipse, Nikon Corporation). The
experiments were quantified using ImageJ software1.53t (National
Institutes of Health). The results were expressed in relation to
the control (C=100%) and plotted (mean ± SEM) from three to six
independent experiments, in duplicate. Images are representative of
three to six independent experiments performed in duplicate.
MTT cell viability assay
The DU-145 cells were incubated in the absence
(control) or presence of VA03 (20 and 200 µM) for 24 h at 37°C.
Cell viability assay was performed using MTT assay (Thermo Fisher
Scientific Inc.) as previously described (44). The cells were washed with ice-cold
PBS, replaced with 100 µl of fresh culture medium containing MTT
(2.4 mM), and incubated for 2 h at 37°C. The medium was removed,
and the formazan product was dissolved in DMSO (100 µl to each
well) at room temperature for 10 min with intermittent shaking.
Each sample was mixed again and the absorbance at 595 nm was read
using the ELx800 absorbance microplate reader (Biotek ELX800,
BioTek Instruments, Inc.). Each assay was repeated at least three
times in triplicate. The negative control was supplemented with 100
µl DMSO (Sigma Chemical Co.; without cells). Each sample from the
control and treated cells was subtracted from the negative control,
and the results were plotted as the mean ± SEM.
Statistical analysis
Data are expressed as the mean ± SEM. Statistical
analysis was performed using one-way ANOVA followed by the
Newman-Keuls test or Tukey's post hoc test for multiple
comparisons. P-values <0.05 were considered to indicate
statistically significant differences.
Results
The activation of ERα and ERβ promotes
the migration and invasion of DU-145 cells
The present study analyzed various cellular
characteristics of tumor development in vitro, using DU-145
cells. At 24 h of treatment E2 (10 nM), the ERα-selective agonist,
PPT (10 nM), or the ERβ-selective agonist, DPN (10 nM), increased
the migration of the DU-145 cells (2.0-, 1.5- and 1.5-fold,
respectively) compared to the control (Fig. 1A), suggesting the involvement of the
ERs, ERα and ERβ, in this process. Of note, the increase in DU-145
cell migration induced by E2 (10 nM) at 24 h was blocked by the
ERα-selective antagonist (MPP, 10 nM), the ERβ-selective antagonist
(PHTPP, 10 nM) or simultaneous pre-treatment with both MPP (10 nM)
and PHTPP (10 nM) (Fig. 1B).
Pre-treatment with MPP, PHTPP or both MPP and PHTPP, in the absence
of the E2, yielded results similar to those of the control (data
not shown).
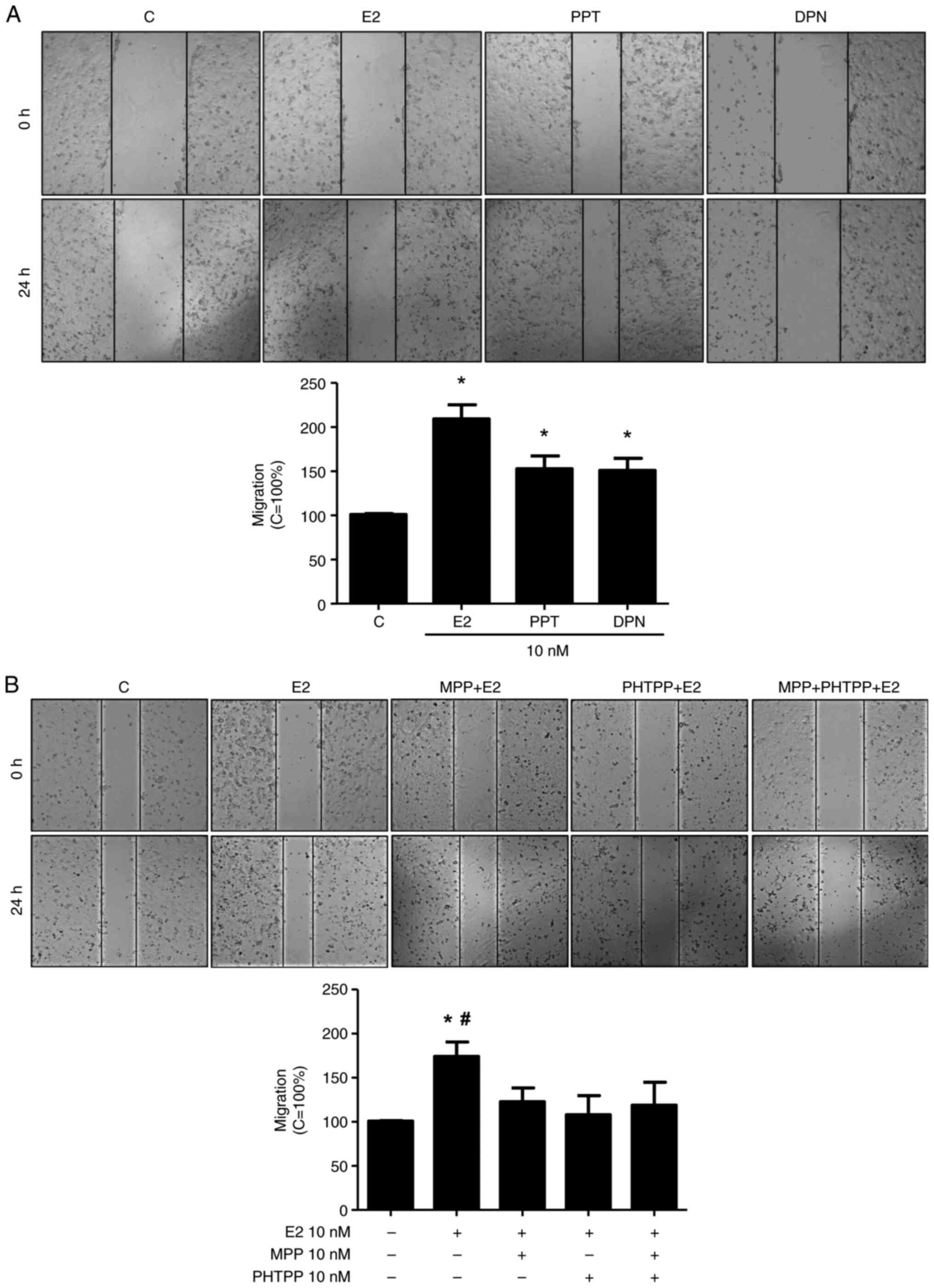 | Figure 1.Effects of E2, the ERα-selective
agonist, PPT, and the ERβ-selective agonist, DPN, on the migration
of the DU-145 cells. (A) Cells, in the same culture plate in
different wells, were wounded and then incubated in the absence (C,
control) or presence of E2 (10 nM), ERα-selective agonist PPT (10
nM) or ERβ-selective agonist DPN (B) for 24 h at 37°C. (B) Cells
were also untreated or pre-treated with the ERα-selective
antagonist MPP (10 nM), ERβ-selective antagonist PHTPP (10 nM) or
with both antagonists, MPP (10 nM) and PHTPP (10 nM) for 30 min.
Incubation was continued in the presence of E2 (10 nM) for 24 h at
37°C. Wound healing assay was performed as described in the
Materials and methods. The results are expressed in relation to the
control (C=100%) and plotted (mean ± SEM) from four to five
independent experiments, in duplicate (bar graphs). Images (×100
magnification) are representative of four to five independent
experiments performed in duplicate. *P<0.05, significantly
different from the control; #P<0.05, significantly
different from the MPP + E2, PHTPP + E2, or MPP + PHTPP + E2 groups
(determined using ANOVA and Tukey's post hoc test). E2,
17β-estradiol; ER, estrogen receptor; PPT, 4,4′,4”-(4-propyl-
(1H)-pyrazole-1,3,5-triyl)trisphenol; DPN,
2,3-bis(4-hydroxyphenyl)-propionitrile; MPP,
1,3-bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole
dihydrochloride; PHTPP,
4-[2-phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]phenol. |
Treatment with E2 (10 nM), PPT (10 nM) or DPN (10
nM) for 24 h led to an enhancement of the invasion of the DU-145
cells (5-, 3,5- and 4-fold, respectively) (Fig. 2). The increase in DU-145 cell
invasion induced by E2 was blocked by simultaneous pre-treatment
with both MPP and PHTPP (Fig.
S2A), suggesting that ERα and ERβ may play a role in the DU-145
cells invasion. To confirm the involvement of these receptors, the
DU-145 cells were also untreated or pre-treated with MPP (10 nM) or
PHTPP (10 nM) and the incubation was continued in the absence or
presence of PPT (10 nM) or DPN (10 nM). The increase in DU-145 cell
invasion induced by PPT or DPN was blocked, respectively, by MPP or
PHTPP (Fig. S2B and C), confirming
that ERα and ERβ are upstream receptors regulating this process.
Pre-treatment with MPP, PHTPP or simultaneous pre-treatment with
both antagonists, in the absence of E2, PPT or DPN, yielded results
similar to those of the control (Fig.
1).
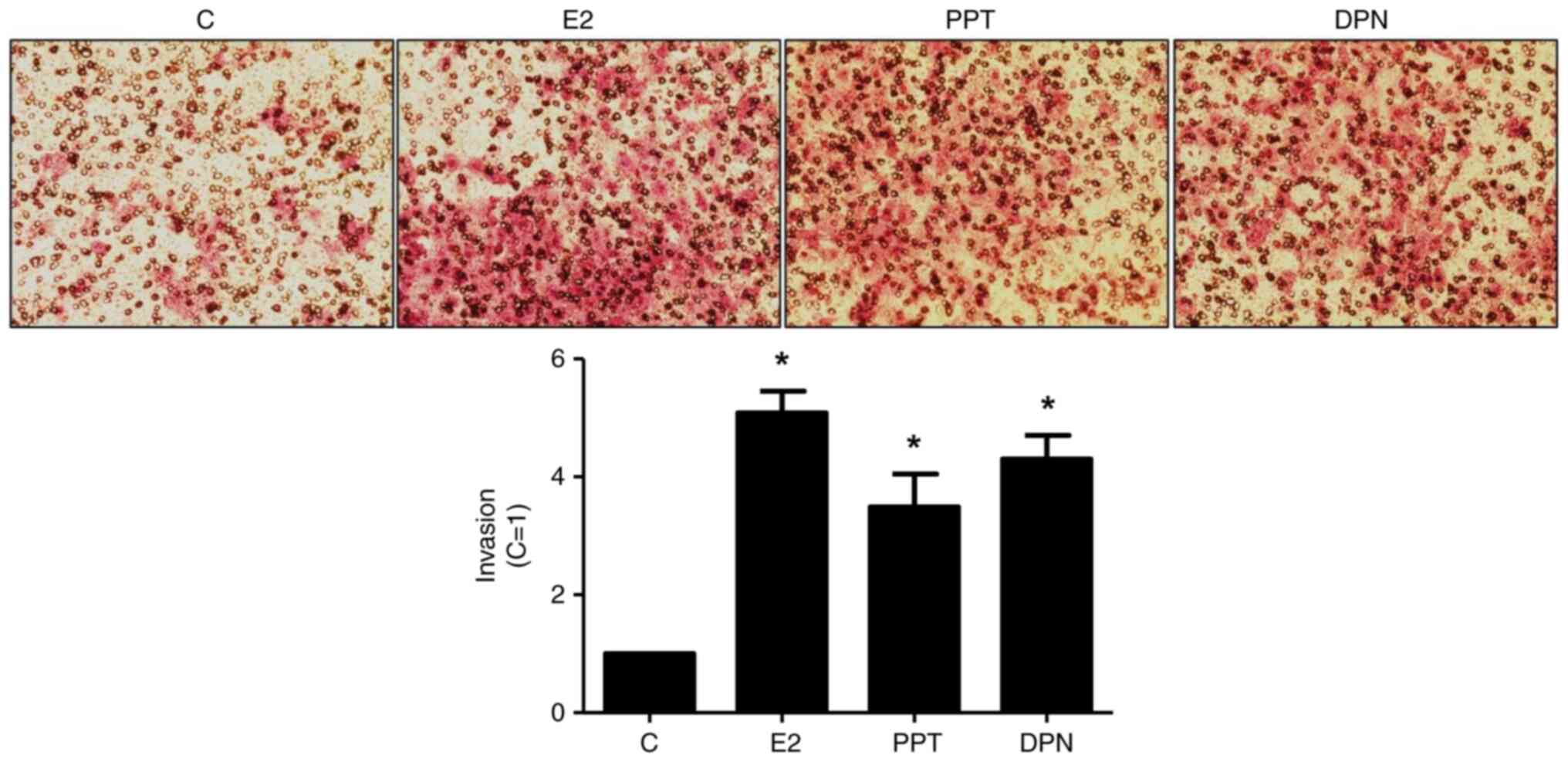 | Figure 2.Effects of E2, the ERα-selective
agonist, PPT, and the ERβ-selective agonist, DPN, on the invasion
of the DU-145 cells. Cells in culture medium without serum were
seeded in ThincertR chambers with polyethylene terephthalate
membranes pre-coated with phenol red-free Matrigel. These chambers
were placed in 24-well plates containing culture medium with 10%
FBS in the lower chamber. Cells in upper chambers of the same
culture plate were incubated in the absence (C, control) and the
presence of E2 (10 nM), ERα-selective agonist PPT (10 nM) or
ERβ-selective agonist DPN (10 nM) for 24 h at 37°C. Cell invasion
assay was performed as described in the Materials and methods. The
results are expressed in relation to control (C=1) and plotted
(mean ± SEM) from five to six independent experiments, in duplicate
(bar graphs). Images (×200 magnification) are representative of
five to six independent experiments performed in duplicate.
*P<0.05, significantly different from the control (determined
using ANOVA and Tukey's post hoc test). E2, 17β-estradiol; ER,
estrogen receptor; PPT,
4,4′,4”-(4-propyl-(1H)-pyrazole-1,3,5-triyl)trisphenol; DPN,
2,3-bis(4-hydroxyphenyl)-propionitrile. |
The activation of ERα and ERβ for 24 h
increases the expression of GAL-3 in DU-145 cells
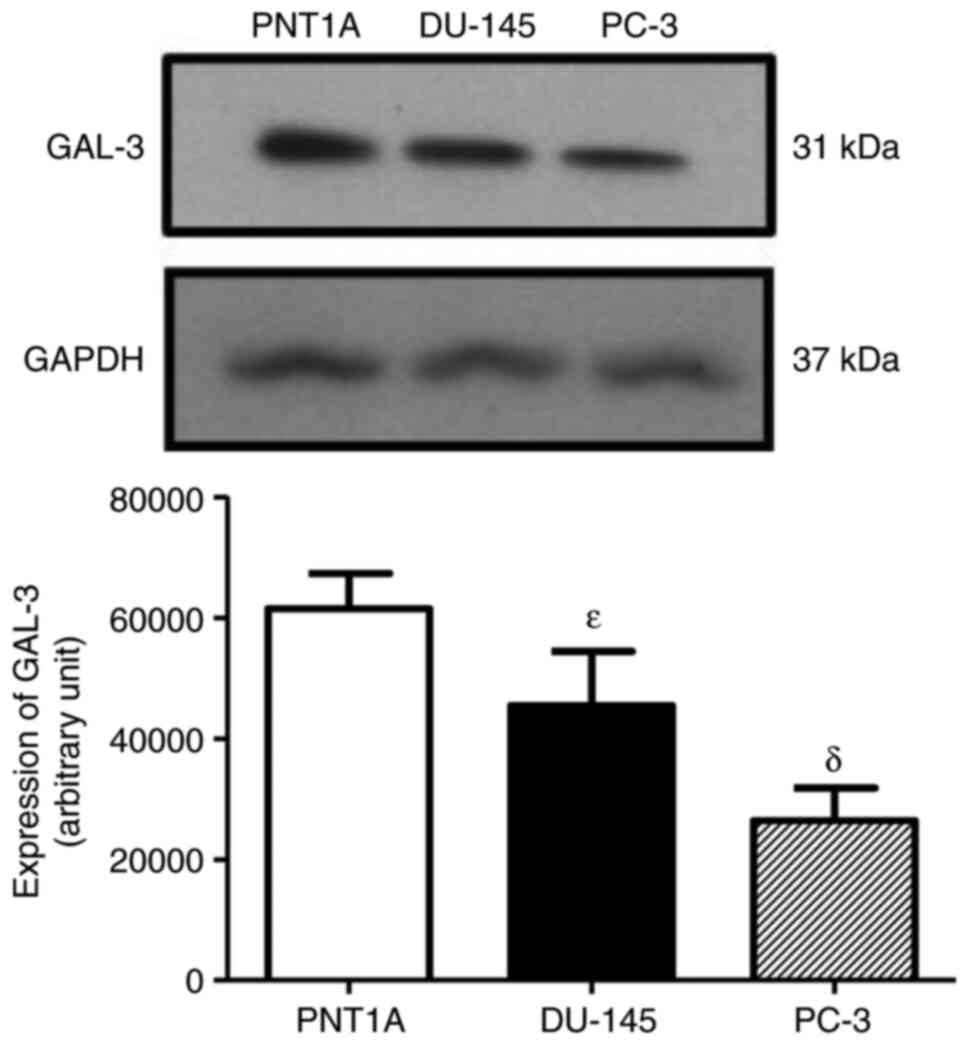
GAL-3 was detected as a single protein band of 31
kDa in the total cell extracts of the PNT1A, PC-3 and DU-145 cells
(Fig. 3). The expression of GAL-3
was higher in the DU-145 and PNT1A cells than in the PC-3 cells
(Fig. 3), suggesting that the AR is
not involved in the regulation of GAL-3. No difference was observed
in the expression of the GAPDH among the three cells, used as
protein loading control (Fig. 3).
Thus, the androgen-independent prostate cancer cells, DU-145, were
used in the analyses of the regulatory effects of the activation of
the ERs on the expression of GAL-3.
Treatment of the DU-145 cells with E2 (10 nM) for 4
h or with PPT (10 nM) for 1 and 2 h increased the expression of
GAL-3 compared to the control DU-145 cells (untreated cells)
(Fig. 4A and B). On the other hand,
treatment of the DU-145 cells with DPN (10 nM) for 30 min, 1, 2 and
4 h did not have any marked effects on the expression of GAL-3
compared to the control (Fig. 4C).
No difference was observed in the expression of β-tubulin under any
of these conditions, used as the protein loading control (Fig. 4).
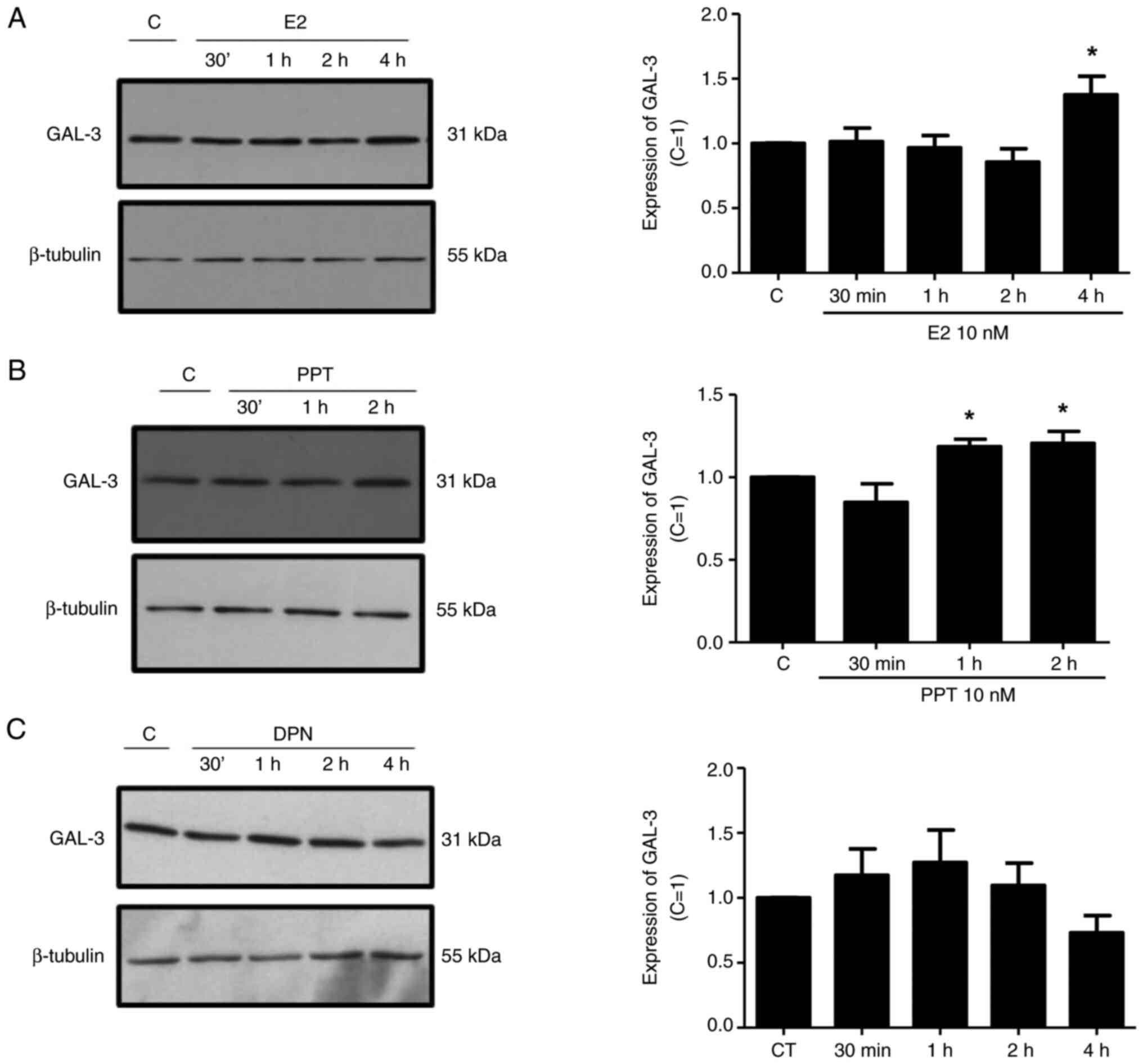 | Figure 4.Effects of E2, the ERα-selective
agonist, PPT, and the ERβ-selective agonist, DPN, on the expression
of GAL-3 in DU-145 cells. Cells were incubated in the absence
(control, C) or presence of (A) E2 (10 nM), (B) ER α-selective
agonist PPT (10 nM) or (C) ERβ-selective agonist DPN (10 nM) for
different periods of time at 37°C. Western blot analysis for the
detection of the GAL-3 in PNT1A, DU-145 and PC-3 cells was
performed as described in the Materials and methods, using 20 µg of
protein/lane and antibody specific for GAL-3 (top row) or antibody
that recognizes β-tubulin (bottom row). The protein sizes of GAL-3
and β-tubulin proteins are shown at the right. The data shown are
representative of three to five independent experiments. Results of
the densitometric analysis of the western blots were normalized to
the respective expression of β-tubulin, expressed in relation to
the control (C=1) and plotted (mean ± SEM) from three to five
independent experiments (bar graphs). *P<0.05, significantly
different from the control (determined using ANOVA and Tukey's post
hoc test). E2, 17β-estradiol; ER, estrogen receptor; PPT,
4,4′,4”-(4-propyl-(1H)-pyrazole-1,3,5-triyl)trisphenol; DPN,
2,3-bis(4-hydroxyphenyl)-propionitrile. |
The localization and expression of GAL-3 were
determined using immunofluorescence assays. In the control DU-145
cells (Fig. 5, Fig. 6, Fig.
7 and S3), the immunostaining
of GAL-3 was predominantly found in the cytoplasm, although
immunostaining in some nuclei was also observed. Treatment of these
cells with E2 (10 nM) for 4 h (Figs.
5 and S4) or PPT (10 nM) for 2
and 4 h (Figs. 6 and S4) increased the expression of GAL-3 in
the cytoplasm and nuclei. On the other hand, treatment of the
DU-145 cells with DPN (10 nM) for 2 and 4 h did not have any marked
effects on the expression of GAL-3 compared to the control
(Fig. S3).
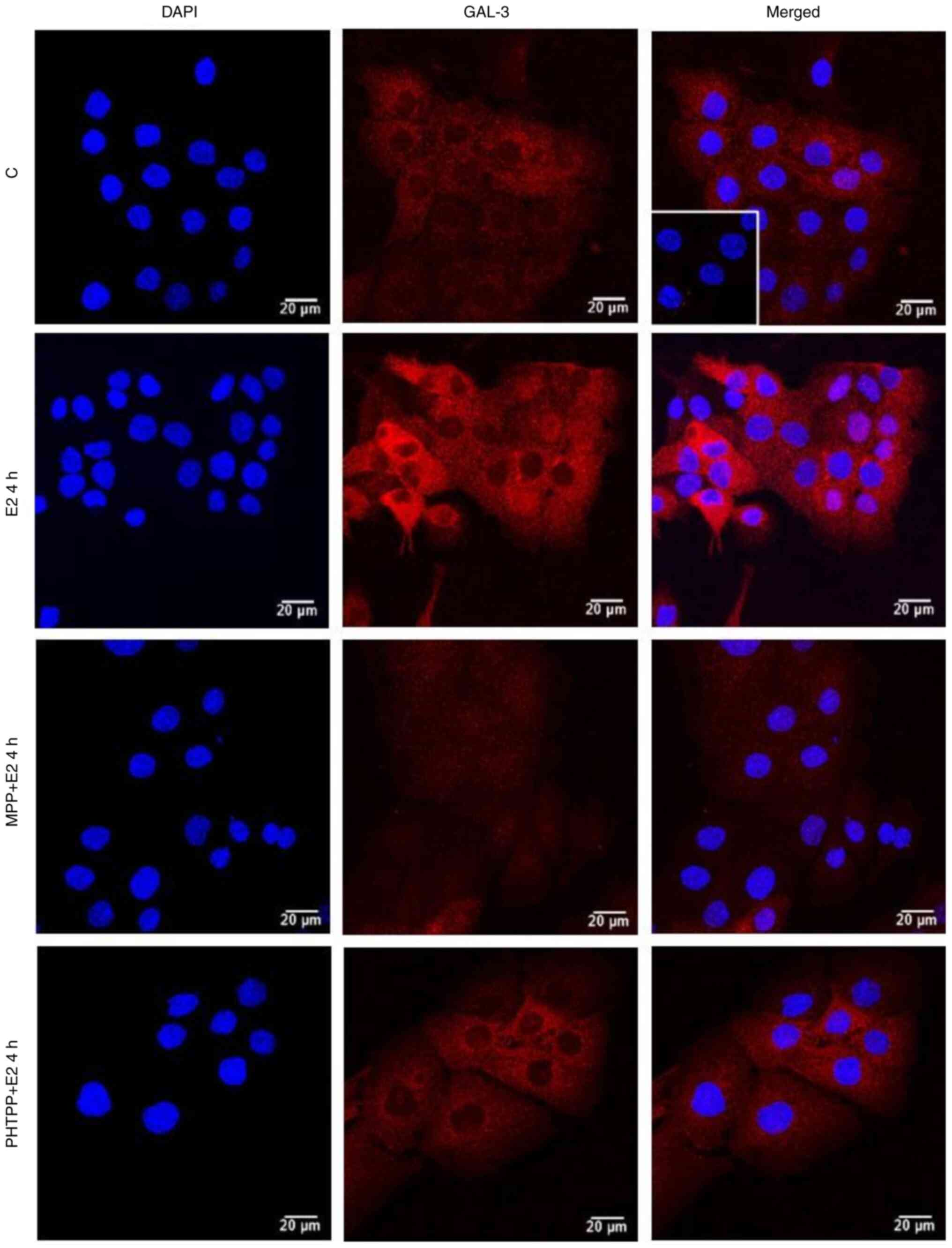 | Figure 5.Effects of treatment with E2 for 4 h
on the expression and localization of the GAL-3 in DU-145 cells.
Cells were incubated in the absence (control, C) or presence of E2
(10 nM) for 4 h at 37°C. Cells were also untreated or pre-treated
with the ERα-selective antagonist, MPP, (10 nM) or the
ERβ-selective antagonist, PHTPP (10 nM), for 30 min. Incubation was
continued in the absence or presence of E2 (10 nM) for 4 h at 37°C.
Immunostaining for GAL-3 (red) was detected as described in the
Materials and methods. Nuclei were stained with DAPI (blue).
Negative control was performed in the absence of primary antibody
(insert). Scale bars, 20 µm. Images are representative of two
independent experiments. E2, 17β-estradiol; GAL-3, galectin-3; ER,
estrogen receptor; MPP,
1,3-bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole
dihydrochloride; PHTPP,
4-[2-phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]phenol. |
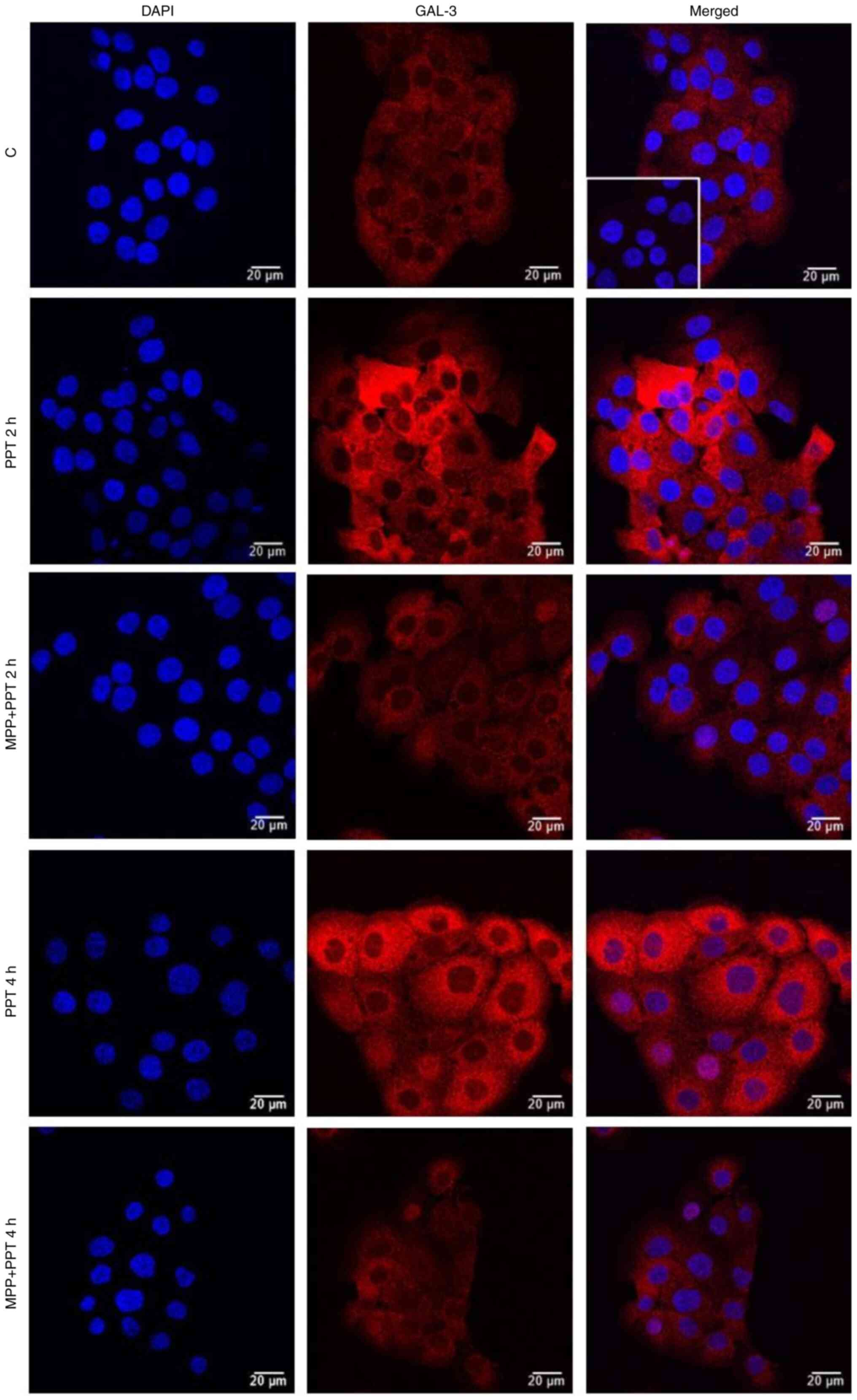 | Figure 6.Effects of the ERα-selective agonist,
PPT, for 2 and 4 h on the expression and localization of GAL-3 in
DU-145 cells. Cells were incubated in the absence (control, C) or
presence of ERα-selective agonist PPT (10 nM) for 2 h and 4 h at
37°C. Cells were also untreated or pre-treated with ERα-selective
antagonist MPP (10 nM) for 30 min. Incubation was continued in the
absence or presence of PPT (10 nM) for 4 h at 37°C. Immunostaining
for GAL-3 (red) was detected as described in the Materials and
methods. Nuclei were stained with DAPI (blue). Negative control was
performed in the absence of primary antibody (insert). Scale bars,
20 µm. Images are representative of four independent experiments.
ER, estrogen receptor; PPT, 4,4′,4”-(4-propyl-(1H)-pyrazole-
1,3,5-triyl)trisphenol; DPN,
2,3-bis(4-hydroxyphenyl)-propionitrile; GAL-3, galectin-3; MPP,
1,3-bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole
dihydrochloride. |
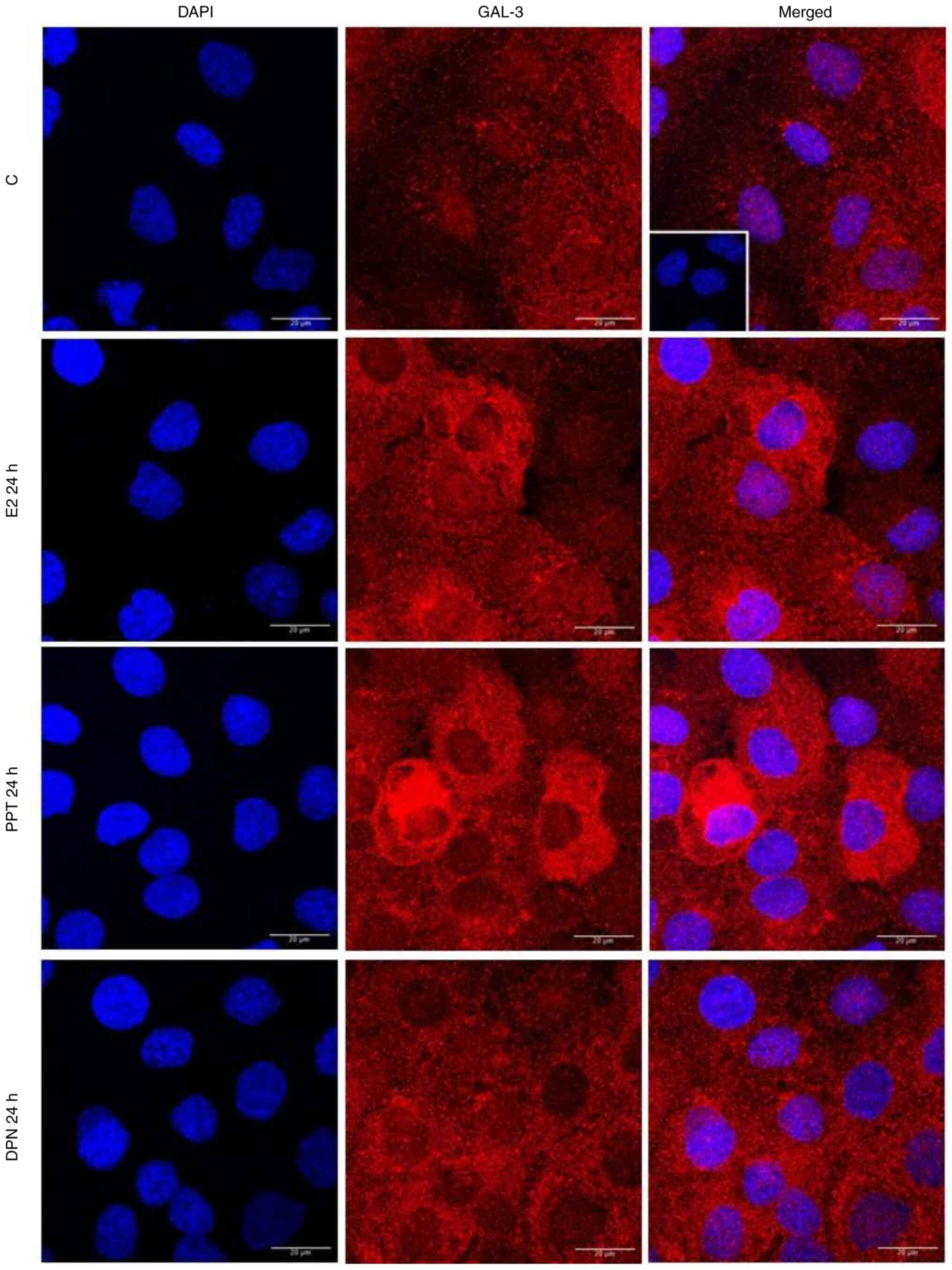 | Figure 7.Effects of treatment with E2, the
ERα-selective agonist, PPT, or the ERβ-selective agonist, DPN, for
24 h on the expression and localization of the GAL-3 in DU-145
cells. Cells were incubated in the absence (control, C) or presence
of E2 (10 nM), ERα-selective agonist PPT (10 nM) or ERβ-selective
agonist DPN (10 nM) for 24 h at 37°C. Immunostaining for GAL-3
(red) was detected as described in the Materials and methods.
Nuclei were stained with DAPI (blue). Negative control was
performed in the absence of primary antibody (insert). Scale bars,
20 µm. Images are representative of two independent experiments.
E2, 17β-estradiol; ER, estrogen receptor; GAL-3, galectin-3; PPT,
4,4′,4”-(4-propyl-(1H)-pyrazole-1,3,5-triyl)trisphenol; DPN,
2,3-bis(4-hydroxyphenyl)-propionitrile. |
The effects of treatment of the DU-145 cells with E2
(10 nM) for 2 h were blocked by pre-treatment with MPP (10 nM) and
partially blocked by PHTPP (10 nM) (Figs. 5 and S4). The expression of GAL-3 induced by 2
or 4 h of treatment with PPT (10 nM) was blocked by pre-treatment
with MPP (10 nM) (Figs. 6 and
S4). Treatment with MPP or PHTPP
alone did not have any marked effects on the expression of the
GAL-3, and the effects were similar to those of the control (data
not shown).
It is important to mention that at 24 h of treatment
with E2, PPT or DPN, the expression of GAL-3 increased compared to
the control (Fig. 7). The analysis
of these findings using ImageJ software revealed that treatment
with E2, PPT and DPN for 24 h increased the fluorescence intensity
of GAL-3 by 25, 39 and 28%, respectively in the whole DU-145 cells
compared to the control (Fig. S4).
No immunostaining was observed in the negative control, performed
in the absence of primary antibodies for GAL-3 (Fig. 5, Fig.
6, Fig. 7 and S2, inserts).
GAL-3 is involved in the migration and
invasion of DU-145 cells
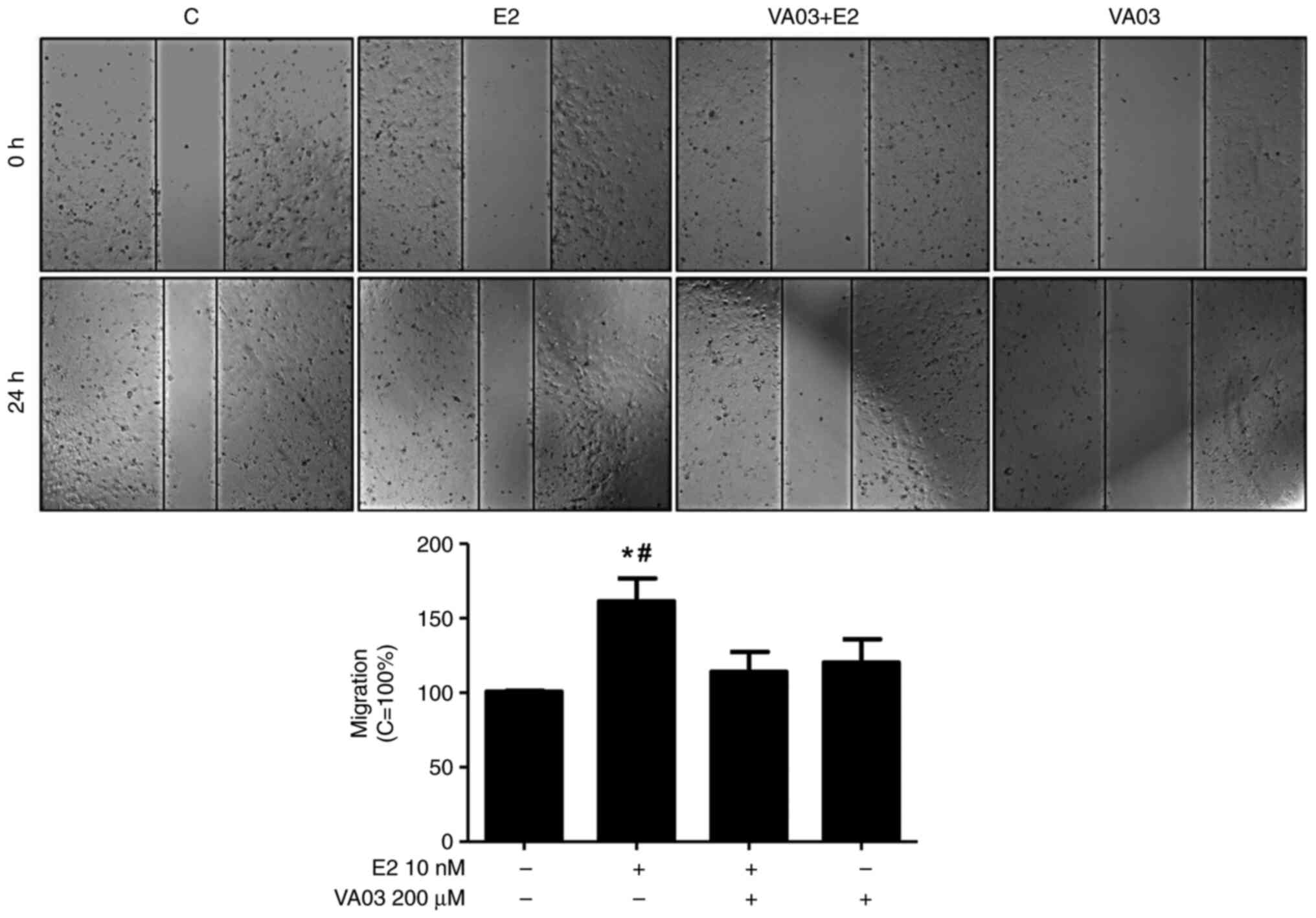
To explore the involvement of GAL-3 in the migration
and invasion of DU-145 cells, VA03 (a specific inhibitor of GAL-3)
was used at 200 µM (Figs. 8 and
9). Pre-treatment with VA03
inhibited the migration of the DU-145 cells induced by E2 (10 nM)
(Fig. 8), PPT (10 nM) or DPN (10
nM) (data not shown). Pre-treatment with VA03 inhibited the
invasion of the DU-145 cells induced by E2 (10 nM), PPT (10 nM) or
DPN (10 nM) (Fig. 9), indicating
the involvement of the complex ERα/GAL-3 and ERβ/GAL-3 in the
regulation of the migration and invasion of DU-145 cells. Treatment
with VA03 alone did not have any marked effects on the migration or
invasion of DU-145 cells (Figs. 8
and 9). In addition, treatment with
VA03 (20 and 200 µM) for 24 h did not have any notable effects on
the number and viability of the DU-145 cells compared to the
control cells (Fig. S5).
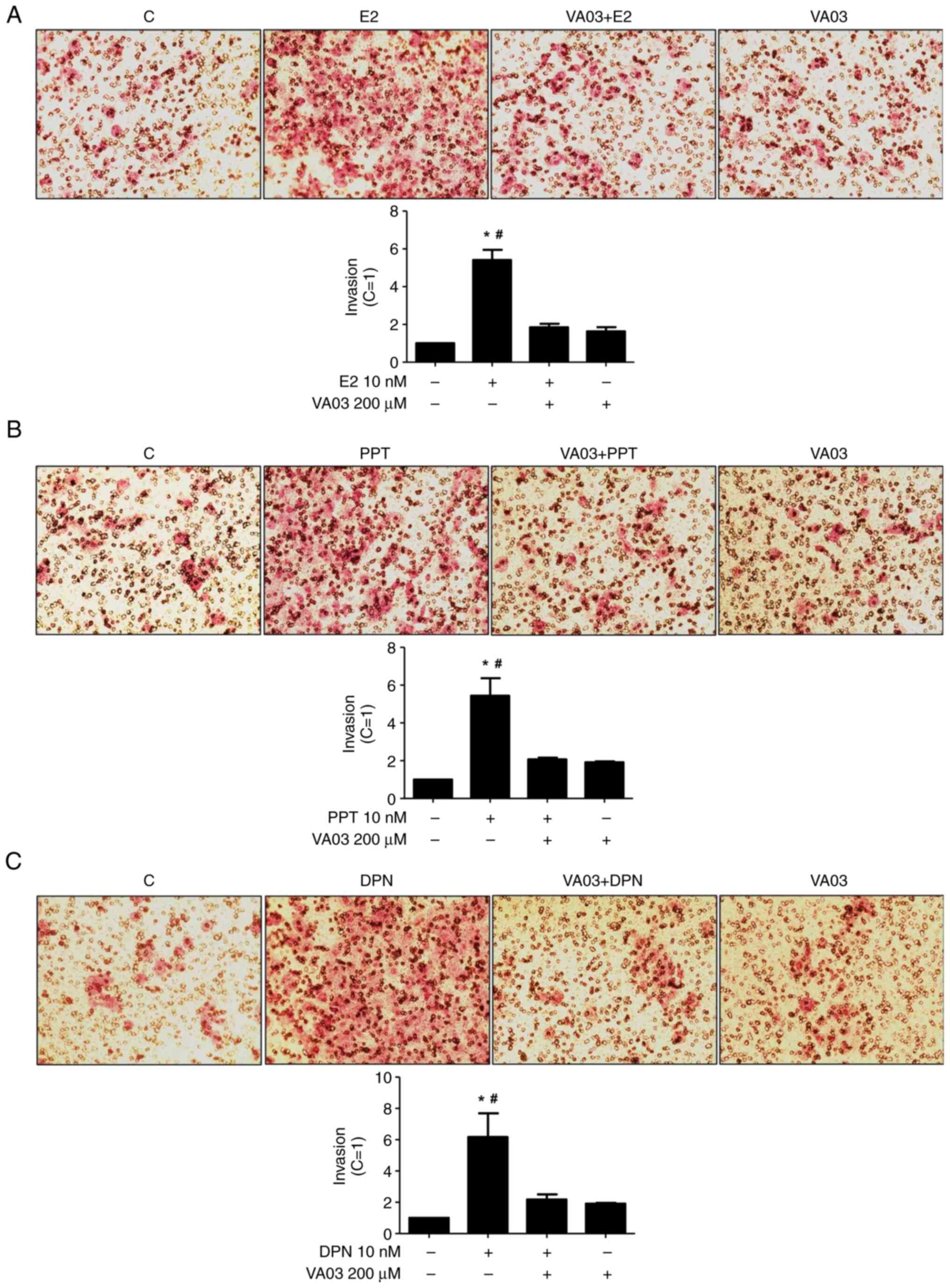 | Figure 9.Effects of a specific inhibitor of
GAL-3 (VA03) on the invasion of DU-145 cells induced by E2, PPT and
DPN. Cells in culture medium without serum were seeded in ThincertR
chambers with polyethylene terephthalate membranes pre-coated with
phenol red-free Matrigel. These chambers were placed in 24-well
plates containing culture medium with 10% FBS in the lower chamber.
Cells in the upper chambers of the same culture plate were
incubated in the absence (C, control) or presence of (A) E2 (10
nM), (B) the ERα-selective agonist, PPT (10 nM), or (C) the
ERβ-selective agonist, DPN (10 nM), for 24 h at 37°C. Cells were
also untreated or pre-treated with a specific inhibitor of GAL-3
(VA03, 200 µM) for 30 min. Incubation was continued in the absence
or presence of (A) E2, (B) PPT or (C) DPN for 24 h at 37°C. Cell
invasion analysis was performed as described in the Materials and
methods. Results are expressed in relation to the control (C=1) and
plotted (mean ± SEM) from three to four independent experiments, in
duplicate (bar graphs). Images (×200 magnification) are
representative of three to four different experiments. *P<0.05,
significantly different from the control; #P<0.05,
significantly different from the VA03 + E2; VA03 + PPT, VA03 + DPN
and VA03 groups (determined using ANOVA and Tukey's post hoc test).
GAL-3, galectin-3; E2, 17β-estradiol; ER, estrogen receptor; PPT,
4,4′,4”-(4-propyl-(1H)-pyrazole-1,3,5-triyl)trisphenol; DPN,
2,3-bis(4-hydroxyphenyl)-propionitrile. |
Discussion
The expression of the ERs, ERα and ERβ, changes in
the different stages of prostate cancer and conflicting findings on
the expression, regulation and roles of these receptors in prostate
cancer development have been found (45–48).
It is recognized that there is wide variability in the sensitivity
and specificity of ERβ antibodies, which may contribute to the
uncertainties surrounding its molecular action and tissue
expression. Nelson et al (49) published a study advising about which
antibodies are acceptable against ERβ. Using the antibody
previously reported (49), the
authors previously demonstrated the presence of ERβ in PNT1A, PC-3
and DU-145 cells (33). The
expression of ERα also was shown in these cells (32,33,
Fig. S1). Taken together, these
results confirm that the expression of ERα and ERβ is higher in the
DU-145 cells than in the PNT1A and PC-3 cells, suggesting that
distinct androgen-independent mechanisms are involved in the
regulation of these receptors. These mechanisms remain to be
explored.
The activation of ERα and ERβ promoted an increase
in the migration and invasion of the DU-145 cells. In the PC-3
cells, the activation of ERβ and ERα increased the invasion and
anchorage-independent growth of these cells (40,50).
The activation of ERβ by DPN has also been shown to promote the
survival and migration of the CPEC cell line (cells expressing
prostate-specific antigens), established from patients with
prostate cancer (51). Furthermore,
the expression of the ERβ5 (ERβ splice variant) in PC-3 cells
increased the cell migration, and the expression of ERβ2 and ERβ5
increased the invasion, but did not affect the proliferation of the
cells (52). It is important to
emphasize that ERβ splice variants do not bind ligands (53), although dimers may be observed with
ERβ (ERβ1). ERα/β heterodimers formation was observed in DU-145
cells (33). Taken together, these
results support an oncogenic role for ERα and ERβ in DU-145
cells.
In the present study, the activation of ERα by PPT,
but not ERβ by DPN, for 2 h increased the expression of the GAL-3
in DU-145 cells. However, at 24 h of treatment, DPN also increased
the expression of GAL-3 compared to the control (basal level of
cellular function). Taken together, these results indicate that ERα
and ERβ are involved in the regulation of the expression of the
GAL-3. The promoter region of the human LGALS3 gene contains
several regulatory elements: Five putative Sp1 binding sites (GC
boxes), five cAMP-dependent response element motifs, four AP-1- and
one AP-4-like sites, two NF-κB-like sites, one sis-inducible
element and a consensus basic helix-loop-helix core sequence
(27,54). Several of these transcription
factors interact with ERs (30) and
induce the genomic signaling. Furthermore, ERs also activate two
major pathways regulating cell proliferation and survival, SRC/MAPK
and PI3K/AKT pathways (rapid or non-genomic signaling) (31). Indeed, in the DU-145 cells, the
activation of ERα and ERβ can activate rapid cell signaling
pathways in these cells, including an increase in the
phosphorylation of ERK1/2 (33).
Thus, the transcriptional regulation (genomic activity) combined
with direct activation of signaling cascades (non-genomic activity)
by ERs may be involved in the expression of the GAL-3 in DU-145
cells. These mechanisms remain to be explored in DU-145 and other
prostate cancer cells.
It is important to mention that E2 and progesterone
have also been shown to induce the upregulation of GAL-3 expression
in RL95-2 epithelial cells from the human endometrium, which in
turn decreases the apoptotic rate of these cells (55). Furthermore, in a preliminary study,
ERα and GAL-3 were shown as markers of aggressiveness and prognosis
in prolactinoma (56).
In the present study, in the DU-145 cells (control),
the immunostaining of GAL-3 was predominantly found in the
cytoplasm, although immunostaining in some nuclei was also
observed. Treatment of these cells for 24 h with E2, PPT or DPN
increased the expression of the GAL-3 in the cytoplasm and nuclei.
Several cytosolic molecules were identified, as GAL-3 ligands and
these proteins are involved in the regulation of cell
proliferation, differentiation, survival and death (27). In addition, GAL-3 interacts with
nuclear factors to regulate the expression of multiple genes
related to tumor plasticity (57).
For example, GAL-3 interacts with the factor activator protein 1
(AP1); this GAL-3-AP1 complex binds to the matrix
metalloproteinase-1 promoter and mediates its transcription, which
facilitates the migration and invasion of melanoma cells (58).
Herein, to explore the involvement of GAL-3 in
migration and invasion of the DU-145 cells induced by activation of
ERs, VA03 (a specific inhibitor of GAL-3) was used. Pre-treatment
with VA03 inhibited the migration and invasion of the DU-145 cells
induced by the activation of ERs, indicating that the complex
ERα/GAL-3 and ERβ/GAL-3 plays a role in these processes.
The regulation of the expression of GAL-3 by ERs in
other prostate cancer cell lines and in different stages of the
prostate cancer remains to be explored. It is important to mention
that in PC-3 cells, the activation of ERs induces an increase of
the active non-phosphorylated β-catenin, and these proteins are
involved in the proliferation, migration, invasion and colony
formation of these cells (34,40).
It has been shown that β-catenin can co-localize with GAL-3 in
other cancer cells (59). Taken
together, these results indicate that the process is more complex
that should be addressed in near future using different prostate
cancer cell lines and prostate cancer tissues.
GAL-3 inhibitors have shown promising results in
preclinical studies (22,60). Notably, TFD100, a GAL3-binding
glycopeptide, has been shown to block GAL3-induced T-cell
apoptosis, and to impair angiogenesis and metastasis in xenograft
models (22). In addition,
G3-C12-modified copolymers (targeting GAL-3) have been shown to
improve the antitumor activity of 5-fluorouracil in prostate cancer
xenograft mouse models (60), and
modified citrus pectin, a natural dietary fiber soluble
polysaccharide, that plays a role as an antagonist of extracellular
GAL-3 (61), sensitizes prostate
cancer cells to radiotherapy, and reduces their migratory and
invasive capabilities (62).
Overall, although GAL-3 levels in the tumor are
decreased during prostate cancer progression, the cytoplasmic
overexpression of this protein has been reported during
progression, and in vitro data and preclinical xenograft
models have shown that strategies targeting GAL-3 may be effective
in impairing prostate cancer progression in vivo. However,
several important challenges need to be addressed before
anti-GAL-based strategies can be translated to clinical
settings.
In conclusion, the activation of both ERs increases
the expression and signaling of the GAL-3, and induces the
migration and invasion of DU-145 cells. The findings of the present
study provide novel insight into the signatures and molecular
mechanisms of ERα and ERβ in DU-145 cells.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Elizabeth
Kanashiro and Dr Caroline Zito Romera [Paulista School of Medicine
(EPM), Federal University of São Paulo (UNIFESP)] for providing
technical assistance. The Leica Microsystems TCSSP8 confocal
microscope is a facility from the Instituto Nacional de
Farmacologia e Biologia Molecular (INFAR), EPM-UNIFESP, and it was
supported by Financiadora de Estudos e Projetos (FINEP) and
Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP). The
research fellowship of CSP and doctoral fellowship of CM were
supported by Conselho Nacional de Desenvolvimento Científico e
Tecnológico (CNPq). The doctoral fellowship of DSS was supported by
Coordenação de Aperfeiçoamento de Pessoal de Nível Superior
(CAPES).
Funding
The present study was supported by Fundação de Amparo à Pesquisa
do Estado de São Paulo (FAPESP, grant no. 2020/01285-2).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DSS conceived and designed the study, collected the
data, performed the analysis and wrote the manuscript. CM and RPC
contributed to data analysis. CMV conceived and designed the study,
contributed data or analysis tools and performed the analyses. VLC
performed the synthesis of the galectin-3 inhibitor. CSP conceived
and designed the study, and performed the revision of the
manuscript. DSS and CSP confirm the authenticity of all the raw
data. All authors have read and approved the submitted version of
the manuscript.
Ethics approval and consent to
participate
All experimental procedures were approved by the
Research Ethical Committee at EPM-UNIFESP (#3527220917).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sathianathen NJ, Konety BR, Crook J, Saad
F and Lawrentschuk N: Landmarks in prostate cancer. Nat Rev Urol.
15:627–642. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baciarello G, Gizzi M and Fizazi K:
Advancing therapies in metastatic castration-resistant prostate
cancer. Expert Opin Pharmacother. 19:1797–1804. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu FT and Rabinovich GA: Galectins as
modulators of tumour progression. Nat Rev Cancer. 5:29–41. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thijssen VL, Heusschen R, Caers J and
Griffioen AW: Galectin expression in cancer diagnosis and
prognosis: A systematic review. Biochim Biophys Acta. 1855:235–247.
2015.PubMed/NCBI
|
|
5
|
Laderach DJ, Gentilini LD, Giribaldi L,
Delgado VC, Nugnes L, Croci DO, Al Nakouzi N, Sacca P, Casas G,
Mazza O, et al: A unique galectin signature in human prostate
cancer progression suggests galectin-1 as a key target for
treatment of advanced disease. Cancer Res. 73:86–96. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Compagno D, Gentilini LD, Jaworski FM,
Pérez IG, Contrufo G and Laderach DJ: Glycans and galectins in
prostate cancer biology, angiogenesis, and metastasis.
Glycobiology. 24:899–906. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Martínez-Bosch N, Rodriguez-Vida A,
Juanpere N, Lloreta J, Rovira A, Albanell J, Bellmunt J and Navarro
P: Galectins in prostate and bladder cancer: Tumorigenic roles and
clinical opportunities. Nat Rev Urol. 16:433–445. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ahmed H, Cappello F, Rodolico V and Vasta
GR: Evidence of heavy methylation in the galectin 3 promoter in
early stages of prostate adenocarcinoma: Development and validation
of a methylated marker for early diagnosis of prostate cancer.
Transl Oncol. 2:146–156. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ellerhorst J, Troncoso P, Xu XC, Lee J and
Lotan R: Galectin-1 and galectin-3 expression in human prostate
tissue and prostate cancer. Urol Res. 27:362–367. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pacis RA, Pilat MJ, Pienta KJ, Wojno K,
Raz A, Hogan V and Cooper CR: Decreased galectin-3 expression in
prostate cancer. Prostate. 44:118–123. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
van den Brûle FA, Waltregny D, Liu FT and
Castronovo V: Alteration of the cytoplasmic/nuclear expression
pattern of galectin-3 correlates with prostate carcinoma
progression. Int J Cancer. 89:361–367. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Merseburger AS, Kramer MW, Hennenlotter J,
Simon P, Knapp J, Hartmann JT, Stenzl A, Serth J and Kuczyk MA:
Involvement of decreased Galectin-3 expression in the pathogenesis
and progression of prostate cancer. Prostate. 68:72–77. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Y, Nangia-Makker P, Tait L, Balan V,
Hogan V, Pienta KJ and Raz A: Regulation of prostate cancer
progression by galectin-3. Am J Pathol. 174:1515–1523. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
de Melo-Júnior MR, Araújo-Filho JL, Lins
CA, de Pontes-Filho NT and de Carvalho LB Jr: Immobilization of
anti-galectin-3 onto polysiloxane-polyvinyl alcohol disks for tumor
prostatic diseases diagnosis. Appl Biochem Biotechnol.
160:2198–2207. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Knapp JS, Lokeshwar SD, Vogel U,
Hennenlotter J, Schwentner C, Kramer MW, Stenzl A and Merseburger
AS: Galectin-3 expression in prostate cancer and benign prostate
tissues: Correlation with biochemical recurrence. World J Urol.
31:351–358. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Geisler C, Gaisa NT, Pfister D, Fuessel S,
Kristiansen G, Braunschweig T, Gostek S, Beine B, Diehl HC, Jackson
AM, et al: Identification and validation of potential new
biomarkers for prostate cancer diagnosis and prognosis using
2D-DIGE and MS. Biomed Res Int. 2015:4542562015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ahmed H, Banerjee PP and Vasta GR:
Differential expression of galectins in normal, benign, and
malignant prostate epithelial cells: Silencing of galectin-3
expression in prostate cancer by its promoter methylation. Biochem
Biophys Res Commun. 358:241–246. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Califice S, Castronovo V, Bracke M and van
den Brûle F: Dual activities of galectin-3 in human prostate
cancer: Tumor suppression of nuclear galectin-3 vs tumor promotion
of cytoplasmic galectin-3. Oncogene. 23:7527–7536. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fukumori T, Oka N, Takenaka Y,
Nangia-Makker P, Elsamman E, Kasai T, Shono M, Kanayama HO,
Ellerhorst J, Lotan R and Raz A: Galectin-3 regulates mitochondrial
stability and antiapoptotic function in response to anticancer drug
in prostate cancer. Cancer Res. 66:3114–3119. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Y, Nangia-Makker P, Balan V, Hogan V
and Raz A: Calpain activation through galectin-3 inhibition
sensitizes prostate cancer cells to cisplatin treatment. Cell Death
Dis. 1:e1012010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Y, Balan V, Gao X, Reddy PG, Kho D,
Tait L and Raz A: The significance of galectin-3 as a new basal
cell marker in prostate cancer. Cell Death Dis. 4:e7532013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guha P, Kaptan E, Bandyopadhyaya G,
Kaczanowska S, Davila E, Thompson K, Martin SS, Kalvakolanu DV,
Vasta GR and Ahmed H: Cod glycopeptide with picomolar affinity to
galectin-3 suppresses T-cell apoptosis and prostate cancer
metastasis. Proc Natl Acad Sci USA. 110:5052–5057. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Glinsky VV, Glinsky GV, Rittenhouse-Olson
K, Huflejt ME, Glinskii OV, Deutscher SL and Quinn TP: The role of
Thomsen-Friedenreich antigen in adhesion of human breast and
prostate cancer cells to the endothelium. Cancer Res. 61:4851–4857.
2001.PubMed/NCBI
|
|
24
|
Glinsky VV, Glinsky GV, Glinskii OV,
Huxley VH, Turk JR, Mossine VV, Deutscher SL, Pienta KJ and Quinn
TP: Intravascular metastatic cancer cell homotypic aggregation at
the sites of primary attachment to the endothelium. Cancer Res.
63:3805–3811. 2003.PubMed/NCBI
|
|
25
|
Dondoo TO, Fukumori T, Daizumoto K, Fukawa
T, Kohzuki M, Kowada M, Kusuhara Y, Mori H, Nakatsuji H, Takahashi
M and Kanayama HO: Galectin-3 is implicated in tumor progression
and resistance to anti-androgen drug through regulation of androgen
receptor signaling in prostate cancer. Anticancer Res. 37:125–134.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Farhad M, Rolig AS and Redmond WL: The
role of Galectin-3 in modulating tumor growth and immunosuppression
within the tumor microenvironment. Oncoimmunology. 7:e14344672018.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dumic J, Dabelic S and Flögel M:
Galectin-3: An open-ended story. Biochim Biophys Acta.
1760:616–635. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang L and Guo XL: Molecular regulation of
galectin-3 expression and therapeutic implication in cancer
progression. Biomed Pharmacother. 78:165–171. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ruebel KH, Jin L, Qian X, Scheithauer BW,
Kovacs K, Nakamura N, Zhang H, Raz A and Lloyd RV: Effects of DNA
methylation on galectin-3 expression in pituitary tumors. Cancer
Res. 65:1136–1140. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Heldring N, Pike A, Andersson S, Matthews
J, Cheng G, Hartman J, Tujague M, Ström A, Treuter E, Warner M and
Gustafsson JA: Estrogen receptors: How do they signal and what are
their targets. Physiol Rev. 87:905–931. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Thiebaut C, Vlaeminck-Guillem V, Trédan O,
Poulard C and Le Romancer M: Non genomic signaling of steroid
receptors in cancer. Mol Cell Endocrinol. 538:1114532021.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pisolato R, Lombardi AP, Vicente CM, Lucas
TF, Lazari MF and Porto CS: Expression and regulation of the
estrogen receptors in PC-3 human prostate cancer cells. Steroids.
107:74–86. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Souza DS, Lombardi A, Vicente CM, Lucas T,
Erustes AG, Pereira G and Porto CS: Estrogen receptors localization
and signaling pathways in DU-145 human prostate cancer cells. Mol
Cell Endocrinol. 483:11–23. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lombardi AP, Pisolato R, Vicente CM,
Lazari MF, Lucas TF and Porto CS: Estrogen receptor beta (ERβ)
mediates expression of β-catenin and proliferation in prostate
cancer cell line PC-3. Mol Cell Endocrinol. 430:12–24. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lombardi A, Cavalheiro RP, Porto CS and
Vicente CM: Estrogen receptor signaling pathways involved in
invasion and colony formation of androgen-independent prostate
cancer cells PC-3. Int J Mol Sci. 22:11532021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Silva RS, Lombardi A, Souza DS, Vicente CM
and Porto CS: Activation of estrogen receptor beta (ERβ) regulates
the expression of N-cadherin, E-cadherin and β-catenin in
androgen-independent prostate cancer cells. Int J Biochem Cell
Biol. 96:40–50. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Stauffer SR, Coletta CJ, Tedesco R,
Nishiguchi G, Carlson K, Sun J, Katzenellenbogen BS and
Katzenellenbogen JA: Pyrazole ligands: Structure-affinity/activity
relationships and estrogen receptor-alpha-selective agonists. J Med
Chem. 43:4934–4947. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Meyers MJ, Sun J, CarlsonK E, Marriner GA,
Katzenellenbogen BS and Katzenellenbogen JA: Estrogen receptor-beta
potency-selective ligands: Structure-activity relationship studies
of diarylpropionitriles and their acetylene and polar analogues. J
Med Chem. 44:4230–4251. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bustos SO, da Silva Pereira GJ, de Freitas
Saito R, Gil CD, Zanatta DB, Smaili SS and Chammas R: Galectin-3
sensitized melanoma cell lines to vemurafenib (PLX4032) induced
cell death through prevention of autophagy. Oncotarget.
9:14567–14579. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lombardi A, Vicente CM and Porto CS:
Estrogen receptors promote migration, invasion and colony formation
of the androgen-independent prostate cancer cells PC-3 through
β-catenin pathway. Front Endocrinol. 11:1842020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vicente CM, Lima MA, Yates EA, Nader HB
and Toma L: Enhanced tumorigenic potential of colorectal cancer
cells by extracellular sulfatases. Mol Cancer Res. 13:510–523.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Marchiori MF, Riul TB, Oliveira Bortot L,
Andrade P, Junqueira GG, Foca G, Doti N, Ruvo M, Dias-Baruffi M,
Carvalho I and Campo VL: Binding of triazole-linked galactosyl
arylsulfonamides to galectin-3 affects Trypanosoma cruzi cell
invasion. Bioorg Med Chem. 25:6049–6059. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang Y, Jing Y, Ding L, Zhang X, Song Y,
Chen S, Zhao X, Huang X, Pu Y, Wang Z, et al: Epiregulin reprograms
cancer-associated fibroblasts and facilitates oral squamous cell
carcinoma invasion via JAK2-STAT3 pathway. J Exp Clin Cancer Res.
38:2742019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Macheroni C, Gameiro Lucas TF, Souza DS,
Vicente CM, Pereira GJDS, Junior IDSV, Juliano MA and Porto CS:
Activation of estrogen receptor ESR1 and ESR2 induces proliferation
of the human testicular embryonal carcinoma NT2/D1 cells. Mol Cell
Endocrinol. 554:1117082022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nelson AW, Tilley WD, Neal DE and Carroll
JS: Estrogen receptor beta in prostate cancer: Friend or foe?
Endocr Relat Cancer. 21:T219–T234. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lau KM and To KF: Importance of estrogenic
signaling and its mediated receptors in prostate cancer. Int J Mol
Sci. 17:14342016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kowalska K and Piastowska-Ciesielska AW:
Oestrogens and oestrogen receptors in prostate cancer. Springer
Plus. 5:5222016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Warner M, Fan X, Strom A, Wu W and
Gustafsson JÅ: 25 years of ERβ: A personal journey. J Mol
Endocrinol. 68:R1–R9. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Nelson AW, Groen AJ, Miller JL, Warren AY,
Holmes KA, Tarulli GA, Tilley WD, Katzenellenbogen BS, Hawse JR,
Gnanapragasam VJ and Carroll JS: Comprehensive assessment of
estrogen receptor beta antibodies in cancer cell line models and
tissue reveals critical limitations in reagent specificity. Mol
Cell Endocrinol. 440:138–150. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Semenas J, Wang T, Sajid Syed Khaja A,
Firoj Mahmud A, Simoulis A, Grundström T, Fällman M and Persson JL:
Targeted inhibition of ERα signaling and PIP5K1α/Akt pathways in
castration-resistant prostate cancer. Mol Oncol. 15:968–986. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Rossi V, Di Zazzo E, Galasso G, De Rosa C,
Abbondanza C, Sinisi AA, Altucci L, Migliaccio A and Castoria G:
Estrogens modulate somatostatin receptors expression and synergize
with the somatostatin analog pasireotide in prostate cells. Front
Pharmacol. 10:282019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Leung YK, Lam HM, Wu S, Song D, Levin L,
Cheng L, Wu CL and Ho SM: Estrogen receptor beta2 and beta5 are
associated with poor prognosis in prostate cancer and promote
cancer cell migration and invasion. Endocr Relat Cancer.
17:675–689. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gustafsson JA, Strom A and Warner M:
Update on ERbeta. J Steroid Biochem Mol Biol. 191:1053122019.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kadrofske MM, Openo KP and Wang JL: The
human LGALS3 (galectin 3) gene: Determination of the gene structure
and functional characterization of the promoter. Arch Biochem
Biophys. 349:7–20. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yang H, Lei C, Cheng C, Feng Y, Zhang W,
Petracco RG and Sak S: The Antiapoptotic effect of Galectin-3 in
human endometrial cells under the regulation of estrogen and
progesterone. Biol Reprod. 87:392012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Bima C, Chiloiro S, Giampietro A, Gessi M,
Mattogno PP, Lauretti L, Anile C, Rindi G, Pontecorvi A, De Marinis
L and Bianchi A: Galectin-3 and estrogen receptor alpha as
prognostic markers in prolactinoma: Preliminary results from a
pilot study. Front Endocrinol (Lausanne). 12:6840552021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Mourad-Zeidan AA, Melnikova VO, Wang H,
Raz A and Bar-Eli M: Expression profiling of Galectin-3-depleted
melanoma cells reveals its major role in melanoma cell plasticity
and vasculogenic mimicry. Am J Pathol. 173:1839–1852. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang YG, Kim SJ, Baek JH, Lee HW, Jeong SY
and Chun KH: Galectin-3 increases the motility of mouse melanoma
cells by regulating matrix metalloproteinase-1 expression. Exp Mol
Med. 44:387–393. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Shimura T, Takenaka Y, Tsutsumi S, Hogan
V, Kikuchi A and Raz A: Galectin-3, a novel binding partner of
beta-catenin. Cancer Res. 64:6363–6367. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yang Y, Zhou Z, He S, Fan T, Jin Y, Zhu X,
Chen C, Zhang ZR and Huang Y: Treatment of prostate carcinoma with
(galectin-3)-targeted HPMA copolymer-(G3-C12)-5-Fluorouracil
conjugates. Biomaterials. 33:2260–2271. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Glinsky VV and Raz A: Modified citrus
pectin anti-metastatic properties: One bullet, multiple targets.
Carbohydr Res. 344:1788–1791. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Conti S, Vexler A, Hagoel L,
Kalich-Philosoph L, Corn BW, Honig N, Shtraus N, Meir Y, Ron I,
Eliaz I and Lev-Ari S: Modified citrus pectin as a potential
sensitizer for radiotherapy in prostate cancer. Integr Cancer Ther.
17:1225–1234. 2018. View Article : Google Scholar : PubMed/NCBI
|